

Abstract
A large portion of the human genome is transcribed into RNAs without known protein-coding functions, far outnumbering coding transcription units. Extensive studies of long noncoding RNAs (lncRNAs) have clearly demonstrated that they can play critical roles in regulating gene expression, development and disease, acting both as transcriptional activators and repressors. More recently, enhancers have been found to be broadly transcribed, resulting in the production of enhancer-derived RNAs, or eRNAs. Here, we review emerging evidence suggesting that at least some eRNAs contribute to enhancer function. We discuss these findings with respect to potential mechanisms of action of eRNAs and other ncRNAs in regulated gene expression.
Keywords: non-coding RNA, enhancer, eRNA, transcription
Enhancers in development and diseases
Functional specialization of cell and tissue types is vital for all metazoans. This requires cells to respond to developmental and environmental cues by generating specific gene expression patterns on the basis of an identical set of genetic material. Enhancers are the principle regulatory components of the genome that enable such cell-type and cell-state specificities of gene expression. Enhancers were initially defined as DNA elements that act over a distance to positively regulate expression of protein encoding target genes [1]. Enhancers contain specific recognition sequences required for binding of transcription factors (TF) that regulate gene expression in a spatial and temporal fashion (Fig 1A). An estimated 400K to 4 million putative enhancers exist in the human genome[2, 3], vastly outnumbering protein-coding genes, therefore suggesting a high complexity of enhancer utilization in gene regulation. Indeed, a precise pattern of activation of specific cohorts of enhancers is critical for cell-type development and cell lineage determination, as well as cellular responses to stimuli (Fig. 1A,B). By contrast, genetic variance in enhancer sequences can alter TF binding, predisposing the organism to ‘improper’ gene expression and ultimately susceptibility to diseases (Fig. 1C) [4, 5].
Figure 1. Roles of enhancers in development, signaling events and diseases.
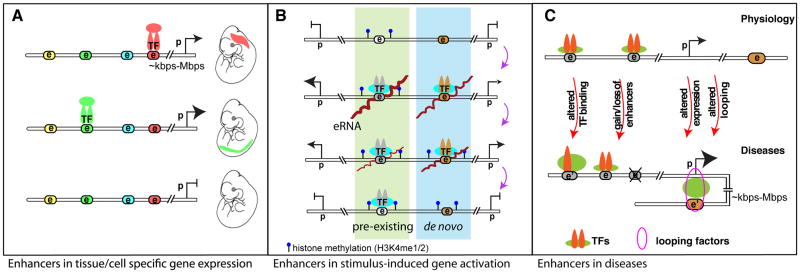
A. Differential enhancer binding patterns dictate temporal and spatial gene regulations during development. Colored nodes with “e” indicate potential enhancer elements, which can be activated during specific developmental windows upon recruitment of certain transcription factors (TF) for coordinated tissue or cell-type specific gene expression patterns. Tissue specific enhancers are color coordinated. Relative expression levels are denoted by the proportional sizes of the arrowheads. “e”, “p” stand for enhancer and promoter, respectively; double-line represents linear genomic distance between enhancer and promoter that can range from several kilo base pairs (kbps) to mega base pairs (Mbps). B. During gene transcriptional activation events in responding to differentiation or environmental cues (e.g. LPS (lipopolysaccharide) stimulation in macrophages), a group of pre-existing enhancers (highlighted green) bearing mono- or di-methylation of histone lysine 4 (H3K4me1/2) modification is readily activated. A small subset of enhancers is generated de novo in response to LPS stimulation resulting in deposition of H3K4me1/2 enhancer marks [Kaikkonen 2013]. This group of enhancers, highlighted in blue, will be readily activated upon repeated stimulation [Otsuni 2013]. The order from the top – stimulus activation – to the bottom - restoration to resting state – indicates the sequential cascade of events; the distance between two histone methylation represents nucleosome spacing and chromatin accessibility; and brown lines on “e” indicates eRNAs. C. Dysregulated enhancer functions are involved in human diseases, as exemplified in breast cancer and coronary artery disease (CAD) [5, 78, 79]. Genetic variation in enhancer (denoted as e’) could alter TF binding, gain or loss of functional enhancers, resulting in differential gene regulation. This is also often paralleled by differential chromosomal looping. Some of these scenarios can be directly linked to disease-associated genetic variants that occur at the DNA level of enhancers (e.g. single nucleotide polymorphism, insertion, deletion, and copy number variants etc.). The sizes of arrowheads of genes or symbols of TFs are proportional to their expression levels or binding intensities, respectively.
Recent genomic and epigenomic advances have contributed many insights into the properties, activity and selection of enhancer elements. Interestingly, the selection of a large fraction of cis-acting regulatory elements for enhancer activity within a cell appears to depend on relatively simple combinations of the Lineage Determining Transcription Factors (LDTFs) for cell type identity. Through cooperative binding to closely spaced recognition motifs, LDTFs initiate binding to otherwise closed chromatin and facilitate chromatin accessibility by recruitment of ATP-dependent nucleosome remodeling complexes. Histone modifiers are recruited to deposit histone marks that demarcate enhancer regions (e.g. monomethylation of histone 3 lysine 4 H3K4me1) [6]. While a large fraction of enhancers requires cooperative binding of transcription factors [Heinz 2010; Heinz 2013], another group of enhancers is activated by sequential binding of transcription factors at different development stages [Gualdi 1996]. A group of nucleosome-avid ‘pioneering transcription factors, exemplified by Forkhead Box A (FoxA) and GATA Binding Protein (GATA), initiated this sequential binding to condensed chromatin to prime a ‘soon-to-be’ enhancer [Zaret 2011]. Either LDTFs or pioneering factors permits the binding of other transcription factors and cofactors upon signal transduction events resulting in cell-type specific, signal-dependent gene expression (Fig. 3A). For more information on the physiology, clinical implication, biochemical and molecular perspectives of enhancers, we refer our readers to several excellent reviews [2, 7–9]
Figure 3. Proposed functional mechanisms of enhancer transcription and transcripts.

A. Collaborative binding of pioneer or lineage determining transcription factors LDTFs (red and dark blue) leads to nucleosome remodeling, increased chromatin accessibility, histone modifications (mono- and di-methylation on H3K4, blue circles on histone tails), and assembly of basal transcription machinery. Upon stimulation, signal-dependent transcription factors (SDTFs, light blue) bind to recognition sequences at enhancers and recruit coactivator complexes (green). This leads to further epigenetic modifications (e.g. histone acetylation, red circles on histone tails) and transcriptional activation of the enhancer. B–C. The enhancer transcription and transcripts, under different circumstances, may have independent functional roles. B. (A proposed model) eRNA functionally contributes to enhancer-mediated coding gene expression. Signal dependent activation of enhancers leads to increased production of eRNAs, which interacts with looping factors (e.g. cohesin complex) and facilitates/stabilizes chromosomal looping between enhancer and the promoter(s) of cognate target gene(s). eRNA mediates the loading of RNA PolII, and likely the transcription initiation complex (denoted by TFIID) at promoter of target gene [Mousavi 2013]. Whether chromosomal looping facilitates RNA PolII loading is unknown. C. Transcription elongation is required for the deposition of di-methylation of histone lysine 4, or H3K4me2, also a histone mark for enhancer, during signal-dependent activation of de novo enhancers. The coactivator complexes generate acetylation of histones, which recruits the positive transcription elongation factor (pTEFb, orange) to promote elongation of RNA Pol II. Subsequently, the elongating Pol II cargos histone methyl transferases, Mixed Lineage Leukemia complexes (MLLs), for di-methylation deposition on H3K4 at enhancers [Kaikkonen, 2013].
Recent findings that enhancers can be transcribed added a new layer of complexity to gene regulation [10]. In addition to messenger RNAs, non-coding RNAs represent a highly functional class of molecules, as they can possess enzymatic activity (e.g. spliceosomes, ribozyme), have structural roles (e.g. tRNA, ribosomes), and transcriptional functions (e.g. lncRNAs) [11, 12]. Understanding how RNAs contribute to enhancer function has thus become an area of active interest. Here we will discuss some of the historical aspects of enhancers as transcription units and recent studies suggesting functional roles of enhancer-derived RNAs, noting similarities and differences with other classes of ncRNAs that can exert positive effects on gene regulation.
Evidence of RNA transcription from enhancers
Evidence of pervasive RNA transcription at active enhancer elements became apparent with recent advances in sequencing technology. Interestingly, several reports over the past half a century had already hinted at the existence of short-lived nuclear RNAs (Table 1). Using pulse-chase labelling of RNA (Table 2), Harris found that the turnover rates of RNA in the nucleus of macrophages or fibroblasts were too high to account for the relatively steady level of cytoplasmic RNA [13]. This study provided the first evidence that the majority of the nascent RNA produced in the nucleus is rapidly turned over and does not contribute to mRNAs. The discovery of pervasive enhancer transcription suggests that eRNAs, in addition to intronic RNA, are quantitatively important contributors to this rapidly degraded pool of nuclear RNA [14]. Reports of specific enhancer-derived transcription were first documented in the Locus Control Region (LCR) of the beta-globin gene clusters [15–17]. Located 10–15kb upstream of the gene cluster, the beta-globin LCR orchestrates temporal and spatial expression of globin genes during development. It consists of five erythroid specific DNAse-I hypersensitivity sites (HS) (Table 2), and binding elements for erythroid LDTFs such as GATA Binding Protein 1 (GATA1), suggesting enhancer-like properties of these regions. Importantly, transcriptional initiation sites were found in several of these DNAse-I hypersensitivity regions [15–17]. Importantly they are distinct from alternative start sites of the globin gene itself [17, 18]. Description of enhancer RNA transcripts was subsequently extended to the LCR of MHC class II [19] and human Growth Hormone (hGH) [20] loci.
Table 1.
Timeline of enhancer and eRNA discovery and functional characterization
| Year | Brief description of discovery | Significance |
|---|---|---|
| 1958, Pardee, Jacob and Monod [72] | Using genetics models of E. coli, the PaJaMa studies proposed a general mechanism for transcriptional regulation via a repressor system to control protein production of beta-galactosidase. This led to the finding of lac operator, or ‘lac operon’ regulatory element. [73] | This landmark study laid the groundwork for the concept of regulatory elements and the heritability of gene regulation. |
| 1959, Harris H. [13] | Using pulse chase of RNA with labelled nucleotide, a large fraction of nascent RNA was shown to retain in the nucleus. In addition, half-lives of nuclear RNA is significantly shorter compared to cytoplasmic RNA. | Harris deduced that most nuclear RNAs are likely non protein-coding. In a recent Nature Correspondence, Harris connected his 1959 finding with the recent studies of ncRNA [Harris, 2013]. Given the high number of transcribing enhancer units, the comparable frequency of transcription initiation at enhancers, and short eRNA half-lives, this 1959 study is consistent with the observation of pervasive transcription at enhancer elements. |
| 1981, Banerji, J., et al. [1] | Banerji et al showed that a 72 repeat sequence motif from SV40 early gene is sufficient to increase expression of ectopic beta-globin gene by 200 fold. This DNA element is functional over long distance in an orientation independent fashion relative to the beta-globin gene. | Discovery of cis-acting enhancer elements. |
| 1990, Collis et al. [15] | Using nuclear run off assays, Collis et al identified transcription at the LCR of the beta globin region, both in the endogenous locus in MEL cells, as well as a mini-gene construct containing the LCR and beta-globing gene. | One of the first studies demonstrating transcription from enhancer regions. |
| 1992, Tuan et al. [16] | Using RNA protection assays in transfected recombinant plasmids, the authors identified that a non-coding RNA is transcribed from the HS2 enhancer. Only the active enhancer can direct synthesis of enhancer-derived RNA; the same group later showed that disruption of intact HS2-initiated long RNA leads to loss of HS2 enhancer activity and target gene silencing [Ling, 2004]. | This study suggested that enhancer ncRNA is only generated from active enhancer and is needed for proper enhancer function. |
| 1997, Ashe et al. [17] | Using nuclear run-on assays and in situ hybridization analysis, the authors found a novel intergenic transcription from the human β-globin locus in K562 but not in HeLa cells. Exogenous expression of the β-globin gene in HeLa cells was sufficient to induce the expression of intergenic ncRNAs from the otherwise silent endogenous chromatin. | Intergenic ncRNA transcript displayed cell line specificity. A ‘trans’ effect is implicated given exogenous plasmid-driven transcription of the globin genes can induce the intergenic ncRNA transcription at the endogenous locus. This mechanism, however, is not well understood. |
| 2000, Gribnau et al. [75] | RNA FISH and DNase I sensitivity assays led to the finding that transcription activity of the human β-globin locus correlates to its sensitivity to DNase I. | Intergenic ncRNA transcription could play a role in maintaining open and active chromatin. |
| 2006, Feng et al. [45] | The ultraconserved region between DLX5/6 was identified to transcribe into Evf-2 ncRNA. This Evf-2 then interacts with DLX2 in vivo to activate the transcription of DLX5 and DLX6 genes. | As ultraconserved regions are frequently enhancers (Pennacchio LA, 2006 Nature), this study implicated that enhancer-derived RNAs can be functionally regulatory. |
| ‘Genomic’ era | ||
| 2010, Kim et al. [10], 2010 De Santa et al. [26] 2011, Koch et al [Koch, 2011] |
Enhancer transcription and transcripts are genome-wide phenomena. Enhancer-templated non-coding RNAs (eRNAs) were usually non-polyadenylated and of lower abundance compared to coding genes. Expression level of eRNAs positively correlated to expression of nearby protein coding genes. | Pervasive genome wide enhancer transcription. The cognate promoter near the enhancer may be required for proper eRNA transcription. |
| 2011, Wang et al. [70] 2011, Hah et al. [22] 2011, Melgar et al. [76] 2013, Hah et al. [35] |
Using GRO-seq data, eRNA transcription was found to be induced by stimuli and widely distributed in the genome; their transcription correlated well with the activity of active enhancers. | eRNA transcription serves as a marker of active enhancers. Pharmacological inhibition of eRNA transcription does not inhibit enhancer-promoter looping with 3C. |
| 2010, Orom et al 2013, Lai et al. | From transcripts annotated in GENCODE, the authors identified a cohort of long non-coding RNAs (lncRNAs) that exhibit enhancer-like properties. Unlike eRNAs, this group of ncRNAs, termed enhancer-like lncRNAs or later as ncRNA-activating (ncRNA-a) are spliced, polyadenlyated transcripts expressed from promoter-like regions (i.e. high H3K4me3, low H3K4me1). They activate gene(s) in their vicinity. | The first description of lncRNAs having enhancer-like properties. Later the same group illustrated that ncRNA-a mediated gene regulation via modulation of Mediator complex and chromatin looping [Lai, 2013]. Recent studies of other lncRNA demonstrate novel mechanisms of gene activation, including physical interaction of lncRNA HOTTIP with methyltransferase to drive H3K4 trimethylation and gene expression in the HoxA cluster [KC Wang, 2011] |
| 2013, Melo et al. [38] 2013, Lam et al. [27] 2013, Li et al. [40] 2013, Mousavi et al. [30] |
Using siRNA, antisense oligonucleotides and reporter assays, with characterization using qRT-PCR, GRO-seq, and 3D-DSLthese studies found that eRNA transcripts have functional roles in regulating the transcription of neighboring coding genes. | The eRNA transcript per se plays a functional role in regulating gene transcription. A stimulus-induced eRNA transcript is needed for proper enhancer- promoter looping formation and gene activation. |
| 2013, Kaikkonen et al. [31] | Using genomic tools, the authors found that pharmacological inhibition of eRNA transcriptional elongation impaired mono- and dimethylation of histone H3K4 on signal-induced de novo enhancers. | RNA PolII elongation has a role independent of the eRNA transcript in modulating chromatin structure and deposition of enhancer histone marks. |
Table 2.
| Method | Description | |
|---|---|---|
| Nascent RNA analysis | Pulse-chase |
A method to examine properties of cellular processes over time. The cells are first shortly exposed to a labeled compound (pulse, e.g. radioactivity), and then to the same compound in an unlabeled form (chase). The amount of the labeled compound will change over time to reflect the effects from the interrogated cellular processes. In the context of this review, Harris measured half-lives of nuclear and cytoplasmic RNA [Harris 1959]. |
| Global Run On sequencing (GRO-seq) [Core 2008; Wang 2011] | A method to identify and quantify the genome-wide transcription of nascent RNA. At a given time in cells, nascent RNAs are synthesized from actively engaged RNA Polymerases. GRO-seq directly measures the frequency of transcription initiation and transcriptional rate and is largely independent of the effect of RNA stability. | |
| Precision global Run On sequencing (PRO-seq) [Kwok, 2013] | This method is an adapted version of GRO-seq, in which biotinylated nucleotides are used during in vitro transcription, enabling the mapping of the positions of the engaged RNA polymerases more precisely by sequencing. | |
| RNA 5′ cap site analysis 5′GRO-seq [Lam 2103] PRO-cap [Kwok 2013] | These methods introduced modifications based on GRO-seq and PRO-seq protocols, allowing for mapping the transcription initiation site and assessing the transcription frequency by measuring 5′ capped nascent RNAs at a given time in cells. | |
| RNA sequencing | Total RNA-seq |
This method measures the overall amounts of cellular RNA species by high throughput sequencing. Different categories of RNAs are represented proportionally to their expression levels in the final sequencing results. Since most cellular RNA consisted of ribosomal RNA, rRNA removal steps are often incorporate to increase signals for mRNAs and other non-coding RNAs (ncRNAs) of interest. RNA-seq can also provide other information about splicing, exon inclusion and exclusion etc. |
| Poly-A RNA-seq | A modified version of total RNA-seq based on using oligo-dT primer during cDNA generation and library preparation to enrich RNA species with poly A tails (e.g. most mRNAs and many long ncRNAs). | |
| Chromosomal accessibility | DNAseI-Hypersentitivity assay [Gross, 1988] | A method to assess open chromatin status by measuring the susceptibility to DNaseI cleavage locally (e.g. beta-globin locus control region) or globally when coupled with high throughput sequencing. |
| Genomic localization | Chromatin Immunoprecipitation coupled with high throughput sequencing (ChIP-seq) [Barski 2007; Robertson 2007] |
A method to study genomic localization of transcription factors, co-regulatory complexes, histones and variants with or without post-translational modifications by sequencing DNA fragments associated with these factors. Utility of this method depends on the availability of high-quality antibodies for the proteins of interest. Alternatively, a high affinity tag (i.e. a flag tag) could be fused to the protein of interest to facilitate affinity purification of the protein:chromatin complexes. |
| Chromosomal interaction | Chromosome conformation capture (3C) [Dekker 2002] |
A method to analyze chromosomal interactions to understand the spatial organization of the chromosomes. Because interacting genomic regions are in close three-dimensional proximity, the likelihood of DNA ligation between two interacting regions after chemical fixation and fragmentation (e.g. restriction enzyme digestion) will be significantly higher than that between any non-interacting regions of similar linear distance. Interactions could be quantified using PCR or qPCR. Control experiments are crucial to minimize misinterpretation due to random ligations. |
| Chromosome Interaction Analysis with Pair End Taq sequencing (ChIA-PET) [Fullwood 2009] |
A method to identify a subgroup of chromosomal interactions mediated by a specific transcription factor of interest. Analogous to 3C, ChIA-PET detects regions of chromosomal interactions that are bound by a factor of interest (e.g. RNA PolII) by ChIP followed by 3C. The selected interactions are processed for high throughput sequencing, which enables an unbiased, genome-wide approach for detecting long-range chromosomal interactions mediated by this factor. |
|
| 3C with DNA Selection and Ligation (3D-DSL) [Harismendy, 2011] |
A method to identify chromosomal interactions at pre-selected genomic regions of interest. On the basis of 3C, the digested and ligated chromatins are hybridized with a pool of oligonucleotides pre-designed to detect chromatin interactions at selected genomic regions. The limitation of resolution is dependent on the distribution of the restriction sites used for 3C and positions of the pre-designed oligonucleotides. |
|
| Flurorescence in-situ hybridization [Rudkin 1977] | A cytogenetic technique to detect and localize specific DNA sequences on chromosomes. A fluorescent probe (e.g. oligonucleotides or long stretches of DNAs) was pre-designed to specifically hybridize to DNA/chromatin regions of interest based on sequence complementarity. Using multiple probes conjugated to multi-color fluorophores, interactions of two or more genomic regions could be approximate by tabulating the frequency of these probes being in close physical proximity by fluorescence microscopy. The resolution of this method is limited by the wavelength of the lower light spectrum (~200-250nm), which could be in the range of ~105 to 106 base pairs depending on the chromatin state. | |
| RNA-chromatin interaction | Chromatin Isolation by RNA Purification coupled with sequencing (ChIRP-seq) [Chu 2011] |
A method to identify chromatin sites bound by an RNA of interest. The target RNA of interest is hybridized and retrieved by biotin-tagged oligonucleotides by sequence complementarity. The RNA-interacting chromatin regions can be recovered by streptavidin affinity purification and determined by high-throughput sequencing. ChIRP-seq usually uses a tiling array of oligonucleotides to enrich one RNA target and requires glutaradehyde to irreversibly crosslink RNA targets and chromatin. |
| Capture Hybridization Analysis of RNA Targets coupled with sequencing (CHART-seq) [Simon 2011] |
Another method to capture the chromatin binding sites of an RNA of interest. Similar to ChIRP, CHART used similar hybridization-based strategy to enrich RNA target and its associated chromatin DNA and proteins, but using only one complementary DNA oligonucleotide, which is pre-designed based on analysis of the target RNA structure. Also, CHART allows using mild and reversible crosslinking method. Since RNA can interact with chromatin by direct binding to DNA or indirectly through a protein intermediate, the sensitivity of CHART-seq and ChIRP-seq to these different modalities needs to be assessed. Whether the differences in these modalities correlate with biological function is not well known. |
A demonstration of a broad pattern of transcription at active enhancers was uncovered by deep sequencing approaches, with total-RNA sequencing (Table 2) revealing enhancer transcription in neurons and T-cells [10, 21]. Similarly, genome-wide detection of nascent RNA transcripts using Global Run-On sequencing (GRO-seq, Table 2) demonstrated robust expression and regulation of eRNA in macrophages, breast or prostate cancer cells [22–25]. RNA Polymerase II (RNA PolII) complexes were noted to be enriched at enhancer elements [21] and to respond dynamically to signal transduction events [10, 26]. Thus, enhancer transcription is a widespread phenomenon observed across multiple cell types in different species.
Properties of eRNAs compared to mRNAs and other ncRNAs
RNA-Seq, chromatin immunoprecipitation (ChIP)-Seq, and chromatin-conformation-capture studies (Table 2) have collectively defined the following characteristics of eRNA transcripts (Figure 2): (i) eRNAs are transcribed from putative enhancer regions characterized by high levels of H3K4me1 and H3K4me2 relative to the H3K4me3 [Heintzman 2007; He 2011]. (ii) These genomic regions are bound by LDTFs and associated transcriptional co-regulators including mediator subunits and histone acetyltransferase p300 and CREB Binding Protein (CBP). (iii) Expression of eRNAs is positively correlated with an enrichment of activated enhancer histone marks, particularly H3K27ac, but the lack of the repressive H3K27me3 mark [27]. (iv) eRNA-expressing enhancers are also enriched with the transcriptional initiation complex (e.g. TBP, TFIIs) and serine 5 phosphorylated RNA PolII, characteristics of coding gene promoter regions. However, in contrast to gene bodies of protein-coding genes, enhancers are less enriched for serine 2 phosphorylated RNA PolII [21]. (v) eRNAs exhibit a 5′ cap [14, 27] but are generally not spliced or polyadenylated. Polyadenylated eRNAs are generally unidirectionally transcribed from enhancers (1D-eRNA) [21]; however, enhancers with bidirectional transcription and non-polyadenylated transcripts (2D-eRNA) are more common [21, 28]. (vi) eRNAs generally exhibit shorter half-lives compared to mRNAs and lncRNAs, but the frequency of transcription initiation of eRNAs appears comparable to that of protein coding genes [27]. (vii) eRNAs are dynamically regulated upon signal transduction events orchestrated by signal-dependent transcription factors such as Nuclear Factor kappa-light-chain-enhancer of Activated B cells (NFκB), p53, or nuclear receptors [10, 22–27, 29, 30]. (viii) Of particular interest, signal-dependent changes in eRNA expression are highly correlated with corresponding signal dependent changes in promoters of nearby genes [10, 25, 27, 31, 32]. (ix) In addition, enhancer transcripts are preferentially enriched at enhancers engaged in chromatin-looping with promoter of protein-coding gene and other enhancers, which is a feature correlated with enhancer activity. [33, 34]. Collectively, eRNA transcription is highly correlated to other parameters of active enhancer elements [23, 24, 35].
Figure 2. Molecular characteristics of protein-coding RNAs, long non-coding RNAs, and enhancer RNAs.

Schematic diagrams of chromatin immunoprecipitation coupled with high-throughput sequencing (ChIP-seq) or RNA-seq analysis centered on the transcription start sites (TSS) to depict the differences and similarities between these four categories of transcription units. Briefly, protein-coding genes have a higher level of H3K4me3, lower level of H3K4me1, relatively lower enrichment of lineage determining transcription factors (LDTF), and higher CpG islands at TSSs; transcription orientation is predominantly uni-directional, transcripts are polyadenylated, and gene bodies demarcated with elongating histone mark H3K36me3. Transcripts of long non-coding RNAs (lncRNAs) resemble protein-coding genes—H3K4me3hi/H3K4me1low promoters with polyadenylated and predominately unidirectional transcripts [42]. Compared to protein coding genes, lncRNAs possess higher H3K4me1, higher LDTF enrichment, and lower CpG density [Koch 2011]. For enhancer derived non-coding RNAs (eRNAs), their TSSs are demarcated with H3K4me1high/H3K4me3low modifications and low CpG density [21]. eRNAs, in a majority, are non-polyadenylated bidirectional transcripts (2D-eRNAs), but also comprise of a less common group of unidirectional transcripts (1D-eRNAs) [26,28]. Both 1D- and 2D-eRNAs often bear shorter half-lives (low signal from total RNA-sequencing), but comparable initiation rates of transcription compared to protein coding genes and lncRNAs, as measured by total RNA Polymerase II (RNA-PolII) enrichment or nascent RNA transcripts (GRO-seq). The elongating form of RNA-PolII with serine 2 phosphorylation (PolII-Ser2p) mainly enriches at protein-coding genes. Chromosomal interaction studies indicated higher frequency of looping events, either enhancer:promoter, enhancer:enhancer, promoter:promoter, at actively transcribed enhancers and protein coding genes [Lin, 2012; Zhang, 2013].
Functional roles of enhancer transcription and transcripts
Whether enhancer transcript is merely a correlation or a functional component of enhancer activity generated an active area of research. Three possibilities have been considered with respect to the physiological roles of enhancer transcription. The first possibility considers enhancer transcription as “noise” from the spurious engagement of RNA PolII complexes to the open chromatin environment of enhancers. The second possibility hypothesizes that it is the process of transcription, not the features of the eRNA transcript itself, that is necessary for the activating functions of enhancers. The third possibility is that the RNA transcripts per se functionally contribute to enhancer activity [28]. These possibilities are not mutually exclusive.
The early investigations of enhancer transcripts from the beta-globin LCR implicated functional significance of enhancer transcription. HS2, a hypersensitivity site within the beta-globin LCR, was sufficient for erythroid-specific enhancer activity when cloned into minigene constructs [16]. The transcription start site for an enhancer transcript was found within HS2 of both the endogenous genomic locus and plasmid constructs [15, 16]. Interestingly, termination of HS2-mediated transcription by inserting a lac operator/R repressor complex downstream of the enhancer led to decreased promoter activity in a reporter construct [36]. This suggested that the transcription from HS2 is important for its neighboring promoter activity. A similar result was observed in the hGH locus when a transcription termination sequence TerF was inserted between the LCR and the promoter of hGH. Transgenic mice with this TerF insertion showed decreased expression of hGH [20]. In another line of investigation, analysis of RNA PolII localization was performed in the beta-globin locus. Expectedly, the authors found RNA PolII at the gene promoter. Surprisingly, RNA PolII was also found at the HS2 enhancer, consistent with the production of enhancer-derived RNA transcript therein. To study the role of RNA transcription in RNA PolII recruitment, cells were treated with RNA PolII elongation inhibitor 5,6-dichlorobenzimidazole (DRB). This resulted in decreased recruitment of RNA PolII to the beta-globin promoter, but not at the HS2 enhancer [Johnson, 2013]. This implies that enhancer recruitment of RNA PolII preceded RNA PolII loading at target gene promoter. This also raised the possibilities that enhancer transcription is functionally significant for regulating RNA PolII “loading” to target gene promoter. This experiment, however, could not differentiate whether the RNA PolII loading was mediated by the act of enhancer transcription (i.e. RNA PolII elongation) or by the eRNA transcript itself, since both processes were inhibited by DRB.
Recently, several reports have taken new approaches to test functions of enhancer transcripts. Targeted degradation of eRNA using either RNA interference (siRNA) or DNA-RNA hybrid induced degradation via RNase-H (i.e. antisense oligonucleotide or locked nucleic acids) proved sufficient to reduce expression of nearby protein-coding genes [25, 27, 29, 30]. To further discriminate the effects of RNA PolII transcription at the enhancer versus the eRNA transcript itself, Li et al. and Melo et al. used “tethering” strategies [23, 38], where eRNA transcripts were fused with RNA tags (i.e. MS2 or BoxB repeats) to generate chimera RNAs that can be bound by a recombinant bridging adaptor protein on one end (i.e. Gal4 fused with MS2-coating protein or λN) and the reporter construct on the other (i.e. UAS sites). This strategy enables an eRNA transcript to be localized to a specific target region for testing of transcriptional activity. Tethering of eRNA transcripts to the promoter [29] or to the enhancer [25] was sufficient to increase transcriptional activity of the reporter gene. Alternative experimental design also supported the function of eRNA to enhancer activity. By cloning different sizes of genomic fragments from an endogenous enhancer locus, Lam et al. showed that, while the ‘core’ enhancer fragments containing LDTF binding sites were sufficient for enhancer activity, enhancer construct containing eRNA-coding sequence has higher transcriptional activity. Importantly, the ‘added’ effect from the eRNA was abolished when the orientation of its coding sequence was reversed relative to the ‘core’ enhancer [27]. Because this inversion completely changes the sequence of the eRNA product but retains any putative transcription factor binding sites, these results implied that the sequence of the eRNA is important for its function. Collectively, these reports suggest that, at least for some enhancers, sequence-specific eRNA transcripts can contribute to enhancer-mediated transcriptional activation of neighboring coding genes.
Mechanisms of eRNA actions
A question immediately emerges as to how eRNAs might contribute to enhancer function. Chromatin interaction studies demonstrated that enhancers engaged in looping with promoters of protein coding genes possess higher expression of eRNAs [33, 34]. These studies suggested a potential role of eRNAs in the process of proper formation of chromosomal looping between enhancers and transcription start sites (TSSs). Indeed, in nuclear receptor regulated gene activation events, ligand treatment induces formation of chromatin looping between enhancer and the cognate TSS, which could be measured by ChIA-PET [39] and three-dimensional DNA selection and ligation (3D-DSL) (Table 2) [40]. More importantly, knockdown of eRNA from the Estrogen Receptor alpha (ERα)-bound enhancers at NR1P1 or GREB1 loci reduced enhancer-promoter interaction and concomitantly decreased coding gene activation [40]. The potential role of estrogen-induced eRNA in the modulation of chromosomal looping was suggested by the observation that eRNAs could interact with the SMC3 and RAD21 subunits of the cohesin complex, which has been shown to control enhancer-promoter looping in stem cells [41]. Targeted degradation of eRNAs attenuated estrogen-induced cohesin increment to several ERα-bound enhancers, and knockdown of cohesin almost completely abolished both the observed induced looping and gene activation events [40] (Figure 3B). These data suggested that eRNA may play a role in the initiation or stabilization of enhancer-promoter looping, but its quantitative effect on cohesin recruitment to the enhancers remains unclear. However, Hah et al. found that reducing the amount of eRNA and coding gene transcripts through chemical inhibition of RNA PolII elongation (i.e. flavopiridol) had no significant effect on estrodiol-induced enhancer-promoter looping at the P2RY2 or the GREB1 loci, as measured by chromosomal conformation capture (3C) (Table 2) [35]. This difference may be explained by the different experimental techniques used (eRNA knockdown vs. pharmacological inhibition of RNA PolII elongation), or it may reflect different mechanisms at specific gene loci or enhancers. In another recent study, Mousavi et al. knocked down eRNA transcripts produced in each MyoD-bound enhancers across the MyoD locus, showing that only the eRNA from the core enhancer (CE) was critical for MyoD expression [30]. Furthermore, knockdown of CEeRNA decreased RNA PolII recruitment at the promoter and gene body of MyoD, but not at the core enhancer itself. The consequences of eRNA knockdown in MyoD locus, as well as RNA synthesis inhibition at HS2 of the beta-globin LCR mentioned earlier [37], suggest that eRNA transcripts facilitate RNA PolII recruitment to the promoter of the target gene (Figure 3B). It seems that targeted down-regulation of eRNA by knockdown or inhibition of RNA PolII elongation did not affect the pre-established “enhancer assembly”, in terms of binding of transcription factors, recruitment of RNA PolII or enrichment in histone marks (e.g. H3K4me1), suggesting that production of eRNA probably represents one of the final steps during activation of pre-established enhancers (Figure 3A).
However, inhibition of enhancer transcription (e.g. by pharmacological inhibitor of RNA PolII elongation) impairs signal-induced de novo activation of enhancers, indicated by decreased acetylation of H3K9 [26] or attenuated mono- and di-methylation of H3K4 [24]. In the latter case, deposition of H3K4 mono- and di-methylation to enhancers is dependent on histone methyltransferases (i.e. Mixed Lineage Leukemia-1 (MLL1), MLL2/4, and MLL3) coupled to RNA PolII elongation, but independent of the eRNA transcript[24], suggesting a key functional role of the process of enhancer transcription, at least for some enhancer cohorts. Collectively, these findings suggest that both enhancer transcription and the resultant enhancer RNAs can contribute to enhancer function.
Mechanism of other ncRNAs that positively regulate gene expression
In addition to eRNAs, a plethora of long non-coding RNAs (lncRNAs) are already reported to play activating roles in gene transcription regulation [42]. These lncRNAs are generally transcribed from regions exhibiting high H3K4me3 at the promoter and H3K36me3 at gene body (a histone mark of elongation), resembling the chromatin properties of protein coding genes [42]. They are also often spliced, polyadenylated, and unidirectionally transcribed. By these criteria, most eRNAs and activating lncRNAs are very distinct (Box 1). However, as noted above, some transcripts derived from enhancer-like regions of the genome are polyadenylated and transcribed unidirectionally [21, 35], and both eRNAs and lncRNAs tend to be cell type specific [14]. Given this overlap in genomic characteristics of these two species of non-coding nuclear RNAs, there may also be overlap in mechanisms by which they influence gene expression.
BOX 1. Conceptual distinctions between eRNAs, 1D-eRNAs, 2D-eRNAs, lncRNAs and ncRNA-a.
eRNAs are non-coding RNA transcripts produced from genomic regions that are marked by high H3k4me1 and low H3k4me3 histone modifications that are presumably enhancer DNA elements. eRNAs can be either polyadenylated or non-polyadenylated and appear as unidirectional (1D-eRNA) or bidirectional transcripts (2D-eRNA) in RNA-seq or GRO-seq profiles [10, 21, 22, 70]. lncRNAs are non-coding RNA transcripts longer than 200nt [71]. ncRNA-a (non-coding RNAs activating) were transcripts annotated in the GENCODE database with an average size of ~800nt, with subsequent studies showing enhancer-like property by activating neighboring genes [42]. Though many ncRNA-a transcripts might be more suitably classified as lncRNAs as they are often spliced and frequently bear H3K4me3 and H3K36me3 histone marks [42], some ncRNA-a transcripts are very similar to eRNAs, especially unidirectional 1D-eRNAs. Functional distinctions between these groups are not fully clarified. Refer to Figure 2 for more information.
Although each lncRNA activator studied to date appears to function through distinct mechanisms, three general themes have emerged: 1) lncRNAs recruit activating proteins and protein complexes, 2) they mediate chromatin interactions, and 3) they have roles in eviction of repressive machineries (Figure 4).
Figure 4. Three mechanistic models underlying functions of activating ncRNAs.

Three general categories of mechanisms for ncRNAs in transcription activation: (A) by directly recruiting transcriptional activator or activator complex; (B) via mediating chromatin looping; (C) and through evicting transcriptional repressors. A. Several ncRNAs (e.g. HOTTIP [KC Wang 2011], Mira [Bertani 2011], SRA [Lanz 1999], and Evf2 [Feng 2006].) physically interacts to recruit transcriptional activators (e.g. WDR5 subunit of MLL complex) to promote target gene activation. This type of activation theme is achieved, till now, only by polyadenylated ncRNAs, and can be thought either in cis (e.g. HOTTIP) or in trans (e.g. SRA) actions. B. Another group of ncRNAs (e.g. ncRNA-a) can modulate chromatin looping between enhancer and promoter through interacting with and recruiting/stabilizing the looping factors (e.g. Mediator and cohesin complexes). This is similar to the mechanism of some estrogen-induced eRNAs (Figure 3B) for gene activation. C. Under some circumstances, some ncRNAs (Braveheart [Klettenhoff 2013], Jpx [Tian 2010 and Sun 2013]), can abolish the effects of transcriptional repressors by titrating them away from the promoters of target genes.
The first category constitutes the most common mechanism of activating ncRNAs (Figure 4A). SRA was the first ncRNA discovered to act as a transcriptional coactivator; it functions in the transcriptional activator complex Steroid Receptor Coactivator-1 (SRC1) to enhance transcription mediated by steroid hormone receptors [43]. Further, Evf-2, a ncRNA generated from an ultraconserved intergenic region, was discovered to recruit transcription factor Distal-less Homeobox 2 (DLX2) and Methyl CpG Binding Protein 2 (MECP2) to regulate expression of the neighboring Dlx5/6 genes [44, 45]. Another recent example is HOTTIP, which drives the transcriptional activation of the Homeobox A (HOXA) gene cluster by directly interacting with the WD Repeat-containing Protein 5 (WDR5) subunit of the MLL complex, leading to deposition of the H3K4me3, a histone mark associated with promoter of protein-coding gene [32]. Consistent with this, MLL complex can also be recruited by Mistral ncRNA to activate Homeobox A Cluster 6 (HOXa6) and HOXa7 during stem cell differentiation [46] and by NeST ncRNA to activate the interferon-gamma locus [47].
The second category of activating ncRNAs is exemplified by ncRNA-a (Figure 4B) [42]. This group of ncRNAs regulates gene activation through a distance by recruiting the Mediator complex to, and controlling the phosphorylation of, histone H3S10 at its target gene promoter [48]. Intriguingly, ncRNAs might, in specific circumstances, indirectly recruit complexes that exert functions in enhancer:promoter looping [49].
Finally, the third type of activating mechanism can be summarized as an “eviction” model (Figure 4C), which is exemplified by X chromosome inactivation (XCI). The Xist gene initiates XCI, and is only expressed when more than one X chromosome is present. In pre-XCI stage, Xist is silenced by the binding of CCCTC-binding Factor (CTCF) at its promoter. At the onset of XCI, a ncRNA transcript Jpx is upregulated from both X chromosomes, which directly binds to CTCF and titrates it away from the Xist promoter. This allows expression of Xist to initiate XCI on one of the X-chromosomes [50, 51]. Similarly, activation of lineage-specific genes during cardiovascular development requires the removal of Polycomb Repressive Complexes (PRC2) and H3K27me3 marks from those genes’ promoters. This process depends on the ncRNA Braveheart, which binds to the Suppressor of Zeste 12 (SUZ12) subunit of PRC2 and titrates the whole PRC2 complex away [52].
Together these data suggest a complex network of interaction between ncRNA and regulatory protein machineries to precisely control gene transcription programs. Though the list of proteins that interact with ncRNAs is still limited (e.g. PRC1/2 and MLL), significant growth of this list is very likely in the near future as many transcriptional regulators contain RNA binding domains [53].
Concluding remarks
A detailed understanding of the molecular mechanisms by which enhancers become activated as transcription units remains an important question. For example, what is the mechanism underlying biogenesis and regulation of eRNAs? Is the transcription machinery on enhancers identical to that on promoters? The precise initiation site of enhancer transcription, and the mechanisms that determine elongation and termination, can be approached with contemporary technologies. For example, 5′ RNA cap site analysis by GRO-seq and PRO-seq [27, 54, 55]could help delineate distinctive features of the TSSs of protein coding genes, ncRNAs, 1D-eRNAs, and 2D-eRNAs.
A corollary issue is whether eRNAs play general roles in enhancer function. For those enhancers in which it exerts a functional role, we also need a deeper understanding of their mechanism of action. In view of the multiple mechanisms of action described thus far for lncRNAs, similar complexity may be expected for the action mechanisms of eRNAs. Given the initial evidence for roles of eRNAs in enhancer/promoter interactions, it will be of particular interest to systematically examine the consequences of loss of eRNA function on short- and long-range interactions of enhancers with coding gene promoters and other enhancers [56].
Although evidences suggested that eRNA transcripts per se and eRNA transcription can both play certain functional roles, it is currently not clear whether these functions are primarily performed in trans or in cis. Current findings regarding eRNA – their low copy numbers per cell [10, 22], the predominant absence of eRNA localization at other genomic regions, and the minimal effect of eRNA knockdown on expression of genes on different chromosomes [40] – are most consistent with function in cis (Figure 5A). Similar conclusions were made for activating lncRNA [Orom 2010; KC Wang, 2011]. While depletions of lncRNAs were sufficient to decrease expression of their target genes, ectopic expressions of lncRNAs had minimal effect on target gene expression at the endogenous loci [Orom 2010; KC Wang, 2011]. This points to activity in cis, where lncRNAs likely exert function within a domain consisted of interactions between neighboring and distal genomic regions (i.e. interchromasomal interaction) refined within a close three-dimensional space.
Figure 5. In trans versus in cis action of ncRNAs in gene transcriptional regulation.

(A,B) Classic in cis (A) or in trans (B) actions of regulatory ncRNAs in transcriptional activation. (C) A scenario that one ncRNA can be assumed to regulate a target gene of a long distance (in trans), which is actually mediated indirectly through its primary effect on a cis-target gene. (D) The postulated dual in cis and in trans role of ncRNA within domain of interacting chromatins. ncRNAs (e.g. ncRNA-a and eRNAs) can regulate chromatin looping formation raised a possibility that ncRNAs can stay at where they are produced (in cis) but exert long-distance regulations on a target gene (i.e. interchromosomal interactions), mimicking a trans effect.
Whether lncRNA and eRNA mediate expression of other genes in trans within the ‘interacting domain’ has not been systemically addressed (Figure 5D). Several observations suggested this possibility. ncRNA-a depletion resulted in greater number of changes in gene expression than would be accounted by its target protein-coding gene alone [Orom 2010]. This suggests trans activity, although the result could be confounded by secondary/indirect effects (i.e. changes in other unforeseen cis-regulated direct target genes) (Figure 5C). The observations that eRNA mediates chromatin looping [Li 2013] and RNA PolII loading at target gene promoters [Mousavi 2013] raised an intriguing possibility of trans function within the ‘interacting domain’ that has not been ‘ruled-out’ by current studies.
Elucidating the cis vs. trans mechanism of eRNA and lncRNA helps tease out allelic specificity in gene regulation, which is an important component of understanding genetic disease and therapeutic applicability. Further understanding of the three-dimensional organization of the genome and genomic localization of eRNA or lncRNA by identifying RNA:chromatin association will be insightful (Table 2). Given the rapid technological advances in genome editing [Mali 2013; Qi 2013], it is now feasible to investigate in cis and in trans mechanism by examining allelic-specificity with carefully designed genetic experiments or genome editing experiment coupled with sequencing modalities.
eRNAs that contribute to enhancer activity presumably due so by interacting with other effector molecules. One approach to investigating the function of a particular eRNA will be to identify the factors it interacts with biochemically. At a more general level, efforts have been made to delineate activating ncRNAs’ dependence on substructure to interact with cofactors. For instance, the RNA motifs of the RNA co-activator SRA are critical for its coactivation function [60]. Case studies of other RNA species indicate that simple secondary structures (e.g. tandem stem loops) enable protein:RNA complex formation [61, 62]. Additionally, repeat elements or GC-rich sequences in ncRNAs have been implicated in gene regulation in trans thru RNA:DNA:DNA triplex formation with similar elements at promoters of target genes [62–64]. As progress is made in understanding protein-RNA binding specificity and RNA secondary structures [53, 65], a major challenge will be to elucidate hidden sequence or structural codes underlying the behaviours of different types of ncRNAs in interacting with their protein partners.
Finally, given increasing evidence that a majority of disease-associated loci represent sequence variations in enhancers [4, 5, 66–68], a major question is whether it will be feasible to alter enhancer function for therapeutic purposes. Advances in antisense oligonucleotide-based methods that target nuclear RNAs in vivo could potentially be applied to knockdown cell-specific eRNAs as a means to modulate coding gene expression in a cell-specific manner [27, 69]. In addition, enhancers are likely to be major targets of small molecules that inhibit specific classes of epigenetic regulators, including histone deacetylases, methyltransferases, demethylases and histone tail mimetics. The further exploration of enhancer transcription and functional roles of eRNAs will therefore not only be of central importance for the further understanding of regulation of gene expression in homeostasis and development, but also for advancing novel therapeutic interventions for human diseases.
Highlights.
Active enhancers generate eRNAs
At least some eRNAs contribute to enhancer function
eRNAs may play roles in enhancer/promoter interactions
References
- 1.Banerji J, et al. Expression of a beta-globin gene is enhanced by remote SV40 DNA sequences. Cell. 1981;27:299–308. doi: 10.1016/0092-8674(81)90413-x. [DOI] [PubMed] [Google Scholar]
- 2.Buecker C, Wysocka J. Enhancers as information integration hubs in development: lessons from genomics. Trends in Genetics. 2012;28:276–284. doi: 10.1016/j.tig.2012.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xie W, Ren B. Developmental biology. Enhancing pluripotency and lineage specification. Science. 2013;341:245–247. doi: 10.1126/science.1236254. [DOI] [PubMed] [Google Scholar]
- 4.Musunuru K, et al. From noncoding variant to phenotype via SORT1 at the 1p13 cholesterol locus. Nature. 2010;466:714–719. doi: 10.1038/nature09266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Harismendy O, et al. 9p21 DNA variants associated with coronary artery disease impair interferon-γ signalling response. Nature. 2011;470:264–268. doi: 10.1038/nature09753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Heinz S, et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bulger M, Groudine M. Functional and mechanistic diversity of distal transcription enhancers. Cell. 2011;144:327–339. doi: 10.1016/j.cell.2011.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Calo E, Wysocka J. Modification of enhancer chromatin: what, how, and why? Molecular cell. 2013;49:825–837. doi: 10.1016/j.molcel.2013.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Smallwood A, Ren B. Genome organization and long-range regulation of gene expression by enhancers. Current Opinion in Cell Biology. 2013;25:387–394. doi: 10.1016/j.ceb.2013.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim TK, et al. Widespread transcription at neuronal activity-regulated enhancers. Nature. 2010;465:182. doi: 10.1038/nature09033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wan Y, et al. Understanding the transcriptome through RNA structure. Nature reviews Genetics. 2011;12:641–655. doi: 10.1038/nrg3049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dethoff EA, et al. Functional complexity and regulation through RNA dynamics. Nature. 2012;482:322–330. doi: 10.1038/nature10885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harris H. Turnover of nuclear and cytoplasmic ribonucleic acid in two types of animal cell, with some further observations on the nucleolus. Biochemical Journal. 1959;73:362. doi: 10.1042/bj0730362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Djebali S, et al. Landscape of transcription in human cells. Nature. 2012;489:101–108. doi: 10.1038/nature11233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Collis P, et al. Definition of the minimal requirements within the human beta-globin gene and the dominant control region for high level expression. EMBO J. 1990;9:233–240. doi: 10.1002/j.1460-2075.1990.tb08100.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tuan D, et al. Transcription of the hypersensitive site HS2 enhancer in erythroid cells. Proc Natl Acad Sci USA. 1992;89:11219–11223. doi: 10.1073/pnas.89.23.11219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ashe HL, et al. Intergenic transcription and transinduction of the human beta-globin locus. Genes Dev. 1997;11:2494–2509. doi: 10.1101/gad.11.19.2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Allan M, et al. Multiple origins of transcription in the 4.5 Kb upstream of the epsilon-globin gene. Cell. 1983;35:187–197. doi: 10.1016/0092-8674(83)90221-0. [DOI] [PubMed] [Google Scholar]
- 19.Masternak K, et al. Chromatin remodeling and extragenic transcription at the MHC class II locus control region. Nature Immunology. 2003;4:132–137. doi: 10.1038/ni883. [DOI] [PubMed] [Google Scholar]
- 20.Ho Y, et al. Locus control region transcription plays an active role in long-range gene activation. Mol Cell. 2006;23:365–375. doi: 10.1016/j.molcel.2006.05.041. [DOI] [PubMed] [Google Scholar]
- 21.Koch F, et al. Transcription initiation platforms and GTF recruitment at tissue-specific enhancers and promoters. Nat Struct Mol Biol. 2011;18:956–963. doi: 10.1038/nsmb.2085. [DOI] [PubMed] [Google Scholar]
- 22.Hah N, et al. A rapid, extensive, and transient transcriptional response to estrogen signaling in breast cancer cells. Cell. 2011;145:622–634. doi: 10.1016/j.cell.2011.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang D, et al. Reprogramming transcription by distinct classes of enhancers functionally defined by eRNA. Nature. 2011;474:390–394. doi: 10.1038/nature10006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kaikkonen MU, et al. Remodeling of the enhancer landscape during macrophage activation is coupled to enhancer transcription. Mol Cell. 2013;51:310–325. doi: 10.1016/j.molcel.2013.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li W, et al. Functional roles of enhancer RNAs for oestrogen-dependent transcriptional activation. Nature. 2013;498:516–520. doi: 10.1038/nature12210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.De Santa F, et al. A large fraction of extragenic RNA pol II transcription sites overlap enhancers. PLoS Biol. 2010;8:e1000384. doi: 10.1371/journal.pbio.1000384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lam MTY, et al. Rev-Erbs repress macrophage gene expression by inhibiting enhancer-directed transcription. Nature. 2013;498:511–515. doi: 10.1038/nature12209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Natoli G, Andrau JC. Noncoding transcription at enhancers: general principles and functional models. Annu Rev Genet. 2012;46:1–19. doi: 10.1146/annurev-genet-110711-155459. [DOI] [PubMed] [Google Scholar]
- 29.Melo CA, et al. eRNAs Are Required for p53-Dependent Enhancer Activity and Gene Transcription. Molecular Cell. 2013;49:524–535. doi: 10.1016/j.molcel.2012.11.021. [DOI] [PubMed] [Google Scholar]
- 30.Mousavi K, et al. eRNAs promote transcription by establishing chromatin accessibility at defined genomic loci. Molecular Cell. 2013;51:606–617. doi: 10.1016/j.molcel.2013.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kaikkonen MU, et al. Remodeling of the Enhancer Landscape during Macrophage Activation Is Coupled to Enhancer Transcription. Mol Cell. 2013;51:310–325. doi: 10.1016/j.molcel.2013.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang KC, et al. A long noncoding RNA maintains active chromatin to coordinate homeotic gene expression. Nature. 2011 doi: 10.1038/nature09819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lin YC, et al. Global changes in the nuclear positioning of genes and intra- and interdomain genomic interactions that orchestrate B cell fate. Nature Immunology. 2012;13:1196–1204. doi: 10.1038/ni.2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sanyal A, et al. The long-range interaction landscape of gene promoters. Nature. 2012;489:109–113. doi: 10.1038/nature11279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hah N, et al. Enhancer transcripts mark active estrogen receptor binding sites. Genome Res. 2013;23:1210–1223. doi: 10.1101/gr.152306.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ling J, et al. HS2 enhancer function is blocked by a transcriptional terminator inserted between the enhancer and the promoter. J Biol Chem. 2004;279:51704–51713. doi: 10.1074/jbc.M404039200. [DOI] [PubMed] [Google Scholar]
- 37.Johnson KD, et al. Highly restricted localization of RNA polymerase II within a locus control region of a tissue-specific chromatin domain. Molecular and Cellular Biology. 2003;23:6484–6493. doi: 10.1128/MCB.23.18.6484-6493.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Melo Carlos A, et al. eRNAs Are Required for p53-Dependent Enhancer Activity and Gene Transcription. Molecular Cell. 2012:1–12. doi: 10.1016/j.molcel.2012.11.021. [DOI] [PubMed] [Google Scholar]
- 39.Fullwood MJ, et al. An oestrogen-receptor-α-bound human chromatin interactome. Nature. 2009;461:58–64. doi: 10.1038/nature08497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li W, et al. Functional roles of enhancer RNAs for oestrogen-dependent transcriptional activation. Nature. 2013;498:516–520. doi: 10.1038/nature12210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kagey MH, et al. Mediator and cohesin connect gene expression and chromatin architecture. Nature. 2010;467:430. doi: 10.1038/nature09380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Orom UA, et al. Long Noncoding RNAs with Enhancer-like Function in Human Cells. Cell. 2010;143:46–58. doi: 10.1016/j.cell.2010.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lanz RB, et al. A steroid receptor coactivator, SRA, functions as an RNA and is present in an SRC-1 complex. Cell. 1999;97:17–27. doi: 10.1016/s0092-8674(00)80711-4. [DOI] [PubMed] [Google Scholar]
- 44.Bond AM, et al. Balanced gene regulation by an embryonic brain ncRNA is critical for adult hippocampal GABA circuitry. Nature Neuroscience. 2009;12:1020–1027. doi: 10.1038/nn.2371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Feng J, et al. The Evf-2 noncoding RNA is transcribed from the Dlx-5/6 ultraconserved region and functions as a Dlx-2 transcriptional coactivator. Genes Dev. 2006;20:1470–1484. doi: 10.1101/gad.1416106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bertani S, et al. The noncoding RNA Mistral activates Hoxa6 and Hoxa7 expression and stem cell differentiation by recruiting MLL1 to chromatin. Molecular cell. 2011;43:1040–1046. doi: 10.1016/j.molcel.2011.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 47.Gomez JA, et al. The NeST long ncRNA controls microbial susceptibility and epigenetic activation of the interferon-γ locus. Cell. 2013;152:743–754. doi: 10.1016/j.cell.2013.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lai F, et al. Activating RNAs associate with Mediator to enhance chromatin architecture and transcription. Nature. 2013:1–7. doi: 10.1038/nature11884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang L, et al. lncRNA-dependent mechanisms of androgen-receptor-regulated gene activation programs. Nature. 2013 doi: 10.1038/nature12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tian D, et al. The long noncoding RNA, Jpx, is a molecular switch for X chromosome inactivation. Cell. 2010;143:390–403. doi: 10.1016/j.cell.2010.09.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sun S, et al. Jpx RNA Activates Xist by Evicting CTCF. Cell. 2013;153:1537–1551. doi: 10.1016/j.cell.2013.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Klattenhoff CA, et al. Braveheart, a long noncoding RNA required for cardiovascular lineage commitment. Cell. 2013;152:570–583. doi: 10.1016/j.cell.2013.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Guenther UP, et al. Hidden specificity in an apparently nonspecific RNA-binding protein. Nature. 2013;502:385. doi: 10.1038/nature12543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nechaev S, et al. Global analysis of short RNAs reveals widespread promoter-proximal stalling and arrest of Pol II in Drosophila. Science (New York, NY) 2010;327:335–338. doi: 10.1126/science.1181421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kwak H, et al. Precise maps of RNA polymerase reveal how promoters direct initiation and pausing. Science (New York, NY) 2013;339:950–953. doi: 10.1126/science.1229386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li G, et al. Extensive promoter-centered chromatin interactions provide a topological basis for transcription regulation. Cell. 2012;148:84–98. doi: 10.1016/j.cell.2011.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mali P, et al. RNA-Guided Human Genome Engineering via Cas9. Science. 2013 doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Qi LS, et al. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell. 2013;152:1173–1183. doi: 10.1016/j.cell.2013.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Heinz S, et al. Effect of natural genetic variation on enhancer selection and function. Nature. 2013;503:487–492. doi: 10.1038/nature12615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lanz RB, et al. Distinct RNA motifs are important for coactivation of steroid hormone receptors by steroid receptor RNA activator (SRA) Proc Natl Acad Sci USA. 2002;99:16081–16086. doi: 10.1073/pnas.192571399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ilik IA, et al. Tandem stem-loops in roX RNAs act together to mediate X chromosome dosage compensation in Drosophila. Molecular cell. 2013;51:156–173. doi: 10.1016/j.molcel.2013.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Holdt LM, et al. Alu elements in ANRIL non-coding RNA at chromosome 9p21 modulate atherogenic cell functions through trans-regulation of gene networks. PLoS Genetics. 2013;9:e1003588. doi: 10.1371/journal.pgen.1003588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Martianov I, et al. Repression of the human dihydrofolate reductase gene by a non-coding interfering transcript. Nature. 2007;445:666. doi: 10.1038/nature05519. [DOI] [PubMed] [Google Scholar]
- 64.Buske FA, et al. Triplexator: detecting nucleic acid triple helices in genomic and transcriptomic data. Genome research. 2012;22:1372–1381. doi: 10.1101/gr.130237.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ding Y, et al. In vivo genome-wide profiling of RNA secondary structure reveals novel regulatory features. Nature. 2013 doi: 10.1038/nature12756. [DOI] [PubMed] [Google Scholar]
- 66.Altshuler D, et al. Genetic mapping in human disease. Science. 2008;322:881–888. doi: 10.1126/science.1156409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Stitzel ML, et al. Global epigenomic analysis of primary human pancreatic islets provides insights into type 2 diabetes susceptibility loci. Cell Metab. 2010;12:443–455. doi: 10.1016/j.cmet.2010.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lee JC, et al. Human SNP links differential outcomes in inflammatory and infectious disease to a FOXO3-regulated pathway. Cell. 2013;155:57–69. doi: 10.1016/j.cell.2013.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wahlestedt C. Targeting long non-coding RNA to therapeutically upregulate gene expression. Nature Reviews Drug Discovery. 2013;12:433–446. doi: 10.1038/nrd4018. [DOI] [PubMed] [Google Scholar]
- 70.Wang D, et al. Reprogramming transcription by distinct classes of enhancers functionally defined by eRNA. Nature. 2011;474:390–394. doi: 10.1038/nature10006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Guttman M, Rinn JL. Modular regulatory principles of large non-coding RNAs. Nature. 2012;482:339–346. doi: 10.1038/nature10887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.PARDEE AB, et al. [The role of the inducible alleles and the constrtutive alleles in the synthesis of beta-galactosidase in zygotes of Escherichia coli] Comptes rendus hebdomadaires des séances de l’Académie des sciences. 1958;246:3125–3128. [PubMed] [Google Scholar]
- 73.JACOB F, MONOD J. Genetic regulatory mechanisms in the synthesis of proteins. Journal of molecular biology. 1961;3:318–356. doi: 10.1016/s0022-2836(61)80072-7. [DOI] [PubMed] [Google Scholar]
- 74.Boseley P, et al. Sequence organization of the spacer DNA in a ribosomal gene unit of Xenopus laevis. Cell. 1979;17:19–31. doi: 10.1016/0092-8674(79)90291-5. [DOI] [PubMed] [Google Scholar]
- 75.Gribnau J, et al. Intergenic transcription and developmental remodeling of chromatin subdomains in the human beta-globin locus. Molecular cell. 2000;5:377–386. doi: 10.1016/s1097-2765(00)80432-3. [DOI] [PubMed] [Google Scholar]
- 76.Melgar M, et al. Discovery of active enhancers through bidirectional expression of short transcripts. Genome Biology. 2011;12:R113–R113. doi: 10.1186/gb-2011-12-11-r113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ostuni R, et al. Latent enhancers activated by stimulation in differentiated cells. Cell. 2013;152:157–171. doi: 10.1016/j.cell.2012.12.018. [DOI] [PubMed] [Google Scholar]
- 78.Cowper-Sal·Lari R, et al. Breast cancer risk-associated SNPs modulate the affinity of chromatin for FOXA1 and alter gene expression. Nature Genetics. 2012;44:1191. doi: 10.1038/ng.2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ross-Innes CS, et al. Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature. 2012;481:389. doi: 10.1038/nature10730. [DOI] [PMC free article] [PubMed] [Google Scholar]