

Abstract
Liver fibrosis is a risk factor for hepatoma. Activated hepatic stellate cells (HSCs) play a critical role in progression of hepatoma. Resected hepatoma patients with high α-SMA+HSCs infiltration had worse survival, OR: 2.2 and p=0.0434. We hypothesized that HSCs could increase the epithelial-mesenchymal transition (EMT) ability of hepatoma cells. In murine model of liver fibrosis with injection of ML1 mice HCC cell line, E-cadherin was lost at the margin of tumor nodule around α-SMA+HSC sites. In subcutaneous tumor model, HSCs could increase the metastatic nodules in the lung, and the expression of E-cadherin was decreased and the Slug was induced. To elucidate the effect of HSCs on hepatoma cells, HSC-T6 was co-cultured with ML1 and the condition medium of HSC-T6 can trigger ML1 cell morphological change, down-expression of E-cadherin, induction of Slug expression, and cell migration. Proteomic analysis of the condition medium showed that collagen I was the target molecule. Collagen type I alone also induced EMT of ML1 cells. Knockdown of collagen type I in HSC-T6 could decrease its induction of EMT on ML1 cells. In conclusion, HSC can secrete collagen type I to trigger hepatoma cells to undergo EMT for metastasis.
Keywords: Hepatic stellate cell, collagen type I, epithelial-mesenchymal transition
Introduction
Hepatocellular carcinoma (HCC) is one of the most severe cancers in the world [1]. Liver fibrosis is considered as a risk factor correlated with HCC formation [2]. An animal model using dimethylnitrosamine (DMN) to induce liver fibrosis demonstrates that long term fibrosis can promote carcinogenesis [3]. However, the connection between liver fibrosis and hepatoma is not fully studied. The fibrotic environment can enhance hepatoma growth through downregulation of immune surveillance of host or increasing angiogenesis for tumor growth [4,5]. Furthermore, the metastatic property of hepatoma cells was increased after transplanted into a fibrotic liver [6], suggesting multiple impacts of fibrotic environment on behavior of hepatoma cells.
During liver damage, hepatic stellate cells (HSCs) become activated and promote liver fibrosis formation. The activated HSCs will trans-differentiate into myofibroblasts and help tissue repair [7-9]. By secreting various factors including extracellular matrix, cytokines, and growth factors, HSCs have great influence on tumor behavior. The cultured supernatant of HSCs increases proliferation and migration activity of hepatoma cells [10,11]. HSCs can also contribute to HCC stroma formation [12], and the existences of HSCs is related with high recurrence and death rate in clinical practice [13]. Various factors could be involved in the process of HSC promoted tumor progression, some of them are potent epithelial-mesenchymal transition (EMT) inducer [14,15]. After induction of EMT, the ability of invasion, metastasis, and chemoresistance of tumor cells will increase, which in turn increases the severity of disease [16,17]. The characteristics of cells undergoing EMT include morphological change, transcription factor activation, and specific protein expression. The presence of transition from epithelial markers to the mesenchymal markers is the evidence of cell conversion [18].
The interaction between HSCs and hepatoma cells remains unclear. Understanding the mehanism underlying liver fibrosis and tumor progression may help in the design of therapeutic strategies in the future. Therefore, this study is aimed to investigate how the HSCs promote hepatoma progression through EMT induction. At first, we analyzed the clinical significance of presence of activated HSCs in HCC patients. Secondly, we evaluate the role of HSCs on EMT ability of murine hepatoma cell ML1 in vitro and in vivo with or without candidate protein.
Material and methods
Clinical outcome analysis
The paraffin block of 92 resectable HCC patients, included 54 male patients and 38 female patients, was included for staining of HSCs marker. The correlation between clinical characteristics and recurrence after resection of these patients was analyzed.
Cell culture and collection of conditioned medium
The rat hepatic stellate cell line, HSC-T6 was cultured in DMEM medium with 10% FBS. The culture supernatant was collected after 3 days culture as a conditioned medium. The supernatant was centrifuged at 2000 rpm for 10 min to deplete cell debris and then stored at -20°C. For the serum free conditioned medium, HSC-T6 cells were subcultured in Waymouth medium with 10% FBS, then transferred into serum free Waymouth medium after attachment, and the supernatant was collected 2 days later.
Immunohistochemistry and immunofluorescence staining
Immunohistochemistry staining was performed as previously described [4]. The primary antibody were anti-E-cadherin, anti-Slug (Cell Signaling Technology, USA), anti-CD31 (BD, USA), anti-α-SMA (Sigma Aldrich, USA), and anti-collagen type I (Abcam, USA). The slides were further stained with DAPI for 15 min to show the cell nucleus position and then observed by confocal microscopy. The percentage of positive cells in each sample was analyzed by Tissue Quest software.
Transwell migration assay
The migration activity of ML1 cells was evaluated by Transwell migration assay using 12-well transwell insert with 5 μm pore size (Corning, USA). Approximately 5x104 ML1 cells were seeded onto the insert, after overnight incubation, the conditioned medium from HSC-T6 cells were added to new 12-well plate and put in the inserts. After 24 h incubation, the cells on upper side of transwell insert were removed by cotton swaps. The cells remained on transwell inserts were further fixed by cold methanol and stained with DAPI. The cell numbers were observed and calculated under fluorescence microscopy. Each treatment were duplicated and repeated once with similar result.
Reverse transcription-PCR
RNA was extracted with TRIZOL reagent (Invitrogen Life Technologies, USA). The cDNA was synthesized by MMLV-reverse transcriptase (Promega, USA) following the manufacturer’s description. Primer sequences were β-Actin-F: 5’-TGA ACC CTA AGG CCA ACC GTG-3’, β-Actin-R: 5’-GCT CAT AGC TCT TCT CCA GGG-3’, Snail-F: 5’-ACC CCC GCC GGA AGC CCA ACT-3’, Snail-R: 5’-AGC GGC GGG GTT GAG GAC CTC-3’; Slug-F: 5’-CTC ACC TCG GGA GCA TAC AGC-3’, Slug-R: 5’-TGA AGT GTC AGA GGA AGG CGG G-3’; Twist-F: 5’-CGG GTC ATG GCT AAC GTG-3’, Twist-R: 5’-CAG CTT GCC ATC TTG GAG TC-3’. PCR products from cDNA were further electrophoresed in 2% agarose gel and stained with ethidium bromide.
Proteomic analysis of conditioned medium
The SF-CM was collected to run 12% SDS-PAGE. After Coomassie blue staining, the gel bands were sliced and incubated in 10 mM of DTT at 65°C for 45 min for reduction, followed by alkylation with 55 mM of iodoacetamide in 25 mM ammonium bicarbonate at room temperature for 1 hr. To digest the proteins, 0.1 μg of trypsin (Promega, USA) was added and incubated at 37°C overnight. Targeted LC-MS/MS analysis was performed on a nano-HPLC system coupled to an ion trap mass spectrometer (LCQ DECAXP Plus, ThermoFinnigan, USA). All MS/MS files (DTA files) were generated using Bioworks Browser 3.1 (ThermoElectron, USA). The resulting peak lists were searched against the Swiss-Prot database via an in-house Mascot server (Matrix Science Ltd.,U.K.) [19].
Western blotting
Total protein extracts from ML1 cells or tissues were obtained by incubating with lysis buffer (Cell Signaling, USA). Western blot was performed as described previously [4]. The primary antibodies were E-cadherin, N-cadherin, Slug (Cell Signaling Technology, USA), Twist (Santa Cruz, USA), and β-actin (Abcam, USA),
Animal model
Two mice models were used to evaluate the effect of HSC on the metastasis of HCC cells. The animals were raised and cared according to the guidelines set up by the National Science Council, ROC. The mouse experiments were approved by the Institutional Animal Care and Use Committee.
Hepatoma in liver fibrosis mice model
A liver fibrosis combined with hepatoma animal model was induced as described in our previous study [4]. In brief, liver fibrosis was induced by an intraperitoneal injection of 200 μg/kg thioacetamide (TAA) 3 times a week for a total of 6 weeks in 8-10 week old male BALB/c mice. After liver fibrosis established, 1x106 mouse hepatoma cells, ML1, was then injected into each mouse spleen, few days later ML1 could migrate to liver to form hepatoma nodules. The liver was harvested at 2 weeks after injection of hepatoma cells.
Subcutaneous hepatoma mice model
The metastatic ability of ML-1 with or without HSCs was evaluated in 8-10 week old male NOD/SCID mice. 1x106 ML1 cells per mice were injected subcutaneously, with or without equal amount of rat hepatic stellate cell line HSC-T6. Six weeks later, the mice were sacrificed and the lungs were harvested to evaluate the tumor status of metastasis.
Statistics
The clinicopathological variables were included as adjusters in the analysis of resected HCC patients. Cox proportional hazards model was used for multivariate analysis while led parameters were significant at the P < 0.1 level on univariate analysis using log-rank test. Results were presented as hazard ratio (HR) and 95% CI. Laboratory data are expressed as mean±SE of three independent experiments. Statistical analysis in this study was performed by applying a student’s t test using Sigma Plot 8.0 software. Statistical significance was considered at p < 0.05.
Results
The impact of HSCs expression on the recurrence of HCC after resection
The α-SMA was used to stain the activated HSCs in hepatoma specimen (Figure 1A). After calculated by Tissue Quest software, high expression meant higher than mean number expression of staining cells. The patients with high expression of α-SMA had higher recurrence rate, p=0.0434 (Figure 1B). Multivariate analysis showed the high expression of HSCs in the tumor was significantly correlated with the recurrence after resection, hazard ratio 2.2 with 95% CI ranged from 1.2 to 3.2, p=0.0434. (Table 1)
Figure 1.
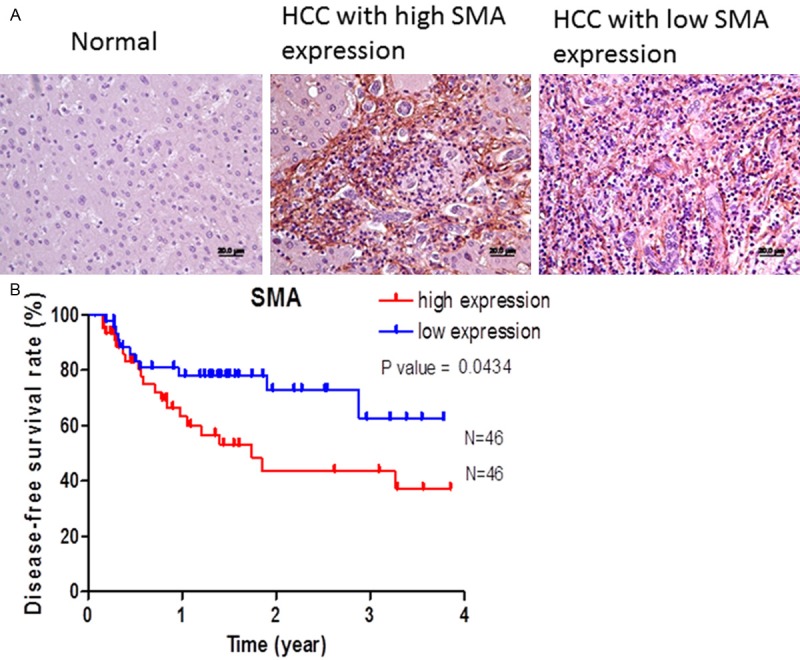
The clinical significance of infiltrative HSCs in HCC on recurrence after resection. A. The active HSCs were stained with SMA, no SMA+ was observed in normal liver, and active HSCs were found within tumor area; B. patients with high expression of SMA+ HSCs had poor disease-free period, p=0.0434.
Table 1.
The impact of HSCs expression on disease-free survival of HCC patients after resection
| Characteristics | Patients (n=92) | Hazard ratio (95% CI) | p-value |
|---|---|---|---|
| Gender | |||
| Male: female | 54:38 | 1:0.6 (0.4-1.2) | 0.768 |
| Underlying liver disease | |||
| Cirrhosis: non-cirrhosis | 39:53 | 1:0.5 (0.3-1.2) | 0.867 |
| Differentiation | |||
| Well: Moderate & poor | 30:52 | 1:1.2 (0.7-1.6) | 0.312 |
| Vascular invasion | |||
| Yes: no | 48:44 | 1:0.6 (0.4-1.3) | 0.464 |
| AFP level (ng/ml) | |||
| AFP ≥ 20: AFP < 20 | 58:34 | 1:0.8 (0.6-1.5) | 0.356 |
| Staging (AJCC) | |||
| I: II & III | 38:54 | 1:1.3 (0.5-1.6) | 0.393 |
| SMA expression | |||
| Low: high | 46:46 | 1:2.2 (1.2-3.2) | 0.0434 |
Hepatic stellate cells enhance ML1 metastasis
We used a subcutaneous tumor model to demonstrate the ability of stellate cells to promote hepatoma metastasis. When injecting ML1, either with or without an equivalent amount of HSC-T6, into NOD/SCID mice, the tumor cells can migrate into the lung after 6-8 weeks. The tumor nodule numbers in the lung were significantly higher in the mice with injection of ML1 with HSC-T6 group (Figure 2A). In these metastatic lung nodules, the immunohistochemistry staining showed decrease expression of E-cadherin and increased Slug (Figure 2B). In the Western blot analysis, E-cadherin was also down-regulated and Slug was induced in the extract of lung metastatic nodules (Figure 2C). However, tumor volume of the primary tumor nodule was slightly larger in the mice with injection of ML1 with HSC-T6 than that in injection of ML-1 only, but no significant differences (Figure 2D), suggesting that stellate cells can enhance metastasis of hepatoma cell (ML-1). A similar effect was observed in the immunohistochemistry staining of primary tu-mor site showed decrease expression of E-cadherin and increased Slug (Figure 2E, 2F) in the mice with injection of ML1 with HSC-T6 compared to those with injection ML-1 only. These results suggest that stellate cells can enhance the metastatic ability of ML1 via induction of EMT.
Figure 2.
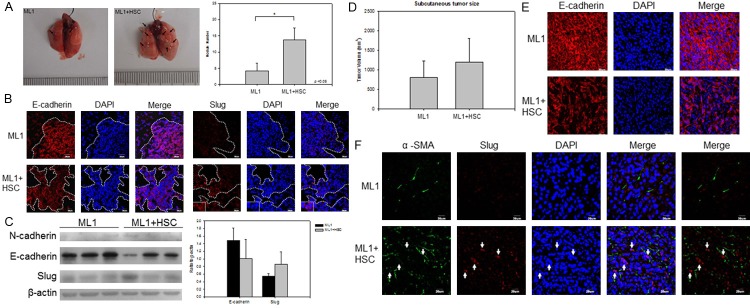
HSCs enhance ML1 metastasis to lung in NOD/SCID mice with EMT induction. The hepatoma cell line ML1 was subcutaneously injected (1x106) into NOD/SCID mice with or without same amount (1x106) of HSC-T6 cells. The mice were sacrificed at 6 weeks post injection. A. A representative lung tissue picture and the number of lung metastatic nodules was significantly higher in the mice of ML1 with HSC; B. the expression of E-cadherin (red) was down-expressed while the expression Slug (red) was increased in the mice with of ML1 with HSC when compared to the mice with of ML1 only; C. The immunoblotting of E-cadherin, N-cadherin, and Slug protein in lung tissue showed similar result; D. The subcutaneous tumor size was measured at sacrifice time point; E and F. The immunohistochemistry staining of E-cadherin (red), α-SMA (green), and Slug (red) in subcutaneous tumor sites. The cell nuclei were stained with DAPI (blue). Each group contains 6-8 mice. P < 0.05 were considered significant.
HSC-T6 conditioned medium induces ML1 to undergo EMT
We collected culture supernatant from the hepatic stellate cell line HSC-T6 as conditioned medium (CM). After treated with CM for 24 ho-urs, the morphology of ML1 cell transformed into a more stretched out shape. A similar morphological change was also observed with serum free conditioned medium (SF-CM) or serum free conditioned medium with 10% fetal bovine serum added (SF-CM+FBS). Since hepatic stellate cells are known to secret TGF-β, it was included as another control. However, TGF-β induces ML1 to transform into a spindle-like morphology that is different from that of CM. The Waymouth control medium was used as a negative control that does not induce any morphological change (Figure 3A).
Figure 3.

HSC-T6 conditioned medium can trigger ML1 undergo EMT. A. The morphological change of ML1 after treated with different medium; 1. DMEM with 10% FBS (control); 2. HSC-T6 conditioned medium (CM), 3. Serum free conditioned medium (SF-CM), 4. serum free conditioned medium with 10% FBS (SF-CM+FBS). 5. 10 ng/ml TGF-b. 6. HSC-T6 culturing medium Waymouth. All treatments were added to ML1 cells for 24 h. The group 1-6 represent the same treatment through this figure; B. The immunoblotting result of E-cadherin, N-cadherin, slug, and twist under different conditioned medium to ML1 cells after 12 h treatment; C. The mRNA expression of EMT related transcription factor snail, twist, and slug in ML1 cells with different treatment for 12 h; D. The migration ability of ML1 cells was determined by transwell migration assay at 24 h post treatment, where E shows the quantitative result; F. Immunofluorescence staining of E-cadherin, N-cadherin, α-SMA, vimentin, and Slug in ML1 cells treated for 12 h; G. The cell number was counted by Eosin Y staining after 12-48 h treatment; H. The hepatoma cell line ML1 were treated with HSC-T6 CM for 12-48 h. Cells were fixed with 70% alcohol and stained with 20 μg/ml propidion indium, result showed percentage in each cell cycle stage. Each treatment was triplicated and repeated once with similar result. The expression level was compared to the control group by analyzing band intensity with Image J software.
The immunoblotting showed that E-cadherin was down-regulated in the CM, SF-CM, and SF-CM+FBS groups, while N-cadherin was up-regulated in the TGF-β treatment group (Figure 3B). The transcription factor Slug was induced under the CM and SF-CM+FBS treatment, and Twist was up-regulated with TGF-β treatment (Figure 3C). The migration activity was detected by the transwell migration assay under CM treatment (Figure 3D, 3E). The similar results were also observed in immunofluorescence staining of Slug expression in the cell nucleus (Figure 3F). There were no differences between the control and the Waymouth medium group, but in the CM with sufficient nutrient or TGF-β treatment, the migration activities were enhanced around 1.5 fold compared to the control group. The cell cycle analysis showed a decreased population in S-phase at 12 and 24 h (Figure 3H). The proliferation status was also evaluated by Eosin Y staining after 12-48 h treatment. In the CM, SF-CM and SF-CM+FBS treated group, the cell proliferation was inhibited, while the TGF-β and Waymouth group showed normal proliferation status (Figure 3G). According to these results, we suggested that the HSC-T6 CM can induce ML1 undergo EMT and further enhance its migration ability.
Collagen type I in HSC-T6 conditioned medium induces EMT of ML1
We further investigated the component in CM that might be responsible for EMT by using the serum free HSC-T6 culture supernatant (SF-CM) to run SDS-PAGE and make identification using a mass spectrometer. Extracellular matrix protein collagen type I is one of the major components in conditioned medium (Figure 4).
Figure 4.

Protein identification in HSC-T6 conditioned medium. The SF-CM was collected and SDS-PAGE was conducted. The proteins on SDS-PAGE were further identified by mass spectrometer. Three regions of different proteins were identified.
To test the effect of collagen type I on the EMT of ML1 cells, pure collagen type I was added to treat ML1 cells. After 12 h of treatment, collagen type I can induce ML1 morphological change in a dose dependent manner (Figure 5A). The immunoblotting results also showed the EMT markers were induced, from 100 μg/ml to 300 μg/ml on the expression of E-cadherin, N-cadherin, and slug (Figure 5B). When the expression of collagen type I was knockdown in HSC-T6 cells, the E-cadherin loss was reversed and the slug expression was reduced, but this phenomenon was not observed after knockdown the expression of fibronectin (Figure 5C).
Figure 5.
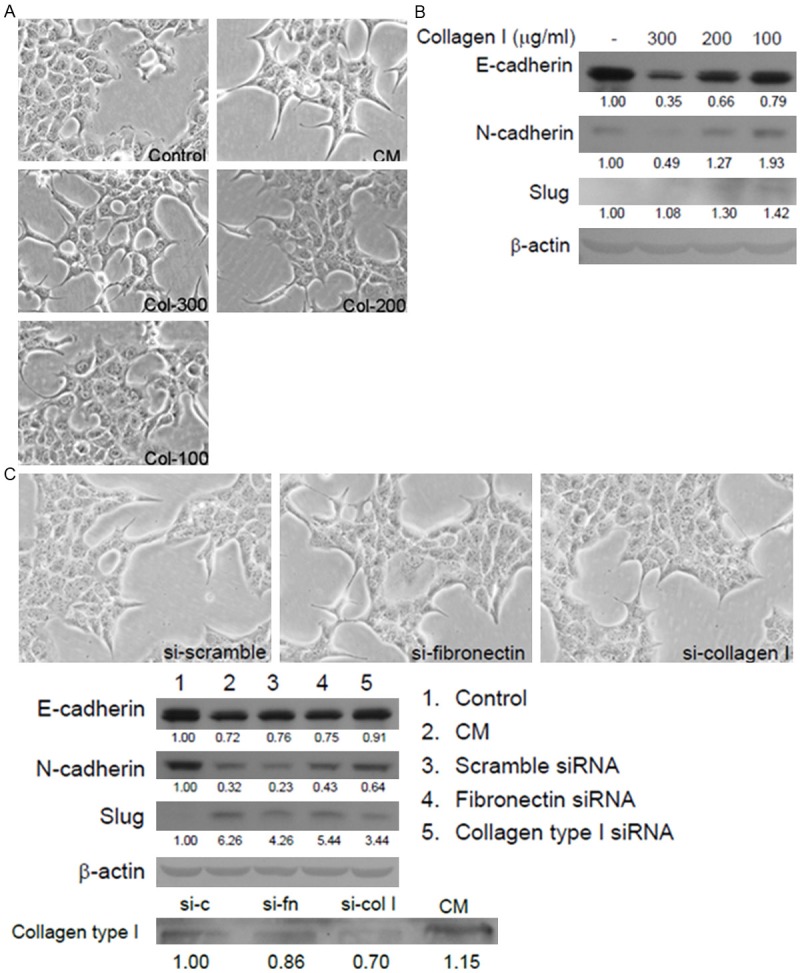
The collagen type I in HSC conditioned medium induced EMT. A. Morphological change of ML1 cells after 24 h of 100-300 μg/ml collagen type I treatment; B. Immunobloting of E-cadherin, N-cadherin, Slug and b-actin in ML1 cells. C. siRNA was used to knockdown fibronectin or collagen type I expression in HSC-T6 cells, and culture supernatant was collected to treat ML1 cells for 24 h. The morphological changes of ML1 cells are shown in 200x magnification, and immunobloting of E-cadherin, N-cadherin, Slug and collagen type I. The knockdown efficiency was confirmed by western blotting of collagen type I expression in conditioned medium.
Hepatic stellate cells were localized around the E-cadherin downregulated tumor region in the liver fibrosis combined with hepatoma model
In order to clarify the effect of naturally activated hepatic stellate cells on the hepatoma cells, we established a liver fibrosis combined with hepatoma model in BALB/c mice4. In the mice bearing hepatoma alone, a high expression level of E-cadherin was found in the whole tumor nodule. However, in the fibrosis combined with hepatoma mice, E-cadherin was down-expressed at the marginal area of some tumor nodules (Figure 6A). The α-smooth muscle actin (α-SMA) was used as a marker of hepatic stellate cells, and the immunohistochemistry staining showed that hepatic stellate cells were localized at these E-cadherin down-expressed sites (Figure 6B). These α-SMA+ cells were desmin+ (Figure 6C) but not CD31+, which therefore excludes the possibility of intratumor endothelial cells. This suggests that hepatic stellate cells can enhance hepatoma cells to undergo EMT in a fibrotic liver. To further confirm the role of collagen type I in vivo, its deposition in the hepatoma mice with or without fibrosis model was stained. In the TAA-ML1 group (lower rows), the E-cadherin expression was loss in the area with high expression of α-SMA+ hepatic stellate cell at the margin of tumor, and the collagen type I deposition in the area with the expression of α-SMA+ hepatic stellate cells (Figure 6C). In the ML1 group, α-SMA+ cells were also observed in the tumor nodules, but the expression level was lower than that observed in TAA-ML1 mice. The E-cadherin expression was mostly expressed within entire tumor nodule, and the α-SMA+ cells did not express E-cadherin and located inside the tumor, with co- expression of collagen type I. Based on these data, we concluded that collagen type I released from activated hepatic stellate cells during fibrosis can contribute to EMT induction in hepatoma.
Figure 6.

Hepatic stellate cells were localized around E-cadherin down-expressed tumor region in the liver fibrosis combined with hepatoma model. A. E-cadherin (red) is highly expressed in DAPI (blue) condensed tumor region (dot line) harvested from hepatoma bearing (ML1) BALB/c mice. In the fibrosis combined with hepatoma (TAA-ML1) mice liver, E-cadherin is down-expressed in front parts of the tumor region as indicated by an arrow. (200x); B. The hepatic stellate cells in tumor were stained with α-smooth muscle actin (α-SMA, green). Anti-E-cadherin (red), anti-CD31 (purple, triangle), and DAPI (blue) were also stained. The tumor regions with high α-SMA expressed cells were E-cadherin down-expressed; C. These cells were desmin+ but CD31-. (600x) The images were representative to 5-7 mice in each group; D. The mice liver with hepatoma (ML1) or hepatoma combined with fibrosis (TAA-ML1) were collected and sliced into 5 μm sections. Immunofluorescence staining of α-smooth muscle actin (α-SMA, green), E-cadherin (red), collagen type I (red), and DAPI (blue) in liver tissue section. The dotted line shows the tumor area. Arrowhead indicates the α-SMA highly expressed cells, and arrow indicates the α-SMA+ cells with lower expression.
Discussion
The HSCs are important in liver fibrosis formation, but their interaction with tumor cells is still unclear. In this study we found that HSCs can trigger hepatoma cells to undergo EMT through the effect of collagen type I, and subsequently enhance tumor metastasis. Using the hepatic stellate cell line HSC-T6 conditioned medium that also can induce morphological change in the hepatoma cell line ML1, down-expression of epithelial marker E-cadherin, and induction of transcription factor Slug. In liver fibrosis and hepatoma coexisting model, the E-cadherin was down expressed around HSCs within tumors. In NOD/SCID mice subcutaneous tumor model, we demonstrated the metastatic effect of HSCs. We further identify that collagen type I released from HSCs during fibrosis is contributed to EMT in enhancing metastasis of hepatoma.
HSCs presenting around tumors have been reported in several studies. The conditioned medium of hepatocellular carcinoma cell can trigger HSCs activation, proliferation, and cell migration [20,21]. This indicates that tumors can stimulate HSCs to form stoma which then help tumor growth. On the other hand, the activated HSCs can also affect tumor behavior. The B7-H1 expressed on HSCs can inhibit the immune response against tumors, and contributes to tumor progression [11,22]. The culture supernatant of HSCs has been reported to enhance hepatoma cell proliferation and migration activity [10]. The activated HSCs can secrete pro-angiogenic factor, such as angioprotein-1, to promote tumor metastasis [23], and they also can promote focal adhesion formation of tumor cells to enhance tumor growth [24]. In clinical practice, the presence of activated HSCs near a tumor area is related to poor patient survival [13]. Our data shows that increased infiltrative active HSCs are correlated with higher postoperative recurrence rate. These studies reveal the importance of the interaction between HSCs and tumor cells.
The CM from cultured HSC-T6 cells can trigger ML1 cell morphological change, down-expression of epithelial marker E-cadherin and up-expression of transcription factor Slug, which implies induction of EMT. However, the mesenchymal marker N-cadherin did not up-express under conditioned medium treatment, suggesting a partial EMT (p-EMT) response. This phenomenon has been reported in several studies, such as hepatocyte growth factor (HGF) could induce MDCK cells to express Slug without down-expression of E-cadherin [25]. TGF-β also could induce keratinocyte to undergo partial EMT, showing down-expression of epithelial marker without up-expression of mesenchymal marker [26]. Although the mesenchymal features did not increase in the partial EMT response, the migration activity of transformed cells was still enhanced.
Activated HSC can secrete TGF-β to promote liver fibrosis, while TGF-β is known as a potent EMT inducer. Both TGF-β and CM of HSC-T6 can induce EMT of ML1 cells, but the effects on ML1 were different. First, in the morphological change of ML1, CM could induce a more stretched out spindle-like shape and these cells still grouped together. In contrast, TGF-b resulted in being an elongated shape with each cell separated from each other. Secondly, the CM of HSC-T6 could induce up-expression of Slug, while TGF-β mainly induced expression of Twist. Thirdly, the proliferation status of ML1 cells was inhibited after CM of HSC-T6 treatment, but not with TGF-β. These results suggest that HSC-T6 can trigger hepatoma cells to undergo EMT different from the influence of TGF-β.
When ML1 cells were treated with CM of HSC-T6, the cell proliferation was inhibited. This similar result was observed by using TGF-β to induce EMT in mammary epithelial cells [27]. The cells tended to delay the proliferation rate during the transition process. Despite the proliferation being inhibited in the in vitro model, the nodule number was increased in the TAA-ML1 mice in our previous report [4]. When using a subcutaneous tumor model, the mixing of HSC-T6 with ML1 showed an enhanced migration activity. Both of the in vivo models showed similar results. We suggest that the hepatoma acquired invasiveness through EMT induction is more important than temporal proliferation inhibition.
Several proteins secreted from activated HSCs are related to EMT induction, including TGF-b, hepatocyte growth factor (HGF) [28], epidermal growth factor (EGF) [29], and collagen type I [30]. The proteomic analysis revealed that some extracellular matrix elements were major components in the HSC-T6 conditioned medium, including collagen type I and fibronectin. We found that treating ML1 cells with collagen type I could trigger a similar effect to CM, when using siRNA to down-express the collagen type I expression in HSC-T6, the EMT induction of CM was reversed, the E-cadherin expression level was increased and Slug induction was decreased. In collection, our study suggests that collagen type I contributes to HSC induced EMT.
The HSCs share several features with pancreatic stellate cells (PSCs), such as extracellular matrix and growth factor production. Both kinds of stellate cells promote tissue fibrosis formation. Previous reports showed that PSCs could induce EMT of pancreatic cancer cells. PSCs harvested from patients co-cultured with tumor cells could induce snail expression and down expression of E-cadherin, and enhance tumor migration ability [31,32]. Interestingly, the PSC induction of tumor cell EMT was not TGF-β dependent, and this result is similar to our observation. Therefore, the interaction between stellate cells and tumor is not specific for liver tissue.
The EMT phenomenon was detected in HCC patients. The malignance of HCC is correlated with snail or twist expression, and patients with snail or twist expression had poor survival rate [33]. The EMT induction is also detected with Slug expression in HBV, HCV, and alcohol related tumor cases [34], indicating that EMT induction is not limited to a particular type of HCC patient. There are more metastasis in HBV related HCC in clinical observations [35], HBV infection can induce more activated HSCs via paracrine effect and causes more collagen production [36]. In conclusion, the activated HSCs can enhance metastasis through EMT induction. To further elucidate the interaction between HSCs and tumor cells could help for future therapy of HCC.
Acknowledgements
In this study, we thank Professor Lei HY’s encouragement and assistance in the designing and completion of experiment.
Disclosure of conflict of interest
None to declare.
Abbreviations
- HCC
hepatocellular carcinomas
- HSC
hepatic stellate cells
- TAA
thioacetamide
References
- 1.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 2.El-Serag HB, Rudolph KL. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology. 2007;132:2557–2576. doi: 10.1053/j.gastro.2007.04.061. [DOI] [PubMed] [Google Scholar]
- 3.Gang Z, Qi Q, Jing C, Wang C. Measuring microenvironment mechanical stress of rat liver during diethylnitrosamine induced hepatocarcinogenesis by atomic force microscope. Microsc Res Tech. 2009;72:672–678. doi: 10.1002/jemt.20716. [DOI] [PubMed] [Google Scholar]
- 4.Yang MC, Chang CP, Lei HY. Induction of liver fibrosis in a murine hepatoma model by thioacetamide is associated with enhanced tumor growth and suppressed antitumor immunity. Lab Invest. 2010;90:1782–1793. doi: 10.1038/labinvest.2010.139. [DOI] [PubMed] [Google Scholar]
- 5.Kornek M, Raskopf E, Tolba R, Becker U, Klockner M, Sauerbruch T, Schmitz V. Accelerated orthotopic hepatocellular carcinomas growth is linked to increased expression of pro-angiogenic and prometastatic factors in murine liver fibrosis. Liver Int. 2008;28:509–518. doi: 10.1111/j.1478-3231.2008.01670.x. [DOI] [PubMed] [Google Scholar]
- 6.Kuriyama S, Yamazaki M, Mitoro A, Tsujimoto T, Kikukawa M, Tsujinoue H, Nakatani T, Toyokawa Y, Yoshiji H, Fukui H. Hepatocellular carcinoma in an orthotopic mouse model metastasizes intrahepatically in cirrhotic but not in normal liver. Int J Cancer. 1999;80:471–476. doi: 10.1002/(sici)1097-0215(19990129)80:3<471::aid-ijc22>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 7.Bataller R, Brenner DA. Liver fibrosis. J Clin Invest. 2005;115:209–218. doi: 10.1172/JCI24282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Friedman SL. Mechanisms of hepatic fibrogenesis. Gastroenterology. 2008;134:1655–1669. doi: 10.1053/j.gastro.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maher JJ. Interactions between hepatic stellate cells and the immune system. Semin Liver Dis. 2001;21:417–426. doi: 10.1055/s-2001-17555. [DOI] [PubMed] [Google Scholar]
- 10.Amann T, Bataille F, Spruss T, Muhlbauer M, Gabele E, Scholmerich J, Kiefer P, Bosserhoff AK, Hellerbrand C. Activated hepatic stellate cells promote tumorigenicity of hepatocellular carcinoma. Cancer Sci. 2009;100:646–653. doi: 10.1111/j.1349-7006.2009.01087.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhao W, Zhang L, Yin Z, Su W, Ren G, Zhou C, You J, Fan J, Wang X. Activated hepatic stellate cells promote hepatocellular carcinoma development in immunocompetent mice. Int J Cancer. 2011;129:2651–61. doi: 10.1002/ijc.25920. [DOI] [PubMed] [Google Scholar]
- 12.Yang JD, Nakamura I, Roberts LR. The tumor microenvironment in hepatocellular carcinoma: Current status and therapeutic targets. Semin Cancer Biol. 2010;21:35–43. doi: 10.1016/j.semcancer.2010.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ju MJ, Qiu SJ, Fan J, Xiao YS, Gao Q, Zhou J, Li YW, Tang ZY. Peritumoral activated hepatic stellate cells predict poor clinical outcome in hepatocellular carcinoma after curative resection. Am J Clin Pathol. 2009;131:498–510. doi: 10.1309/AJCP86PPBNGOHNNL. [DOI] [PubMed] [Google Scholar]
- 14.Mikula M, Proell V, Fischer AN, Mikulits W. Activated hepatic stellate cells induce tumor progression of neoplastic hepatocytes in a TGF-beta dependent fashion. J Cell Physiol. 2006;209:560–567. doi: 10.1002/jcp.20772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xu J, Lamouille S, Derynck R. TGF-beta-induced epithelial to mesenchymal transition. Cell Res. 2009;19:156–172. doi: 10.1038/cr.2009.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee JM, Dedhar S, Kalluri R, Thompson EW. The epithelial-mesenchymal transition: new insights in signaling, development, and disease. J Cell Biol. 2006;172:973–981. doi: 10.1083/jcb.200601018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871–890. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 18.Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119:1420–1428. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu HY, Tseng VS, Chen LC, Chang HY, Chuang IC, Tsay YG, Liao PC. Identification of tyrosine-phosphorylated proteins associated with lung cancer metastasis using label-free quantitative analyses. J Proteome Res. 2010;9:4102–4112. doi: 10.1021/pr1006153. [DOI] [PubMed] [Google Scholar]
- 20.Faouzi S, Lepreux S, Bedin C, Dubuisson L, Balabaud C, Bioulac-Sage P, Desmouliere A, Rosenbaum J. Activation of cultured rat hepatic stellate cells by tumoral hepatocytes. Lab Invest. 1999;79:485–493. [PubMed] [Google Scholar]
- 21.Sancho-Bru P, Juez E, Moreno M, Khurdayan V, Morales-Ruiz M, Colmenero J, Arroyo V, Brenner DA, Gines P, Bataller R. Hepatocarcinoma cells stimulate the growth, migration and expression of pro-angiogenic genes in human hepatic stellate cells. Liver Int. 2010;30:31–41. doi: 10.1111/j.1478-3231.2009.02161.x. [DOI] [PubMed] [Google Scholar]
- 22.Yu MC, Chen CH, Liang X, Wang L, Gandhi CR, Fung JJ, Lu L, Qian S. Inhibition of T-cell responses by hepatic stellate cells via B7-H1-mediated T-cell apoptosis in mice. Hepatology. 2004;40:1312–1321. doi: 10.1002/hep.20488. [DOI] [PubMed] [Google Scholar]
- 23.Taura K, De Minicis S, Seki E, Hatano E, Iwaisako K, Osterreicher CH, Kodama Y, Miura K, Ikai I, Uemoto S, Brenner DA. Hepatic stellate cells secrete angiopoietin 1 that induces angiogenesis in liver fibrosis. Gastroenterology. 2008;135:1729–1738. doi: 10.1053/j.gastro.2008.07.065. [DOI] [PubMed] [Google Scholar]
- 24.Kang N, Yaqoob U, Geng Z, Bloch K, Liu C, Gomez T, Billadeau D, Shah V. Focal adhesion assembly in myofibroblasts fosters a microenvironment that promotes tumor growth. Am J Pathol. 2010;177:1888–1900. doi: 10.2353/ajpath.2010.100187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Leroy P, Mostov KE. Slug is required for cell survival during partial epithelial-mesenchymal transition of HGF-induced tubulogenesis. Mol Biol Cell. 2007;18:1943–1952. doi: 10.1091/mbc.E06-09-0823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rasanen K, Vaheri A. TGF-beta1 causes epithelial-mesenchymal transition in HaCaT derivatives, but induces expression of COX-2 and migration only in benign, not in malignant keratinocytes. J Dermatol Sci. 2010;58:97–104. doi: 10.1016/j.jdermsci.2010.03.002. [DOI] [PubMed] [Google Scholar]
- 27.Thuault S, Valcourt U, Petersen M, Manfioletti G, Heldin CH, Moustakas A. Transforming growth factor-beta employs HMGA2 to elicit epithelial-mesenchymal transition. J Cell Biol. 2006;174:175–183. doi: 10.1083/jcb.200512110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cheng H, Fukushima T, Takahashi N, Tanaka H, Kataoka H. Hepatocyte growth factor activator inhibitor type 1 regulates epithelial to mesenchymal transition through membrane-bound serine proteinases. Cancer Res. 2009;69:1828–1835. doi: 10.1158/0008-5472.CAN-08-3728. [DOI] [PubMed] [Google Scholar]
- 29.Cai Z, Wang Q, Zhou Y, Zheng L, Chiu JF, He QY. Epidermal growth factor-induced epithelial-mesenchymal transition in human esophageal carcinoma cells--a model for the study of metastasis. Cancer Lett. 2010;296:88–95. doi: 10.1016/j.canlet.2010.03.020. [DOI] [PubMed] [Google Scholar]
- 30.Medici D, Nawshad A. Type I collagen promotes epithelial-mesenchymal transition through ILK-dependent activation of NF-kappaB and LEF-1. Matrix Biol. 2010;29:161–165. doi: 10.1016/j.matbio.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kikuta K, Masamune A, Watanabe T, Ariga H, Itoh H, Hamada S, Satoh K, Egawa S, Unno M, Shimosegawa T. Pancreatic stellate cells promote epithelial-mesenchymal transition in pancreatic cancer cells. Biochem Biophys Res Commun. 2010;403:380–384. doi: 10.1016/j.bbrc.2010.11.040. [DOI] [PubMed] [Google Scholar]
- 32.Duner S, Lopatko Lindman J, Ansari D, Gundewar C, Andersson R. Pancreatic cancer: the role of pancreatic stellate cells in tumor progression. Pancreatology. 2010;10:673–681. doi: 10.1159/000320711. [DOI] [PubMed] [Google Scholar]
- 33.Yang MH, Chen CL, Chau GY, Chiou SH, Su CW, Chou TY, Peng WL, Wu JC. Comprehensive analysis of the independent effect of twist and snail in promoting metastasis of hepatocellular carcinoma. Hepatology. 2009;50:1464–1474. doi: 10.1002/hep.23221. [DOI] [PubMed] [Google Scholar]
- 34.Giannelli G, Bergamini C, Fransvea E, Sgarra C, Antonaci S. Laminin-5 with transforming growth factor-beta1 induces epithelial to mesenchymal transition in hepatocellular carcinoma. Gastroenterology. 2005;129:1375–1383. doi: 10.1053/j.gastro.2005.09.055. [DOI] [PubMed] [Google Scholar]
- 35.Ou DP, Tao YM, Tang FQ, Yang LY. The hepatitis B virus X protein promotes hepatocellular carcinoma metastasis by upregulation of matrix metalloproteinases. Int J Cancer. 2007;120:1208–1214. doi: 10.1002/ijc.22452. [DOI] [PubMed] [Google Scholar]
- 36.Martin-Vilchez S, Sanz-Cameno P, Rodriguez-Munoz Y, Majano PL, Molina-Jimenez F, Lopez-Cabrera M, Moreno-Otero R, Lara-Pezzi E. The hepatitis B virus X protein induces paracrine activation of human hepatic stellate cells. Hepatology. 2008;47:1872–1883. doi: 10.1002/hep.22265. [DOI] [PubMed] [Google Scholar]