

Abstract
A protective effect of interferon-gamma (IFNγ) has been described in a number of models of autoimmune disease, including experimental autoimmune myocarditis (EAM). Some reports have suggested that regulation of apoptosis in autoreactive lymphocytes mediate these protective functions. We examined the potential of IFNγ to regulate apoptotic mechanisms in detail, both in vitro and in vivo in EAM. We observed multiple apoptotic defects in caspase activity, and the expression of TNF superfamily members on CD4+ T cells. In addition, we observed selective defects in CD4+ T cell activation in response to antigenic stimulation. These activation and apoptotic defects were CD4+ cell autonomous, independent of the genotype of APCs. Inhibition of nitric oxide production in vivo did not reproduce the severe form of EAM of IFNγ-deficient mice, indicating that this pathway does not mediate the protective effect of IFNγ. Crosswise adoptive transfer of wild type, IFNγ−/−, and IFNγR−/− EAM demonstrated that IFNγ signaling was critical in CD4+ cells, but that non-CD4+ sources of IFNγ production were also involved in the control of disease. Together, these data indicate multiple mechanisms of autonomous and non-autonomous CD4+ T cell regulation mediated by IFNγ in the control of autoimmune heart disease.
Keywords: myocarditis, interferon-gamma, apoptosis, caspase 8, nitric oxide, autoimmune disease
Introduction
Interferon gamma (IFNγ), the prototypic cytokine associated with Th1 immune responses, was formerly considered essential for the development of autoimmune disease. Yet, in some well-studied animal models, IFNγ has been shown to possess protective functions. Mice deficient in IFNγ (IFNγ−/−) or its receptor, IFNγR, are hypersusceptible to a wide variety of autoimmune disease models, including experimental autoimmune myocarditis (EAM). This unexpected protective effect of IFNγ has been ascribed to dysregulation of IL17-producing Th17 cells, which participate in autoimmune pathology in many of these disease models, through regulation of the Th17-associated transcription factor RORγt.
However, work in several models suggests that suppression of pathogenic Th17 differentiation is not the sole function of IFNγ; Th1 cells are purported to also carry distinct pathogenic roles, independent of Th17 cells, resulting in qualitatively distinct disease phenotypes. These data raise the possibility that additional functions of IFNγ may participate in the disease process, exerting both pathogenic and protective roles.
Defects in lymphocyte apoptosis in IFNγ−/− mice have been reported in several autoimmune and infectious disease models, and have been hypothesized to mediate protective functions. IFNγ−/− CD4+ T cells are resistant to activation-induced cell death, through an autonomous mechanism dependent on the regulation of the proximal proapoptotic enzyme caspase 8. Inducible nitric oxide synthase (iNOS, Nos2) is one of the canonical transcriptional targets of IFNγ, and is purported to mediate apoptotic control of autoreactive lymphocytes in EAM. Together, these data suggest a paradoxical role for IFNγ in the regulation of the progression or contraction of an autoaggressive lymphocyte response.
Our goal in this study has been to characterize the extent of the apoptotic defects in the severe EAM of IFNγ-deficient mice. If defects were limited to specific apoptotic pathways, these pathways may represent attractive rational targets for future intervention. It was also important to determine whether these apoptotic defects resulted from altered activation thresholds; if fewer precursor cells were activated in IFNγ−/− mice, the apoptotic defect may reflect diminished turnover. We next examined the contribution of nitric oxide to this mode of regulation of the autoimmune disease process in EAM. Finally, we determined whether these apoptotic defects were autonomously regulated by autocrine or paracrine signaling in CD4+ T cells, or if they required additional cells, such as antigen-presenting cells (APC), to mediate these effects.
Materials and Methods
Mice
IFNγ−/−, IFNγR−/−, and WT BALB/cJ mice were obtained from the Jackson Laboratory (Bar Harbor, ME) and maintained in the Johns Hopkins University School of Medicine specific-pathogen free vivarium. Experiments were conducted on 6–12 week old female mice. All methods and protocols involving mice were approved by the AALAC-certified Animal Care and Use Committee of Johns Hopkins University, in accordance with PHS and NIH guidelines.
Induction of EAM
Active EAM was induced in mice as previously described. For the adoptive transfer of EAM, donor mice were immunized and lymphocytes harvested from spleen and lymph node at day 14. CD4+ cells were isolated to >90% purity by paramagnetic negative selection according to manufacturers' instructions (Miltenyi Biotec) and transferred iv in sterile 1x PBS into naïve recipients sublethally irradiated with <500 rads (Nordion).
Assessment of EAM
Mice were evaluated for the development of EAM at the peak of disease on day 21. Recipient mice in adoptive transfer experiments were examined at day 14 following transfer. Heart tissues were fixed in 10% formalin. Serial sections (5 μm) were cut longitudinally and stained with hematoxylin and eosin (HistoServ). Myocarditis severity was evaluated by histopathologic microscopic approximation of the percent area of myocardium infiltrated with mononuclear cells or fibrosis determined from five sections per heart according to a scoring system described previously. Slides were graded independently in a masked fashion by two observers.
In vitro stimulation
Viable mononuclear splenocytes from mice were ACK-lysed or Lymphocyte-selected (Cedarlane) into DMEM + 10% FBS. For selected experiments, CD4+ T cells were isolated by MACS™ negative selection to >90% purity, according to manufacturer's instructions (Miltenyi Biotec). Nonspecific primary stimulations were performed on naïve CD4+ T cells with plates coated with 1.0 μg/mL αCD3 and 2.5 μg/mL αCD28 (BD Pharmingen). For restimulation experiments, cells were stimulated as above, washed, replated to rest for 24–48h in fresh medium supplemented with 50 U/mL rmIL2 (R&D Systems), washed, and recultured on plates coated with 1.0 μg/mL αCD3 for 24h. For MyHCα614-629-specific recall stimulations, stimulator APCs were harvested from naïve mice, and gamma-irradiated with ~500 rads. 2 × 106 responders from mice with EAM were cocultured with 4 × 106 APCs in the presence of 10 μg/mL MyHCα614-629 in 24-well format, or unstimulated for control. Proliferation experiments were performed by labeling cells with 5–10 μM CFDA-SE (Molecular Probes) on ice for 10 minutes, followed by generous quenching and washing, prior to stimulation.
Flow cytometry
Splenocytes were extracted into single cell suspension in 1x PBS + 0.5% FBS, and RBCs lysed by <5 minute incubation in ACK lysis buffer (Biofluids). Cells were washed and FcγRII/III blocked with αCD16/32 (eBiosciences). Surface markers were stained with fluorochrome-conjugated mAbs to CD4, CD8α, CD11c, CD44, CD45RB, CD62L, CD69, CD95 (Fas), CD178 (FasL), CD197 (CCR7), CD253 (TRAIL), CD262 (DR5/TRAIL-R2) (eBiosciences, BD Pharmingen, BioLegend). CaspGLOW™ caspase substrate (BioVision), Annexin V (BD Pharmingen), and DAF-FM (Molecular Probes) staining were performed according to manufacturer's instructions. Samples were acquired on a four-color dual-laser FACScalibur cytometer running CellQuest, or the LSR II quad-laser cytometer running FACSDiva (BD Immunocytometry). Data were analyzed with FlowJo 7 (Treestar).
ELISA
Supernatants from stimulated cells were stored at −80° C. Linco multiplex cytokine assays (Millipore) were used according to manufacturer's instructions, and acquired on a Luminex XMAP reader. For nitric oxide assays, supernatants were cleared of protein by centrifugal filtration through a 10 kDA MWCO filter (Millipore) Total nitrate/nitrite was determined by a Greiss reaction-based kit, according to manufacturer's instructions (R&D Systems).
Statistics
Normally-distributed data on continuous parametric axes were analyzed with the two-tailed Student's t-test. The Mann-Whitney U-test was used to compare EAM severity scores between treatment groups, and other non-normally distributed data. Multiple group analyses were analyzed by ANOVA. Values of p < 0.05 were considered statistically significant.
Results
IFNγ protects from MyHCα614-629-induced EAM, associated with defects in apoptosis
First, we determined whether immunization with MyHCα614-629 induced severe EAM in IFNγ−/− mice, confirming previous findings with whole cardiac myosin. As shown in Figure 1, substantial myocardial inflammation was already observed in IFNγ−/− mice by day 14 post-immunization (Fig 1a), continuing to day 21 (Fig 1b), consistent with disease induced by whole cardiac myosin, characterized by extensive mononuclear and polymorphonuclear cell infiltration (Fig 1c), myocyte necrosis, and fibrosis. Control unimmunized or mock-immunized (CFA alone) IFNγ−/− mice did not develop this pathology.
Figure 1.
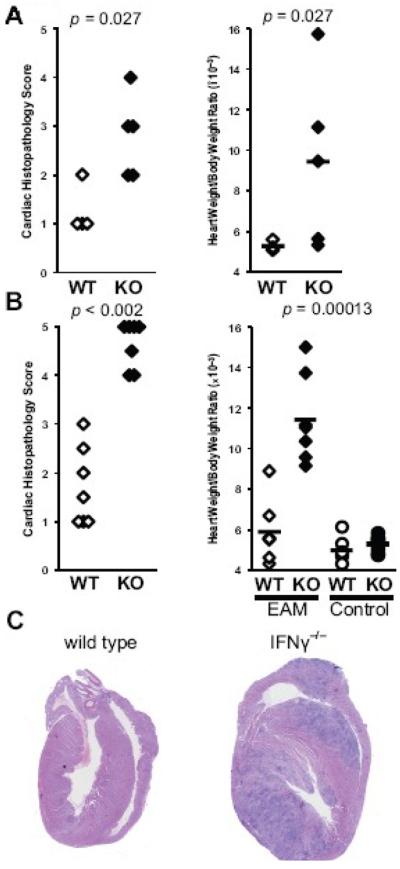
Severe MyHCα614-629-induced EAM in IFNγ−/− mice. A) Relative heart weight and B) histopathologic severity of hearts of WT (open) and IFNγ−/− (filled) mice at day 14 of EAM. C) Relative heart weight and D) histopathologic severity of hearts of WT and IFNγ−/− mice at day 21 of EAM. Control unimmunized or mock-immunized (circles) WT (open) and IFNγ−/− (filled) heart weights are shown. Representative histopathology of MyHCα614-629-induced EAM at day 21 from E) WT and F) IFNγ−/− mice, 200× magnification. Statistics are by two-tailed Students' t-test for HW/BW ratios, and Mann-Whitney U-test for EAM scores.
Apoptotic defects have been reported in several autoimmune disease models in IFNγ-deficient mice, and have been thought to account for the protective effect of IFNγ in these models. In order to characterize these defects, we examined Annexin V staining in CD4+ T cells over the course of the severe EAM of IFNγ−/− mice. Whereas Annexin V staining peaks in WT CD4+ T cells at day 21 pi, defective apoptosis is observed in IFNγ−/− CD4+ cells as early as day 10, continuing onto day 42 pi (Fig 2a). We observed apoptotic defects in other cell populations in IFNγ−/− mice, including CD8α+ T cells and CD11chi dendritic cells, although with unchanging kinetics (data not shown).
Figure 2.

Apoptotic defects of CD4+ T cells in IFNγ−/− EAM. Time course of A) Annexin V staining for apoptosis and B) caspase 8 staining of CD4+7AAD−-gated spleen cells at days 10, 21 and 42 pi from WT (open) and IFNγ−/− (filled) mice. Fluorescence intensity analysis for C) caspase 3 and D) caspase 9 substrate staining of CD4+7AAD−-gated spleen cells at day 21 pi. Surface expression of E) FasL and TRAIL and F) Fas, and DR5/TRAIL-R2 on spleen CD4+7AAD−-gated spleen cells at day 21 pi. Individual data points represent individual animals; bars represent arithmetic mean of each group.
The proapoptotic mechanism of IFNγ has been ascribed to regulation of caspase 8 transcription. We examined caspase activity by cytometric staining of lymphocytes with fluorogenic substrate peptides based on known cleavage specificities. Defects in caspase 8 activation are observed in IFNγ−/− CD4+ cells at day 10 pi, continuing to day 21 pi (Fig 2b). At day 21, we further observed diminished activity of caspases 3 and 9 in IFNγ−/− CD4+ cells (Figs 2c–d). Extrinsic apoptotic signaling is accomplished primarily through members of the TNF and TNF receptor superfamilies, which are known to be regulated on CD4+ cells upon activation. Therefore, we examined activated CD44hi CD4+ cells for the expression of several of these apoptotic mediators, and observed diminished surface expression of the TNFSF ligands Fas ligand and TRAIL (Fig 2e), but not the TNFRSF receptors Fas or DR5/TRAIL-R2 (Fig 2f).
IFNγ−/− CD4+ cells exhibit apoptotic defects in recall response in vitro
Next, we investigated the in vitro behavior of IFNγ−/− CD4+ T cells in recall responses to MyHCα614-629. In comparing the stimulation of splenocytes from IFNγ−/− and WT mice with EAM, we observed diminished Annexin V staining (Fig 3a) and caspase 8 activity (Fig 3b) in IFNγ−/− CD4+ cells, similar to that seen in freshly isolated cells. We also observed an apoptotic defect in unstimulated IFNγ−/− cells, albeit to a lesser degree.
Figure 3.

Apoptotic and activation defects of IFNγ−/− CD4+ T cells in vitro. A) Annexin V staining and B) caspase 8 substrate staining of CD4+7AAD−-gated spleen cells from day 21 pi of EAM following 48h restimulation with 10 μg/mL MyHCα614-629 or unstimulated. C) Cell death by 7AAD staining and D) caspase 8 substrate staining of purified naive CD4+ cells following restimulation with αCD3. E) After primary stimulation, surface expression of CD69 (left) and CD44 (right) on CD4+-gated cells. F) After secondary stimulation, surface expression of CD69 (left) and CD44 (right). Cells were additionally supplemented with 50 U/mL rmIFNγ. Bars represent mean of each group, plus standard error.
We turned to a more robust assay of T cell activation-induced cell death, stimulating purified CD4+ cells with αCD3 + αCD28, in order to further dissect specific mechanisms. We observed similar diminution of cell death and caspase 8 activity (Figs 3c–d), indicating the apoptotic defect is cell-autonomous. In order to confirm the dependency of this effect on IFNγ, we added exogenous recombinant IFNγ to the CD4+ T cells from IFNγ−/− mice. While we failed to reverse the cell death defect (Fig 3c), we did observe rescue of the activity of caspase 8 (Fig 3d). These data suggested that while IFNγ was important for caspase 8 activation, direct signaling through this pathway is not critical for the control of apoptosis in autoreactive lymphocytes.
IFNγ−/− CD4+ T cells exhibit selective activation defects in vitro
Because caspase 8 is implicated in early signaling events in T cell activation, we examined markers of T cell activation in vitro, in order to determine whether IFNγ-driven, caspase 8-associated defects in T cell activation may help to explain the selective apoptotic defects. Following 72 hours of primary activation in vitro, we observed diminished expression of the early activation marker CD69 (Fig 3e, left). This defect was reversed by supplementation with exogenous recombinant IFNγ. We also observed increased expression of the late activation marker CD44 in cells that were left unstimulated for 72 hours; stimulated cells did not show changes in CD44 expression (Fig 3e, right).
These alterations of primary activation, coupled with selective apoptotic defects, suggested that IFNγ−/− CD4+ cells may be differentially sensitive to antigen restimulation. Following primary stimulation, cells were rested, then restimulated. Differences in CD69 expression were not observed in restimulation (Fig 3f, left). However, increased proportions of CD4+ cells were CD44hi if left unrestimulated, indicating that a greater proportion of IFNγ−/− CD4+ T cells retained their activated phenotype through the rest step (Fig 3f, right). This increased expression of CD44 persisted through restimulation, and was partially reversed by addition of exogenous IFNγ.
Cells were labeled with CFDA-SE to observe proliferation. After primary stimulation, fewer IFNγ−/− CD4+ T cells had proliferated than WT (data not shown). However, inollowing rest, this pattern was markedly reversed in secondary stimulation. Fluorescence intensity analysis was used to examine the distribution of proliferative peaks in CFSE dilution. IFNγ−/− CD4+ T cells proliferated more than WT cells following restimulation, as shown by less intense mean fluorescence (data not shown). However, this effect could be reversed by the addition of recombinant IFNγ during primary stimulation.
IFNγ−/− CD4+ T cells exhibit selective activation defects during EAM
The in vitro activation experiments described above suggest that IFNγ control of caspase expression may also affect the development of the myocarditogenic, autoaggressive CD4+ T cell repertoire in EAM by controlling cell activation. As shown in Figure 4a, we observed diminished expression of CD69 on spleen IFNγ−/− CD4+ T cells at day 21 pi of EAM. In the supernatants of purified IFNγ−/− CD4+ cells stimulated with MyHCα614-629, we observed increased production of IL2 (Fig 4b), further suggesting regulatory interactions between IFNγ signaling and IL2 production were involved in the control of T cell proliferation and apoptosis.
Figure 4.

Apoptotic and activation defects of CD4+ T cells in IFNγ−/− EAM. A) Surface expression of CD69 on CD4+-gated spleen cells at day 21 pi from WT (open) and IFNγ−/− (filled) mice. B) Supernatant IL2 levels after 48h of MyHCα614-629 restimulation. Proportions of C) CD45RBloCD44hiCD62LhiCCR7hi central memory (left) and CD45RBloCD44hiCD62LloCCR7lo effector memory (right) spleen CD4+ T cells at day 21 pi. D) Absolute enumeration of MyHCα614-629-stimulated CD4+CD69+-gated cells in 24-well culture, from day 21 pi of EAM. E–G) Responder CD4+ T cells were purified from mice at day 21 of EAM, and cultured with irradiated stimulator APC splenocytes from naïve WT or IFNγ−/− mice for 48h. Cell death was assayed by staining with E) caspase 8 substrate or F) 7AAD. G) Proliferation was assayed by normalized CFSE dilution. Individual data points represent individual animals; bars represent arithmetic mean of each group. Crosswise stimulation comparisons across stimulator conditions were by paired Students' t-test.
Because of the severe form of EAM by day 21, we expected to find significant elicitation of autoaggressive memory T cell subsets. In order to assess the development of memory in the CD4+ compartment, we examined phenotypic markers associated with T cell memory. We observed a selective diminution of CD4+ cells bearing an effector memory CD45RBloCD44hiCD62LloCCR7lo phenotype in IFNγ−/− mice, with no corresponding decrease in CD45RBloCD44hiCD62LhiCCR7hi central memory cells (Fig 4c). In contrast, CD8α+ memory subpopulations are decreased in IFNγ−/− spleens at day 21 of EAM (data not shown). In the whole splenocyte in vitro recall stimulation described above (Figs 3a–b), we found a dramatic expansion of total IFNγ−/− cells following MyHCα614-629 stimulation (data not shown), including CD4+ T cells expressing CD69 (Fig 4d).
The apoptotic defect of IFNγ−/− CD4+ T cells is cell-autonomous in vitro
The presence of non-CD4+ cells in some (Figs 3a–b), but not all of the experiments described above, suggested the possibility that non-CD4+ sources of IFNγ, such as CD8α+ T cells, NK cells, monocytes, or macrophages may have be relevant to the control of apoptosis in autoreactive lymphocytes. Therefore, we purified CD4+ cells at day 21 of EAM from IFNγ−/− and WT spleens. We used APCs from the spleens of naïve IFNγ−/− and WT mice, in a crosswise recall stimulation assay, to determine the dependence of altered activation and apoptosis in IFNγ−/− CD4+ upon autocrine or paracrine signaling.
Defective cell death and caspase 8 activity in IFNγ−/− CD4+ cells were entirely dependent on the genotype of responding CD4+ T cells, and not on the genotype of the stimulator APC (Figs 4e–f). The dilution of CFSE label, used to study cell proliferation, was increased in IFNγ−/− CD4+ cells, independent of the genotype of stimulator APCs (Figure 4g). The production of effector cytokines by CD4+ cells was also not dependent upon the genotype of the APC (data not shown). Together, these data indicate that the control of CD4+ activation, apoptosis, and effector function directly mediated by IFNγ depend on autocrine or paracrine signaling, and not upon APC-generated IFNγ.
iNOS-mediated nitric oxide production does not mediate protection from EAM
It has been suggested that IFNγ-mediated control of nitric oxide production in macrophages mediates the protective effects of IFNγ in myocarditis through control of T cell outgrowth. To address this possibility, we examined nitric oxide production by macrophages by staining with the intracellular NO probe DAF-FM. As shown in Figure 5a, IFNγ−/− CD11b+F4/80+ macrophages expressed less nitric oxide, compared to WT controls.
Figure 5.
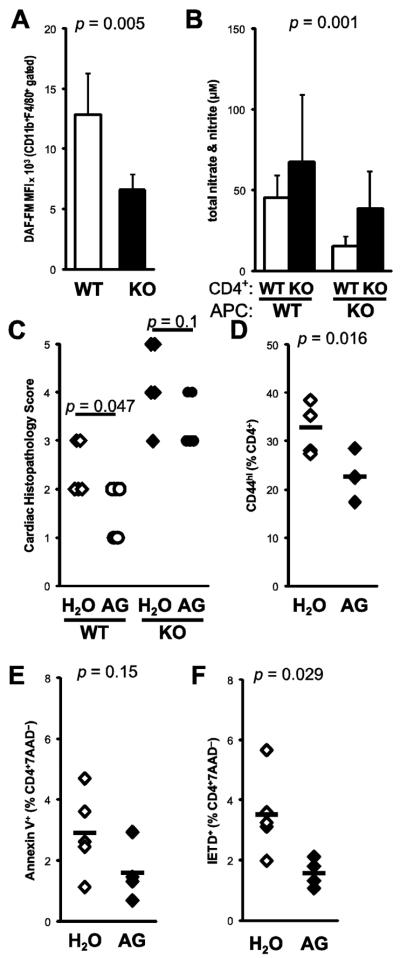
Nitric oxide does not mediate protection from EAM. A) Nitric oxide production from macrophages was determined by cytometric staining with DAF-FM at day 21 of EAM from WT (open) and IFNγ−/− (filled) mice. B) Total nitrate/nitrite were determined from supernatants of MyHCα614-629-stimulated CD4+ cells cocultured with WT or IFNγ−/− stimulator APCs. To selectively suppress iNOS, WT BALB/cJ mice were fed 5.6 g/L aminoguanidine in drinking water ad libitum, beginning at day −1 of EAM until sacrifice. C) Histopathologic severity of EAM at day 21 pi in both wild-type (open) and IFNγ−/− (filled) mice D) CD44 expression, E) Annexin V staining, and F) caspase 8 activity in WT spleen CD4+ T cells at day 21 pi. Individual data points represent individual animals; bars represent arithmetic mean of each group.
To examine the relevant sources of IFNγ necessary for the control of nitric oxide production, we assayed nitric oxide levels in the supernatants of our crosswise stimulation assay. As shown in Figure 5b, nitrite/nitrate levels were increased in the supernatants of IFNγ−/− CD4+ T cells, but diminished when IFNγ−/− APCs were used. These data indicate that autocrine IFNγ signaling is important for iNOS expression in macrophages, but that pathways independent of IFNγ mediate CD4+ control of iNOS expression.
To further address the possibility that IFNγ-driven nitric oxide is the critical mediator of protection in EAM, we pharmacologically suppressed iNOS function in vivo by treating mice with EAM with the NOS2-selective inhibitor aminoguanidine. WT BALB/cJ mice were protected from EAM by continuous treatment with aminoguanidine. Notably, aminoguanidine-treated IFNγ−/− mice were also mildly protected from EAM (Fig 5c). We observed fewer CD44hi activated CD4+ cells in spleen following aminoguanidine treatment (Fig 5d). In addition, the Annexin V staining and caspase 8 activity of spleen CD4+ T cells were diminished by aminoguanidine treatment (Figs 5e–f), indicating that iNOS control of lymphocyte apoptosis was not responsible for protection from EAM.
Non-CD4+ cells are critical for the adoptive transfer of severe IFNγ−/− EAM
To determine the autonomy of IFNγ−/− CD4+ T cells in mediating severe EAM, IFNγ−/− and WT BALB/cJ mice were immunized, and used as donors for the adoptive transfer of EAM to naïve irradiated IFNγ−/− and WT in a crosswise manner. As shown in Figure 6a, KO→KO transfers resulted in more severe EAM than WT→WT transfers in recipients at day 14 post-transfer (p = 0.018). However, both WT→KO and KO→WT crosswise transfers resulted in low-grade disease, comparable to WT→WT controls. Comparison of the difference in disease severity between KO→KO and KO→WT transfers (p = 0.00025) indicated that non-CD4+ sources of IFNγ production were relevant in controlling the severe EAM of IFNγ−/− mice.
Figure 6.
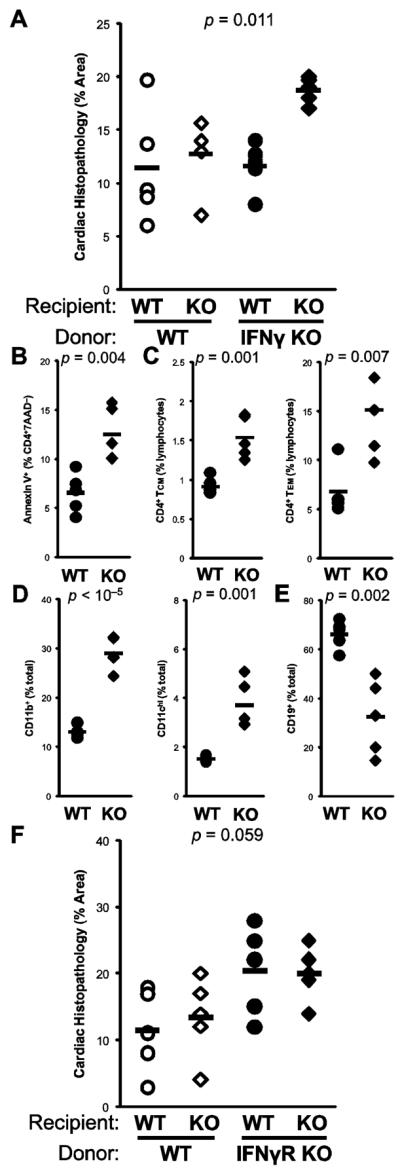
The severe EAM of IFNγ−/− mice does not transfer to WT with CD4+ cells. Cells were harvested from donor WT (open) and IFNγ−/− (filled) mice at day 14 of EAM, and transferred into irradiated WT (circle) and IFNγ−/− (diamond) recipients. A) Histopathologic severity of EAM in recipients at day 14 post-transfer. In spleens of recipients of IFNγ−/−CD4+ donor cells, B) apoptosis by Annexin V staining, C) central memory (CD4+CD45RBloCD62Lhi) and effector memory (CD4+CD45RBloCD62Llo)T cell subsets, D) proportions of CD11b+ monocyte/macrophages and CD11chi dendritic cells, and E) proportions of CD19+ B cells. F) Crosswise adoptive transfer of donor WT (open) and IFNγR−/− (filled) mice into WT (circle) or IFNγR−/− recipients (diamond); data depict histopathologic severity of EAM in recipients at day 14 post-transfer. Individual data points represent individual animals; bars represent arithmetic mean of each group
In order to understand mechanisms underlying this effect, we examined cell populations and phenotypes in the spleens of recipient mice at day 14 post-transfer. For cellular analyses, the most relevant comparison was in mice that had received IFNγ−/− CD4+ donor cells. Interestingly, IFNγ−/− CD4+ T cells were protected from apoptosis when transferred into WT recipients (p = 0.008), consistent with the notion that the apoptotic defect in IFNγ−/− CD4+ cells is cell-autonomous. Moreover, this finding also indicated that non-CD4+ sources of IFNγ operated in opposition to CD4+-derived IFNγ in the control of lymphocyte apoptosis (Figure 6b).
Our in vivo and in vitro data suggested that IFNγ controls the activation, differentiation, and outgrowth of autoaggressive memory T cells. At day 14 post-transfer, spleen CD4+ cells with central or effector memory surface marker phenotypes were more numerous in KO→KO transferred animals, compared to KO→WT transfers (Figure 6c). We further examined non-CD4+ cell populations in transferred spleens, to determine effects of non-CD4+ sources of IFNγ on the reconstitution or outgrowth of these cell populations. Myeloid cell populations, including CD11b+ monocytes and macrophages were increased, as were CD11chi dendritic cells in KO→KO transfers (Figure 6d). Both CD4+ and CD8α+ T cells were more numerous in KO→KO recipients (data not shown); however, fewer CD19+ B cells were observed (Figure 6e).
We further recapitulated these findings by utilizing donor CD4+ cells from mice deficient in the receptor for IFNγ (IFNγR−/−). As shown in Figure 6f, disease was more severe in mice that receive IFNγ receptor-deficient MyHCα614–629-primed CD4+ cells. Together these data argue that in vivo, IFNγ-mediated control of apoptosis is not cell-autonomous, but instead relies upon additional physiologic sources of IFNγ for the control of CD4+ T cell outgrowth.
Discussion
The immunologic mechanisms underlying the pathogenesis of organ-directed autoimmune disease remain elusive. In many of these models, including EAM, the centrality of CD4+-mediated effector mechanisms has been well-established. However, the prevailing paradigm of Th1 and Th2 differentiation has been insufficient to account for autoimmune pathogenesis. IFNγ appears to have protective functions in EAM, while IL13 also limits the progression of disease. The discovery of the Th17 lineage of CD4+ cells has resolved some, but not all of the inconsistencies regarding the roles of the p35 and p40 subunits of IL12, and IFNγ in the progression of autoimmune disease.
Complex interactions between Th1 and Th17 CD4+ T cells mediate disease pathogenesis in EAM. Th17-polarized CD4+ cells are sufficient to transfer myocarditis, while IL17A itself is dispensable for myocardial inflammatory infiltration. Evidence from other autoimmune disease models indicate that Th1 and Th17 cells mediate discrete features of the disease process. The diverse functions of IFNγ suggest the possibility that deviation or suppression of pathogenic Th17 cells may not be its sole protective role. Investigations into the specific pathophysiologic functions of the Th17 lineage indicate that while important, they are not the only mediator of severe disease in IFNγ-deficient animals..
It is currently understood that Th17 cells mediate EAE in the specific absence of IL12p35. However, our data indicate that other factors, including control of lymphocyte apoptosis, also contribute to the protective effect of IFNγ in autoimmune disease models. IFNγ has been reported to mediate control of activation-induced cell death in CD4+ cells through an autonomous mechanism involving caspase 8. IFNγ has been shown to directly regulate caspase 8 expression, as well as Fas, TRAIL, DR5/TRAIL-R2, and TNFR1 in CD4+ cells.
In addition to effecting apoptotic pathways, caspase 8 signaling has also been shown to be important in TCR signaling events critical to T cell activation. IFNγ regulation of caspase 8 may be a homeostatic regulator of both the initiation and the resolution of CD4+ T cell responses. These findings are consistent with our demonstration of selective early activation defects in CD4+ T cells during primary stimulation, in the absence of IFNγ. It should be noted that early activation defects may agree with later apoptotic defects, reflecting the accumulation of activated cells.
The demonstration of selective activation defects in vitro in primary stimulation of IFNγ−/− defects may be helpful in this regard. Fewer IFNγ−/−\CD4+ T cells acquired CD69 expression following primary stimulation, a defect that could be corrected by supplementation with exogenous IFNγ. In contrast, CD44 expression was actually increased in secondary stimulation, and surprisingly, in IFNγ−/− CD4+ Tcells that had gone unstimulated or unrestimulated following rest. Together, these data indicate that IFNγ signaling modulates activation signaling in a complex manner, with a kinetically-delimited dual effect on T cell function. During early activation events, IFNγ-directed caspase 8 expression may be important in initial CARMA/Bcl10/MALT1 signal integration into TCR activation signaling, upstream of NFκB activation. Following activation, IFNγ regulation of caspase activity may potentiate clearance of responding cells by enabling activation-induced cell death (AICD)-like apoptosis. In IFNγ−/− EAM, this may account for the diminution of CD69-expressing cells, and the selective accumulation of CD44hi activated CD4+ T cells. These data may point to a form of apoptosis that resembles cytokine withdrawal more closely than activation-induced cell death, due to the defects in cell death seen in vitro during rest, following primary stimulation. This interpretation may also be consistent with the increased production of IL2 by IFNγ−/− CD4+ cells, as well as the selective increase of CD4+ effector memory phenotype cells in vivo.
Nitric oxide has been purported to mediate the protective effect of IFNγ in EAM, a finding inconsistent with previous reports that suppression of iNOS ameliorates EAM in rats. In addition to confirming a pathogenic role for iNOS-dependent nitric oxide in mice, we further observed diminished T cell activation and apoptosis in vivo, following aminoguanidine suppression of iNOS. These data support the view that nitric oxide also participates in the control of T cell activation and apoptosis, independent of EAM. Macrophage-sourced iNOS appears to participate in pathogenic, rather than protective, mechanisms in vivo.
By using APC-independent anti-CD3 stimulation, our data indicate that IFNγ-mediated control of activation and apoptosis in CD4+ cells can be mediated autonomously in an auto- or paracrine manner. In EAM, we confirmed this independent control of CD4+ cell survival by IFNγ in vitro, as well as in vivo, through the transfer of IFNγR1−/− CD4+ T cells. However, in transfer experiments with IFNγ−/− CD4+ cells, additional cellular sources of IFNγ appear to participate in the control of lymphocyte activation, expansion and survival – as well as the eventual control of autoimmune heart disease. Importantly, the autonomy of the activation and apoptotic defects of IFNγ−/− CD4+ cells did not track with the transfer of disease severity; IFNγ−/− CD4+ cells transferred less severe disease to WT recipients than to IFNγ−/− recipients. These data compel us to conclude that control of apoptosis is not the predominant mode of regulation in EAM.
The finding that non-CD4+ sources of IFNγ are sufficient for the control of disease implicates several potential candidates, including CD8+ cells, NK cells, macrophages, and dendritic cell subsets. T-bet expression in CD8+ cells has been reported to be protective in EAM. The induction of FoxP3+ Tregs, believed to participate in peripheral tolerance to autoantigen, has been reported to be mediated in part by IFNγ signaling. These mechanisms need not be exclusive of mechanisms mediated by CD4+-derived IFNγ. Our adoptive transfer experiments point towards a collaborative effect of IFNγ derived from CD4+ cells and other sources in mediating protection from autoimmune pathology in vivo.
The regulation of autoimmune pathophysiology by CD4+ cells may not be simply mediated by countersuppression between Th1 and Th17 subsets. Additional regulatory mechanisms, probably involving trans-acting factors, appear to participate in vivo. Evidence from other models suggests that IL9, IL22, and GM-CSF may be important mediators of Th17-dependent pathology, more so than IL17 itself. There may be collaborative effects directly mediated by IFNγ upon the differentiation of pathogenic CD4+ effector subsets, and indirectly through other cell types.
Together, these data indicate that IFNγ controls a broad program of activation and apoptosis regulation in CD4+ cells that is mediated through caspase signaling. While IFNγ-dependent regulation of CD4+ activation and apoptosis can be autonomously effected in vitro, additional trans-actors participate in these processes in vivo. Inducible nitric oxide production appeared to be autonomously IFNγ-driven in the APC compartment, and was not responsible for mediating protection in EAM. CD4+-derived IFNγ-controlled apoptosis, by itself, was sufficient for protection from EAM, although additional cellular sources of IFNγ participated in the protective effect.
Acknowledgements
The authors wish to express their gratitude to Ophelia Rogers, Tonya Webb, Aaron Selya, and the laboratories of Jonathan Schneck for assistance with paramagnetic selections; R. Lee Blosser and Ada Tam for expert assistance with flow cytometric analyses; Nicole Barat and the laboratories of J. Steven Dumler for assistance with Luminex XMAP multiplex cytokine assays; Norman J. Barker for assistance with photomicrography; Dongfeng Zheng and Erin Medley for technical assistance.
This work was supported by NIH/NHLBI grants R01 HL70729, R01 HL67290, and HL87033. JGB is the Mary Renner Fellow in Autoimmune Disease Research, at the Johns Hopkins Autoimmune Disease Research Center. DC is supported by a research fellowship grant from the Myocarditis Foundation.
Abbreviations
- 7AAD
7-amino actinomycin
- AG
aminoguanidine
- DAF-FM
4-amino-5-methylamino-2',7'-difluorofluorescein
- EAM
experimental autoimmune myocarditis
- iNOS
inducible nitric oxide synthase
- KO
knockout
- MO
monocyte
- Mφ
macrophage
- WT
wild type
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures The authors have no conflicting interests to report.