

Abstract
Glioblastoma is the most aggressive adult primary brain tumor. Although progress has been made in understanding the molecular mechanisms underlying these tumors, current treatments are ineffective. Recent studies have identified iNOS as a critical regulator of glial transformation downstream of EGFRvIII/STAT3 signaling, a key oncogenic pathway in glioblastoma. STAT3 directly binds the promoter of the iNOS gene and thereby stimulates its expression. Importantly, inhibition of iNOS by genetic and pharmacological approaches impedes glial cell proliferation, invasiveness, and tumor growth in vivo. iNOS expression is also elevated in a population of human brain tumor stem cells (BTSCs), and iNOS is required for BTSC proliferation and tumorigenesis. Together, these findings suggest that development of iNOS-targeted therapies may prove valuable in the treatment of glioblastoma. Here, we review our current understanding of iNOS signaling in the regulation of glioblastoma pathogenesis and the potential mechanisms by which iNOS inhibition might suppress the malignant behavior of these devastating tumors.
Keywords: Astrocytes, brain tumor stem cells, EGFRvIII, glioblastoma, invasiveness, iNOS, NO, proliferation, STAT3, signaling
INTRODUCTION
Gliomas are the most common primary tumors in the brain [1, 2]. Based on the glial cell type that predominates in glioma, these tumors are classified as astrocytoma, ependymoma, or oligodendroglioma. Astrocytomas are classified into different grades according to histological criteria [1, 2]. Grade 4 astrocytoma, known as glioblastoma, is currently incurable. Despite multimodal treatment including surgery, chemotherapy, and radiation, patients with glioblastoma have a median survival rate of less than 2 years [3-7]. Glioblastoma may arise from the transformation of mature astrocytes or neural stem cells (NSC) [1, 8-11]. Regardless of the cell of origin, the resulting tumors are heterogeneous and composed of cells with variable differentiation.
Multiple genetic alterations have been identified in malignant glioma, which have been reviewed [1, 6, 10, 12, 13]. Overexpression of growth factors and of their receptors, amplification of cyclin dependent kinase 4 (Cdk4), mutations of p53, and loss of phosphatase and tensin homology (PTEN), INK4A, and retinoblastoma (RB) are among frequent genetic alterations in glioma. Activating mutations or amplifications of the epidermal growth factor receptor (EGFR), a member of the ErbB family of receptor tyrosine kinases, occurs in nearly 40 percent of all glioblastoma tumors [6, 14].
The most common active mutant of EGFR in glioblastoma is a mutant allele leading to deletions of exons 2-7 (EGFRvIII) [15]. EGFRvIII is a constitutively active receptor in the absence of epidermal growth factor (EGF) ligand. Activation of EGFR transforms murine Ink4/Arf-null neural stem cell (NSC) or astrocytes in the mouse brain [6, 8]. EGFR signaling is also deregulated in other human cancers [16]. The major pathways downstream of plasma membrane-bound EGFR include PLC-γ-PKC, Ras-Raf-MEK, PI3K-AKT-mTOR and JAK-STAT3 signaling [1, 16, 17]. The net effect of these pathways is enhanced cell cycle progression and cell survival. On the other hand, EGFR can be shuttled into the cell nucleus and mitochondrion [18-23]. Nuclear EGFR behaves as a transcriptional regulator, whereas mitochondrial EGFR might inhibit cell death. Nuclear EGFR has been implicated in a number of biological and pathological processes, including cell proliferation, inflammation, metastasis, DNA repair, and resistance to radiation and alkylating anti-cancer agents [24, 25]. Nuclear EGFR also appears to be an indicator of poor clinical outcomes in cancer patients [16]. EGFR forms a complex with and directly phosphorylates the transcription factor STAT3 [22, 23, 26].
STAT3 has multiple functions in the mammalian brain [27]. STAT3 drives astrocyte differentiation in the developing central nervous system [28, 29]. However, in the early stages of embryonic development, STAT3 promotes the self-renewal of neural stem cells [30]. In a similar vein, STAT3 has a dual role in the context of brain tumor biology depending on the mutational background of the tumor [23, 26, 31]. Whereas STAT3 functions as a tumor suppressor in PTEN-deficient astrocytes, it promotes oncogenesis in EGFRvIII-expressing astrocytes. Interestingly, in glial cells, STAT3 functions as a transcriptional activator or repressor. STAT3 represses IL8 in PTEN-deficient glioblastoma cells to inhibit their proliferation and invasiveness [31]. By contrast, STAT3 stimulates iNOS expression in EGFRvIII-expressing astrocytes to induce their transformation [32]. Further, STAT3 can act on the same target gene to either repress or induce its expression depending on cell type and context. For instance, in endothelial cells, STAT3 represses iNOS expression by inhibiting NF-KB activity [33, 34]. The DNA binding domain of STAT3 directly interacts with NF-KB to inhibit iNOS transcription. In EGFRvIII-expressing astrocytes, however, STAT3 directly activates iNOS transcription.
STAT3 activation occurs in the subset of human glioblastoma tumors that express EGFRvIII [23], and STAT3 promotes survival of a distinct set of glioblastoma cells in vitro [26, 35-37]. Activation of a STAT3-dependent transcriptional network also appears to be associated with mesenchymal transformation in glioblastoma [38]. Interestingly, treatment of glioma cell lines with STAT3 decoy leads to decreased proliferation, increased apoptosis and cell cycle arrest in vitro [39]. Since STAT3 plays a crucial role in the malignant biology of brain tumor cells [23], the development of STAT3 inhibitors for treatment of glioblastoma remains an active area of research. However, in view of the opposing functions of STAT3 in the pathogenesis of glioblastoma depending on the mutational profile of the tumor, STAT3 inhibitors will require a patient-tailored approach. A greater understanding of the roles and mechanisms of STAT3 in glioblastoma is essential to ensure the success of potential STAT3-based therapeutics in the future.
An alternative to STAT3 modulators in the treatment of glioblastoma would be to identify downstream targets of EGFRvIII/STAT3 signaling and assess their therapeutic value. We have recently identified iNOS as a direct transcriptional target of STAT3 in EGFRvIII-expressing astrocytes [32]. iNOS plays a critical role in transformation of mouse astrocytes as well as human BTSCs [32, 40]. Thus, iNOS represents an attractive candidate for therapeutic intervention. Here, we review our current understanding of iNOS signaling in the regulation of brain tumor biology and highlight the potential for novel iNOS-based treatments for malignant glioma.
Nitric Oxide (NO)
Nitric Oxide (NO) is an uncharged molecule critical to numerous physiological processes including vasodilation, neurotransmission, and immunity [41]. Within the central nervous system, NO is a key component of signaling pathways that regulate memory, sensory processing, and cerebral blood flow [42-44]. The role of NO in tumor biology has been the subject of scrutiny, where it is thought to exhibit pro- or anti- tumor activities. For example, NO triggers the accumulation of p53 [45] which may lead to apoptosis of tumor cells. However, excess NO can also lead to the generation of peroxynitrite (ONOO-), which inhibits p53 in malignant glioma cells [46]. Several mechanisms may explain NO’s dual role in cancer biology [44, 47-53]. Briefly, NO can react with a wide range of molecules from proteins to transition metals. This can result in the modification of proteins, lipids, and DNA. Reactive intermediates of NO also regulate DNA damage and DNA repair. In addition, the mode of NO production within each cell type may result in different outcomes. At high concentrations, NO induces apoptosis and inhibits cancer growth, whereas at physiological concentrations similar to those in tumor samples, NO favors cell proliferation and tumor growth.
Three NO synthases (NOS) are responsible for the production of NO from the amino acid L-arginine. The NOS1, NOS2, and NOS3 genes encode, respectively, neuronal NOS (nNOS), endothelial NOS (eNOS), and inducible NOS (iNOS). The mechanism of NO production by each NOS isoform appears to be directly correlated with the amount of NO produced, which can in turn influence the biological outcome [54-57]. iNOS is induced in a calcium/calmodulin-independent manner and generates NO in a sustained manner, whereas nNOS and eNOS generate low quantities of NO in a calcium/calmodulin-dependent manner. Growing evidence suggests that iNOS harbors tumor-promoting activity in glioblastoma.
Inducible Nitric Oxide Synthase (iNOS)
iNOS is inducible in many types of cells including epithelial, mesenchymal, and myeloid cells [58]. Induction of iNOS expression varies depending on cell type and species [59]. The inflammatory cytokines interleukin-1s (IL-1s), tumor necrosis factor-α (TNF- α), and interferon-γ (IFN-γ) induce iNOS expression in most murine and rat cells [60]. iNOS is also induced by EGF, colony stimulating factor 1 (CSF1), hypoxia, and WNT signaling [61-63]. EGF induces the accumulation of EGFR in the nucleus, where it interacts with STAT3 leading to the upregulation of iNOS in human breast cancer cells [22].
Aberrant expression of iNOS has been documented in different human tumors including head and neck, breast, colon, stomach, and lung cancer [64-69]. Increased iNOS expression correlates with tumor grade and angiogenesis in breast cancer patients [65, 66, 70]. A positive correlation between iNOS expression and tumor grade also holds for brain tumors. iNOS appears to be highly expressed in glioblastoma and grade III astrocytoma compared to normal brain tissue and grade II astrocytoma [71].
iNOS SIGNALING IN GLIOBLASTOMA
Although multiple studies emphasize the significance of iNOS and iNOS-mediated NO production in tumor progression, the biological significance of these molecules in the regulation of glioblastoma remained unexplored until recently. New studies have identified iNOS as a potential target for therapeutic design in glioblastoma [32, 40].
The EGFRvIII/STAT3 Oncogenic Pathway Operates via iNOS
Using a mouse genetics approach, an oncogenic function for STAT3 has been identified in astrocytes that express the major oncogenic stimulus EGFRvIII [23]. Using a rational approach, iNOS has been identified as a novel target gene of STAT3 in these cells [32]. iNOS is specifically downregulated upon STAT3 knockout, and endogenous STAT3 occupies the promoter of the iNOS gene in EGFRvIII-expressing astrocytes to regulate its transcription [32]. In contrast, STAT3 does not seem to regulate iNOS transcription in astrocytes that are deficient in the major glioblastoma tumor suppressor protein PTEN [32]. These findings suggest that in glial cells, STAT3 regulates iNOS transcription specifically in EGFRvIII-expressing astrocytes. STAT3 induces the expression of a reporter gene driven by the iNOS promoter, and mutation of a conserved STAT3 binding site within the iNOS promoter blocks the STAT3-induced expression of the reporter gene [32]. These findings suggest that STAT3 directly activates iNOS transcription downstream of EGFRvIII (Fig. 1A). The selective iNOS inhibitors 1400W and S-MIU [72-75] reduce the population growth of both astrocytes and U87 glioblastoma cells that express EGFRvIII (Fig. 1B). The NO scavenger c-PTIO, which converts free NO to NO2 [76-78], also reduces the population growth of EGFRvIII-expressing astrocytes. Knockdown of iNOS by RNA interference (RNAi) mimics the effect of pharmacological inhibition of iNOS on the population growth of these cells. Notably, iNOS knockdown or pharmacological inhibition of iNOS with 1400W significantly reduces their invasiveness. Importantly, iNOS knockdown EGFRvIII-expressing astrocytes fail to produce tumors or lead to significantly smaller tumors than control EGFRvIII-expressing astrocytes (Fig. 1B). Thus, iNOS plays a critical role in malignant glial transformation in vivo.
Fig. (1). Inhibitors of iNOS pathway may provide the basis for treatment of human glioblastoma tumors.
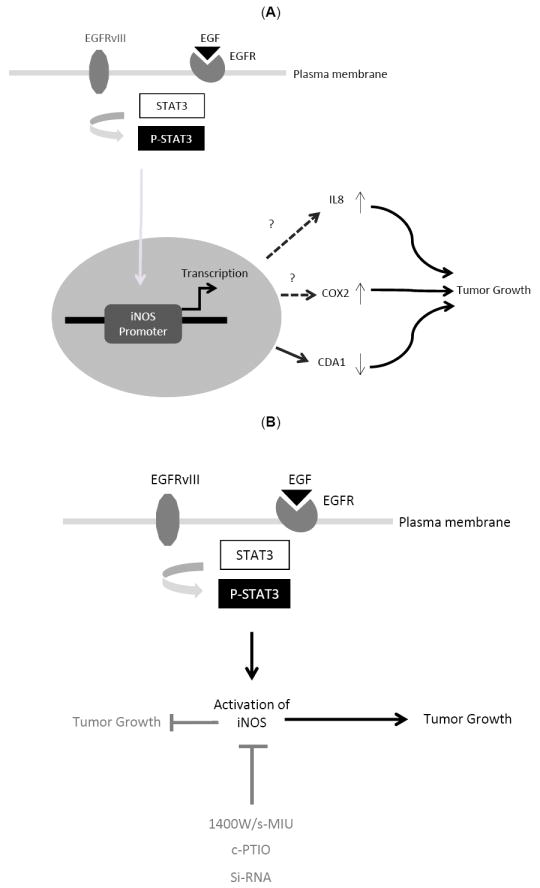
(A). STAT3-iNOS signaling regulates EGFRvIII-mediated glial transformation. In EGFRvIII-expressing astrocytes, endogenous STAT3 occupies the promoter of the iNOS gene to regulate its transcription. STAT3 induces the expression of a reporter gene driven by the iNOS promoter, and mutation of a conserved STAT3 binding site within the iNOS promoter blocks the STAT3-induced expression of the reporter gene. The downstream signaling mechanisms by which iNOS regulates tumorigenesis remain to be studied. Here, we propose upregulation of cytokines such as IL8 and COX2, or changes in cell cycle kinetics as potential mechanism by which iNOS may regulate tumorigenesis. (B) Pharmacological inhibition of iNOS pathway or knockdown (KD) of iNOS by RNA interference (RNAi) suppresses tumor growth in vivo. The selective iNOS inhibitors 1400W and S-MIU and the NO scavenger c-PTIO reduce the population growth of EGFRvIII-expressing astrocytes. Knockdown of iNOS by RNA interference (Si-RNA) also reduces the population growth of these cells. iNOS knockdown EGFRvIII-expressing astrocytes fail to produce tumors or produce much smaller tumors than control EGFRvIII-expressing astrocytes. iNOS inhibition may therefore have potential therapeutic value in the treatment of glioblastoma.
Although, iNOS appears to be a key regulator of the proliferation, invasiveness, and transformation of EGFRvIII-expressing astrocytes, it remains to be determined whether iNOS inhibition might impact glioma recurrence. To address this question, it would be important to first determine the efficacy and toxicity of different iNOS inhibitors on relevant models and follow up on the recurrence pattern of glioma treated with specific iNOS inhibitors.
iNOS -Regulation of Oncogenesis in BTSCs
Identification of brain tumor stem cells (BTSCs) has opened new avenues in the study of glioblastoma. BTSCs constitute a subpopulation of cells in human brain tumors that are capable of self-renewal and tumor formation [79-85]. These cells show remarkable similarities to neuronal stem cells (NSC) [13, 85-87]. For example, BTSCs express neuronal stem/progenitor markers such as Olig2, Sox2, and nestin, and can differentiate to glial or neuronal cells. The precise mechanisms that give rise to BTSCs in the adult brain remain to be elucidated. The production of NO has been recently compared in CD133+ BTSCs and CD133− glioma cells (non-BTSC) by quantification of nitrite (NO2-), a stable byproduct of NO. BTSCs produce more NO2- than non-BTSC cells, suggesting that elevated NO synthesis might be a distinct feature of BTSCs [40]. In addition, endogenous NO depletion by expression of flavohemoglobin (FlavoHb), a potent NO-consuming enzyme [88, 89], impairs BTSC growth and neurosphere formation. These results suggest a role for NO synthesis in BTSC proliferation. Among the different NOS isoforms, iNOS appears to be the only isoform that exhibits elevated expression in BTSCs, suggesting that iNOS regulates NO production in these cells and consequently their proliferation. iNOS levels are also elevated in CD15+ BTSCs relative to CD15− non-BTSCs, suggesting that iNOS correlates with BTSC phenotypes in glioma regardless of whether CD133 or CD15 are used for BTSC enrichment. RNAi-mediated knockdown of iNOS or pharmacological inhibition of iNOS by 1400W both results in decreased BTSC proliferation and neurosphere formation. However, iNOS inhibitors do not appear to have a significant effect on growth rate of normal neuronal progenitor cell (NPCs), suggesting that iNOS targeted therapy might specifically target BTSCs. Further, application of iNOS inhibitors in mice bearing human glioma xenografts leads to reduced tumor volumes. It appears that the decrease in tumor growth in these animals correlates with a decrease in the number of BTSCs and their ability to self-renew and form spheres [40]. Together, just as with studies of the iNOS pathway in mouse astrocytes, these findings in human BTSCs suggest that iNOS inhibitors may hold promise for the treatment of glioblastoma.
MECHANISMS OF iNOS FUNCTION IN GLIOBLASTOMA
The two recent studies suggest that iNOS may play a critical role in the pathogenesis of glioblastoma [32, 40] and may therefore provide a promising target for therapeutic intervention in the treatment of this devastating disease. iNOS inhibitors have been shown to be beneficial in animal models of other types of cancer. For example, NG-nitro-L-arginine-methyl ester (L-NNA) and 1400W reduce tumor growth and angiogenesis in mice bearing mammary tumors [90, 91]. Also, the administration of L-NNA reduces tumor blood flow in BD9 rats harboring P22 carcinosarcoma [92] suggesting the involvement of iNOS in tumor angiogenesis. In support of this conclusion, iNOS knockout mice have reduced tumor growth and vascularization [93, 94]. Importantly, L-NNA was assessed in a phase one clinical study and found to reduce tumor blood volume in cervical and non-small-cell lung cancers [95]. Thus, iNOS inhibitors should be considered in the treatment of glioblastoma. Establishing the in vivo efficacy of iNOS inhibitors is the first step toward the possibility of ultimately using these inhibitors in the treatment of glioblastoma. However, it is also important to gain mechanistic insights into how iNOS regulates tumorigenesis. This will facilitate the design of better therapeutic strategies. Here, we review potential mechanisms by which iNOS may regulate glioblastoma pathogenesis.
iNOS Signaling: Regulation of Cell Cycle
iNOS-dependent tumor cell proliferation appears to be regulated through components of cell cycle machinery. Cell division autoantigen1 (CDA1) is a cell cycle inhibitor that acts in a tumor-suppressive manner [96-98]. iNOS negatively regulates CDA1 expression in BTSCs [40]. Interestingly, suppression of CDA1 correlates with decreased survival in patients. Inhibition of iNOS by RNAi or pharmacologically reduces the rate of cell cycle transit of these cells. Overexpression of CDA1 reduces BTSC numbers and neurosphere formation, phenocopying the effects of iNOS RNAi. These results suggest that iNOS may influence cell cycle progression.
NO activates the AKT signaling pathway in breast cancer cells [50, 99]. AKT suppresses apoptotic signaling by inhibiting pro-apoptotic proteins [100]. In addition, AKT activates eNOS, which contributes to tumor maintenance [101, 102]. Importantly the PI3K/AKT pathway is also required for the regulation of G2/M transition [103]. Although these studies suggest iNOS may promote oncogenesis via distinct mechanisms, iNOS signaling appears to induce the proliferation of glioblastoma cells by influencing cell cycle kinetics. In support of this conclusion, iNOS appears to promote the transformation of EGFRvIII-expressing astrocytes in the absence of changes in apoptosis [32].
iNOS Signaling: The Cytokine Connection
NO regulates the expression of IL8 [104-106], which is associated with increased cell invasion in cancer and poor patient survival [107]. Like other cytokines, IL8 might alter the tumor microenvironment and lead to a pro-inflammatory state and increased oxygen radical formation [108]. This raises the question of whether iNOS-mediated NO production regulates oncogenesis in brain tumors by upregulation of IL8. IL8 is upregulated in higher grade astrocytoma [109]. IL8 has also been implicated in the regulation of angiogenesis in tumors including glioma [110, 111]. In addition, iNOS regulates angiogenesis in C6 rat glioma cells [112]. Therefore, iNOS may promote glioblasotma angiogenesis through upregulation of IL8.
Notably, IL8 is upregulated in a subset of glioblastoma, though in these tumors IL8 upregulation correlates with inhibition of STAT3 and loss of PTEN [31]. In PTEN-deficient human glioblastoma cells, STAT3 represses IL8 and repression of IL8 mediates the ability of STAT3 to inhibit glioblastoma cell proliferation and invasiveness in PTEN-deficient glioblastoma cells. IL8 expression is also increased in an invasive glioblastoma mouse model [113]. Whether iNOS-mediated NO production downstream of EGFRvIII/STAT3 signaling induce glial transformation via upregulation of IL8 remains an open question.
NO activates cyclooxygenase 2 (COX-2) [114]. Overexpression of COX2 in rat intestinal epithelial cells reduces the rate of apoptosis and increases the expression of the anti-apoptotic protein Bcl-2 [115]. Conversely, the inhibition of COX2 induces apoptosis and inhibits cell proliferation [116-118]. Interestingly, the levels of COX2 are elevated in high-grade glioma [119, 120]. In addition, the selective COX2 inhibitor, NS398, reduces the proliferation of human glioblastoma cells [119]. Whether iNOS promotes oncogenesis of glioblastoma tumors via upregulation of COX2 remains to be investigated. Interestingly, COX2 expression is increased in glioblastoma cell lines in response to EGF or EGFRvIII expression [121]. Nuclear EGFR and STAT3 occupy the promoter of the COX-2 gene and thereby activate its expression [121]. Therefore, it is tempting to hypothesize that in EGFRvIII-expressing astrocytes, COX2 is either directly activated by the nuclear EGFRvIII/STAT3 or via STAT3-mediated iNOS production.
PERSPECTIVES
iNOS has been recently identified as a key player in the transformation of mouse astrocytes and human BTSCs. Notably, iNOS inhibition by genetic and pharmacological approaches inhibits tumor growth in both of these models in vivo. Development of novel iNOS-based therapeutics may therefore prove valuable in the treatment of glioblastoma. In particular, iNOS is a direct target of STAT3 in EGFRvIII-expressing astrocytes and mediates STAT3-induced glial proliferation and transformation. Thus, patients with activating EGFR mutations may benefit from pharmacological agents that inhibit iNOS.
Although identification of iNOS as a pathophysio-logically relevant STAT3 target in glial transformation has shed light on an important underlying mechanism of STAT3 function in brain tumor pathogenesis, many unresolved questions remain to be addressed. In view of the highly complex interactive nature of EGFR signaling and the dual role of STAT3 in the pathogenesis of glioblastoma, identification of genome-wide STAT3 targets in patients with different genetic backgrounds should provide important insights on how STAT3 operates in a specific tumor (Fig. 2). This can be achieved by genetic manipulation of brain tumor stem cell cultures following surgical excision, and subjecting them to a combination of genome wide binding site analyses and gene expression data (Fig. 2). Identified targets can be further assessed in functional experiments in vitro and in vivo. In this manner, classification of global STAT3 networks and assigning of molecular signatures to each patient may provide the means for patient-tailored approaches. Recent advances in next generation sequencing platforms should facilitate the discovery of oncogenic and tumor suppressive STAT3 targets. This will provide a strong foundation for better treatments in which an array of drugs and inhibitors can be assessed to identify the unique drug susceptibilities of a given tumor.
Fig. (2). Future STAT3-based-patient-tailored therapies for malignant glioma.
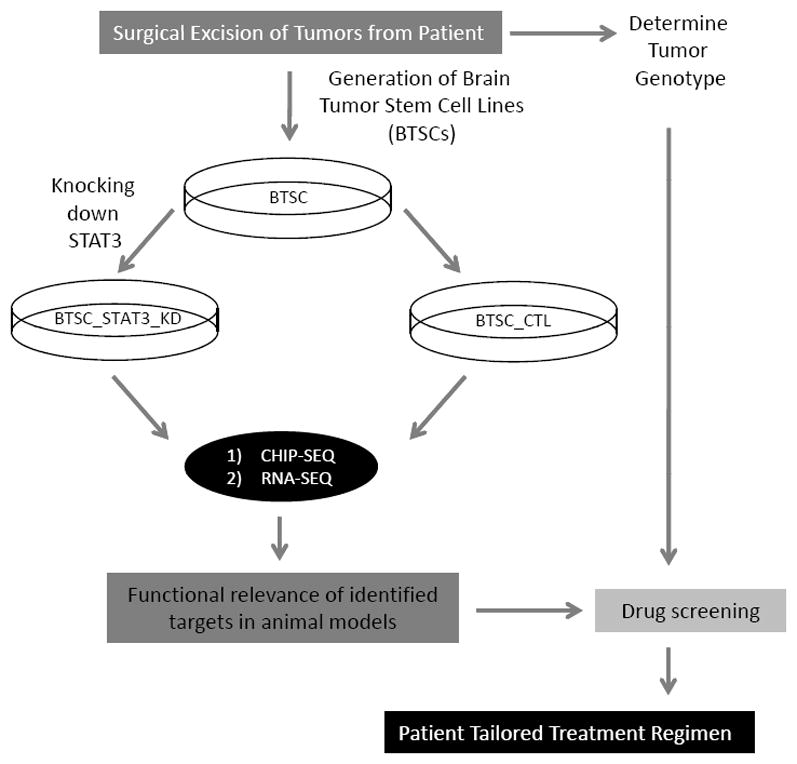
Following surgical excision of tumors, tumor cells can be dissociated and cultured. Cells can be enriched for tumor stem cells based on culture conditions. Brain tumor stem cells (BTSCs) can be genetically manipulated by lentiviral-based system. To identify STAT3 global networks, STAT3-KD-BTSCs and control counterparts can be subjected to chromatin immunoprecipitation with a STAT3 antibody followed by massive parallel sequencing (ChIP-Seq). To derive the functional relevance of those targets, BTSCs will be subjected to RNA-Seq and whole genome analyses. Identified targets will be assessed in functional experiments in animal models. An array of drugs and inhibitors can be assessed to identify the unique drug susceptibilities of a given tumor.
Acknowledgments
We thank members of Bonni laboratory for critical reading of this manuscript. Supported by NIH grant NS064007 (A.B.) and a fellowship from Canadian Institute of Health Research (A.J-A.).
Footnotes
CONFLICT OF INTEREST
The authors confirm that this article content has no conflicts of interest.
References
- 1.Holland EC. Gliomagenesis: genetic alterations and mouse models. Nat Rev Genet. 2001;2:120–129. doi: 10.1038/35052535. [DOI] [PubMed] [Google Scholar]
- 2.Louis DN, Ohgaki H, Wiestler OD, et al. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007;114:97–109. doi: 10.1007/s00401-007-0243-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 4.Robins HI, Chang S, Butowski N, Mehta M. Therapeutic advances for glioblastoma multiforme: current status and future prospects. Curr Oncol Reports. 2007;9:66–70. doi: 10.1007/BF02951428. [DOI] [PubMed] [Google Scholar]
- 5.Maher EA, Furnari FB, Bachoo RM, et al. Malignant glioma: genetics and biology of a grave matter. Genes Dev. 2001;15:1311–1333. doi: 10.1101/gad.891601. [DOI] [PubMed] [Google Scholar]
- 6.Furnari FB, Fenton T, Bachoo RM, et al. Malignant astrocytic glioma: genetics, biology, and paths to treatment. Genes Dev. 2007;21:2683–2710. doi: 10.1101/gad.1596707. [DOI] [PubMed] [Google Scholar]
- 7.Wen PY, Kesari S. Malignant gliomas in adults. N Engl J Med. 2008;359:492–507. doi: 10.1056/NEJMra0708126. [DOI] [PubMed] [Google Scholar]
- 8.Bachoo RM, Maher EA, Ligon KL, et al. Epidermal growth factor receptor and Ink4a/Arf: convergent mechanisms governing terminal differentiation and transformation along the neural stem cell to astrocyte axis. Cancer Cell. 2002;1:269–277. doi: 10.1016/s1535-6108(02)00046-6. [DOI] [PubMed] [Google Scholar]
- 9.Uhrbom L, Dai C, Celestino JC, et al. Ink4a-Arf loss cooperates with KRas activation in astrocytes and neural progenitors to generate glioblastomas of various morphologies depending on activated Akt. Cancer Res. 2002;62:5551–5558. [PubMed] [Google Scholar]
- 10.Louis DN. Molecular pathology of malignant gliomas. Annu Rev Pathol. 2006;1:97–117. doi: 10.1146/annurev.pathol.1.110304.100043. [DOI] [PubMed] [Google Scholar]
- 11.Bajenaru ML, Hernandez MR, Perry A, et al. Optic nerve glioma in mice requires astrocyte Nf1 gene inactivation and Nf1 brain heterozygosity. Cancer Res. 2003;63:8573–8577. [PubMed] [Google Scholar]
- 12.Konopka G, Bonni A. Signaling pathways regulating gliomagenesis. Curr Mol Med. 2003;3:73–84. doi: 10.2174/1566524033361609. [DOI] [PubMed] [Google Scholar]
- 13.Chen J, McKay RM, Parada LF. Malignant glioma: lessons from genomics, mouse models, and stem cells. Cell. 2012;149:36–47. doi: 10.1016/j.cell.2012.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wong AJ, Ruppert JM, Bigner SH, et al. Structural alterations of the epidermal growth factor receptor gene in human gliomas. Proc Natl Acad Sci USA. 1992;89:2965–2969. doi: 10.1073/pnas.89.7.2965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nagane M, Lin H, Cavenee WK, Huang HJ. Aberrant receptor signaling in human malignant gliomas: mechanisms and therapeutic implications. Cancer Lett. 2001;162:S17–S21. doi: 10.1016/s0304-3835(00)00648-0. [DOI] [PubMed] [Google Scholar]
- 16.Han W, Lo HW. Landscape of EGFR signaling network in human cancers: biology and therapeutic response in relation to receptor subcellular locations. Cancer Lett. 2012;318:124–134. doi: 10.1016/j.canlet.2012.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mizoguchi M, Betensky RA, Batchelor TT, et al. Activation of STAT3, MAPK, and AKT in malignant astrocytic gliomas: correlation with EGFR status, tumor grade, and survival. J Neuropath Exp Neur. 2006;65:1181–1188. doi: 10.1097/01.jnen.0000248549.14962.b2. [DOI] [PubMed] [Google Scholar]
- 18.Boerner JL, Demory ML, Silva C, Parsons SJ. Phosphorylation of Y845 on the epidermal growth factor receptor mediates binding to the mitochondrial protein cytochrome c oxidase subunit II. Mol Cell Biol. 2004;24:7059–7071. doi: 10.1128/MCB.24.16.7059-7071.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Demory ML, Boerner JL, Davidson R, et al. Epidermal growth factor receptor translocation to the mitochondria: regulation and effect. J Biol Chem. 2009;284:36592–36604. doi: 10.1074/jbc.M109.000760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yue X, Song W, Zhang W, et al. Mitochondrially localized EGFR is subjected to autophagic regulation and implicated in cell survival. Autophagy. 2008;4:641–649. doi: 10.4161/auto.5971. [DOI] [PubMed] [Google Scholar]
- 21.Lin SY, Makino K, Xia W, et al. Nuclear localization of EGF receptor and its potential new role as a transcription factor. Nat Cell Biol. 2001;3:802–808. doi: 10.1038/ncb0901-802. [DOI] [PubMed] [Google Scholar]
- 22.Lo HW, Hsu SC, Ali-Seyed M, et al. Bartholomeusz G, Shih JY, Hung MC: Nuclear interaction of EGFR and STAT3 in the activation of the iNOS/NO pathway. Cancer Cell. 2005;7:575–589. doi: 10.1016/j.ccr.2005.05.007. [DOI] [PubMed] [Google Scholar]
- 23.de la Iglesia N, Konopka G, Puram SV, et al. Identification of a PTEN-regulated STAT3 brain tumor suppressor pathway. Genes Dev. 2008;22:449–462. doi: 10.1101/gad.1606508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang YN, Yamaguchi H, Hsu JM, Hung MC. Nuclear trafficking of the epidermal growth factor receptor family membrane proteins. Oncogene. 2010;29:3997–4006. doi: 10.1038/onc.2010.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lo HW. Nuclear mode of the EGFR signaling network: biology, prognostic value, and therapeutic implications. Discovery Med. 2010;10:44–51. [PMC free article] [PubMed] [Google Scholar]
- 26.de la Iglesia N, Puram SV, Bonni A. STAT3 regulation of glioblastoma pathogenesis. Curr Mol Med. 2009;9:580–590. doi: 10.2174/156652409788488739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bonni A. STATs in the central nervous system. 2003 [Google Scholar]
- 28.Rajan P, McKay RD. Multiple routes to astrocytic differentiation in the CNS. J Neurosci. 1998;18:3620–3629. doi: 10.1523/JNEUROSCI.18-10-03620.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bonni A, Sun Y, Nadal-Vicens M, et al. Regulation of gliogenesis in the central nervous system by the JAK-STAT signaling pathway. Science. 1997;278:477–483. doi: 10.1126/science.278.5337.477. [DOI] [PubMed] [Google Scholar]
- 30.Yoshimatsu T, Kawaguchi D, Oishi K, et al. Non-cell-autonomous action of STAT3 in maintenance of neural precursor cells in the mouse neocortex. Development. 2006;133:2553–2563. doi: 10.1242/dev.02419. [DOI] [PubMed] [Google Scholar]
- 31.de la Iglesia N, Konopka G, Lim KL, et al. Deregulation of a STAT3-interleukin 8 signaling pathway promotes human glioblastoma cell proliferation and invasiveness. J Neurosci. 2008;28:5870–5878. doi: 10.1523/JNEUROSCI.5385-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Puram SV, Yeung CM, Jahani-Asl A, et al. STAT3-iNOS Signaling Mediates EGFRvIII-Induced Glial Proliferation and Transformation. Neurosci. 2012;32:7806–7818. doi: 10.1523/JNEUROSCI.3243-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yu Z, Kone BC. The STAT3 DNA-binding domain mediates interaction with NF-kappaB p65 and inducible nitric oxide synthase transrepression in mesangial cells. J Am Soc Nephrol (JASN) 2004;15:585–591. doi: 10.1097/01.asn.0000114556.19556.f9. [DOI] [PubMed] [Google Scholar]
- 34.Yu Z, Zhang W, Kone BC. Signal transducers and activators of transcription 3 (STAT3) inhibits transcription of the inducible nitric oxide synthase gene by interacting with nuclear factor kappaB. Biochem J. 2002;367:97–105. doi: 10.1042/BJ20020588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Konnikova L, Kotecki M, Kruger MM, Cochran BH. Knockdown of STAT3 expression by RNAi induces apoptosis in astrocytoma cells. BMC Cancer. 2003;3:23. doi: 10.1186/1471-2407-3-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rahaman SO, Harbor PC, Chernova O, et al. Inhibition of constitutively active Stat3 suppresses proliferation and induces apoptosis in glioblastoma multiforme cells. Oncogene. 2002;21:8404–8413. doi: 10.1038/sj.onc.1206047. [DOI] [PubMed] [Google Scholar]
- 37.Dasgupta A, Raychaudhuri B, Haqqi T, et al. Stat3 activation is required for the growth of U87 cell-derived tumours in mice. Eur J Cancer. 2009;45:677–684. doi: 10.1016/j.ejca.2008.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Carro MS, Lim WK, Alvarez MJ, et al. The transcriptional network for mesenchymal transformation of brain tumours. Nature. 2010;463:318–325. doi: 10.1038/nature08712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gu J, Li G, Sun T, et al. Blockage of the STAT3 signaling pathway with a decoy oligonucleotide suppresses growth of human malignant glioma cells. J Neuro-Oncol. 2008;89:9–17. doi: 10.1007/s11060-008-9590-9. [DOI] [PubMed] [Google Scholar]
- 40.Eyler CE, Wu Q, Yan K, et al. Glioma stem cell proliferation and tumor growth are promoted by nitric oxide synthase-2. Cell. 2011;146:53–66. doi: 10.1016/j.cell.2011.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Moncada S, Palmer RM, Higgs EA. Nitric oxide: physiology, pathophysiology, and pharmacology. Pharmacol Rev. 1991;43:109–142. [PubMed] [Google Scholar]
- 42.Esplugues JV. NO as a signalling molecule in the nervous system. Brit J Pharmacol. 2002;135:1079–1095. doi: 10.1038/sj.bjp.0704569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Toda N, Ayajiki K, Okamura T. Cerebral blood flow regulation by nitric oxide in neurological disorders. Can J Physiol Pharm. 2009;87:581–594. doi: 10.1139/y09-048. [DOI] [PubMed] [Google Scholar]
- 44.Schmidt HH, Walter U. NO at work. Cell. 1994;78:919–925. doi: 10.1016/0092-8674(94)90267-4. [DOI] [PubMed] [Google Scholar]
- 45.Forrester K, Ambs S, Lupold SE, et al. Nitric oxide-induced p53 accumulation and regulation of inducible nitric oxide synthase expression by wild-type p53. Proc Natl Acad Sci USA. 1996;93:2442–2447. doi: 10.1073/pnas.93.6.2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cobbs CS, Whisenhunt TR, Wesemann DR, et al. Inactivation of wild-type p53 protein function by reactive oxygen and nitrogen species in malignant glioma cells. Cancer Res. 2003;63:8670–8673. [PubMed] [Google Scholar]
- 47.Singh S, Gupta AK. Nitric oxide: role in tumour biology and iNOS/NO-based anticancer therapies. Cancer Chemother Pharmacol. 2011;67:1211–1224. doi: 10.1007/s00280-011-1654-4. [DOI] [PubMed] [Google Scholar]
- 48.Pervin S, Singh R, Chaudhuri G. Nitric oxide-induced cytostasis and cell cycle arrest of a human breast cancer cell line (MDA-MB-231): potential role of cyclin D1. Proc Natl Acad Sci USA. 2001;98:3583–3588. doi: 10.1073/pnas.041603998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pervin S, Singh R, Gau CL, et al. Potentiation of nitric oxide-induced apoptosis of MDA-MB-468 cells by farnesyltransferase inhibitor: implications in breast cancer. Cancer Res. 2001;61:4701–4706. [PubMed] [Google Scholar]
- 50.Pervin S, Singh R, Hernandez E, Wu G, Chaudhuri G. Nitric oxide in physiologic concentrations targets the translational machinery to increase the proliferation of human breast cancer cells: involvement of mammalian target of rapamycin/eIF4E pathway. Cancer Res. 2007;67:289–299. doi: 10.1158/0008-5472.CAN-05-4623. [DOI] [PubMed] [Google Scholar]
- 51.Thomas DD, Espey MG, Ridnour LA, et al. Hypoxic inducible factor 1alpha, extracellular signal-regulated kinase, and p53 are regulated by distinct threshold concentrations of nitric oxide. Proc Natl Acad Sci USA. 2004;101:8894–8899. doi: 10.1073/pnas.0400453101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ridnour LA, Windhausen AN, Isenberg JS, et al. Nitric oxide regulates matrix metalloproteinase-9 activity by guanylyl-cyclase-dependent and -independent pathways. Proc Natl Acad Sci USA. 2007;104:16898–16903. doi: 10.1073/pnas.0702761104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jenkins DC, Charles IG, Thomsen LL, et al. Roles of nitric oxide in tumor growth. Proc Natl Acad Sci USA. 1995;92:4392–4396. doi: 10.1073/pnas.92.10.4392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Aktan F. iNOS-mediated nitric oxide production and its regulation. Life Sci. 2004;75:639–653. doi: 10.1016/j.lfs.2003.10.042. [DOI] [PubMed] [Google Scholar]
- 55.Rosselli M. Nitric oxide and reproduction. Mol Hum Reprod. 1997;3:639–641. doi: 10.1093/molehr/3.8.639. [DOI] [PubMed] [Google Scholar]
- 56.Forstermann U, Sessa WC. Nitric oxide synthases: regulation and function. Eur Heart J. 2012;33:829–837. doi: 10.1093/eurheartj/ehr304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kroncke KD, Fehsel K, Kolb-Bachofen V. Inducible nitric oxide synthase in human diseases. Clin Exp Immunol. 1998;113:147–156. doi: 10.1046/j.1365-2249.1998.00648.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Xie Q, Nathan C. The high-output nitric oxide pathway: role and regulation. J Leukoc Biol. 1994;56:576–582. doi: 10.1002/jlb.56.5.576. [DOI] [PubMed] [Google Scholar]
- 59.Pautz A, Art J, Hahn S, Nowag S, Voss C, Kleinert H. Regulation of the expression of inducible nitric oxide synthase. Nitric Oxide. 2010;23:75–93. doi: 10.1016/j.niox.2010.04.007. [DOI] [PubMed] [Google Scholar]
- 60.Kleinert H, Schwarz PM, Forstermann U. Regulation of the expression of inducible nitric oxide synthase. Biol Chem. 2003;384:1343–1364. doi: 10.1515/BC.2003.152. [DOI] [PubMed] [Google Scholar]
- 61.Lin CW, Shen SC, Ko CH, Lin HY, Chen YC. Reciprocal activation of macrophages and breast carcinoma cells by nitric oxide and colony-stimulating factor-1. Carcinogenesis. 2010;31:2039–2048. doi: 10.1093/carcin/bgq172. [DOI] [PubMed] [Google Scholar]
- 62.Melillo G, Musso T, Sica A, et al. A hypoxia-responsive element mediates a novel pathway of activation of the inducible nitric oxide synthase promoter. J Exp Med. 1995;182:1683–1693. doi: 10.1084/jem.182.6.1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Du Q, Park KS, Guo Z, et al. Regulation of human nitric oxide synthase 2 expression by Wnt beta-catenin signaling. Cancer Res. 2006;66:7024–7031. doi: 10.1158/0008-5472.CAN-05-4110. [DOI] [PubMed] [Google Scholar]
- 64.Brennan PA, Dennis S, Poller D, et al. Inducible nitric oxide synthase: correlation with extracapsular spread and enhancement of tumor cell invasion in head and neck squamous cell carcinoma. Head Neck. 2008;30:208–214. doi: 10.1002/hed.20675. [DOI] [PubMed] [Google Scholar]
- 65.Thomsen LL, Miles DW, Happerfield L, et al. Nitric oxide synthase activity in human breast cancer. Brit J Cancer. 1995;72:41–44. doi: 10.1038/bjc.1995.274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Vakkala M, Kahlos K, Lakari E, et al. Inducible nitric oxide synthase expression, apoptosis, and angiogenesis in in situ and invasive breast carcinomas. Clin Cancer Res. 2000;6:2408–2416. [PubMed] [Google Scholar]
- 67.Ambs S, Merriam WG, Bennett WP, et al. Frequent nitric oxide synthase-2 expression in human colon adenomas: implication for tumor angiogenesis and colon cancer progression. Cancer Res. 1998;58:334–341. [PubMed] [Google Scholar]
- 68.Chen CN, Hsieh FJ, Cheng YM, Chang KJ, Lee PH. Expression of inducible nitric oxide synthase and cyclooxygenase-2 in angiogenesis and clinical outcome of human gastric cancer. J Surg Oncol. 2006;94:226–233. doi: 10.1002/jso.20372. [DOI] [PubMed] [Google Scholar]
- 69.Marrogi AJ, Travis WD, Welsh JA, et al. Nitric oxide synthase, cyclooxygenase 2, and vascular endothelial growth factor in the angiogenesis of non-small cell lung carcinoma. Clin Cancer Res. 2000;6:4739–4744. [PubMed] [Google Scholar]
- 70.Glynn SA, Boersma BJ, Dorsey TH, et al. Increased NOS2 predicts poor survival in estrogen receptor-negative breast cancer patients. J Clin Invest. 2010;120:3843–3854. doi: 10.1172/JCI42059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cobbs CS, Brenman JE, Aldape KD, Bredt DS, Israel MA. Expression of nitric oxide synthase in human central nervous system tumors. Cancer Res. 1995;55:727–730. [PubMed] [Google Scholar]
- 72.Garvey EP, Oplinger JA, Tanoury GJ, et al. Potent and selective inhibition of human nitric oxide synthases. Inhibition by non-amino acid isothioureas. J Biol Chem. 1994;269:26669–26676. [PubMed] [Google Scholar]
- 73.Jafarian-Tehrani M, Louin G, Royo NC, et al. 1400W, a potent selective inducible NOS inhibitor, improves histopathological outcome following traumatic brain injury in rats. Nitric Oxide. 2005;12:61–69. doi: 10.1016/j.niox.2004.12.001. [DOI] [PubMed] [Google Scholar]
- 74.Southan GJ, Szabo C, Thiemermann C. Isothioureas. potent inhibitors of nitric oxide synthases with variable isoform selectivity. Brit J Pharmacol. 1995;114:510–516. doi: 10.1111/j.1476-5381.1995.tb13256.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Iuvone T, Van Osselaer N, D’Acquisto F, Carnuccio R, Herman AG. Differential effect of L-NAME and S-methyl-isothiourea on leukocyte emigration in carrageenin-soaked sponge implants in rat. Brit J Pharm. 1997;121:1637–1644. doi: 10.1038/sj.bjp.0701317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Akaike T, Yoshida M, Miyamoto Y, et al. Antagonistic action of imidazolineoxyl N-oxides against endothelium-derived relaxing factor NO through a radical reaction. Biochemistry. 1993;3:827–832. doi: 10.1021/bi00054a013. [DOI] [PubMed] [Google Scholar]
- 77.Tsunoda R, Okumura K, Ishizaka H, et al. Vasodilator effect of carboxy-2-phenyl-4,4,5,5-tetramethylimidazoline-1-oxyl in the coronary circulation: in vivo and in vitro studies. Eur J Pharm. 1994;262:55–63. doi: 10.1016/0014-2999(94)90028-0. [DOI] [PubMed] [Google Scholar]
- 78.Rand MJ, Li CG. Discrimination by the NO-trapping agent, carboxy-PTIO, between NO and the nitrergic transmitter but not between NO and EDRF. Brit J Pharmacol. 1995;116:1906–1910. doi: 10.1111/j.1476-5381.1995.tb16681.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Dirks PB. Brain tumour stem cells: the undercurrents of human brain cancer and their relationship to neural stem cells. Philos Trans R Soc Lond B Biol Sci. 2008;363:139–152. doi: 10.1098/rstb.2006.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Singh S, Dirks PB. Brain tumor stem cells: identification and concepts. Neurosurg Clin N Am. 2007;18:31–38. doi: 10.1016/j.nec.2006.10.014. [DOI] [PubMed] [Google Scholar]
- 81.Singh SK, Hawkins C, Clarke ID, et al. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- 82.Clarke MF, Dick JE, Dirks PB, et al. Cancer stem cells--perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res. 2006;66:9339–9344. doi: 10.1158/0008-5472.CAN-06-3126. [DOI] [PubMed] [Google Scholar]
- 83.Lee J, Kotliarova S, Kotliarov Y, et al. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell. 2006;9:391–403. doi: 10.1016/j.ccr.2006.03.030. [DOI] [PubMed] [Google Scholar]
- 84.Park DM, Rich JN. Biology of glioma cancer stem cells. Mol Cells. 2009;28:7–12. doi: 10.1007/s10059-009-0111-2. [DOI] [PubMed] [Google Scholar]
- 85.Vescovi AL, Galli R. Reynolds BA. Brain tumour stem cells. Nat Rev Cancer. 2006;6:425–436. doi: 10.1038/nrc1889. [DOI] [PubMed] [Google Scholar]
- 86.Calabrese C, Poppleton H, Kocak M, et al. A perivascular niche for brain tumor stem cells. Cancer Cell. 2007;11:69–82. doi: 10.1016/j.ccr.2006.11.020. [DOI] [PubMed] [Google Scholar]
- 87.Gilbertson RJ, Rich JN. Making a tumour’s bed: glioblastoma stem cells and the vascular niche. Nat Rev Cancer. 2007;7:733–736. doi: 10.1038/nrc2246. [DOI] [PubMed] [Google Scholar]
- 88.Gardner PR, Gardner AM, Martin LA, Salzman AL. Nitric oxide dioxygenase: an enzymic function for flavohemoglobin. Proc Natl Acad Sci USA. 1998;95:10378–10383. doi: 10.1073/pnas.95.18.10378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hausladen A, Gow AJ, Stamler JS. Nitrosative stress: metabolic pathway involving the flavohemoglobin. Proc Natl Acad Sci USA. 1998;95:14100–14105. doi: 10.1073/pnas.95.24.14100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Thomsen LL, Scott JM, Topley P, et al. Selective inhibition of inducible nitric oxide synthase inhibits tumor growth in vivo: studies with 1400W, a novel inhibitor. Cancer Res. 1997;57:3300–3304. [PubMed] [Google Scholar]
- 91.Jadeski LC, Lala PK. Nitric oxide synthase inhibition by N(G)-nitro-L-arginine methyl ester inhibits tumor-induced angiogenesis in mammary tumors. Am J Pathol. 1999;155:1381–1390. doi: 10.1016/S0002-9440(10)65240-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Tozer GM, Prise VE, Chaplin DJ. Inhibition of nitric oxide synthase induces a selective reduction in tumor blood flow that is reversible with L-arginine. Cancer Res. 1997;57:948–955. [PubMed] [Google Scholar]
- 93.Wang B, Xiong Q, Shi Q, et al. Genetic disruption of host nitric oxide synthase II gene impairs melanoma-induced angiogenesis and suppresses pleural effusion. Int J Cancer. 2001;91:607–611. [PubMed] [Google Scholar]
- 94.Konopka TE, Barker JE, Bamford TL, et al. Nitric oxide synthase II gene disruption: implications for tumor growth and vascular endothelial growth factor production. Cancer Res. 2001;61:3182–3187. [PubMed] [Google Scholar]
- 95.Ng QS, Goh V, Milner J, et al. Effect of nitric-oxide synthesis on tumour blood volume and vascular activity: a phase I study. Lancet Oncol. 2007;8:111–118. doi: 10.1016/S1470-2045(07)70001-3. [DOI] [PubMed] [Google Scholar]
- 96.Chai Z, Sarcevic B, Mawson A, Toh BH. SET-related cell division autoantigen-1 (CDA1) arrests cell growth. J Biol Chem. 2001;276:33665–33674. doi: 10.1074/jbc.M007681200. [DOI] [PubMed] [Google Scholar]
- 97.Kandalaft LE, Zudaire E, Portal-Nunez S, Cuttitta F, Jakowlew SB. Differentially expressed nucleolar transforming growth factor-beta1 target (DENTT) exhibits an inhibitory role on tumorigenesis. Carcinogenesis. 2008;29:1282–1289. doi: 10.1093/carcin/bgn087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Tu Y, Wu W, Wu T, et al. Antiproliferative autoantigen CDA1 transcriptionally up-regulates p21(Waf1/Cip1) by activating p53 and MEK/ERK1/2 MAPK pathways. J Biol Chem. 2007;282:11722–11731. doi: 10.1074/jbc.M609623200. [DOI] [PubMed] [Google Scholar]
- 99.Prueitt RL, Boersma BJ, Howe TM, et al. Inflammation and IGF-I activate the Akt pathway in breast cancer. Int J Cancer. 2007;120:796–805. doi: 10.1002/ijc.22336. [DOI] [PubMed] [Google Scholar]
- 100.McDowell KA, Riggins GJ, Gallia GL. Targeting the AKT pathway in glioblastoma. Curr Pharm Des. 2011;17:2411–2420. doi: 10.2174/138161211797249224. [DOI] [PubMed] [Google Scholar]
- 101.Lim KH, Ancrile BB, Kashatus DF, Counter CM. Tumour maintenance is mediated by eNOS. Nature. 2008;452:646–649. doi: 10.1038/nature06778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Dimmeler S, Fleming I, Fisslthaler B, et al. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature. 1999;399:601–605. doi: 10.1038/21224. [DOI] [PubMed] [Google Scholar]
- 103.Kandel ES, Skeen J, Majewski N, et al. Activation of Akt/protein kinase B overcomes a G(2)/m cell cycle checkpoint induced by DNA damage. Mol Cell Biol. 2002;22:7831–7841. doi: 10.1128/MCB.22.22.7831-7841.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Xiong Q, Shi Q, Le X, Wang B, Xie K. Regulation of interleukin-8 expression by nitric oxide in human pancreatic adenocarcinoma. J Interferon Cytokine Res. 2001;21:529–537. doi: 10.1089/10799900152434411. [DOI] [PubMed] [Google Scholar]
- 105.Ma P, Cui X, Wang S, et al. Nitric oxide post-transcriptionally up-regulates LPS-induced IL-8 expression through p38 MAPK activation. J Leukoc Biol 2004. 76:278–287. doi: 10.1189/jlb.1203653. [DOI] [PubMed] [Google Scholar]
- 106.Sparkman L, Boggaram V. Nitric oxide increases IL-8 gene transcription and mRNA stability to enhance IL-8 gene expression in lung epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2004;287:L764–773. doi: 10.1152/ajplung.00165.2004. [DOI] [PubMed] [Google Scholar]
- 107.Benoy IH, Salgado R, Van Dam P, et al. Increased serum interleukin-8 in patients with early and metastatic breast cancer correlates with early dissemination and survival. Clin Cancer Res. 2004;10:7157–7162. doi: 10.1158/1078-0432.CCR-04-0812. [DOI] [PubMed] [Google Scholar]
- 108.Ambs S, Glynn SA. Candidate pathways linking inducible nitric oxide synthase to a basal-like transcription pattern and tumor progression in human breast cancer. Cell Cycle. 2011;10:619–624. doi: 10.4161/cc.10.4.14864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Yamanaka R, Tanaka R, Saitoh T, Okoshi S. Cytokine gene expression on glioma cell lines and specimens. J Neuro-oncol. 1994;21:243–247. doi: 10.1007/BF01063773. [DOI] [PubMed] [Google Scholar]
- 110.Garkavtsev I, Kozin SV, Chernova O, et al. The candidate tumour suppressor protein ING4 regulates brain tumour growth and angiogenesis. Nature. 2004;428:328–332. doi: 10.1038/nature02329. [DOI] [PubMed] [Google Scholar]
- 111.Heidemann J, Ogawa H, Dwinell MB, et al. Angiogenic effects of interleukin 8 (CXCL8) in human intestinal microvascular endothelial cells are mediated by CXCR2. J Biol Chem. 2003;278:8508–8515. doi: 10.1074/jbc.M208231200. [DOI] [PubMed] [Google Scholar]
- 112.Kostourou V, Cartwright JE, Johnstone AP, et al. The role of tumour-derived iNOS in tumour progression and angiogenesis. Br J Cancer. 2011;104:83–90. doi: 10.1038/sj.bjc.6606034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Xie Q, Thompson R, Hardy K, et al. A highly invasive human glioblastoma pre-clinical model for testing therapeutics. J Transl Med. 2008;6:77. doi: 10.1186/1479-5876-6-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Salvemini D, Misko TP, Masferrer JL, et al. Nitric oxide activates cyclooxygenase enzymes. Proc Natl Acad Sci USA. 1993;90:7240–7244. doi: 10.1073/pnas.90.15.7240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Tsujii M, DuBois RN. Alterations in cellular adhesion and apoptosis in epithelial cells overexpressing prostaglandin endoperoxide synthase 2. Cell. 1995;83:493–501. doi: 10.1016/0092-8674(95)90127-2. [DOI] [PubMed] [Google Scholar]
- 116.Sharma V, Dixit D, Ghosh S, Sen E. COX-2 regulates the proliferation of glioma stem like cells. Neurochem Int. 2011;59:567–571. doi: 10.1016/j.neuint.2011.06.018. [DOI] [PubMed] [Google Scholar]
- 117.Elder DJ, Halton DE, Hague A, Paraskeva C. Induction of apoptotic cell death in human colorectal carcinoma cell lines by a cyclooxygenase-2 (COX-2)-selective nonsteroidal anti-inflammatory drug: independence from COX-2 protein expression. Clin Cancer Res. 1997;3:1679–1683. [PubMed] [Google Scholar]
- 118.Liu XH, Yao S, Kirschenbaum A, Levine AC. NS398, a selective cyclooxygenase-2 inhibitor, induces apoptosis and down-regulates bcl-2 expression in LNCaP cells. Cancer Res. 1998;58:4245–4249. [PubMed] [Google Scholar]
- 119.Joki T, Heese O, Nikas DC, et al. Expression of cyclooxygenase 2 (COX-2) in human glioma and in vitro inhibition by a specific COX-2 inhibitor, NS-398. Cancer Res. 2000;60:4926–4931. [PubMed] [Google Scholar]
- 120.Deininger MH, Weller M, Streffer J, Mittelbronn M, Meyermann R. Patterns of cyclooxygenase-1 and -2 expression in human gliomas in vivo. Acta Neuropathol. 1999;98:240–244. doi: 10.1007/s004010051075. [DOI] [PubMed] [Google Scholar]
- 121.Lo HW, Cao X, Zhu H, Ali-Osman F. Cyclooxygenase-2 is a novel transcriptional target of the nuclear EGFR-STAT3 and EGFRvIII-STAT3 signaling axes. Mol Cancer Res. 2010;8:232–245. doi: 10.1158/1541-7786.MCR-09-0391. [DOI] [PMC free article] [PubMed] [Google Scholar]