

Abstract
A general method for the formation of fused dihydroazepine derivatives from 1-sulfonyl-1,2,3-triazoles bearing a tethered diene is reported. The process involves an intramolecular cyclopropanation of an α-imino Rh(II)-carbenoid, leading to a transient 1-imino-2-vinylcyclopropane intermediate which rapidly undergoes a 1-aza-Cope rearrangement to generate fused dihydroazepine derivatives in moderate to excellent yields. The reaction proceeds with similar efficiency on gram-scale. The use of catalyst-free conditions leads to the formation of a novel [4.4.0] bicyclic heterocycle.
Keywords: α-Iminocarbene, Rh catalysis, Dihydroazepine, Triazole, N-Heterocycle
A zepine and azepane derivatives are found in numerous bioactive natural products and other compounds of pharmaceutical interest (Figure 1).[1–2] While a vastarray of methods have been developed through the years for their synthesis,[1] only a few approaches exist for the preparation of their ring-fused analogs.[1b] The ubiquity of these units in a variety of biologically relevant compounds such as alkaloids (see Figure 1) makes the development of new stereoselective strategies to access polycyclic, substitute dazepines and azepanes in a single operation from readily available acyclic precursors highly desirable. The [3,3]-sigmatropic rearrangement of 1,2-divinylcyclopropane derivatives is a well-established strategy to access 7-membered ring compounds in a stereospecific manner.[3] The analogous transformation where one of the vinyl groups is replaced by a C=N group is known to lead to azepine derivatives through a similar mechanism. While 2-aza-Cope rearrangements of this type are quite common for 2-azepinone synthesis using an isocyanate intermediate (formed in situ),[4] examples of the corresponding 1-aza-Cope rearrangement, which directly leads to 2,5-dihydro[1H] azepines, remain scarce.[5] Indeed, the preparation of the cis-1-imino-2-vinylcyclopropanes (IVC) required for such a rearrangement to occur is hampered by the multiple steps needed for their synthesis, discouraging the use of this approach for the formation of azepine derivatives.[5b,6]
Figure 1.
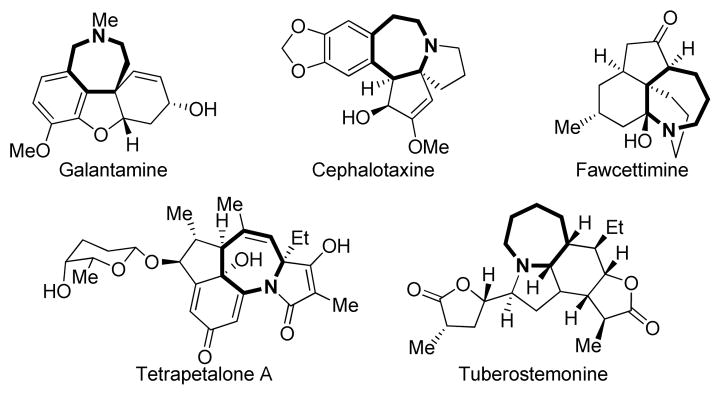
Natural products containing fused azepine derivatives.
In recent years, 1-sulfonyl-1,2,3-triazoles have emerged as stable and readily available azavinyl carbene equivalents for a variety of useful transformations.[7–8] We have recently shown that these carbenes can be utilized in the synthesis of 3,4-fused pyrroles upon reaction with a tethered allene functionality, through a Rh-TMM intermediate (Scheme 1a).[9] To access fused azepine derivatives through an analogous approach, we envisioned the reaction of an α-iminometallocarbene with a tethered E,E-1,3-diene would instead generate a cis-1-imino-2-vinylcyclopropane (IVC), ideally substituted for a subsequent 1-aza-Cope rearrangement (Scheme 1b). Herein, we report our studies on the synthesis of 3,4-fused dihydroazepines, which is complementary to a recent report by Tang et al. that appeared during the completion of this work.[10] In their report, Tang et al. achieve an elegant divergent synthesis of nitrogen heterocycles by intermolecular reaction of triazoles with 1,3-dienes to provide access to azepine or pyrroline derivatives, with the pyrroline products being favored over extended reaction times.
Scheme 1.

Synthesis of fused heterocycles using azavinyl carbene equivalents
In this Communication, we report a general method for the expedient synthesis of fused azepine derivatives as the sole products from dienyltriazoles. Our mechanistic studies strongly suggest that the reaction proceeds by a sequential intramolecular Rh(II)-catalyzed cyclopropanation / 1-aza-Cope rearrangement. The stereospecific nature of each step in this transformation is critical and allows for a highly diastereoselective process, leading to fused 2,5-dihydro [1H] azepines in good to excellent yields.
Applying the conditions we previously developed for the formation of 3,4-fused pyrroles from allenyltriazoles to dienyltriazole 1a (see Table 1),[9a] we were delighted to observe the formation of dihydroazepine 2a in 54% yield (entry 1). A major byproduct observed in the reaction was α,β-unsaturated N-tosylimine 3, resulting from a competing 1,2-hydride shift from the Rh(II)-carbenoid intermediate.[11] To combat this challenge, we investigated a variety of more sterically encumbered Rh(II) complexes (entries 3–6).[12] Gratifyingly, we found that Rh2(Adc)4 affords the desired dihydroazepine in increased yield (entry 6). Varying the nature of the solvent and the concentration of the reactants did not significantly improve the yield.[13] Decreasing the temperature to 60 °C and increasing the reaction time to 16 h afforded dihydroazepine 2a in 74% isolated yield (entry 7), along with a minimal amount of the α,β-unsaturated N-tosylimine (3).
Table 1.
Catalyst optimization for the intramolecular Rh(II)-catalyzed dihydroazepine formation[a]
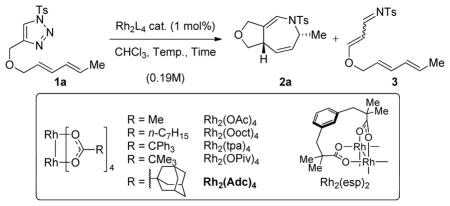
| ||||
|---|---|---|---|---|
| Entry | Rh2L4 | Temp.(°C) | Time | Yield 2a(%)[b],[c] |
| 1 | Rh2(Ooct)4 | 140[d] | 0.25 h | 54 (34) |
| 2 | Rh2(OAc)4 | 140[d] | 0.25 h | 47 (48) |
| 3 | Rh2(tpa)4 | 140[d] | 0.25 h | 18 (70) |
| 4 | Rh2(OPiv)4 | 140[d] | 0.25 h | 58 (18) |
| 5 | Rh2(esp)2 | 140[d] | 0.25 h | 66 (33) |
| 6 | Rh2(Adc)4 | 140[d] | 0.25 h | 68 (17) |
| 7 | Rh2(Adc)4 | 60 | 16 h | 74 (14) |
Only one diastereomer of the dihydroazepine product 2a was observed by 1H NMR in all cases.
Isolated yield.
NMR yield of imine 3 in parentheses.
Reaction was performed in a microwave apparatus.
Using the optimized conditions, a range of dienyltriazole substrates is transformed into the corresponding 3,4-fused dihydroazepines (Table 2). A variety of aryl-substituted substrates, including phenyl, electron-rich and electron-poor dienyls are tolerated (entries 2–4). In addition, internal substitution of the dieneportion is compatible in the transformation, albeit proceeding in slightly lower yield to the dihydroazepine (entry 2 vs 5). Furthermore, dienyltriazole substrates bearing N-tosylamine or diester groups instead of the ether tether furnish the corresponding 3,4-fused dihydroazepines in moderate to good yields (entries 6–8). In the case of 1h, the observed yield of the corresponding product (2h) was significantly lower as a result of side-reactions, presumably arising from intramolecular attack of the proximal ester groups on the Rh-carbene intermediate. Importantly, all-carbon tethered dihydroazepine 2i, corresponding to a portion of tetrapetalone A (see Figure 1), could be accessed in excellent yield using the optimized reaction conditions (entry 9). In all cases, only one diastereomer of the dihydroazepine product is observed. Notably, dienyltriazole substrates that would yield a 6–7 fused dihydroazepine product afford only the corresponding 1,2-hydride shift product. It is noteworthy that the 1-aza-Cope rearrangement was found to proceed significantly more slowly for substrates 1d and 1e, where the IVC intermediates were observed by NMR after 0.5 h, and complete conversion was only achieved after 16 h.
Table 2.
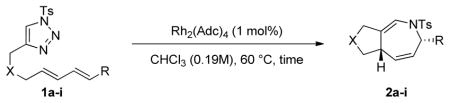
| ||||
|---|---|---|---|---|
| Entry | Substrate | Product | Time | Yield(%)[c] |
| 1 |
 1a |
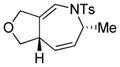 2a |
16 h | 74 |
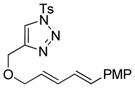
| 2 |
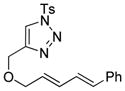 1b |
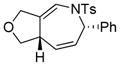 2b |
0.5 h | 92 |
| 3 |
 1c |
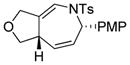 2c |
0.5 h | 61 |
| 4 |
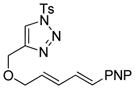 1d |
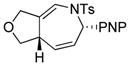 2d |
16 h | 69 |
| 5 |
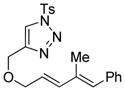 1e |
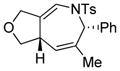 2e |
16 h | 63 |
| 6 |
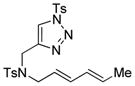 1f |
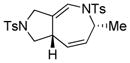 2f |
0.5 h | 72 |
| 7 |
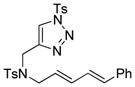 1g |
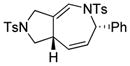 2g |
0.5 h | 72 |
| 8 |
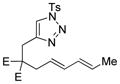 1h |
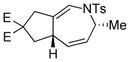 2h |
0.5 h | 45(60)[d] |
| 9 |
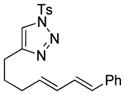 1i |
 2i |
0.5 h | 92 |
PMP=4-MeO-C6H4-, PNP=4-O2N-C6H4-, E=CO2Me.
Only one diastereomer of the dihydroazepine product was observed by 1H NMR in all cases.
Isolated yields.
Yield in parentheses obtained when Rh2(Ooct)4 was used at 140 °C in the microwave for 0.25 h.
Finally, the reaction is amenable to a gram-scale synthesis of dihydroazepines, as exemplified with 2a (Scheme 2a). Interestingly, we also found that in the absence of a Rh(II) catalyst under more forcing conditions, a distinct heterocyclic product (4) was formed in 50% yield, presumably arising from aketenimine intermediate (Scheme 2b).[14] For both types of products (i.e., 2a and 4), the structure and relative configuration was confirmed by X-ray analysis.[15]
Scheme 2.
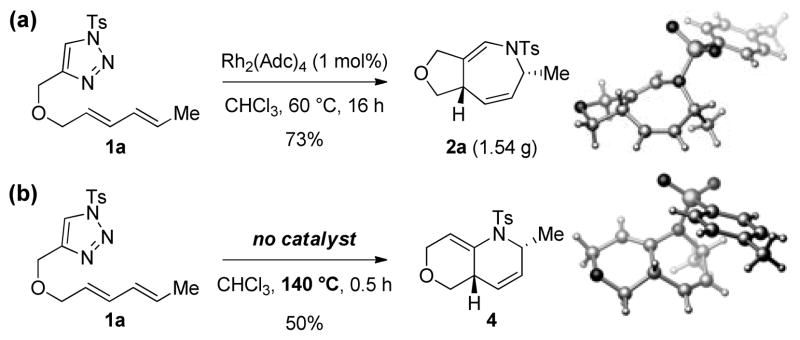
Gram-scale synthesis of dihydroazepine 2a and metal-free access to [4.4.0] bicycle 4
The Rh(II)-catalyzed dihydroazepine formation can, in principle, occur through three distinct mechanistic pathways (Scheme 3). First, the azavinyl-substituted Rh-carbenoid species formed by reaction of the dienyltriazole with the Rh catalyst could undergo a [2+1] cycloaddition with the proximal alkenyl group to generate a cis-1-imino-2-vinylcyclopropane (IVC), which could undergo a [3,3] sigmatropic rearrangement to directly afford the dihydroazepine product (Path A). Alternatively, the IVC intermediate could undergo ring-opening to a zwitterionic intermediate, generating an allylcation capable of ring-closure by an intramolecular N-attack of the azanide thus formed on the distal alkenyl moiety (Path B). Similarly, the Rh-carbenoid that is formed initially could be attacked by the dienyl moiety to generate a Rh-bound zwitterion, able to cyclize via an analogous mechanism (Path C). While the stereospecificity of Path A should lead to a single diastereomer of the product bearing the predicted stereochemistry as shown, Path B or C could give rise to a mixture of both isomers. Notably, the single diastereomer2a obtained from the reaction, as determined by X-ray analysis, is consistent with the [2+1]/[3,3] sequence depicted in Path A (see Scheme 2a).
Scheme 3.

Different possible mechanistic pathways for the dihydroazepine formation
To obtain further insight, several other triazoles were synthesized and evaluated (Scheme 4). To gain support for the intermediacy of iminocyclopropanes, alkenyltriazole 1j (Scheme 4a), which lacks the distal alkenyl group required for the subsequent 1-aza-Cope to occur, was submitted to the standard reaction conditions. Iminocyclopropane 5a was obtained as a single diastereomer in 66% yield (determined by NMR). Although 1-sulfonyl-1,2,3-triazoles are known to lead to iminocyclopropanes by intermolecular cyclopropanation with alkenes,[7–8a,b] to our knowledge, the analogous intramolecular cyclopropanation has never been reported. Thus the viability and stereo selectivity of such a mechanistic step is confirmed by this observation. Sterically encumbered dienyltriazoles 1k and 1l (Scheme 4b) lead to the formation of IVC intermediates (5b and 5c, respectively), which do not undergo the subsequent 1-aza-Cope rearrangement. This is likely due to an unfavorable interaction with the cis-R group in the transition state of the [3,3] rearrangement, significantly slowing the rate of this pathway.[16–17] The fact that none of the dihydroazepine was observed in this particular case strongly suggests that a concerted mechanism is operative for the rearrangement, as the increased flexibility of a ring-opened zwitterionic intermediate (as shown in Path B or C) should still allow for the cyclization to occur. Notably, the formation of such zwitterionic intermediates should not be hampered by the steric hinderance of the distal alkenyl moiety in 1k or 1l. Possibly due to a similar steric effect, it is noteworthy that 1,1-disubstituted dienes, which would generate a quartenarycenter in the product, only reacted sluggishly. Finally, Z,E-dienyltriazole 1m (Scheme 4c), which leads to a trans-IVC (5d), was also found to be unreactive toward dihydroazepine formation. This observation can be attributed to the inability of such an intermediate to engage in a concerted 1-aza-Cope rearrangement. The corresponding zwitterionic intermediate formed from 1m should be identical to the case of E,E-dienyltriazole 1b, which in contrast to 1m, leads to dihydroazepine formation in excellent yield in only 0.5 h (see Table 2, entry 2). These results strongly support a sequential intramolecular Rh(II)-catalyzed cyclopropanation / 1-aza-Cope rearrangement as the operative pathway for dihydroazepine formation (see Scheme 3, Path A). Moreover, if a zwitterionic intermediate is involved in the process, the formation of the corresponding 5-membered heterocycle (pyrroline) would be expected to be competitive, as the generation of 5-membered ring compounds from this type of zwitterionic intermediate is typically a fast process.[3f,8g,10] Notably, this type of product was not observed in any case throughout this study, again lending support to Path A.
Scheme 4.

Experimental mechanistic insights: [3,3] sigmatropic rearrangement vs zwitterionic pathways
In summary, a general approach for the synthesis of fused dihydroazepines from dienyltriazoles is reported. A range of substrates have been found to participate in the transformation, and several mechanistic investigations strongly support a sequential intramolecular Rh(II)-catalyzed cyclopropanation / 1-aza-Cope rearrangement as the operative mechanistic pathway. The reaction can be scaled with similar efficiency, and the use of catalyst-free conditions provides access to a novel [4.4.0] bicyclic heterocycle. Given the ubiquity of these scaffolds in biologically relevant compounds, this work should prove valuable for the synthesis of new and useful fused azepine-based building blocks.
Supplementary Material
Footnotes
This work was supported by the NSF (CAREER 0643264 to R.S.) and a Research Scholar Grant from the American Cancer Society (RSG-09-017-01-CDD to R.S.). R.S. is a Camille Dreyfus Teacher Scholar. We are grateful to the NSF for a graduate fellowship to E.E.S., to the FRQNT (B3) for a postdoctoral scholarship to V.N.G.L., and to Abbott, Eli Lilly, and Roche for financial support. We thank A. Di Pasquale for solving the crystal structures of 2a and 4 (displayed with CYL View), supported by NIH Shared Instrumentation Grant (S10-RR027172).
Supporting information for this article is available on the WWWunder http://dx.doi.org/10.1002/anie.201xxxxxx.
References
- 1.For reviews on the synthesis, properties and natural occurrences of azepine derivatives, see: Vaquero JJ, Cuadro AM, Herradón B. Modern Heterocyclic Chemistry. Wiley-VCH Verlag GmbH & Co. KGaA; 2011. pp. 1865–1988.Bremner JB, Samosorn S. In: Comprehensive Heterocyclic Chemistry III. Katritzky AR, Rees CW, Scriven EFV, Taylor R, editors. Vol. 13. Elsevier; Oxford: 2008. pp. 1–43.Proctor GR, Redpath J. In: The Chemistry of Heterocyclic Compounds. Taylor EC, editor. Vol. 56. Wiley-Interscience Publication; Chichester: 1996.
- 2.For recent reports of bioactive synthetic azepine derivatives, see: Kunick C, Bleeker C, Prühs C, Totzke F, Schächtele C, Kubbutat MHG, Link A. Bioorg Med Chem Lett. 2006;16:2148–2153. doi: 10.1016/j.bmcl.2006.01.071.Habermann J, Capitò E, Ferreira MdRR, Koch U, Narjes F. Bioorg Med Chem Lett. 2009;19:633–638. doi: 10.1016/j.bmcl.2008.12.039.Breitenlechner CB, Wegge T, Berillon L, Graul K, Marzenell K, Friebe WG, Thomas U, Schumacher R, Huber R, Engh RA, Masjost B. J Med Chem. 2004;47:1375–1390. doi: 10.1021/jm0310479.
- 3.a) Vogel E. Angew Chem. 1960;72:4–26. [Google Scholar]; b) Vogel E, Ott KH, Gajek K. Liebigs Ann Chem. 1961;644:172–188. [Google Scholar]; c) Vogel E. Angew Chem Int Ed. 1963;2:1–11. [Google Scholar]; d) Wong HNC, Hon MY, Tse CW, Yip YC, Tanko J, Hudlicky T. Chem Rev. 1989;89:165–198. [Google Scholar]; e) Hudlicky T, Fan R, Reed JW, Gadamasetti KG. Org React. 1992;41:1–133. [Google Scholar]; f) Sperling D, Reißig HU, Fabian J. Eur J Org Chem. 1999:1107–1114. [Google Scholar]
- 4.For examples of 2-aza Cope rearrangements with cyclopropane derivatives, see: von Doering EW, Goldstein MJ. Tetrahedron. 1959;5:53–69.Vogel E, Erb R. Angew Chem Int Ed. 1962;1:53–54.Vogel E, Erb R, Lenz G, Bothner-By AA. Liebigs Ann Chem. 1965;682:1–20.Sasaki T, Eguchi S, Ohno M. J Am Chem Soc. 1970;92:3192–3194.Sasaki T, Eguchi S, Ohno M. J Org Chem. 1972;37:466–469.Müller P, Bernardinelli G, Nury P. Tetrahedron: Asymmetry. 2002;13:551–558.and references therein Reissig HU, Böttcher G, Zimmer R. Can J Chem. 2004;82:166–176.
- 5.For rare examples of 1-aza Cope rearrangements with cyclopropane derivatives, see: Paquette LA, Ewing GD. J Am Chem Soc. 1978;100:2908–2909.Boeckman RK, Shair MD, Vargas JR, Stolz LA. J Org Chem. 1993;58:1295–1297.
- 6.For a review on the preparation and application of cyclopropylimines, see: Soldevilla A, Sampedro D. Org Prep Proced Int. 2007;39:561–590.
- 7.For reviews, see: Davies HML, Alford JS. Chem Soc Rev. 2014:43. doi: 10.1039/c4cs00072b.Chattopadhyay B, Gevorgyan V. Angew Chem, Int Ed. 2012;51:862–872. doi: 10.1002/anie.201104807.Roy M-N, Lindsay VNG, Charette AB. In: Stereoselective Synthesis 1: Stereoselective Reactions of Carbon-Carbon Double Bonds. de Vries J, editor. chap 1.14 Georg Thieme-Verlag; New York: 2011.
- 8.For selected recent examples, see: Horneff T, Chuprakov S, Chernyak N, Gevorgyan V, Fokin VV. J Am Chem Soc. 2008;130:14972–14974. doi: 10.1021/ja805079v.Chuprakov S, Kwok SW, Zhang L, Lercher L, Fokin VV. J Am Chem Soc. 2009;131:18034–18035. doi: 10.1021/ja908075u.Miura T, Biyajima T, Fujii T, Murakami M. J Am Chem Soc. 2012;134:194–196. doi: 10.1021/ja2104203.Parr BT, Davies HML. Angew Chem Int Ed. 2013;52:10044–10047. doi: 10.1002/anie.201304310.Yang J-M, Zhu C-Z, Tang X-Y, Shi M. Angew Chem Int Ed. 2014;53 doi: 10.1002/anie.201400881.Miura T, Funakoshi Y, Murakami M. J Am Chem Soc. 2014;136:2272–2275. doi: 10.1021/ja412663a.Kwok SW, Zhang L, Grimster NP, Fokin VV. Angew Chem Int Ed. 2014;53:3452–3456. doi: 10.1002/anie.201306706.
- 9.Schultz EE, Sarpong R. J Am Chem Soc. 2013;135:4696–4699. doi: 10.1021/ja401380d.For a similar methodology using alkynes instead of allenes, see: Shi Y, Gevorgyan V. Org Lett. 2013;15:5394–5396. doi: 10.1021/ol4027655.For an intermolecular version of this reaction, see: Chattopadhyay B, Gevorgyan V. Org Lett. 2011;13:3746–3749. doi: 10.1021/ol2014347.Miura T, Hiraga K, Biyajima T, Nakamuro T, Murakami M. Org Lett. 2013;15:3298–3301. doi: 10.1021/ol401340u.
- 10.Shang H, Wang Y, Tian Y, Feng J, Tang Y. Angew Chem Int Ed. 2014;53 doi: 10.1002/anie.201400426. [DOI] [PubMed] [Google Scholar]
- 11.Such a 1,2-hydride shift has been shown to be a significant side-reaction for other α-alkyl azavinyl-substituted Rh-carbenoids. For example, see references 8c and 8e.
- 12.Steric interactions between the ligands on the Rh(II) and the metal-carbenoid substituents have been hypothesized to favorcyclopropanation over 1,2-hydride migration for intermolecular reactions: Panne P, Fox JM. J Am Chem Soc. 2007;129:22–23. doi: 10.1021/ja0660195.Panne P, De Angelis A, Fox JM. Org Lett. 2008;10:2987–2989. doi: 10.1021/ol800983y.
- 13.See Supporting Information for details.
- 14.Yoo EJ, Chang S. Curr Org Chem. 2009;13:1766–1776. [Google Scholar]
- 15.CCDC 998199 (2a) and CCDC 998200 (4) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
- 16.Similar substitution effects have recently been demonstrated to significantly slow down the divinylcyclopropane rearrangement with various derivatives, by impeding the adoption of the requisite boat conformation needed for the 3,3-sigmatropic rearrangement to take place. Osler JD, Unsworth WP, Taylor RJK. Org Biomol Chem. 2013;11:7587. doi: 10.1039/c3ob41617h.
- 17.For a related study, see: Su JT, Sarpong R, Stoltz BM, Goddard WA. J Am Chem Soc. 2003;126:24–25. doi: 10.1021/ja037716p.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.