

Abstract
Epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs) have shown dramatic effects against that tumors harboring EGFR activating mutations in the EGFR intracytoplasmic tyrosine kinase domain and resulted in cell apoptosis. Unfortunately, a number of patients ultimately developed resistance by multiple mechanisms. Thus, elucidation of the mechanism of resistance to EGFR-TKIs can provide strategies for blocking or reversing the situation. Recent studies suggested that redundant kinase activation plays pivotal roles in escaping from the effects of EGFR-TKIs. Herein, we aimed to characterize several molecular events involved in the resistance to EGFR-TKIs mediated by redundant kinase activation.
Keywords: EGFR, redundant kinase activation, resistance to EGFR-TKIs
Introduction
Epidermal growth factor receptor (EGFR), a member of a family which consists of at least 4 receptor tyrosine kinases, including EGFR (ErbB1), HER2 (ErbB2), HER3 (ErbB3), and HER4 (ErbB4) (Figure 1). To date, seven ligands for EGFR have been identified: epidermal growth factor (EGF), transforming growth factor (TGF)-α, heparin-binding EGF-like growth factor (HB-EGF), amphiregulin, betacellurin, epiregulin, and epigen [1]. The EGFR family of cell surface-receptor tyrosine kinases controls the intracellular signaling pathways that promote cell growth, proliferation, differentiation, and migration [2]. The important roles of EGFR in the activation of cancer relevant cellular processes, together with the presence of overexpressed or aberrantly activated EGFR in non-small cell lung carcinoma (NSCLC), suggest that targeting the EGFR may provide a strategy for NSCLC.
Figure 1.
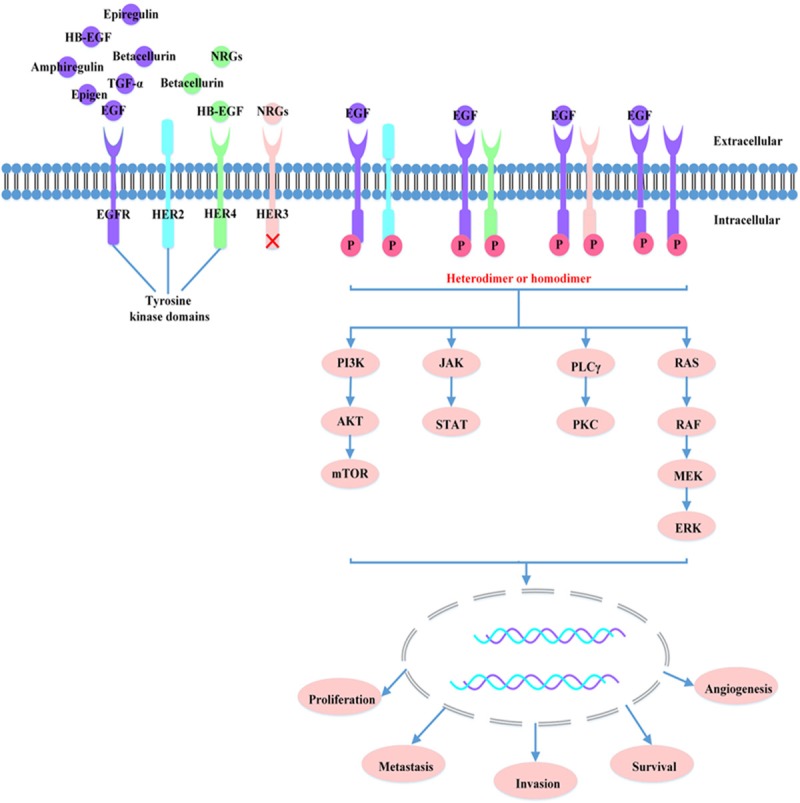
HER family and EGFR signaling pathway. Ligand-bound receptors form functionally active homodimers or heterodimers, resulting in the activation of downstream signaling pathways such as PI3K/AKT, RAS/RAF/MAPK, PLCγ/PKC and JAK/STAT pathway, leading to cell proliferation, invasion, metastasis, survival and angiogenesis.
Two main anti-EGFR strategies are currently in clinical application: low-molecular-weight TKIs that compete with adenosine triphosphate (ATP) for binding to the tyrosine kinase portion of a mutant EGFR receptor, and monoclonal antibodies (mAbs) that are directed at the ligand-binding extracellular domain, thereby preventing ligand binding, and consequently receptor dimerization, and receptor signaling. Among these, gefitinib and erlotinib were the first EGFR-TKIs to be approved by Food and Drug Administration (FDA) for treatment of NSCLC (Table 1). These drugs inhibit kinase activity by competitively bind to the ATP-binding site of EGFR, preventing auto-phosphorylation and consequently blocking downstream signaling cascades of RAS/RAF/MEK/ERK and PI3K/AKT pathway, resulting in proliferation inhibition, cell cycle progression delay, and cell apoptosis [3].
Table 1.
Clinical drugs targeting EGFR approved by FDA
| Drugs | Trade Name | Target | Category | Times | Application |
|---|---|---|---|---|---|
| Erlotinib | Tarceva | EGFR | TKI | 2004 | NSCLC, pancreatic cancer |
| Gefitinib | Iressa | EGFR | TKI | 2003 | NSCLC |
| Lapatinib | Tykerb | EGFR/HER2 | TKI | 2007 | metastatic breast cancer |
| Afatinib | Gilotrif | EGFR/HER2 | TKI | 2013 | NSCLC, metastatic breast cancer |
| Cetuximab | Erbitux | EGFR | Monoclonal antibody | 2004 | colorectal cancer |
| Trastuzumab | Herceptin | HER2 | Monoclonal antibody | 1998 | metastatic breast cancer |
| Panitumumab | Vectibix | EGFR | Monoclonal antibody | 2006 | colorectal cancer |
Although EGFR-TKIs treatment shows good response rates and progression free survival (PFS) in NSCLC patients with EGFR gene mutations, acquired resistance of TKIs therapy is common after a median of 12-16 months [4]. To date, various mechanisms of resistance to erlotinib and gefitinib have been identified, including 1) gatekeeper mutations in EGFR, such as the T790M second mutation which is thought to be responsible in over 50% in patients who acquire secondary resistance [5]; 2) activation of redundant kinase signaling pathway such as c-Met [6], insulin-like growth factor receptor (IGFR) [7], HER family members [8,9], growth arrest specific gene6 (Gas6)-AXL pathway [10], fibroblast growth factor receptor (FGFR) [11], vascular endothelial growth factor (VEGFR) [12], platelet-derived growth factor receptor (PDGFR) [2], and interleukin-6 receptor (IL-6R) signaling pathway [13]; 3) activation of downstream molecules (PTEN loss or K-RAS, PIK3CA mutation) [14,15]; 4) small-cell lung cancer transformation [16] and 5) epithelial-to-mesenchymal transition (EMT) [17]. Therefore, it is essential to understand the mechanisms of resistance to TKIs for the development of new EGFR-targeted drugs. This review focuses on the mechanisms of resistance to EGFR-TKIs mediated by redundant kinase activation.
Redundant kinase pathways as mechanisms for resistance to EGFR-TKIs
A simple explanation for the insensitivity to EGFR inhibitors is through a “redundant effect” mechanism, the dominant activity of redundant receptor tyrosine kinase (RTK) systems distinct from EGFR [18]. In this regard, it has been observed that a large fraction of the tyrosine phosphoproteome was abundant in erlotinib-treated cells [19]. Activation of these receptor tyrosine kinases by growth factors could protect cells against the EGFR-TKIs. Thus, there is no shortage of candidates for RTKs that may function as alternatives to EGFR in signal transduction of growth and transformation in NSCLC.
c-Met pathway
c-Met, a transmembrane tyrosine kinase receptor that binds with HGF, then induces recruitment of the Grb2-associated binder (GAB1) and activation of multiple signaling networks including the phosphoinositide PI3K/AKT/mTOR and RAS/RAF/MEK/ERK pathways independent of EGFR, HER2, HER3, and HER4 [20]. Deregulation of c-Met signaling due to overexpression of c-Met or HGF has been associated with poor prognosis in advanced gastric carcinomas [21]. A well-documented mechanism is c-Met amplification initially reported in 15-20% of resistant patients [6], but recently another reported in 3-5% [5]. Strong HGF expression was observed in > 60% of tumors with secondary EGFR-TKIs resistance [22]. Activation of c-Met pathway in human tumors can be induced by various means, like HGF overexpression, transactivation by other membrane proteins (including EGFR), and mutations [23,24]. Accordingly, a recent study suggests that c-Met activation caused by c-met gene amplification is a suitable surrogate marker of resistance to EGFR-TKIs [25]. Interestingly, c-met gene was also found amplify before drug exposure [26] as well as the c-Met activity and protein levels were elevated in nonexposed NSCLC patients [27]. This suggests c-Met overexpression is associated with primary resistance of EGFR-TKIs in NSCLC.
As for transactivation by other membrane proteins, Engelman et al. [6] found that there is a cross-talk between EGFR and c-Met mediated by phosphorylation and signaling from HER3 to AKT in lung cancer cell lines, which leads to the resistance of EGFR TKIs. Much to their surprise, even if oncogenic EGFR was fully inhibited, activation of the PI3K/AKT/mTOR pathway could continue through the interaction of c-Met and HER3. Phosphorylation of HER3 by c-Met has been shown to occur via direct as well as indirect mechanisms. With respect to direct phosphorylation, the c-Met receptor may homodimerize with HER3 activates the PI3K/AKT pathway independent of EGFR [28]. This mechanism is analogous to the manner in which EGFR itself activates PI3K-driven signal transduction. Indirect phosphorylation of HER3 include up-regulation of EGFR ligands, and activation of other tyrosine kinases (for example, c-Src) [29,30]. Moreover, when this redundant c-Met signaling via HER3 was simultaneously inhibited, apoptosis increased dramatically among resistant cells [30]. Specific short hairpin RNA (shRNA) and small interfering RNA (siRNA) of c-Met could restore the ability of gefitinib in resistant cells [31]. However, Rho et al. [32] found that there was no cross-talk between c-Met and EGFR. These phenomena may be explained by a previous report [27], in which it was shown that mutated and amplified EGFR can activate c-Met. Likewise, enhanced levels of HGF, active the c-Met/PI3K/AKT signaling pathway, thus induce gefitinib resistance of lung cancer cells harboring EGFR-activating mutations [33]. An anti-HGF neutralizing antibody or an HGF antagonist (NK4), when combined with EGFR-TKIs, dramatically reversed HGF-induced resistance in vitro and in vivo [34]. Moreover, transient but intensive inhibition of PI3K/AKT by PI3K inhibitors and gefitinib successfully overcame HGF-induced EGFR-TKIs resistance in vitro and in vivo [35]. 17-DMAG (an HSP90 inhibitor) has efficacy for HGF-triggered erlotinib resistance in cell lines and animal models [36]. Another research found that through promoting c-Met-integrin association, HGF-FN (fibronectin) and HGF-VN (vitronectin) complexes coordinated and enhanced endothelial cell migration through activation of the PI3K pathway [37]. There is also an important cross-talk between c-Met and the α2β1 integrin in mast cell, resulting in the release of the pro-inflammatory cytokine, IL-6 [38], which can activate the IL-6R/JAK/STAT signaling related with EGFR-TKI resistance [13].
Many of these mechanisms above are thought to be critical for the contribution of c-Met to tumorigenesis and may be involved in both primary and acquired resistance to gefitinib, meanwhile provide a rationale for targeting HGF/c-Met pathway. The c-Met inhibitor PHA-665752 [39] and NPS-1034 [40] has great effect against lung cancer cells resistant to EGFR-TKIs. Mueller et al. [25] have shown that inhibiting c-Met kinase activity in breast cancer cell lines with constitutive c-Met activation sensitizes these cells to EGFR-TKIs.
HER pathway
Recent studies have suggested that overexpression of other members of the EGFR receptor family, namely HER2 and HER3 are involved in EGFR-TKIs resistance [41,42]. Activation of HER2 signaling was recently reported to cause resistance to cetuximab alone in patients with colorectal cancer [43]. The recent role of HER2 amplification in the acquisition of resistance to TKIs, reported in 12-13% of patients [5]. HER2 can be actived by IGFR1 through a physical association between the two receptors [44]. Importantly, IGFR1 signaling via the PI3K/AKT pathway is associated with resistance to trastuzumab (an anti-HER2 monoclonal antibody) in breast cancer, it also demonstrate evidence of the existence of a physical interaction between IGFR1 and HER2 [45]. Furthermore, activated IGFR1 can also physically associate with HER3 and HER4 [46]. Cretella et al. [8] found that targeting HER2 with trastuzumab-DM1 can improve the treatment of HER2 positive breast cancer. It offers a new therapeutic approach in lung cancers expressing HER2 even when resistant to EGFR-TKIs. The combination of afatinib plus cetuximab could be efficacious in overcoming acquired resistance in lung cancer [47]. HER2 mutations are present in about 2-4% of NSCLC, especially in women, never-smokers, Asian patients and in adenocarcinomas without EGFR or K-ras mutations [48]. These mutations render the receptors activation, resulting in proliferation and metastasis of tumor cells. Alternatively, through study of receptor down-regulation, data suggests that mutant EGFRs, especially the L858R/T790M variant, have a propensity to heterodimerize with HER2, which allows for evasion of Casitas B-cell lineagelymphoma (CBL) mediated ubiquitinylation and subsequent lysosomal degradation [49].
Likewise, HER3 overexpression was previously reported to be associated with impaired survival in breast cancer [50]. Almost all de novo resistant NSCLC tumors the HER3 receptor is strongly phosphorylated [51]. HER3 lacks tyrosine kinase activity but it can be trans-phosphorylated efficiently by c-Met [6] or other RTKs such as HER2 and HER4 [52]. HER3 interacts with the other HER family members to active intracellular pro-survival signaling due to several tyrosine residues in its intracytoplasmic domain, which can be phosphorylated and become high affinity docking sites for the catalytic subunit of PI3K. High surface HER3 expression correlates with AKT phosphorylation in lung adenocarcinoma primary cultures [10]. Byun et al. [53] reported that genetic silencing of USP8 led to the downregulation of several RTKs including EGFR, HER2, HER3, and c-Met, markedly decreased the viability of gefitinib-resistant and -sensitive NSCLC cells by decreasing RTKs expression while having no effect on normal cells. Furthermore, erlotinib with either HER2 or HER3 knockdown by their cognate siRNAs also inhibited PI3K/AKT activation [54]. This indicates that the loss of addiction to mutant EGFR results in the gain of addiction to both HER2 and HER3. Antibodies against HER3 only work in cells overexpressing surface HER3 [9]. And combination of anti-HER3 antibodies with EGFR-TKIs synergistically affect cell proliferation in vitro, resulting in cell cycle arrest, p21 expression upregulation and tumor growth inhibition in mouse xenografts [9]. Hence surface HER3 may be considered a predictive marker of efficacy if appropriately validated in a more number of cases. In light of these considerations, HER3 might be a central node in the resistance to EGFR-TKIs, and agents targeting this molecule are being developed (NCT01211483) [55].
VEGFR pathway
Vascular endothelial growth factor (VEGF) is an important survival factor of vascular endothelial cells that activates tyrosine kinase after binding to VEGFR. VEGFR2 is the key mediator of VEGF-mediated angiogenesis, and VEGFR1 and VEGFR3 are involved in vasculogenesis, and lymphangiogenesis, respectively [12]. Recent studies showed that VEGF overexpression was associated with clinical response to EGFR-TKIs in patients with lung cancer [56,57]. It suggested the VEGF may play a key role in resistance to EGFR-TKIs. EGFR and VEGFR signaling pathways are independent but are closely interlinked, both EGF and TGF-α can induce VEGF expression via activation of EGFR in cell culture models [58].
HGF is also associated with VEGFR signaling pathway. High serum HGF is relevant to short progression-free survival in a clinical trial of a VEGFR inhibitor, sorafenib, for the treatment of hepatocellular carcinoma [59]. Overexpression of HGF conferred resistance to lenvatinib (a VEGFR inhibitor) and it was cancelled by golvatinib (a c-Met inhibitor) [60]. In renal cell carcinoma, HGF was also reported to induce resistance to sunitinib, an inhibitor of multiple kinases, including VEGFR2, by compensating for inhibited angiogenesis [61]. Previous study showed HGF stimulated VEGF production by activation of the c-Met/Gab1 signaling pathway in EGFR mutant lung cancer cell lines [62]. Silencing of Gab1 successfully canceled HGF-stimulated VEGF production and HGF-induced EGFR-TKIs resistance. These findings suggest that Gab1 may be a novel ideal target for controlling EGFR mutant lung cancer. Though inhibition of VEGFR shrinked the tumor, meanwhile it made the tumor more aggressive with more metastatic behavior in a model of pancreatic neuroendocrine cancer [63]. Maybe the blockade of VEGFR signaling caused hypoxia and that hypoxia is likely to enhance HGF/c-Met pathway that promotes tumor survival and metastasis [64]. Therefore, dual inhibition of HGF and VEGF may be therapeutically useful for EGFR-TKIs resistant lung cancer. Golvatinib is an orally active dual TKI for c-Met and VEGFR2, it exerts effect by inhibiting the c-Met/Gab1/PI3K/AKT pathway [65].
IGFR pathway
IGFR is a member of the class II receptor tyrosine kinase family. It has two distinct ligands (IGF1 and IGF2) plus insulin, and two receptors (IGFR1 and the insulin receptor). The two receptors are capable of homo- and hetero-polymerization, leading to receptor auto-phosphorylation and subsequent phosphorylation of substrate proteins, the insulin receptor substrate-1 (IRS-1) [66]. Similar to the EGFR pathway, IGFR1 activation triggers the RAS/RAF/MAPK pathway and the PI3K/AKT/mTOR pathway [67]. Overexpression of IGFR1 was detected in 33% of HCCs and increased activation of IGFR1 was observed in 52% of tumors [68]. A report indicated that IGF1R activation is a molecular mechanism that confers acquired resistance to erlotinib in lung cancers with the wild-type EGFR [69]. Overexpression of the igfr1 gene constitutes a common theme in many human cancers including NSCLC [70]. Interestingly, IGFR1 expression in NSCLC specimens was associated with a history of tobacco smoking, squamous cell carcinoma histology, mutant (mut) K-Ras, and wild-type (wt) EGFR, all of which have been strongly associated with poor response to EGFR-TKIs [71]. Kim et al. [72] found that activation of IGFR1 caused by IGF1 overexpression led to spontaneous lung tumor development that progressed to adenocarcinoma upon exposure to tobacco carcinogens. It was suppressed by a selective IGFR1 inhibitor, cis-3-[3-(4-methyl-piperazin-l-yl)-cyclobutyl]-1-(2-phenyl-quinolin-7-yl)-imidazo [1, 5-a] pyrazin-8-ylamine (PQIP) on the early stage. Jameson et al. [7] found that IGFR1 activation partially reverses the cell cycle arrest caused by gefitinib in oral squamous cell carcinoma (OSCC) cells. Importantly, IGFR1 stimulation does not eliminate the gefitinib-induced increase in total p27 (cyclin kinase inhibitor), its phosphorylation state and subcellular localization are altered. This suggested that the IGFR1 can rescue OSCC cells from EGFR-TKIs treatment. Knockdown of IGFR1 with siRNAs, mammary tumor growth was strongly delayed in vitro [73]. And lung adenocarcinoma cell lines responded to combined therapy with erlotinib and NVP-AEW541, an IGF1R-TKI [69]. Thus, it has been proposed that reduction of IGFR signaling in some cancer types may have therapeutic benefit to EGFR-TKI treatment.
In addition to the level of IGFR1 and IGF, the degree of IGFR1 activation is dependent on the abundance of insulin like growth factor binding proteins (IGFBPs) [74]. Epidemiological studies have shown that decreasing levels of IGF-1 and increasing levels of IGFBP-3 are independently associated with a high risk of colorectal cancer [75]. IGFBP-3 is a potent negative regulator of IGFR1 activation by binding with IGF-1 and then regulates the mitogenic and anti-apoptotic actions of IGFs independent of IGF [76]. Choi et al. [77] showed significant downregulation of IGFBP-3 expression in resistant cells, and addition of recombinant IGFBP-3 restored the ability of gefitinib to downregulate PI3K/AKT signaling and to inhibit cell growth. On the other hand, adenovirus-mediated overexpression of or recombinant IGFBP-3 slightly inhibited the growth of HCC cells in vitro [78]. A report showed that in breast cancer, trastuzumab regulates IGFBP-2 and -3 expressions, which impacts IGFR1 downstream signaling [79]. Collectively, these results suggest that loss of expression of IGFBPs in tumor cells treated with EGFR-TKIs results in the activation of IGFR1 signaling, which in turn mediates resistance to EGFR-TKIs.
Hurbin et al. [80] observed a cross-talk between EGFR and IGFR1 and their ligands, amphiregulin and IGF1 under gefitinib treatment in resistant mucinous cells. It is reported that amphiregulin and IGFR1 mediate gefitinib resistance through increasing the interaction between the proapoptotic protein BAX and Ku70 [81]. The inhibition of Ku70 acetylation enhances BAX/Ku70 binding and prevents gefitinib-induced apoptosis. In contrast, the acetylation of Ku70 releases BAX from Ku70 and restores the sensitivity to gefitinib. Indeed, amphiregulin is a principal activator of the ligand-receptor autocrine pathway, members of the HER family (HER 1-4) can form heterodimers with IGFR1 and InsR, leading to the formation of hybrid receptors through physical associations between heterologous families [80]. Morgillo et al. [82] also reported that increased levels of EGFR/IGFR1 heterodimers activated IGFR1 and its downstream signaling mediators, leading to acquired resistance to erlotinib. Co-treatment of erlotinib and an IGFR1 inhibitor induced both apoptosis and cell cycle arrest, while either agent or EGFR-TKI alone only induced cell cycle arrest in some EGFR mutant NSCLC cells [83].
FGFR pathway
FGFs bind with members of a family of RTKs (FGFR1-4), then lead to receptor dimerization and activation of the PI3K/AKT/mTOR and RAS/RAF/MEK/ERK pathways [84]. Recently, FGFR was regarded as an important autocrine growth factor pathway for resistance to EGFR-TKIs in NSCLC [85]. FGFR1 amplification is associated with poor prognosis in NSCLC [86]. A plenty of in vitro studies revealed overexpression of FGF2, FGFR1 and FGFR2 mRNA and protein in primary NSCLC specimens [87,88]. Recently, two independent groups reported that FGFR1 was amplified in approximately 10% to 20% of squamous cell lung cancers [89,90]. However, another study found only 3 of 41 NSCLC cell lines showed evidence for activated FGFR1 [91]. And amplification of the fgfr gene has been detected in bladder cancer, albeit at a very low frequency [92]. It is suggested that FGF/FGFR pathway activation is one of the important mechanisms to escape from EGFR-TKIs. Terai et al. [11] found that the expression of FGFR1 and FGF2 were increased in gefitinib-resistant cells and that the phosphorylation status of EGFR itself was not affected by FGF2/FGFR1 activation and completely inhibited by gefitinib. Ware and colleagues reported on rapid acquired resistance to EGFR-TKIs in NSCLC cell lines through derepression of expressions of FGFR2 and FGFR3 [93]. They demonstrated that FGFR2 and FGFR3 can mediate FGF2 and FGF7 stimulated ERK activation as well as FGF stimulated transformed growth in the setting of EGFR-TKIs. It means that FGF2 or FGF7 rescues NSCLC cells from treatment with an EGFR-TKI. Also, co-culture of H322c cells with human fibroblasts rescues EGFR-TKIs induced growth inhibition in an FGFR-dependent manner [93]. Interestingly, FGFR2 and FGFR3 expression was induced in all gefitinib-sensitive NSCLC cells and correlated with cells that co-express EGFR and EGF ligands or bear gain-of-function EGFR. However, NSCLC cells that do not express EGFR or are gefitinib-resistant did not exhibit FGFR2 and FGFR3 mRNA induction in response to gefitinib [94]. Much to surprise, FGFR2 and FGFR3 induction occurs quickly (1-2 days) compared with met gene amplification in response to gefitinib (~6 months) [6]. It suggests that the fgfr2 and fgfr3 gene are not amplified but are being regulated at the transcriptional level. Thus, increased FGFR2 mRNA is partially mediated by transcriptional induction of the fgfr2 gene following gefitinib treatment. Importantly, the application of siRNA and neutralizing FGF antibodies is an efficient therapy against tumor growth [95,96]. Also, RO4383596 (an FGFR inhibitor) inhibited basal fibroblast growth factor receptor substrate-2 (FRS2) and ERK phosphorylation as well as tumor proliferation and growth [94].
In addition to inappropriate expression of FGF ligands and FGFRs, FGFR mutations could participate in oncogenesis. FGFR2 mutations are mainly located within the hinge between Ig-like domains (exon 7), around the 3rd Ig-like domains and within the kinase domain [97]. FGFR2 mutations are observed gain-of-function in 10% of primary endometrial cancers as well as endometrial tumor cell lines [98]. In urothelial cancers, FGFR3 mutations in the ligand binding domain lead to ligand-independent dimerization or stabilization of the active conformation of the receptor while mutations in the kinase domain can render the receptor constitutively active [92]. FGFR4 mutations have been observed in lung adenocarcinoma with a potential contributing role to carcinogenesis [99]. One study [18] suggested that epithelial to mesenchymal transition (EMT) can mediate EGFR-TKIs resistance by kinase switch, such as those activated by FGFR, PDGFR or α5β1 integrin. Their results were provided by primary lung cancer cells without exposure to EGFR-TKIs and cells with wild-type EGFR. FGFRs have also been shown to be physically associated with N-cadherin in mammary cancer cells, resulting in cell survival, invasion, proliferation and metastasis [100]. Maybe the N-cadherin promotes ERK and AKT phosphorylation resulting in sustained signaling.
PDGFR pathway
PDGFR is a member of the class III receptor tyrosine kinase family. PDGFR can activate the PI3K, PLCγ, and mitogen-activated protein kinase (MAPK) signaling pathways [101]. High expression of PDGFRβ is a predictor of poor prognosis [102]. The PDGFRβ isoform has been shown to mediate EGFR transactivation, suggesting this class of receptors may play a role in the response to TKIs. Importantly, phosphorylated PDGFRβ was observed in glioblastoma that lacked of EGFR signaling [103]. The contribution of PDGFRβ signaling to drug resistance remains incompletely understood. PDGFRβ amplifications and/or mutations are exceedingly rare events in glioblastoma [104]. In mouse genetic models, PDGFβ ligand overexpression can promote gliomagenesis by enhancing cellular proliferation [105]. Kassouf et al. [106] revealed that PDGFRβ was undetectable or expressed at very low levels in gefitinib-sensitive cell lines, but was expressed at higher levels in all resistant cell lines. Akhavan et al. [2] first demonstrated that mTORC1 inhibition mediates EGFR-TKIs resistance in glioblastoma through transcriptional regulation of PDGFRβ, a mechanism which could also be active in other cancer types. In mouse embryonic fibroblasts, PDGFRβ was shown to be a target of mTOR-dependent negative transcriptional downregulation [107]. Also, PDGFRβ has been shown to mediate vemurafenib resistance through transcriptional upregulation in melanoma [108]. Akhavan et al. [2] identified that EGFR inhibitors derepress PDGFRβ transcription, providing a potent mechanism underlying RTK switching. Thomson et al. [18] suggested that a switch to PDGFR signaling occurs in concert with EMT. Recently, PDGFRβ has been shown to promote glioma stem cell self-renewal [71], suggesting a more definitive role in tumorigenesis and/or maintenance. Moreover, a PDGFRβ inhibitor significantly reduced PDGFR and MAPK phosphorylation [71]. Although these observations indicate that PDGFRβ can active EGFR/MAPK pathway, there is still not any clinical data suggesting a relationship between PDFGR expression and acquired resistance to EGFR-TKIs.
In addition, Yeh et al. [109] showed that PDGFRα have a functional interaction with c-Met in vitro and in vivo. Previously, Black and his colleagues reported co-expression of c-Met/PDGFRα in all of 9 human bladder cancer cell lines [110]. The interaction between c-Met and PDGFRα was further corroborated by HGF stimulation and siRNA silencing experiments in vitro [109]. The interaction may be initiated by signal regulation. That PD98059 rather than FTI-277 (RAS inhibitor) or PP2 (Src inhibitor) successfully inhibited c-Met activation, suggests transactivation of PDGFRα is independent of RAS or Src. Consistent with this, Kina et al. [111] showed that PDGFα-mediated signaling plays a key role in c-Met upregulation, which in turn is relevant with chemotherapy resistance. And PDGFα receptor inhibition eliminates cisplatin-dependent c-Met expression in cervical cancer cell lines [111]. Future studies are required to explore the mechanism of PDGFR pathway in resistant cancers.
AXL pathway
AXL is a member of the tyro3 tyrosine kinase receptor family of RTKs, which also includes MER and TYRO-3. After binding with growth arrest-specific gene 6 (GAS6), it activates the PI3K/AKT/mTOR and RAS/RAF/MEK/ERK pathways to promote proliferation, survival, and migration of cancer cells in vitro [112] and tu-mor angiogenesis and metastasis in vivo [113]. Recently, the activation of AXL kinase confers acquired resistance mechanism of EGFR-TKIs [10]. Overexpression of AXL and/or GAS6 is the main mechanisms of activation in a wide range of human cancers, and it often correlates with poor prognosis [114]. In a small cohort of NSCLC patients refractory to EGFR-TKIs, higher expression of AXL and GAS6 was detected in 20% and 25% of cases, respectively [10]. High levels of AXL in EGFR mutant lung cancer cell lines induced erlotinib resistance [52,80]. Zhang et al. [10] shows that AXL upregulation is the second most common mechanism of EGFR-TKIs acquired resistance (after EGFR T790M) in EGFR-mutant NSCLCs. In some erlotinib resistance cell lines, GAS6 is not indispensable because AXL overexpression can promote downstream signaling and induce transformation in the absence of GAS6 expression [10].
Resistant cells with AXL overexpression are more inclined to migrate and adhere, which is the same with EMT and c-Met [115]. AXL was also activated in this cell line with T790M and c-Met amplification, whereas a report [10] found increased activation of AXL in EGFR-mutant lung cancer models with erlotinib acquired resistance in the absence of EGFR T790M or c-Met activation. Even so, c-Met was shown to interact with AXL, and promote signal transduction downstream of AXL [116]. Increased expression and coactivation of c-Met and AXL have been described in NSCLC [91]. Both c-Met and AXL was involved in HER2-positive breast cancer resistant to lapatinib [117,118] and resistance to AKT inhibition in preclinical models [119]. Salian-Mehta et al. [120] showed HGF/c-Met signaling modulated neuron migration dependent and independent of AXL co-expression and p38MAPK. Conversely, AXL controls gonadotropin-releasing hormone (GnRH)neuronal survival via HGF/c-Met signaling. When altering the levels of AXL, the bi-directional cross-talk between AXL and c-Met was affected. The kinase dead mutant of AXL expression reduced the phosphorylation of AKT and p38MAPK induced by c-Met, but with no effect on ERK or STAT3 [120]. It confirmed the cell specific pathways downstream of the interaction between AXL and c-Met in GnRH neurons. Similarly, either deletion of the intracytoplasmic domain or mutating the tyrosine kinase domain of AXL reduced HGF- induced activation of c-Met. Interestingly, the AXL pathway was also associated with increased levels of tumor vimentin, thus suggesting that AXL may mediate EMT in EGFR-TKIs resistant patients [16,17]. Consistently, a prior study showed vimentin upregulation was associated with AXL overexpression in breast cancer cells [40]. AXL and MER also regulate tumor stromal cell interactions via secretion of proinflammatory cytokines [114]. Only MER (but not AXL or TYRO-3) inhibits IL-6 secretion by lipopolysaccharide (LPS)-stimulated U937 cells and monocytes/macrophages [121].
Pharmacologically or genetically inhibiting AXL restored erlotinib sensitivity both in vitro and in vivo. Rho et al. [40] investigated the antitumor activity of NPS-1034, a newly developed drug that targets both c-Met and AXL, in gefitinib or erlotinib resistant NSCLC cells. Combining gefitinib or erlotinib with NPS-1034 effectively induced cell proliferation delay and cell apoptosis in both resistant cell lines. Combining AXL siRNA or NPS-1034 with EGFR-TKIs is also effective, suggesting that AXL is a key role in EGFR-TKIs resistance. Importantly, whether GAS6 might induce EGFR-TKIs resistance via AXL pathway and whether somatic alterations (amplifications, rearrangements, point mutations) in AXL or GAS6 occur in human EGFR-mutant NSCLCs needs further study to fully elucidate.
IL-6R pathway
IL-6 was hypothesized to reduce the dependence of EGFR pathway through the IL-6/gp130/STAT3 axis [122]. Serum IL-6 levels are elevated in patients with lung cancer than in normal individuals [123]. Also, IL-6 is detected at higher levels in tumor-associated stroma than in normal bone marrow stroma [124]. Recently, it has been reported that STAT3 activation via IL-6R is relevant with multidrug resistance in cancer cells [125]. Afatinib can promote the secretion of IL-6 by activating a positive feedback loop for IL-6/STAT3 axis. Among soluble factors secreted from stromal cells in tumor microenvironment, IL-6 is the most widely studied factor to induce resistance to anti-cancer drugs in many cancers [126]. Kim et al. [13] found that AKT and ERK were dramatically inactivated due to afatinib treatment, but STAT3 was paradoxically hyperactivated via increase of autocrine IL-6 production. Moreover, overexpression or addition of IL-6 to TKI-sensitive cells induced TKI resistance, which could be overcome by metformin [127]. Finally, metformin-based combinatorial therapy effectively blocked tumor growth in TKI-resistant cancer cells, which was associated with decreased IL-6 secretion and decreased IL-6-signaling activation in vivo. In addition, activation of NF-κB is another possible explanation for the autocrine IL-6 production by afatinib [13]. IL-6 is a well-known downstream target of NF-κB. Recently, it was reported that increased IL-6 production via NF-κB activation mediated resistance to docetaxel in prostate cancer [128]. These reports support the hypothesis that NF-κB activation is involved in autocrine IL-6 production upon afatinib treatment. Because IL-6 is mainly secreted from fibroblasts in vivo [129], there may be a cross-talk between fibroblasts and IL-6, which leads to afatinib resistance through activation of the IL-6R/JAK1/STAT3 signaling pathway in cancer cells. Co-culturing cancer cells and MRC5 fibroblasts (secrete IL-6), afatinib-induced STAT3 activation was enhanced in the presence of MRC5-CM [13]. And treating with IL-6R neutralizing antibody or IL-6R siRNA completely suppressed afatinib-induced STAT3 activation and significantly restored the effect of afatinib. However, the treatment of MRC5-CM did not affect the inactivation of AKT and ERK by afatinib in both cells. These findings indicate that interaction with fibroblasts is important for de novo resistance of NSCLC cells to afatinib through activation of the IL-6R/JAK1/STAT3 signaling pathway.
Other pathways
Several recent studies demonstrated that the FAS-NFκB signaling pathway can promote tumor growth [130,131]. NFκB signaling has been broadly associated with inflammation and cancer [132]. A recent report showed activated NFκB pathway rescued NSCLC cells bearing a mutant EGFR from EGFR inhibitors. Bivona et al. [133] identified activation of NFκB signaling as a new mechanism of de novo resistance to erlotinib treatment. Of the 36 shRNAs recovered from the pooled screen, 18 targeted genes that are involved in NFκB signaling directly or indirectly. Interestingly, one of the top hits in the pooled screen was CD95/FAS, the ligand of the FAS death receptor, it functions upstream of NFκB to promote cell survival and tumor growth [131]. They also observed increased FAS expression and NFκB pathway activation in resistant cells [133]. And knockdown of FAS and several components of the NFκB pathway enhanced cell death in EGFR-mutant lung cancer cells treated with EGFR-TKIs. Low expression of the NFκB inhibitor IκB (high NFκB activation state) was predictive of a poor clinical outcome in patients treated with EGFR-TKIs [133]. IκB status was not a predictive outcome in EGFR mutant lung cancer patients treated with surgery or chemotherapy, indicating NFκB signaling is specific biomarker of EGFR-TKIs response in this patient population. Presumably more data about this pathway and its clinical relevance will become available in the near future.
The echinoderm microtubule-associated protein-like (EML) 4-ALK (anaplastic lymphoma kinase) fusions gene that encodes a cytoplasmic chimeric protein with constitutive kinase activity have been found in 5-7% of NSCLC patients, more frequently in those with young age, adenocarcinoma histology, and never or light smokers [134]. The resulting protein carries a coiled-coil basic domain from the upstream fusion partner, which may promote dimerization to activate the ALK tyrosine kinase [135]. EML4-ALK overexpression activated ERK and STAT3, but not AKT [136]. Moreover, ALK gene rearrangements are often mutually exclusive with EGFR mutations, even if there were cases of patients harboring both EGFR activating mutations and ALK translocation [137]. Activating mutations or translocations of the alk gene have been identified in anaplastic large-cell lymphoma, neuroblastoma, inflammatory myofibroblastic tumor and NSCLC [135]. Activated alk gene initiates signaling mostly through RAS/RAF/ERK and PI3K/AKT pathway. ALK inhibition results in downregulation of both AKT and ERK phosphorylation [138], and cell apoptosis mediated by ERK-dependent BIM upregulation and STAT3-dependent survivin downregulation [136].
Rearrangements of the receptor tyrosine kinase c-ros oncogene 1 (ROS1) appeared to occur in approximately 1% to 2% of NSCLC [139]. ROS1 is located on chromosome 6 and has a high degree of amino acid homology with ALK (49% within the kinase domain and 77% within the ATP-binding site). Clinicopathologic features of ROS1-positive cases are the same as ALK-rearranged NSCLC, including younger age, never smokers, and adenocarcinoma histology [140]. Multi-targeted ALK/MET/ROS1 inhibitors, such as crizotinib, have demonstrated efficacy in this population [141].
Strategies
Targeting redundant kinase and its ligands
There are many inhibitors and anti-bodies targeting both receptors and ligands of these redundant kinases (Figure 2). For example, adding a c-Met inhibitor (PHA-665752 or NPS-1034) may be beneficial to EGFR mutant lung cancer patients whose tumors harbor c-Met amplification as a mechanism of EGFR-TKIs resistance. Antibodies targeting the HGF (NK4) are currently in clinical development. Besides, 17-DMAG (an HSP90 inhibitor) has efficacy for HGF-triggered erlotinib resistance in cell lines and animal models [36]. Likewise, as an inhibitor of IGFR1 and AKT phosphorylation, PQ401 is reported to mimic IGFBP-3 and an IGFR1-blocking antibody that does not bind the InsR [142]. Amphiregulin might also be a therapeutic target. Amphiregulin inhibition combined with gefitinib strongly reduced tumor growth of mucinous cells with wild-type EGFR and mutated K-ras in vivo [81]. It is also noteworthy that inhibition of the InsR along with the IGFR1 may be clinically desirable due to InsR can substitute for IGFR1 when IGFR1 is selectively inhibited [143]. Importantly, targeting multiple receptors with a single agent may potentially overcome molecular heterogeneity and improve efficacy. HKI-272 (neratinib) is an irreversible inhibitor with activity against both EGFR and HER2 [144]. Idacomitinb, a pan-HER inhibitor that irreversibly and covalently binds to the ATP domain of each of three kinase-active member of the HER family (EGFR, HER2 and HER4) [145]. BMS-690514 is a TKI targeting both EGFR and VEGFR that has shown interesting phase II data with patients with NSCLC [146]. Likewise, AZD2171 (cediranib) was developed as a VEGFR inhibitor [147], but exhibits good potency for FGFRs and has been employed as an effective inhibitor of growth of FGFR2-driven gastric cancer cell lines [148]. Additionally, a multi-kinase targeted TKI, dovitinib, has been used to inhibit activated FGFR3 in multiple myeloma [149]. Sorafenib is a multi-targeted tyrosine kinase inhibitor acting on PDGFR, VEGFR, RAF, c-Kit, and fms-like tyrosine kinase-3 (FLT3), and has been shown to inhibit hepatic cellular cancer (HCC)-induced proliferation and angiogenesis [150,151].
Figure 2.

Redundant kinase signaling pathway. The activation of redundant kinase leads to downstream pathway such as PI3K/AKT, RAS/RAF/MEK and JAK/STAT signaling actived, which offsets the blockade of EGFR pathway by TKIs.
Inhibition of downstream molecules
Since a lot of redundant kinase signaling share the same downstream signaling, PI3K/AKT/mTOR or RAS/RAF/MEK/ERK or JAK/STAT pathway, the inhibitors of these downstream molecules may be of a great efficency to block the activation of various redundant kinase (Figure 2). At the present time, several drugs that inhibit activated RAF, MEK, PI3K, AKT and mTOR are available and clinical trials with these agents are actively recruiting patients, some of them selecting therapy based on the genetic profile of the tumor. Addition of PI3K inhibitors to standard treatment is an interesting approach already being explored in multiple phase- I/II trials [152]. Besides, mTOR is a key mediator of PI3K/AKT downstream signaling and is commonly activated in NSCLC. To date, several mTOR inhibitor rapamycin analogs are available, including temsirolimus and everolimus, which show effect in renal cell carcinomas and pancreatic neuroendocrine tumors [153]. Rapamycin and its analogs bind FK506-binding protein-12 (FKBP12) inhibits mTOR activity and halting the translation of proteins critical for cell proliferation and survival [154]. Moreover, mTOR, PI3K, and dual PI3K/mTOR inhibitors are being evaluated in early-stage clinical trials of lung cancer, either alone or in combination with EGFR inhibitors. The MEK inhibitors, such as CI-1040 and AZD6244, reversed the resistance both in vitro and in vivo [155]. Several agents, OPB-31121 (STAT3 decoy oligonucleotides) [156], or AZD1480 (a small molecule inhibitor for JAK) [157] has been developed, it can block the IL-6R and EGFR pathway, may be suitable candidates for future combined therapy with irreversible EGFR-TKIs.
Combination therapy
A number of studies provided increasing evidences supporting the dual inhibition of two or more receptors rather than single receptor targeting. Combining a reversible EGFR-TKI and an anti-EGFR antibody may be a relevant strategy for overcoming EGFR-TKIs resistance. Afatinib (BIBW 2992), an irreversible inhibitor of EGFR, HER2, and HER4 [158], in combination with cetuximab, was reported to have significant activity in patients with acquired resistance to EGFR-TKIs [47]. Due to the cross-talk between EGFR family and other kinase receptors, such as EGFR-VEGFR, HER2-IGFR, HER3-c-Met, as well as the interaction between c-Met and other redundant kinases, combination therapy is indispensable for overcoming the resistant tumors. Dual blockade of the EGFR and VEGFR axes may be valuable for overcoming not only EGFR-TKIs resistance but also angiogenesis inhibitor resistance. Combining drugs targeting HER2 or HER3 with inhibitors of IGFR or c-Met can cause both two pathways blocked, respectively. Dual inhibition of c-Met and VEGFR pathway, resulting in the blockage of two signaling and better effect if combined with EGFR-TKIs. The combination of small molecule kinase inhibitors targeting AXL (XL880 or MP-470) or an AXL neutralizing antibody with an EGFR-TKI is a potential approach to overcome resistance. In addition, NPS-1034 inhibited cell proliferation as well as ROS1 activity in HCC78 cells with ROS1 rearrangement. Rho et al. [40] established the efficacy of NPS-1034 in NSCLC cells resistant to EGFR-TKIs because of AXL activation or ROS1 rearrangement. Combining inhibitions of receptor kinases and downstream molecules is also applicable for treatment. Kim et al. [71] provide a rationale for the therapeutic use of IGF-1R TKIs, either singly or in combination with MAPK/ERK inhibitors, particularly in tumors with K-ras mutations. In patients with resistance to first-generation EGFR-TKIs generated by c-Met, it is unlikely that an irreversible EGFR inhibitor alone would be effective, but the combination of an irreversible EGFR inhibitor and an mTOR inhibitor may be an effective strategy for overcoming resistance [159].
Conclusion
Clinical and biological evidence suggests that the EGFR does not function as a single dominant receptor tyrosine kinase in autocrine growth of NSCLC, but that multiple redundant kinases will participate in (Figure 2). Cancers harboring EGFR mutations depend on constitutive activation of these kinases for survival independent of EGFR. Explanations for the EGFR-TKIs resistance of redundant kinase activation, up to now, have not been fully clarified. Thus, effective blockade of these signaling in primary NSCLC tumors will require precise identification of the active receptor tyrosine kinase pathways through appropriate biomarkers. The development of multi-TKIs with the capacity to inhibit several different receptor tyrosine kinases should also be pursued, as these drugs would represent a more optimal choice than a combination of several different TKIs. It is likely, that specific combinations of selective TKIs will be required to completely inhibit signaling and cell transformation.
Acknowledgements
We thank for the funding support from the Major State Basic Research Development Program of China (973 Program) (NO.2012CB967000).
Disclosure of conflict of interest
Authors have no relevant, potential conflicts of interest to declare.
References
- 1.Yasumoto K, Yamada T, Kawashima A, Wang W, Li Q, Donev IS, Tacheuchi S, Mouri H, Yamashita K, Ohtsubo K, Yano S. The EGFR ligands amphiregulin and heparin-binding egf-like growth factor promote peritoneal carcinomatosis in CXCR4-expressing gastric cancer. Clin Cancer Res. 2011;17:3619–3630. doi: 10.1158/1078-0432.CCR-10-2475. [DOI] [PubMed] [Google Scholar]
- 2.Akhavan D, Pourzia AL, Nourian AA, Williams KJ, Nathanson D, Babic I, Villa GR, Tanaka K, Nael A, Yang H, Dang J, Vinters HV, Yong WH, Flagg M, Tamanoi F, Sasayama T, James CD, Kornblum HI, Cloughesy TF, Cavenee WK, Bensinger SJ, Mischel PS. De-repression of PDGFRbeta transcription promotes acquired resistance to EGFR tyrosine kinase inhibitors in glioblastoma patients. Cancer Discov. 2013;3:534–547. doi: 10.1158/2159-8290.CD-12-0502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pal SK, Figlin RA, Reckamp K. Targeted therapies for non-small cell lung cancer: an evolving landscape. Mol Cancer Ther. 2010;9:1931–1944. doi: 10.1158/1535-7163.MCT-10-0239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Janne PA, Wang X, Socinski MA, Crawford J, Stinchcombe TE, Gu L, Capelletti M, Edelman MJ, Villalona-Calero MA, Kratzke R, Vokes EE, Miller VA. Randomized phase II trial of erlotinib alone or with carboplatin and paclitaxel in patients who were never or light former smokers with advanced lung adenocarcinoma: CALGB 30406 trial. J. Clin. Oncol. 2012;30:2063–2069. doi: 10.1200/JCO.2011.40.1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yu HA, Arcila ME, Rekhtman N, Sima CS, Zakowski MF, Pao W, Kris MG, Miller VA, Ladanyi M, Riely GJ. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin Cancer Res. 2013;19:2240–2247. doi: 10.1158/1078-0432.CCR-12-2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen J, Kosaka T, Holmes AJ, Rogers AM, Cappuzzo F, Mok T, Lee C, Johnson BE, Cantley LC, Janne PA. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316:1039–1043. doi: 10.1126/science.1141478. [DOI] [PubMed] [Google Scholar]
- 7.Jameson MJ, Beckler AD, Taniguchi LE, Allak A, Vanwagner LB, Lee NG, Thomsen WC, Hubbard MA, Thomas CY. Activation of the insulin-like growth factor-1 receptor induces resistance to epidermal growth factor receptor antagonism in head and neck squamous carcinoma cells. Mol Cancer Ther. 2011;10:2124–2134. doi: 10.1158/1535-7163.MCT-11-0294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cretella D, Saccani F, Quaini F, Frati C, Lagrasta C, Bonelli M, Caffarra C, Cavazzoni A, Fumarola C, Galetti M, La Monica S, Ampollini L, Tiseo M, Ardizzoni A, Petronini PG, Alfieri RR. Trastuzumab emtansine is active on HER-2 overexpressing NSCLC cell lines and overcomes gefitinib resistance. Mol Cancer. 2014;13:143. doi: 10.1186/1476-4598-13-143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Noto A, De Vitis C, Roscilli G, Fattore L, Malpicci D, Marra E, Luberto L, D’Andrilli A, Coluccia P, Giovagnoli MR, Normanno N, Ruco L, Aurisicchio L, Mancini R, Ciliberto G. Combination therapy with anti-ErbB3 monoclonal antibodies and EGFR TKIs potently inhibits non-small cell lung cancer. Oncotarget. 2013;4:1253–1265. doi: 10.18632/oncotarget.1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang Z, Lee JC, Lin L, Olivas V, Au V, LaFramboise T, Abdel-Rahman M, Wang X, Levine AD, Rho JK, Choi YJ, Choi CM, Kim SW, Jang SJ, Park YS, Kim WS, Lee DH, Lee JS, Miller VA, Arcila M, Ladanyi M, Moonsamy P, Sawyers C, Boggon TJ, Ma PC, Costa C, Taron M, Rosell R, Halmos B, Bivona TG. Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat Genet. 2012;44:852–860. doi: 10.1038/ng.2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Terai H, Soejima K, Yasuda H, Nakayama S, Hamamoto J, Arai D, Ishioka K, Ohgino K, Ikemura S, Sato T, Yoda S, Satomi R, Naoki K, Betsuyaku T. Activation of the FGF2-FGFR1 autocrine pathway: a novel mechanism of acquired resistance to gefitinib in NSCLC. Mol Cancer Res. 2013;11:759–767. doi: 10.1158/1541-7786.MCR-12-0652. [DOI] [PubMed] [Google Scholar]
- 12.Korpanty G, Smyth E, Carney DN. Update on anti-angiogenic therapy in non-small cell lung cancer: Are we making progress? J Thorac Dis. 2011;3:19–29. doi: 10.3978/j.issn.2072-1439.2010.11.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim SM, Kwon OJ, Hong YK, Kim JH, Solca F, Ha SJ, Soo RA, Christensen JG, Lee JH, Cho BC. Activation of IL-6R/JAK1/STAT3 signaling induces de novo resistance to irreversible EGFR inhibitors in non-small cell lung cancer with T790M resistance mutation. Mol Cancer Ther. 2012;11:2254–2264. doi: 10.1158/1535-7163.MCT-12-0311. [DOI] [PubMed] [Google Scholar]
- 14.Ludovini V, Bianconi F, Pistola L, Chiari R, Minotti V, Colella R, Giuffrida D, Tofanetti FR, Siggillino A, Flacco A, Baldelli E, Iacono D, Mameli MG, Cavaliere A, Crino L. Phosphoi- nositide-3-kinase catalytic alpha and KRAS mutations are important predictors of resistance to therapy with epidermal growth factor receptor tyrosine kinase inhibitors in patients with advanced non-small cell lung cancer. J Thorac Oncol. 2011;6:707–715. doi: 10.1097/JTO.0b013e31820a3a6b. [DOI] [PubMed] [Google Scholar]
- 15.Yamamoto C, Basaki Y, Kawahara A, Nakashima K, Kage M, Izumi H, Kohno K, Uramoto H, Yasumoto K, Kuwano M, Ono M. Loss of PTEN expression by blocking nuclear translocation of EGR1 in gefitinib-resistant lung cancer cells harboring epidermal growth factor receptor-activating mutations. Cancer Res. 2010;70:8715–8725. doi: 10.1158/0008-5472.CAN-10-0043. [DOI] [PubMed] [Google Scholar]
- 16.Sequist LV, Waltman BA, Dias-Santagata D, Digumarthy S, Turke AB, Fidias P, Bergethon K, Shaw AT, Gettinger S, Cosper AK, Akhavanfard S, Heist RS, Temel J, Christensen JG, Wain JC, Lynch TJ, Vernovsky K, Mark EJ, Lanuti M, Iafrate AJ, Mino-Kenudson M, Engelman JA. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med. 2011;3:75ra26. doi: 10.1126/scitranslmed.3002003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Suda K, Tomizawa K, Fujii M, Murakami H, Osada H, Maehara Y, Yatabe Y, Sekido Y, Mitsudomi T. Epithelial to mesenchymal transition in an epidermal growth factor receptor-mutant lung cancer cell line with acquired resistance to erlotinib. J Thorac Oncol. 2011;6:1152–1161. doi: 10.1097/JTO.0b013e318216ee52. [DOI] [PubMed] [Google Scholar]
- 18.Thomson S, Petti F, Sujka-Kwok I, Epstein D, Haley JD. Kinase switching in mesenchymal-like non-small cell lung cancer lines contributes to EGFR inhibitor resistance through pathway redundancy. Clin Exp Metastasis. 2008;25:843–854. doi: 10.1007/s10585-008-9200-4. [DOI] [PubMed] [Google Scholar]
- 19.Yoshida T, Zhang G, Smith MA, Lopez AS, Bai Y, Li J, Fang B, Koomen J, Rawal B, Fisher KJ, Chen AY, Kitano M, Morita Y, Yamaguchi H, Shibata K, Okabe T, Okamoto I, Nakagawa K, Haura EB. Tyrosine Phosphoproteomics Identifies Both Codrivers and Cotargeting Strategies for T790M-Related EGFR-TKI Resistance in Non-Small Cell Lung Cancer. Clin Cancer Res. 2014;20:4059–4074. doi: 10.1158/1078-0432.CCR-13-1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bertotti A, Burbridge MF, Gastaldi S, Galimi F, Torti D, Medico E, Giordano S, Corso S, Rolland-Valognes G, Lockhart BP, Hickman JA, Comoglio PM, Trusolino L. Only a subset of Met-activated pathways are required to sustain oncogene addiction. Sci Signal. 2009;2:er11. doi: 10.1126/scisignal.2000643. [DOI] [PubMed] [Google Scholar]
- 21.Ha SY, Lee J, Kang SY, Do IG, Ahn S, Park JO, Kang WK, Choi MG, Sohn TS, Bae JM, Kim S, Kim M, Kim S, Park CK, Ignatius Ou SH, Kim KM. MET overexpression assessed by new interpretation method predicts gene amplification and poor survival in advanced gastric carcinomas. Mod Pathol. 2013;26:1632–1641. doi: 10.1038/modpathol.2013.108. [DOI] [PubMed] [Google Scholar]
- 22.Yano S, Yamada T, Takeuchi S, Tachibana K, Minami Y, Yatabe Y, Mitsudomi T, Tanaka H, Kimura T, Kudoh S, Nokihara H, Ohe Y, Yokota J, Uramoto H, Yasumoto K, Kiura K, Higashiyama M, Oda M, Saito H, Yoshida J, Kondoh K, Noguchi M. Hepatocyte growth factor expression in EGFR mutant lung cancer with intrinsic and acquired resistance to tyrosine kinase inhibitors in a Japanese cohort. J Thorac Oncol. 2011;6:2011–2017. doi: 10.1097/JTO.0b013e31823ab0dd. [DOI] [PubMed] [Google Scholar]
- 23.Troiani T, Martinelli E, Napolitano S, Vitagliano D, Ciuffreda LP, Costantino S, Morgillo F, Capasso A, Sforza V, Nappi A, De Palma R, D’Aiuto E, Berrino L, Bianco R, Ciardiello F. Incre- ased TGF-alpha as a mechanism of acquired resistance to the anti-EGFR inhibitor cetuximab through EGFR-MET interaction and activation of MET signaling in colon cancer cells. Clin Cancer Res. 2013;19:6751–6765. doi: 10.1158/1078-0432.CCR-13-0423. [DOI] [PubMed] [Google Scholar]
- 24.Yano S, Nakagawa T. The current state of molecularly targeted drugs targeting HGF/Met. Jpn J Clin Oncol. 2014;44:9–12. doi: 10.1093/jjco/hyt188. [DOI] [PubMed] [Google Scholar]
- 25.Mueller KL, Madden JM, Zoratti GL, Kuperwasser C, List K, Boerner JL. Fibroblast-secreted hepatocyte growth factor mediates epidermal growth factor receptor tyrosine kinase inhibitor resistance in triple-negative breast cancers through paracrine activation of Met. Breast Cancer Res. 2012;14:R104. doi: 10.1186/bcr3224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Turke AB, Zejnullahu K, Wu YL, Song Y, Dias-Santagata D, Lifshits E, Toschi L, Rogers A, Mok T, Sequist L, Lindeman NI, Murphy C, Akhavanfard S, Yeap BY, Xiao Y, Capelletti M, Iafrate AJ, Lee C, Christensen JG, Engelman JA, Janne PA. Preexistence and clonal selection of MET amplification in EGFR mutant NSCLC. Cancer Cell. 2010;17:77–88. doi: 10.1016/j.ccr.2009.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kubo T, Yamamoto H, Lockwood WW, Valencia I, Soh J, Peyton M, Jida M, Otani H, Fujii T, Ouchida M, Takigawa N, Kiura K, Shimizu K, Date H, Minna JD, Varella-Garcia M, Lam WL, Gazdar AF, Toyooka S. MET gene amplification or EGFR mutation activate MET in lung cancers untreated with EGFR tyrosine kinase inhibitors. Int J Cancer. 2009;124:1778–1784. doi: 10.1002/ijc.24150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tanizaki J, Okamoto I, Sakai K, Nakagawa K. Differential roles of trans-phosphorylated EGFR, HER2, HER3, and RET as heterodimerisation partners of MET in lung cancer with MET amplification. Br J Cancer. 2011;105:807–813. doi: 10.1038/bjc.2011.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ma J, Lyu H, Huang J, Liu B. Targeting of erbB3 receptor to overcome resistance in cancer treatment. Mol Cancer. 2014;13:105. doi: 10.1186/1476-4598-13-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mueller KL, Yang ZQ, Haddad R, Ethier SP, Boerner JL. EGFR/Met association regulates EGFR TKI resistance in breast cancer. J Mol Signal. 2010;5:8. doi: 10.1186/1750-2187-5-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen G, Noor A, Kronenberger P, Teugels E, Umelo IA, De Greve J. Synergistic effect of afatinib with su11274 in non-small cell lung cancer cells resistant to gefitinib or erlotinib. PLoS One. 2013;8:e59708. doi: 10.1371/journal.pone.0059708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rho JK, Choi YJ, Lee JK, Ryoo BY, Na II, Yang SH, Lee SS, Kim CH, Yoo YD, Lee JC. The role of MET activation in determining the sensitivity to epidermal growth factor receptor tyrosine kinase inhibitors. Mol Cancer Res. 2009;7:1736–1743. doi: 10.1158/1541-7786.MCR-08-0504. [DOI] [PubMed] [Google Scholar]
- 33.Kang XH, Xu ZY, Gong YB, Wang LF, Wang ZQ, Xu L, Cao F, Liao MJ. Bufalin Reverses HGF-Induced Resistance to EGFR-TKIs in EGFR Mutant Lung Cancer Cells via Blockage of Met/PI3k/Akt Pathway and Induction of Apoptosis. Evid Based Complement Alternat Med. 2013;2013:243859. doi: 10.1155/2013/243859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang W, Li Q, Yamada T, Matsumoto K, Matsumoto I, Oda M, Watanabe G, Kayano Y, Nishioka Y, Sone S, Yano S. Crosstalk to stromal fibroblasts induces resistance of lung cancer to epidermal growth factor receptor tyrosine kinase inhibitors. Clin Cancer Res. 2009;15:6630–6638. doi: 10.1158/1078-0432.CCR-09-1001. [DOI] [PubMed] [Google Scholar]
- 35.Donev IS, Wang W, Yamada T, Li Q, Takeuchi S, Matsumoto K, Yamori T, Nishioka Y, Sone S, Yano S. Transient PI3K inhibition induces apoptosis and overcomes HGF-mediated resistance to EGFR-TKIs in EGFR mutant lung cancer. Clin Cancer Res. 2011;17:2260–2269. doi: 10.1158/1078-0432.CCR-10-1993. [DOI] [PubMed] [Google Scholar]
- 36.Koizumi H, Yamada T, Takeuchi S, Nakagawa T, Kita K, Nakamura T, Matsumoto K, Suda K, Mitsudomi T, Yano S. Hsp90 inhibition overcomes HGF-triggering resistance to EGFR-TKIs in EGFR-mutant lung cancer by decreasing client protein expression and angiogenesis. J Thorac Oncol. 2012;7:1078–1085. doi: 10.1097/JTO.0b013e3182519a2c. [DOI] [PubMed] [Google Scholar]
- 37.Ju L, Zhou C. Integrin beta 1 enhances the epithelial-mesenchymal transition in association with gefitinib resistance of non-small cell lung cancer. Cancer Biomark. 2013;13:329–336. doi: 10.3233/CBM-130362. [DOI] [PubMed] [Google Scholar]
- 38.McCall-Culbreath KD, Li Z, Zutter MM. Crosstalk between the alpha2beta1 integrin and c-met/HGF-R regulates innate immunity. Blood. 2008;111:3562–3570. doi: 10.1182/blood-2007-08-107664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu T, Li Q, Sun Q, Zhang Y, Yang H, Wang R, Chen L, Wang W. MET inhibitor PHA-665752 suppresses the hepatocyte growth factor-induced cell proliferation and radioresistance in nasopharyngeal carcinoma cells. Biochem Biophys Res Commun. 2014;449:49–54. doi: 10.1016/j.bbrc.2014.04.147. [DOI] [PubMed] [Google Scholar]
- 40.Rho JK, Choi YJ, Kim SY, Kim TW, Choi EK, Yoon SJ, Park BM, Park E, Bae JH, Choi CM, Lee JC. MET and AXL inhibitor NPS-1034 exerts efficacy against lung cancer cells resistant to EGFR kinase inhibitors because of MET or AXL activation. Cancer Res. 2014;74:253–262. doi: 10.1158/0008-5472.CAN-13-1103. [DOI] [PubMed] [Google Scholar]
- 41.Berghoff AS, Bartsch R, Preusser M, Ricken G, Steger GG, Bago-Horvath Z, Rudas M, Streubel B, Dubsky P, Gnant M, Fitzal F, Zielinski CC, Birner P. Co-overexpression of HER2/HER3 is a predictor of impaired survival in breast cancer patients. Breast. 2014;23:637–43. doi: 10.1016/j.breast.2014.06.011. [DOI] [PubMed] [Google Scholar]
- 42.Takezawa K, Pirazzoli V, Arcila ME, Nebhan CA, Song X, de Stanchina E, Ohashi K, Janjigian YY, Spitzler PJ, Melnick MA, Riely GJ, Kris MG, Miller VA, Ladanyi M, Politi K, Pao W. HER2 amplification: a potential mechanism of acquired resistance to EGFR inhibition in EGFR-mutant lung cancers that lack the second-site EGFRT790M mutation. Cancer Discov. 2012;2:922–933. doi: 10.1158/2159-8290.CD-12-0108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bertotti A, Migliardi G, Galimi F, Sassi F, Torti D, Isella C, Cora D, Di Nicolantonio F, Buscarino M, Petti C, Ribero D, Russolillo N, Muratore A, Massucco P, Pisacane A, Molinaro L, Valtorta E, Sartore-Bianchi A, Risio M, Capussotti L, Gambacorta M, Siena S, Medico E, Sapino A, Marsoni S, Comoglio PM, Bardelli A, Trusolino L. A molecularly annotated platform of patient-derived xenografts (“xenopatients”) identifies HER2 as an effective therapeutic target in cetuximab-resistant colorectal cancer. Cancer Discov. 2011;1:508–523. doi: 10.1158/2159-8290.CD-11-0109. [DOI] [PubMed] [Google Scholar]
- 44.Liu B, Fan Z, Edgerton SM, Yang X, Lind SE, Thor AD. Potent anti-proliferative effects of metformin on trastuzumab-resistant breast cancer cells via inhibition of erbB2/IGF-1 receptor interactions. Cell Cycle. 2011;10:2959–2966. doi: 10.4161/cc.10.17.16359. [DOI] [PubMed] [Google Scholar]
- 45.Gallardo A, Lerma E, Escuin D, Tibau A, Munoz J, Ojeda B, Barnadas A, Adrover E, Sanchez-Tejada L, Giner D, Ortiz-Martinez F, Peiro G. Increased signalling of EGFR and IGF1R, and deregulation of PTEN/PI3K/Akt pathway are related with trastuzumab resistance in HER2 breast carcinomas. Br J Cancer. 2012;106:1367–1373. doi: 10.1038/bjc.2012.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jones HE, Gee JM, Hutcheson IR, Knowlden JM, Barrow D, Nicholson RI. Growth factor receptor interplay and resistance in cancer. Endocr Relat Cancer. 2006;13(Suppl 1):S45–51. doi: 10.1677/erc.1.01275. [DOI] [PubMed] [Google Scholar]
- 47.Janjigian YY, Smit EF, Groen HJ, Horn L, Gettinger S, Camidge DR, Riely GJ, Wang B, Fu Y, Chand VK, Miller VA, Pao W. Dual Inhibition of EGFR with Afatinib and Cetuximab in Kinase Inhibitor-Resistant EGFR-Mutant Lung Cancer with and without T790M Mutations. Cancer Discov. 2014;4:1036–45. doi: 10.1158/2159-8290.CD-14-0326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nicos M, Krawczyk P, Mlak R, Sawicki M, Jarosz B, Powrozek T, Milanowski P, Trojanowski T, Milanowski J. The presence of HER2 exon 20 insertion in patients with central nervous system metastases from non-small lung cancer--a potential application in classification for therapy. Pneumonol Alergol Pol. 2013;81:294–297. [PubMed] [Google Scholar]
- 49.Shtiegman K, Kochupurakkal BS, Zwang Y, Pines G, Starr A, Vexler A, Citri A, Katz M, Lavi S, Ben-Basat Y, Benjamin S, Corso S, Gan J, Yosef RB, Giordano S, Yarden Y. Defective ubiquitinylation of EGFR mutants of lung cancer confers prolonged signaling. Oncogene. 2007;26:6968–6978. doi: 10.1038/sj.onc.1210503. [DOI] [PubMed] [Google Scholar]
- 50.Bae SY, La Choi Y, Kim S, Kim M, Kim J, Jung SP, Choi MY, Lee SK, Kil WH, Lee JE, Nam SJ. HER3 status by immunohistochemistry is correlated with poor prognosis in hormone receptor-negative breast cancer patients. Breast Cancer Res Treat. 2013;139:741–750. doi: 10.1007/s10549-013-2570-6. [DOI] [PubMed] [Google Scholar]
- 51.Bean J, Brennan C, Shih JY, Riely G, Viale A, Wang L, Chitale D, Motoi N, Szoke J, Broderick S, Balak M, Chang WC, Yu CJ, Gazdar A, Pass H, Rusch V, Gerald W, Huang SF, Yang PC, Miller V, Ladanyi M, Yang CH, Pao W. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc Natl Acad Sci U S A. 2007;104:20932–20937. doi: 10.1073/pnas.0710370104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cortot AB, Repellin CE, Shimamura T, Capelletti M, Zejnullahu K, Ercan D, Christensen JG, Wong KK, Gray NS, Janne PA. Resistance to irreversible EGF receptor tyrosine kinase inhibitors through a multistep mechanism involving the IGF1R pathway. Cancer Res. 2013;73:834–843. doi: 10.1158/0008-5472.CAN-12-2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Byun S, Lee SY, Lee J, Jeong CH, Farrand L, Lim S, Reddy K, Kim JY, Lee MH, Lee HJ, Bode AM, Won LK, Dong Z. USP8 is a novel target for overcoming gefitinib resistance in lung cancer. Clin Cancer Res. 2013;19:3894–3904. doi: 10.1158/1078-0432.CCR-12-3696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tabara K, Kanda R, Sonoda K, Kubo T, Murakami Y, Kawahara A, Azuma K, Abe H, Kage M, Yoshinaga A, Tahira T, Hayashi K, Arao T, Nishio K, Rosell R, Kuwano M, Ono M. Loss of activating EGFR mutant gene contributes to acquired resistance to EGFR tyrosine kinase inhibitors in lung cancer cells. PLoS One. 2012;7:e41017. doi: 10.1371/journal.pone.0041017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bar J, Onn A. Overcoming molecular mechanisms of resistance to first-generation epidermal growth factor receptor tyrosine kinase inhibitors. Clin Lung Cancer. 2012;13:267–279. doi: 10.1016/j.cllc.2011.09.001. [DOI] [PubMed] [Google Scholar]
- 56.Kasahara K, Arao T, Sakai K, Matsumoto K, Sakai A, Kimura H, Sone T, Horiike A, Nishio M, Ohira T, Ikeda N, Yamanaka T, Saijo N, Nishio K. Impact of serum hepatocyte growth factor on treatment response to epidermal growth factor receptor tyrosine kinase inhibitors in patients with non-small cell lung adenocarcinoma. Clin Cancer Res. 2010;16:4616–4624. doi: 10.1158/1078-0432.CCR-10-0383. [DOI] [PubMed] [Google Scholar]
- 57.Tanaka H, Kimura T, Kudoh S, Mitsuoka S, Watanabe T, Suzumura T, Tachibana K, Noguchi M, Yano S, Hirata K. Reaction of plasma hepatocyte growth factor levels in non-small cell lung cancer patients treated with EGFR-TKIs. Int J Cancer. 2011;129:1410–1416. doi: 10.1002/ijc.25799. [DOI] [PubMed] [Google Scholar]
- 58.Tabernero J. The role of VEGF and EGFR inhibition: implications for combining anti-VEGF and anti-EGFR agents. Mol Cancer Res. 2007;5:203–220. doi: 10.1158/1541-7786.MCR-06-0404. [DOI] [PubMed] [Google Scholar]
- 59.Miyahara K, Nouso K, Tomoda T, Kobayashi S, Hagihara H, Kuwaki K, Toshimori J, Onishi H, Ikeda F, Miyake Y, Nakamura S, Shiraha H, Takaki A, Yamamoto K. Predicting the treatment effect of sorafenib using serum angiogenesis markers in patients with hepatocellular carcinoma. J Gastroenterol Hepatol. 2011;26:1604–1611. doi: 10.1111/j.1440-1746.2011.06887.x. [DOI] [PubMed] [Google Scholar]
- 60.Nakagawa T, Matsushima T, Kawano S, Nakazawa Y, Kato Y, Adachi Y, Abe T, Semba T, Yokoi A, Matsui J, Tsuruoka A, Funahashi Y. Lenvatinib in combination with golvatinib overcomes hepatocyte growth factor pathway-induced resistance to vascular endothelial growth factor receptor inhibitor. Cancer Sci. 2014;105:723–730. doi: 10.1111/cas.12409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shojaei F, Lee JH, Simmons BH, Wong A, Esparza CO, Plumlee PA, Feng J, Stewart AE, Hu-Lowe DD, Christensen JG. HGF/c-Met acts as an alternative angiogenic pathway in sunitinib-resistant tumors. Cancer Res. 2010;70:10090–10100. doi: 10.1158/0008-5472.CAN-10-0489. [DOI] [PubMed] [Google Scholar]
- 62.Keedy VL, Sandler AB. Inhibition of angiogenesis in the treatment of non-small cell lung cancer. Cancer Sci. 2007;98:1825–1830. doi: 10.1111/j.1349-7006.2007.00620.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Casanovas O, Hicklin DJ, Bergers G, Hanahan D. Drug resistance by evasion of antiangiogenic targeting of VEGF signaling in late-stage pancreatic islet tumors. Cancer Cell. 2005;8:299–309. doi: 10.1016/j.ccr.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 64.Takeuchi S, Wang W, Li Q, Yamada T, Kita K, Donev IS, Nakamura T, Matsumoto K, Shimizu E, Nishioka Y, Sone S, Nakagawa T, Uenaka T, Yano S. Dual inhibition of Met kinase and angiogenesis to overcome HGF-induced EGFR-TKI resistance in EGFR mutant lung cancer. Am J Pathol. 2012;181:1034–1043. doi: 10.1016/j.ajpath.2012.05.023. [DOI] [PubMed] [Google Scholar]
- 65.Nakagawa T, Tohyama O, Yamaguchi A, Matsushima T, Takahashi K, Funasaka S, Shirotori S, Asada M, Obaishi H. E7050: a dual c-Met and VEGFR-2 tyrosine kinase inhibitor promotes tumor regression and prolongs survival in mouse xenograft models. Cancer Sci. 2010;101:210–215. doi: 10.1111/j.1349-7006.2009.01343.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rosenzweig SA, Atreya HS. Defining the pathway to insulin-like growth factor system targeting in cancer. Biochem Pharmacol. 2010;80:1115–1124. doi: 10.1016/j.bcp.2010.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Murphy CT, Hu PJ. Insulin/insulin-like growth factor signaling in C. elegans. WormBook. 2013:1–43. doi: 10.1895/wormbook.1.164.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Desbois-Mouthon C, Baron A, Blivet-Van Eggelpoel MJ, Fartoux L, Venot C, Bladt F, Housset C, Rosmorduc O. Insulin-like growth factor-1 receptor inhibition induces a resistance mechanism via the epidermal growth factor receptor/HER3/AKT signaling pathway: rational basis for cotargeting insulin-like growth factor-1 receptor and epidermal growth factor receptor in hepatocellular carcinoma. Clin Cancer Res. 2009;15:5445–5456. doi: 10.1158/1078-0432.CCR-08-2980. [DOI] [PubMed] [Google Scholar]
- 69.Suda K, Mizuuchi H, Sato K, Takemoto T, Iwasaki T, Mitsudomi T. The insulin-like growth factor 1 receptor causes acquired resistance to erlotinib in lung cancer cells with the wild-type epidermal growth factor receptor. Int J Cancer. 2014;135:1002–1006. doi: 10.1002/ijc.28737. [DOI] [PubMed] [Google Scholar]
- 70.Dziadziuszko R, Merrick DT, Witta SE, Mendoza AD, Szostakiewicz B, Szymanowska A, Rzyman W, Dziadziuszko K, Jassem J, Bunn PA Jr, Varella-Garcia M, Hirsch FR. Insulin-like growth factor receptor 1 (IGF1R) gene copy number is associated with survival in operable non-small-cell lung cancer: a comparison between IGF1R fluorescent in situ hybridization, protein expression, and mRNA expression. J. Clin. Oncol. 2010;28:2174–2180. doi: 10.1200/JCO.2009.24.6611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kim WY, Prudkin L, Feng L, Kim ES, Hennessy B, Lee JS, Lee JJ, Glisson B, Lippman SM, Wistuba II, Hong WK, Lee HY. Epidermal growth factor receptor and K-Ras mutations and resistance of lung cancer to insulin-like growth factor 1 receptor tyrosine kinase inhibitors. Cancer. 2012;118:3993–4003. doi: 10.1002/cncr.26656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kim WY, Jin Q, Oh SH, Kim ES, Yang YJ, Lee DH, Feng L, Behrens C, Prudkin L, Miller YE, Lee JJ, Lippman SM, Hong WK, Wistuba II, Lee HY. Elevated epithelial insulin-like growth factor expression is a risk factor for lung cancer development. Cancer Res. 2009;69:7439–7448. doi: 10.1158/0008-5472.CAN-08-3792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Durfort T, Tkach M, Meschaninova MI, Rivas MA, Elizalde PV, Venyaminova AG, Schillaci R, Francois JC. Small interfering RNA targeted to IGF-IR delays tumor growth and induces proinflammatory cytokines in a mouse breast cancer model. PLoS One. 2012;7:e29213. doi: 10.1371/journal.pone.0029213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tian D, Kreeger PK. Analysis of the quantitative balance between insulin-like growth factor (IGF)-1 ligand, receptor, and binding protein levels to predict cell sensitivity and therapeutic efficacy. BMC Syst Biol. 2014;8:98. doi: 10.1186/s12918-014-0098-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sax AT, Jenkins DG, Devin JL, Hughes GI, Bolam KA, Skinner TL. The insulin-like growth factor axis: A biological mechanism linking physical activity to colorectal cancer survival. Cancer Epidemiol. 2014;38:455–459. doi: 10.1016/j.canep.2014.05.011. [DOI] [PubMed] [Google Scholar]
- 76.Tas F, Karabulut S, Bilgin E, Tastekin D, Duranyildiz D. Clinical significance of serum insulin-like growth factor-1 (IGF-1) and insulin-like growth factor binding protein-3 (IGFBP-3) in patients with breast cancer. Tumour Biol. 2014;35:9303–9. doi: 10.1007/s13277-014-2224-2. [DOI] [PubMed] [Google Scholar]
- 77.Choi YJ, Rho JK, Jeon BS, Choi SJ, Park SC, Lee SS, Kim HR, Kim CH, Lee JC. Combined inhibition of IGFR enhances the effects of gefitinib in H1650: a lung cancer cell line with EGFR mutation and primary resistance to EGFR-TK inhibitors. Cancer Chemother Pharmacol. 2010;66:381–388. doi: 10.1007/s00280-009-1174-7. [DOI] [PubMed] [Google Scholar]
- 78.Choi YJ, Park GM, Rho JK, Kim SY, So GS, Kim HR, Choi CM, Lee JC. Role of IGF-binding protein 3 in the resistance of EGFR mutant lung cancer cells to EGFR-tyrosine kinase inhibitors. PLoS One. 2013;8:e81393. doi: 10.1371/journal.pone.0081393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Dokmanovic M, Shen Y, Bonacci TM, Hirsch DS, Wu WJ. Trastuzumab regulates IGFBP-2 and IGFBP-3 to mediate growth inhibition: implications for the development of predictive biomarkers for trastuzumab resistance. Mol Cancer Ther. 2011;10:917–928. doi: 10.1158/1535-7163.MCT-10-0980. [DOI] [PubMed] [Google Scholar]
- 80.Hurbin A, Wislez M, Busser B, Antoine M, Tenaud C, Rabbe N, Dufort S, de Fraipont F, Moro-Sibilot D, Cadranel J, Coll JL, Brambilla E. Insulin-like growth factor-1 receptor inhibition overcomes gefitinib resistance in mucinous lung adenocarcinoma. J Pathol. 2011;225:83–95. doi: 10.1002/path.2897. [DOI] [PubMed] [Google Scholar]
- 81.Busser B, Sancey L, Josserand V, Niang C, Khochbin S, Favrot MC, Coll JL, Hurbin A. Amphiregulin promotes resistance to gefitinib in nonsmall cell lung cancer cells by regulating Ku70 acetylation. Mol Ther. 2010;18:536–543. doi: 10.1038/mt.2009.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Morgillo F, Kim WY, Kim ES, Ciardiello F, Hong WK, Lee HY. Implication of the insulin-like growth factor-IR pathway in the resistance of non-small cell lung cancer cells to treatment with gefitinib. Clin Cancer Res. 2007;13:2795–2803. doi: 10.1158/1078-0432.CCR-06-2077. [DOI] [PubMed] [Google Scholar]
- 83.Gong Y, Yao E, Shen R, Goel A, Arcila M, Teruya-Feldstein J, Zakowski MF, Frankel S, Peifer M, Thomas RK, Ladanyi M, Pao W. High expression levels of total IGF-1R and sensitivity of NSCLC cells in vitro to an anti-IGF-1R antibody (R1507) PLoS One. 2009;4:e7273. doi: 10.1371/journal.pone.0007273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Brooks AN, Kilgour E, Smith PD. Molecular pathways: fibroblast growth factor signaling: a new therapeutic opportunity in cancer. Clin Cancer Res. 2012;18:1855–1862. doi: 10.1158/1078-0432.CCR-11-0699. [DOI] [PubMed] [Google Scholar]
- 85.Azuma K, Kawahara A, Sonoda K, Nakashima K, Tashiro K, Watari K, Izumi H, Kage M, Kuwano M, Ono M, Hoshino T. FGFR1 activation is an escape mechanism in human lung cancer cells resistant to afatinib, a pan-EGFR family kinase inhibitor. Oncotarget. 2014;5:5908–19. doi: 10.18632/oncotarget.1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Seo AN, Jin Y, Lee HJ, Sun PL, Kim H, Jheon S, Kim K, Lee CT, Chung JH. FGFR1 amplification is associated with poor prognosis and smoking in non-small-cell lung cancer. Virch- ows Arch. 2014 doi: 10.1007/s00428-014-1634-2. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 87.Behrens C, Lin HY, Lee JJ, Raso MG, Hong WK, Wistuba II, Lotan R. Immunohistochemical expression of basic fibroblast growth factor and fibroblast growth factor receptors 1 and 2 in the pathogenesis of lung cancer. Clin Cancer Res. 2008;14:6014–6022. doi: 10.1158/1078-0432.CCR-08-0167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Donnem T, Al-Shibli K, Al-Saad S, Busund LT, Bremnes RM. Prognostic impact of fibroblast growth factor 2 in non-small cell lung cancer: coexpression with VEGFR-3 and PDGF-B predicts poor survival. J Thorac Oncol. 2009;4:578–585. doi: 10.1097/JTO.0b013e31819f2e38. [DOI] [PubMed] [Google Scholar]
- 89.Dutt A, Ramos AH, Hammerman PS, Mermel C, Cho J, Sharifnia T, Chande A, Tanaka KE, Stransky N, Greulich H, Gray NS, Meyerson M. Inhibitor-sensitive FGFR1 amplification in human non-small cell lung cancer. PLoS One. 2011;6:e20351. doi: 10.1371/journal.pone.0020351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Weiss J, Sos ML, Seidel D, Peifer M, Zander T, Heuckmann JM, Ullrich RT, Menon R, Maier S, Soltermann A, Moch H, Wagener P, Fischer F, Heynck S, Koker M, Schöttle J, Leenders F, Gabler F, Dabow I, Querings S, Heukamp LC, Balke-Want H, Ansén S, Rauh D, Baessmann I, Altmüller J, Wainer Z, Conron M, Wright G, Russell P, Solomon B, Brambilla E, Brambilla C, Lorimier P, Sollberg S, Brustugun OT, Engel-Riedel W, Ludwig C, Petersen I, Sänger J, Clement J, Groen H, Timens W, Sietsma H, Thunnissen E, Smit E, Heideman D, Cappuzzo F, Ligorio C, Damiani S, Hallek M, Beroukhim R, Pao W, Klebl B, Baumann M, Buettner R, Ernestus K, Stoelben E, Wolf J, Nürnberg P, Perner S, Thomas RK. Frequent and focal FGFR1 amplification associates with therapeutically tractable FGFR1 dependency in squamous cell lung cancer. Sci Transl Med. 2010;2:62ra93. doi: 10.1126/scitranslmed.3001451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Rikova K, Guo A, Zeng Q, Possemato A, Yu J, Haack H, Nardone J, Lee K, Reeves C, Li Y, Hu Y, Tan Z, Stokes M, Sullivan L, Mitchell J, Wetzel R, Macneill J, Ren JM, Yuan J, Bakalarski CE, Villen J, Kornhauser JM, Smith B, Li D, Zhou X, Gygi SP, Gu TL, Polakiewicz RD, Rush J, Comb MJ. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell. 2007;131:1190–1203. doi: 10.1016/j.cell.2007.11.025. [DOI] [PubMed] [Google Scholar]
- 92.Fischbach A, Rogler A, Erber R, Stoehr R, Poulsom R, Heidenreich A, Schneevoigt BS, Hauke S, Hartmann A, Knuechel R, Veeck J, Gaisa NT. Fibroblast Growth Factor Receptor (FGFR) amplifications are rare events in bladder cancer. Histopathology. 2014 doi: 10.1111/his.12473. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 93.Ware KE, Marshall ME, Heasley LR, Marek L, Hinz TK, Hercule P, Helfrich BA, Doebele RC, Heasley LE. Rapidly acquired resistance to EGFR tyrosine kinase inhibitors in NSCLC cell lines through de-repression of FGFR2 and FGFR3 expression. PLoS One. 2010;5:e14117. doi: 10.1371/journal.pone.0014117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Marek L, Ware KE, Fritzsche A, Hercule P, Helton WR, Smith JE, McDermott LA, Coldren CD, Nemenoff RA, Merrick DT, Helfrich BA, Bunn PJ, Heasley LE. Fibroblast growth factor (FGF) and FGF receptor-mediated autocrine signaling in non-small-cell lung cancer cells. Mol Pharmacol. 2009;75:196–207. doi: 10.1124/mol.108.049544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ronca R, Benzoni P, Leali D, Urbinati C, Belleri M, Corsini M, Alessi P, Coltrini D, Calza S, Presta M, Dell’Era P. Antiangiogenic activity of a neutralizing human single-chain antibody fragment against fibroblast growth factor receptor 1. Mol Cancer Ther. 2010;9:3244–3253. doi: 10.1158/1535-7163.MCT-10-0417. [DOI] [PubMed] [Google Scholar]
- 96.Zhou D, Jiang X, Ding W, Zheng L, Yang L, Zheng C, Lu L. siRNA-participated chemotherapy: an efficient and specific therapeutic against gastric cancer. J Cancer Res Clin Oncol. 2013 doi: 10.1007/s00432-013-1492-3. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 97.Spiegelberg C, Giedl J, Gaisa NT, Rogler A, Riener MO, Filbeck T, Burger M, Ruemmele P, Hartmann A, Stoehr R. Frequency of activating mutations in FGFR2 exon 7 in bladder tumors from patients with early-onset and regular-onset disease. Int J Clin Exp Pathol. 2014;7:1708–1713. [PMC free article] [PubMed] [Google Scholar]
- 98.Dutt A, Salvesen HB, Chen TH, Ramos AH, Onofrio RC, Hatton C, Nicoletti R, Winckler W, Grewal R, Hanna M, Wyhs N, Ziaugra L, Richter DJ, Trovik J, Engelsen IB, Stefansson IM, Fen-nell T, Cibulskis K, Zody MC, Akslen LA, Gabriel S, Wong KK, Sellers WR, Meyerson M, Greulich H. Drug-sensitive FGFR2 mutations in endometrial carcinoma. Proc Natl Acad Sci U S A. 2008;105:8713–8717. doi: 10.1073/pnas.0803379105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ding L, Getz G, Wheeler DA, Mardis ER, McLellan MD, Cibulskis K, Sougnez C, Greulich H, Muzny DM, Morgan MB, Fulton L, Fulton RS, Zhang Q, Wendl MC, Lawrence MS, Larson DE, Chen K, Dooling DJ, Sabo A, Hawes AC, Shen H, Jhangiani SN, Lewis LR, Hall O, Zhu Y, Mathew T, Ren Y, Yao J, Scherer SE, Clerc K, Metcalf GA, Ng B, Milosavljevic A, Gonzalez-Garay ML, Osborne JR, Meyer R, Shi X, Tang Y, Koboldt DC, Lin L, Abbott R, Miner TL, Pohl C, Fewell G, Haipek C, Schmidt H, Dunford-Shore BH, Kraja A, Crosby SD, Sawyer CS, Vickery T, Sander S, Robinson J, Winckler W, Baldwin J, Chirieac LR, Dutt A, Fennell T, Hanna M, Johnson BE, Onofrio RC, Thomas RK, Tonon G, Weir BA, Zhao X, Ziaugra L, Zody MC, Giordano T, Orringer MB, Roth JA, Spitz MR, Wistuba II, Ozenberger B, Good PJ, Chang AC, Beer DG, Watson MA, Ladanyi M, Broderick S, Yoshizawa A, Travis WD, Pao W, Province MA, Weinstock GM, Varmus HE, Gabriel SB, Lander ES, Gibbs RA, Meyerson M, Wilson RK. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 2008;455:1069–1075. doi: 10.1038/nature07423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Qian X, Anzovino A, Kim S, Suyama K, Yao J, Hulit J, Agiostratidou G, Chandiramani N, McDaid HM, Nagi C, Cohen HW, Phillips GR, Norton L, Hazan RB. N-cadherin/FGFR promotes metastasis through epithelial-to-mesenchymal transition and stem/progenitor cell-like properties. Oncogene. 2014;33:3411–3421. doi: 10.1038/onc.2013.310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Andrae J, Gallini R, Betsholtz C. Role of platelet-derived growth factors in physiology and medicine. Genes Dev. 2008;22:1276–1312. doi: 10.1101/gad.1653708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Chu JS, Ge FJ, Zhang B, Wang Y, Silvestris N, Liu LJ, Zhao CH, Lin L, Brunetti AE, Fu YL, Wang J, Paradiso A, Xu JM. Expression and prognostic value of VEGFR-2, PDGFR-beta, and c-Met in advanced hepatocellular carcinoma. J Exp Clin Cancer Res. 2013;32:16. doi: 10.1186/1756-9966-32-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Brennan C, Momota H, Hambardzumyan D, Ozawa T, Tandon A, Pedraza A, Holland E. Glioblastoma subclasses can be defined by activity among signal transduction pathways and associated genomic alterations. PLoS One. 2009;4:e7752. doi: 10.1371/journal.pone.0007752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Assanah MC, Bruce JN, Suzuki SO, Chen A, Goldman JE, Canoll P. PDGF stimulates the massive expansion of glial progenitors in the neonatal forebrain. Glia. 2009;57:1835–1847. doi: 10.1002/glia.20895. [DOI] [PubMed] [Google Scholar]
- 106.Kassouf W, Dinney CP, Brown G, McConkey DJ, Diehl AJ, Bar-Eli M, Adam L. Uncoupling between epidermal growth factor receptor and downstream signals defines resistance to the antiproliferative effect of Gefitinib in bladder cancer cells. Cancer Res. 2005;65:10524–10535. doi: 10.1158/0008-5472.CAN-05-1536. [DOI] [PubMed] [Google Scholar]
- 107.Zhang H, Bajraszewski N, Wu E, Wang H, Moseman AP, Dabora SL, Griffin JD, Kwiatkowski DJ. PDGFRs are critical for PI3K/Akt activation and negatively regulated by mTOR. J Clin Invest. 2007;117:730–738. doi: 10.1172/JCI28984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Nazarian R, Shi H, Wang Q, Kong X, Koya RC, Lee H, Chen Z, Lee MK, Attar N, Sazegar H, Chodon T, Nelson SF, McArthur G, Sosman JA, Ribas A, Lo RS. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature. 2010;468:973–977. doi: 10.1038/nature09626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Yeh CY, Shin SM, Yeh HH, Wu TJ, Shin JW, Chang TY, Raghavaraju G, Lee CT, Chiang JH, Tseng VS, Lee YC, Shen CH, Chow NH, Liu HS. Transcriptional activation of the Axl and PDGFR-alpha by c-Met through a ras- and Src-independent mechanism in human bladder cancer. BMC Cancer. 2011;11:139. doi: 10.1186/1471-2407-11-139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Black PC, Brown GA, Inamoto T, Shrader M, Arora A, Siefker-Radtke AO, Adam L, Theodorescu D, Wu X, Munsell MF, Bar-Eli M, McConkey DJ, Dinney CP. Sensitivity to epidermal growth factor receptor inhibitor requires E-cad- herin expression in urothelial carcinoma cells. Clin Cancer Res. 2008;14:1478–1486. doi: 10.1158/1078-0432.CCR-07-1593. [DOI] [PubMed] [Google Scholar]
- 111.Kina S, Phonaphonh T, Liang F, Kuang H, Arasaki A, Arakaki K, Nakasone T, Sunakawa H. PDGF alpha receptor is a mediator for Cisplatin-induced Met expression. Eur J Pharmacol. 2013;699:227–232. doi: 10.1016/j.ejphar.2012.11.014. [DOI] [PubMed] [Google Scholar]
- 112.Paccez JD, Vasques GJ, Correa RG, Vasconcellos JF, Duncan K, Gu X, Bhasin M, Libermann TA, Zerbini LF. The receptor tyrosine kinase Axl is an essential regulator of prostate cancer proliferation and tumor growth and represents a new therapeutic target. Oncogene. 2013;32:689–698. doi: 10.1038/onc.2012.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Li Y, Ye X, Tan C, Hongo JA, Zha J, Liu J, Kallop D, Ludlam MJ, Pei L. Axl as a potential therapeutic target in cancer: role of Axl in tumor growth, metastasis and angiogenesis. Oncogene. 2009;28:3442–3455. doi: 10.1038/onc.2009.212. [DOI] [PubMed] [Google Scholar]
- 114.Verma A, Warner SL, Vankayalapati H, Bearss DJ, Sharma S. Targeting Axl and Mer kinases in cancer. Mol Cancer Ther. 2011;10:1763–1773. doi: 10.1158/1535-7163.MCT-11-0116. [DOI] [PubMed] [Google Scholar]
- 115.Linger RM, Keating AK, Earp HS, Graham DK. Taking aim at Mer and Axl receptor tyrosine kinases as novel therapeutic targets in solid tumors. Expert Opin Ther Targets. 2010;14:1073–1090. doi: 10.1517/14728222.2010.515980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Gujral TS, Karp RL, Finski A, Chan M, Schwartz PE, MacBeath G, Sorger P. Profiling phospho-signaling networks in breast cancer using reverse-phase protein arrays. Oncogene. 2013;32:3470–3476. doi: 10.1038/onc.2012.378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Liu L, Greger J, Shi H, Liu Y, Greshock J, Annan R, Halsey W, Sathe GM, Martin AM, Gilmer TM. Novel mechanism of lapatinib resistance in HER2-positive breast tumor cells: activation of AXL. Cancer Res. 2009;69:6871–6878. doi: 10.1158/0008-5472.CAN-08-4490. [DOI] [PubMed] [Google Scholar]
- 118.Minuti G, Cappuzzo F, Duchnowska R, Jassem J, Fabi A, O’Brien T, Mendoza AD, Landi L, Biernat W, Czartoryska-Arlukowicz B, Jankowski T, Zuziak D, Zok J, Szostakiewicz B, Foszczynska-Kloda M, Tempinska-Szalach A, Rossi E, Varella-Garcia M. Increased MET and HGF gene copy numbers are associated with trastuzumab failure in HER2-positive metastatic breast cancer. Br J Cancer. 2012;107:793–799. doi: 10.1038/bjc.2012.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Chandarlapaty S, Sawai A, Scaltriti M, Rodrik-Outmezguine V, Grbovic-Huezo O, Serra V, Majumder PK, Baselga J, Rosen N. AKT inhibition relieves feedback suppression of receptor tyrosine kinase expression and activity. Cancer Cell. 2011;19:58–71. doi: 10.1016/j.ccr.2010.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Salian-Mehta S, Xu M, Wierman ME. AXL and MET crosstalk to promote gonadotropin releasing hormone (GnRH) neuronal cell migration and survival. Mol Cell Endocrinol. 2013;374:92–100. doi: 10.1016/j.mce.2013.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Alciato F, Sainaghi PP, Sola D, Castello L, Avanzi GC. TNF-alpha, IL-6, and IL-1 expression is inhibited by GAS6 in monocytes/macrophages. J Leukoc Biol. 2010;87:869–875. doi: 10.1189/jlb.0909610. [DOI] [PubMed] [Google Scholar]
- 122.Yao Z, Fenoglio S, Gao DC, Camiolo M, Stiles B, Lindsted T, Schlederer M, Johns C, Altorki N, Mittal V, Kenner L, Sordella R. TGF-beta IL-6 axis mediates selective and adaptive mechanisms of resistance to molecular targeted therapy in lung cancer. Proc Natl Acad Sci U S A. 2010;107:15535–15540. doi: 10.1073/pnas.1009472107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Yamaguchi N, Lucena-Araujo AR, Nakayama S, de Figueiredo-Pontes LL, Gonzalez DA, Yasuda H, Kobayashi S, Costa DB. Dual ALK and EGFR inhibition targets a mechanism of acquired resistance to the tyrosine kinase inhibitor crizotinib in ALK rearranged lung cancer. Lung Cancer. 2014;83:37–43. doi: 10.1016/j.lungcan.2013.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Arnulf B, Lecourt S, Soulier J, Ternaux B, Lacassagne MN, Crinquette A, Dessoly J, Sciaini AK, Benbunan M, Chomienne C, Fermand JP, Marolleau JP, Larghero J. Phenotypic and functional characterization of bone marrow mesenchymal stem cells derived from patients with multiple myeloma. Leukemia. 2007;21:158–163. doi: 10.1038/sj.leu.2404466. [DOI] [PubMed] [Google Scholar]
- 125.Kim HJ, Kim SM, Park KR, Jang HJ, Na YS, Ahn KS, Kim SH, Ahn KS. Decursin chemosensitizes human multiple myeloma cells through inhibition of STAT3 signaling pathway. Cancer Lett. 2011;301:29–37. doi: 10.1016/j.canlet.2010.11.002. [DOI] [PubMed] [Google Scholar]
- 126.Wang Y, Niu XL, Qu Y, Wu J, Zhu YQ, Sun WJ, Li LZ. Autocrine production of interleukin-6 confers cisplatin and paclitaxel resistance in ovarian cancer cells. Cancer Lett. 2010;295:110–123. doi: 10.1016/j.canlet.2010.02.019. [DOI] [PubMed] [Google Scholar]
- 127.Li L, Han R, Xiao H, Lin C, Wang Y, Liu H, Li K, Chen H, Sun F, Yang Z, Jiang J, He Y. Metformin sensitizes EGFR-TKI-resistant human lung cancer cells in vitro and in vivo through inhibition of IL-6 signaling and EMT reversal. Clin Cancer Res. 2014;20:2714–2726. doi: 10.1158/1078-0432.CCR-13-2613. [DOI] [PubMed] [Google Scholar]
- 128.Domingo-Domenech J, Oliva C, Rovira A, Codony-Servat J, Bosch M, Filella X, Montagut C, Tapia M, Campas C, Dang L, Rolfe M, Ross JS, Gascon P, Albanell J, Mellado B. Interleukin 6, a nuclear factor-kappaB target, predicts resistance to docetaxel in hormone-independent prostate cancer and nuclear factor-kappaB inhibition by PS-1145 enhances docetaxel antitumor activity. Clin Cancer Res. 2006;12:5578–5586. doi: 10.1158/1078-0432.CCR-05-2767. [DOI] [PubMed] [Google Scholar]
- 129.Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–867. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.LA OR, Tai L, Lee L, Kruse EA, Grabow S, Fairlie WD, Haynes NM, Tarlinton DM, Zhang JG, Belz GT, Smyth MJ, Bouillet P, Robb L, Strasser A. Membrane-bound Fas ligand only is essential for Fas-induced apoptosis. Nature. 2009;461:659–663. doi: 10.1038/nature08402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Peter ME, Budd RC, Desbarats J, Hedrick SM, Hueber AO, Newell MK, Owen LB, Pope RM, Tschopp J, Wajant H, Wallach D, Wiltrout RH, Zornig M, Lynch DH. The CD95 receptor: apoptosis revisited. Cell. 2007;129:447–450. doi: 10.1016/j.cell.2007.04.031. [DOI] [PubMed] [Google Scholar]
- 132.Ben-Neriah Y, Karin M. Inflammation meets cancer, with NF-kappaB as the matchmaker. Nat Immunol. 2011;12:715–723. doi: 10.1038/ni.2060. [DOI] [PubMed] [Google Scholar]
- 133.Bivona TG, Hieronymus H, Parker J, Chang K, Taron M, Rosell R, Moonsamy P, Dahlman K, Miller VA, Costa C, Hannon G, Sawyers CL. FAS and NF-kappaB signalling modulate dependence of lung cancers on mutant EGFR. Nature. 2011;471:523–526. doi: 10.1038/nature09870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Shaw AT, Yeap BY, Mino-Kenudson M, Digumarthy SR, Costa DB, Heist RS, Solomon B, Stubbs H, Admane S, McDermott U, Settleman J, Kobayashi S, Mark EJ, Rodig SJ, Chirieac LR, Kwak EL, Lynch TJ, Iafrate AJ. Clinical features and outcome of patients with non-small-cell lung cancer who harbor EML4-ALK. J. Clin. Oncol. 2009;27:4247–4253. doi: 10.1200/JCO.2009.22.6993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Soda M, Choi YL, Enomoto M, Takada S, Yamashita Y, Ishikawa S, Fujiwara S, Watanabe H, Kurashina K, Hatanaka H, Bando M, Ohno S, Ishikawa Y, Aburatani H, Niki T, Sohara Y, Sugiyama Y, Mano H. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007;448:561–566. doi: 10.1038/nature05945. [DOI] [PubMed] [Google Scholar]
- 136.Takezawa K, Okamoto I, Nishio K, Janne PA, Nakagawa K. Role of ERK-BIM and STAT3-survivin signaling pathways in ALK inhibitor-induced apoptosis in EML4-ALK-positive lung cancer. Clin Cancer Res. 2011;17:2140–2148. doi: 10.1158/1078-0432.CCR-10-2798. [DOI] [PubMed] [Google Scholar]
- 137.Tiseo M, Gelsomino F, Boggiani D, Bortesi B, Bartolotti M, Bozzetti C, Sammarelli G, Thai E, Ardizzoni A. EGFR and EML4-ALK gene mutations in NSCLC: a case report of erlotinib-resistant patient with both concomitant mutations. Lung Cancer. 2011;71:241–243. doi: 10.1016/j.lungcan.2010.11.014. [DOI] [PubMed] [Google Scholar]
- 138.McDermott U, Iafrate AJ, Gray NS, Shioda T, Classon M, Maheswaran S, Zhou W, Choi HG, Smith SL, Dowell L, Ulkus LE, Kuhlmann G, Greninger P, Christensen JG, Haber DA, Settleman J. Genomic alterations of anaplastic lymphoma kinase may sensitize tumors to anaplastic lymphoma kinase inhibitors. Cancer Res. 2008;68:3389–3395. doi: 10.1158/0008-5472.CAN-07-6186. [DOI] [PubMed] [Google Scholar]
- 139.Chin LP, Soo RA, Soong R, Ou SH. Targeting ROS1 with anaplastic lymphoma kinase inhibitors: a promising therapeutic strategy for a newly defined molecular subset of non-small-cell lung cancer. J Thorac Oncol. 2012;7:1625–1630. doi: 10.1097/JTO.0b013e31826baf83. [DOI] [PubMed] [Google Scholar]
- 140.Bergethon K, Shaw AT, Ou SH, Katayama R, Lovly CM, McDonald NT, Massion PP, Siwak-Tapp C, Gonzalez A, Fang R, Mark EJ, Batten JM, Chen H, Wilner KD, Kwak EL, Clark JW, Carbone DP, Ji H, Engelman JA, Mino-Kenudson M, Pao W, Iafrate AJ. ROS1 rearrangements define a unique molecular class of lung cancers. J. Clin. Oncol. 2012;30:863–870. doi: 10.1200/JCO.2011.35.6345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Shaw AT, Yeap BY, Solomon BJ, Riely GJ, Gainor J, Engelman JA, Shapiro GI, Costa DB, Ou SH, Butaney M, Salgia R, Maki RG, Varella-Garcia M, Doebele RC, Bang YJ, Kulig K, Selaru P, Tang Y, Wilner KD, Kwak EL, Clark JW, Iafrate AJ, Camidge DR. Effect of crizotinib on overall survival in patients with advanced non-small-cell lung cancer harbouring ALK gene rearrangement: a retrospective analysis. Lancet Oncol. 2011;12:1004–1012. doi: 10.1016/S1470-2045(11)70232-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Sivakumar R, Koga H, Selvendiran K, Maeyama M, Ueno T, Sata M. Autocrine loop for IGF-I receptor signaling in SLUG-mediated epithelial-mesenchymal transition. Int J Oncol. 2009;34:329–338. [PubMed] [Google Scholar]
- 143.Buck E, Mulvihill M. Small molecule inhibitors of the IGF-1R/IR axis for the treatment of cancer. Expert Opin Investig Drugs. 2011;20:605–621. doi: 10.1517/13543784.2011.558501. [DOI] [PubMed] [Google Scholar]
- 144.Schwab CL, English DP, Roque DM, Bellone S, Lopez S, Cocco E, Nicoletti R, Rutherford TJ, Schwartz PE, Santin AD. Neratinib shows efficacy in the treatment of HER2/neu amplified uterine serous carcinoma in vitro and in vivo . Gynecol Oncol. 2014;135:142–8. doi: 10.1016/j.ygyno.2014.08.006. [DOI] [PubMed] [Google Scholar]
- 145.Ramalingam SS, Blackhall F, Krzakowski M, Barrios CH, Park K, Bover I, Seog Heo D, Rosell R, Talbot DC, Frank R, Letrent SP, Ruiz-Garcia A, Taylor I, Liang JQ, Campbell AK, O’Connell J, Boyer M. Randomized phase II study of dacomitinib (PF-00299804), an irreversible pan-human epidermal growth factor receptor inhibitor, versus erlotinib in patients with advanced non-small-cell lung cancer. J. Clin. Oncol. 2012;30:3337–3344. doi: 10.1200/JCO.2011.40.9433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Soria JC, Baselga J, Hanna N, Laurie SA, Bahleda R, Felip E, Calvo E, Armand JP, Shepherd FA, Harbison CT, Berman D, Park JS, Zhang S, Vakkalagadda B, Kurland JF, Pathak AK, Herbst RS. Phase I-IIa study of BMS-690514, an EGFR, HER-2 and -4 and VEGFR-1 to -3 oral tyrosine kinase inhibitor, in patients with advanced or metastatic solid tumours. Eur J Cancer. 2013;49:1815–1824. doi: 10.1016/j.ejca.2013.02.012. [DOI] [PubMed] [Google Scholar]
- 147.Nikolinakos P, Heymach JV. The tyrosine kinase inhibitor cediranib for non-small cell lung cancer and other thoracic malignancies. J Thorac Oncol. 2008;3:S131–134. doi: 10.1097/JTO.0b013e318174e910. [DOI] [PubMed] [Google Scholar]
- 148.Takeda M, Arao T, Yokote H, Komatsu T, Yanagihara K, Sasaki H, Yamada Y, Tamura T, Fukuoka K, Kimura H, Saijo N, Nishio K. AZD2171 shows potent antitumor activity against gastric cancer over-expressing fibroblast growth factor receptor 2/keratinocyte growth factor receptor. Clin Cancer Res. 2007;13:3051–3057. doi: 10.1158/1078-0432.CCR-06-2743. [DOI] [PubMed] [Google Scholar]
- 149.Xin X, Abrams TJ, Hollenbach PW, Rendahl KG, Tang Y, Oei YA, Embry MG, Swinarski DE, Garrett EN, Pryer NK, Trudel S, Jallal B, Mendel DB, Heise CC. CHIR-258 is efficacious in a newly developed fibroblast growth factor receptor 3-expressing orthotopic multiple myeloma model in mice. Clin Cancer Res. 2006;12:4908–4915. doi: 10.1158/1078-0432.CCR-06-0957. [DOI] [PubMed] [Google Scholar]
- 150.Chaparro M, Gonzalez ML, Trapero-Marugan M, Medina J, Moreno-Otero R. Review article: pharmacological therapy for hepatocellular carcinoma with sorafenib and other oral agents. Aliment Pharmacol Ther. 2008;28:1269–1277. doi: 10.1111/j.1365-2036.2008.03857.x. [DOI] [PubMed] [Google Scholar]
- 151.Cheng AL, Kang YK, Chen Z, Tsao CJ, Qin S, Kim JS, Luo R, Feng J, Ye S, Yang TS, Xu J, Sun Y, Liang H, Liu J, Wang J, Tak WY, Pan H, Burock K, Zou J, Voliotis D, Guan Z. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: a phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2009;10:25–34. doi: 10.1016/S1470-2045(08)70285-7. [DOI] [PubMed] [Google Scholar]
- 152.Sano T, Takeuchi S, Nakagawa T, Ishikawa D, Nanjo S, Yamada T, Nakamura T, Matsumoto K, Yano S. The novel phosphoinositide 3-kinase-mammalian target of rapamycin inhibitor, BEZ235, circumvents erlotinib resistance of epidermal growth factor receptor mutant lung cancer cells triggered by hepatocyte growth factor. Int J Cancer. 2013;133:505–513. doi: 10.1002/ijc.28034. [DOI] [PubMed] [Google Scholar]
- 153.Wang X, Sun SY. Enhancing mTOR-targeted cancer therapy. Expert Opin Ther Targets. 2009;13:1193–1203. doi: 10.1517/14728220903225008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Ishikawa D, Takeuchi S, Nakagawa T, Sano T, Nakade J, Nanjo S, Yamada T, Ebi H, Zhao L, Yasumoto K, Nakamura T, Matsumoto K, Kagamu H, Yoshizawa H, Yano S. mTOR inhibitors control the growth of EGFR mutant lung cancer even after acquiring resistance by HGF. PLoS One. 2013;8:e62104. doi: 10.1371/journal.pone.0062104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Huang MH, Lee JH, Chang YJ, Tsai HH, Lin YL, Lin AM, Yang JC. MEK inhibitors reverse resistance in epidermal growth factor receptor mutation lung cancer cells with acquired resistance to gefitinib. Mol Oncol. 2013;7:112–120. doi: 10.1016/j.molonc.2012.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Bendell JC, Hong DS, Burris HR, Naing A, Jones SF, Falchook G, Bricmont P, Elekes A, Rock EP, Kurzrock R. Phase 1, open-label, dose-escalation, and pharmacokinetic study of STAT3 inhibitor OPB-31121 in subjects with advanced solid tumors. Cancer Chemother Pharmacol. 2014;74:125–130. doi: 10.1007/s00280-014-2480-2. [DOI] [PubMed] [Google Scholar]
- 157.Ioannidis S, Lamb ML, Wang T, Almeida L, Block MH, Davies AM, Peng B, Su M, Zhang HJ, Hoffmann E, Rivard C, Green I, Howard T, Pollard H, Read J, Alimzhanov M, Bebernitz G, Bell K, Ye M, Huszar D, Zinda M. Discovery of 5-chloro-N2-[(1S)-1-(5-fluoropyrimidin-2-yl)ethyl] -N4-(5-methyl-1H-pyrazol-3-yl)p yrimidine-2,4-diamine (AZD1480) as a novel inhibitor of the Jak/Stat pathway. J Med Chem. 2011;54:262–276. doi: 10.1021/jm1011319. [DOI] [PubMed] [Google Scholar]
- 158.Li D, Ambrogio L, Shimamura T, Kubo S, Takahashi M, Chirieac LR, Padera RF, Shapiro GI, Baum A, Himmelsbach F, Rettig WJ, Meyerson M, Solca F, Greulich H, Wong KK. BIBW- 2992, an irreversible EGFR/HER2 inhibitor highly effective in preclinical lung cancer models. Oncogene. 2008;27:4702–4711. doi: 10.1038/onc.2008.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Xu L, Kikuchi E, Xu C, Ebi H, Ercan D, Cheng KA, Padera R, Engelman JA, Janne PA, Shapiro GI, Shimamura T, Wong KK. Combined EGFR/ MET or EGFR/HSP90 inhibition is effective in the treatment of lung cancers codriven by mutant EGFR containing T790M and MET. Cancer Res. 2012;72:3302–3311. doi: 10.1158/0008-5472.CAN-11-3720. [DOI] [PMC free article] [PubMed] [Google Scholar]