

Abstract
Urokinase receptor interacts with α5β1-integrin and enhances cancer cell proliferation and metastasis. Activation of α5β1-integrin requires caveolin-1 and is regulated by uPAR, which upregulates persistently the activated ERK necessary for tumor growth. In this study, we show that the ganglioside GT1b induces proapoptotic signaling through two uPAR-ERK signaling pathways in A549 lung cancer cells. GT1b downregulated the expression of α5β1 integrin, caveolin-1, fibronectin, FAK, and ERK, whereas GT1b upregulated the expression of p53 and uPAR, suggesting GT1b mediated depletion of caveolin-1 in uPAR-expressing A549 cells also disrupts uPAR/integrin complexes, resulting in downregulation of fibronectin-α5β1-integrin-ERK signaling. Following p53 siRNA treatment, FAK and ERK expression was recovered, meaning the presence of reentry uPAR-FAK-ERK signaling pathway. These findings reveal that GT1b is involved in both caveolin-1-dependent uPAR-α5β1-integrin-ERK signaling and caveolin-1-independent uPAR-FAK-ERK signaling. These results suggest a novel function of GT1b as a dual regulator of ERK by modulating caveolin-1 and p53.
Keywords: Lung cancer, ganglioside GT1b, uPAR, caveolin-1, ERK, p53
Introduction
Gangliosides are lipids that contain sialic acid modifications. Ganglioside GT1b is the member of the glycosphingolipid family, and is present in the plasma membrane of essentially all mammalian cells. GT1b plays a role in the regulation of several biological processes, including cell migration, differentiation, and apoptosis, and functions as an antitumor agent in various cancers [1,2].
Urokinase, also called urokinase-type plasminogen activator (uPA, PLAU), is a serine protease [3] that regulates several cellular signaling pathways and stimulates a variety of responses. uPA also regulates the viability of human lung cancer cells. Expression of the urokinase receptor (uPAR/CD87, PLAUR) is significantly elevated in several types of tumors, and is frequently associated with poor prognosis [4]. uPAR regulates membrane-associated extracellular proteolysis by binding to the extracellular protease uPA, and also activates many membrane-associated extracellular proteins and signals [5]. uPA-α5β1 integrin interactions play a critical role in regulating cancer cell metastasis and drug resistance. Proper integrin function is required for uPAR signaling. The binding of uPA to uPAR activates signaling via the α5β1 or α3β1 integrins in some cancer cells [5]. GT1b is known to inhibit uPAR/α5β1-integrin signaling. However, the role of the uPAR-α5β1 integrin signaling pathway in apoptosis is poorly understood. Crosstalk between uPAR and p53 [6], and caveolin-induced uPAR-dependent ERK activation [7] have also been reported.
Lung cancer, which accounts for the majority (up to 85%) of non-small-cell lung carcinomas, is the second leading cause of cancer deaths worldwide [8]. Thus, lung cancer is still considered to have a very high mortality rate [9]. Surgical and combined therapies have helped to reduce the mortality and morbidity of lung cancer over the last several decades; however, these therapies have limitations.
The aim of this study was to assess the role of uPAR/ERK signaling in lung cancer cells and its regulation by caveolin-1-dependent and -independent processes. Our data provide novel insights into the mechanisms by which the downregulation of uPAR signaling contributes to proapoptosis. We also identified the proteins responsible for inhibiting uPAR signaling, which resulted in increased expression levels of p53 and uPAR, reduced expression of α5β1-integrin signaling, and decreased caveolin-1, focal adhesion kinase 1 (FAK), and ERK activity. In addition, treatment with GT1b caused significant induction of early apoptosis in A549 lung cancer cells.
Materials and methods
Cell culture
Human lung cancer (A549) cells were obtained from the American Type Culture Collection (Rockville, MD, USA). Cell lines were grown in Dulbecco’s Modified Eagle Medium (Sigma; St. Louis, MO, USA). The cells were incubated at 37°C with 5% (v/v) CO2 for 24 h. The cells were then plated in 96-well plates at a density of 5 × 103 cells/well. After a 24-h incubation, the cells were treated with 50 μg/mL GT1b. The optimal dose was determined using cytotoxicity tests after treatment for 24 h. To assess cell viability, 10 μL of cell-counting kit-8 solution (Dojindo; Kumamoto, Japan) was added, and cell numbers were determined using a microplate reader (Sunrise; Tecan, Switzerland) to measure the absorbance at 450 nm.
Reagents and chemicals
Chemicals such as phosphate-buffered saline (PBS) were purchased from Sigma. GT1b was purchased from Sigma. Annexin-V-FLUOS staining kits were purchased from Roche Diagnostics GmbH (Mannheim, Germany). Whole cell lysis buffer was purchased from iNtRON (Seoul, Korea). Hilymax transfection reagent was purchased from Dojindo.
Microarray analysis
GT1b-treated A549 lung cancer cells were analyzed using microarrays. Total RNA was extracted from control or GT1b-treated cells. The resulting probes were then coupled to Cy3 (control) or Cy5 (GT1b-treated). The mixed Cy3- and Cy5-labeled RNA was hybridized with one side of Twin ChipTM Human 44K (Genocheck; Seoul, Korea), and RNA from GT1b-treated cells was hybridized to the other side of the chip. The resulting fluorescent images were quantified and normalized using software provided by the manufacturer. The experiment was repeated four times. Genes were considered to be differentially expressed when the global M value, log2 (R/G fluorescence), exceeded 1.0 (two-fold change) with a P-value < 0.05 after significance analysis of microarray (SAM).
Gene ontology-related network analysis
Bioinformatic gene network analyses were performed using ingenuity pathways analysis (IPA, http://www.ingenuity.com) to examine the biological functions of the differentially regulated genes and proteins according to ontology-related interaction networks. IPA provides protein interaction networks based on the regularly updated “ingenuity pathways knowledgebase”. The network is optimized to include as many proteins from the input expression profile as possible, and aims to produce highly connected networks.
Fluorescence-assisted cytometric spectroscopy
To detect apoptosis, propidium iodide (PI)-annexin-V staining was performed using an Annexin-V-FLUOS Staining kit according to the manufacturer’s instructions. Briefly, cells were treated with GT1b for 48 h, harvested, and washed twice with PBS. The cell suspension was centrifuged at 2000 rpm for 2 min, and was then incubated with 0.2 mg/mL Annexin-V-FLUOS and 1.4 mg/mL PI for 15 min at room temperature. Data were analyzed using a FACS Caliber flow cytometer (Becton Dickinson; San Jose, CA, USA) with excitation at 488 nm and emission at 530/30 nm (band-pass filter) for annexin-V and 670 nm (high-pass filter) for PI. Data were analyzed using WinMDI V2.9 software.
Western blotting
Thirty micrograms of denatured proteins were separated using 12% polyacrylamide gel electrophoresis and transferred onto nitrocellulose membranes. The membranes were blocked for 1 h with 5% (w/v) skimmed milk in TTBS (0.1% Tween-20 and Tris-buffered saline). They were then incubated with diluted (1:1000) primary antibodies against Bcl-2, Bcl-XL, Bax, caspase 8, PLAUR (uPAR), integrin-α8β1, integrin-α5β1, p-p53, p21 (all from Cell Signaling Technologies), ERK, p-ERK, FAK1, fibronectin (FN), caveolin-1, and actin (all from Santa Cruz). The membranes were washed three times with TTBS for 5 min, followed by incubation with 1:2000 dilutions of horseradish peroxidase-conjugated goat anti-mouse or rabbit anti-goat IgG in TBS containing 5% (w/v) skimmed milk at room temperature for 1 h. The membranes were rinsed three times with TTBS for 5 min, and an enhanced chemiluminescence system (Thermo Scientific; San Jose, CA, USA) was used to visualize the bands on X-ray film (Kodak; Seoul, Korea).
p53 and caveolin-1 small interfering RNA (siRNA)
Non-specific control, p53 siRNAs (sequence: 5’-GCG CAC AGA GGA AGA GAA U-3’) and caveolin-1 siRNAs (sequence; 5’-AGA CGA GCU GAG CGA GAA GCA-3’) were obtained from Bioneer (Daejon, Korea). The transfection of siRNAs into A549 cells was performed using Hilymax reagent following the manufacturer’s instructions. Cells were then treated with 50 μg/mL GT1b for 48 h, and the cell cycle were assessed using a MoFlo Astrios flow cytometer (Beckman Coulter; Miami, FL, USA) with excitation at 488 nm and emission at 670 nm to detect PI.
Statistical analyses
GraphPad Prism software (GraphPad; San Diego, CA, USA) was used for all statistical analyses. Student’s t-tests were used to assess the differences between control and GT1b-treated groups. The values were calculated and presented as means ± SD. P-values < 0.05 were considered to be significant.
Results
GT1b treatment inhibits lung cancer cell proliferation
To investigate the effect of GT1b on the growth of lung cancer cells, A549 cells were pre-treated with GT1b, and growth inhibition was assessed using cell viability assays. As shown in Figure 1A, treatment with 50 μg/mL GT1b for 24 h significantly decreased the growth of A549 cells. To assess whether GT1b induces early apoptosis in A549 cells, the morphology of untreated and GT1b-treated A549 cells was compared. GT1b-treated A549 cells seemed to attach poorly to the plate when visualized under a normal light microscope (Figure 1B).
Figure 1.
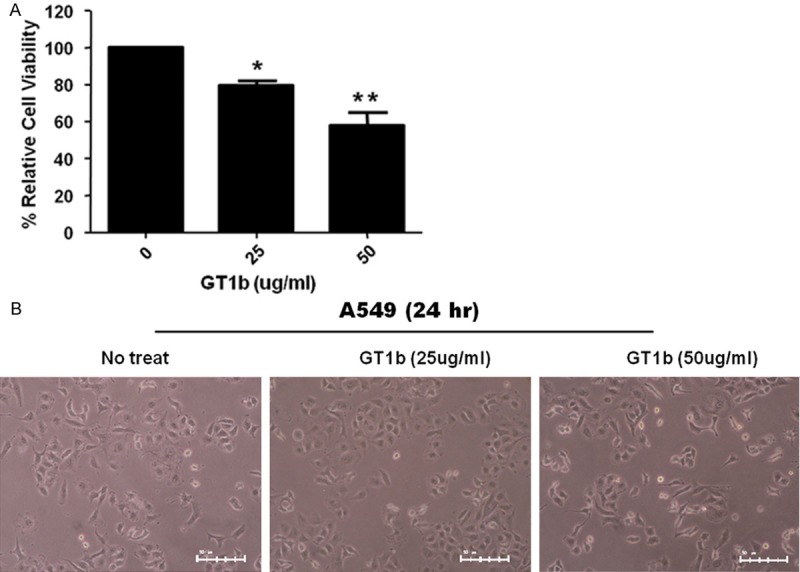
GT1b inhibits the growth of lung cancer cells. Cells were treated (as indicated) with 0 μg/mL, 25 μg/mL, and 50 μg/mL GT1b for 24 h A. The bar graphs show the means ± SDs of triplicate experiments. *, P < 0.05; **, P < 0.01 vs. control. B. Light microscopy images of cells treated with 50 μg/mL GT1b for 24 h (× 200 magnification).
The effect of GT1b on the cell cycle and cell death
The effect of GT1b on the cell cycle and cell death was analyzed quantitatively using PI staining followed by flow cytometry (Figure 2A), which allows for simultaneous assessment of the cell cycle and cell death. Specifically, the relative proportion of viable and apoptotic cells can be measured when co-staining with annexin V is performed. The treatment of A549 cells with 50 μg/mL GT1b led to the induction of pro-apoptosis. The proportion of early apoptotic cells increased dramatically from 16% in the control cells to 52% in GT1b-treated cells. In addition, the respective proportions of late apoptotic cells in the two groups were 3% and 5%. But, another ganglioside GD2, which as a negative control, had been no changes (16% to 14% early apoptic cells, and 3% to 4% late apoptotic cells). Therefore, A549 cells appeared to be very sensitive to only GT1b. In GT1b-treated A549 cells, the expression of Bax (a pro-apoptotic protein) was upregulated, whereas Bcl-2 and Bcl-XL (anti-apoptotic proteins) were down regulated (Figure 2B). However, caspase-8 levels were unchanged.
Figure 2.
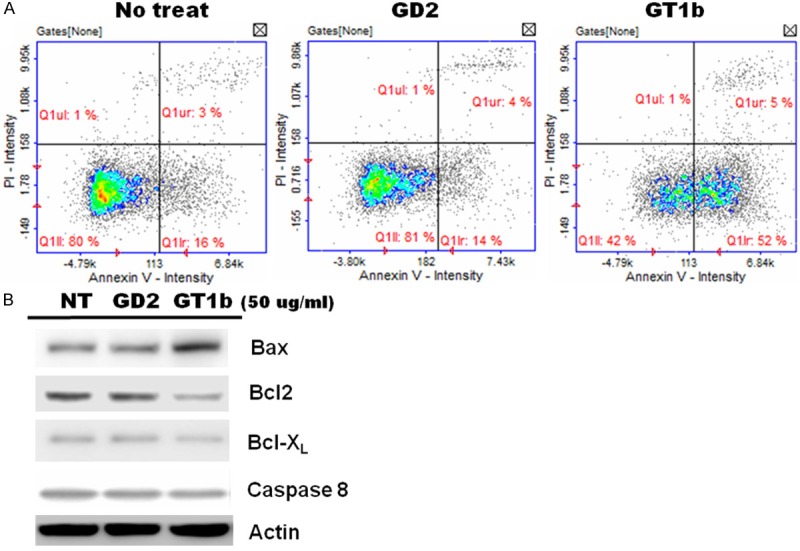
Analysis of apoptosis using double-labeling flow cytometry in A549 cells treated with GT1b. Cells were untreated (A, left panel) or treated with 50 μg/mL GD2 (A, middle panel) and 50 μg/mL GT1b for 24 h (A, right panel). The cells were double-stained with PI and annexin-V. The lower-left quadrant indicates normal cells, the lower-right quadrant contains early apoptotic cells, and the upper-right quadrant contains late apoptotic cells. The data are representative of three independent experiments. B. Western blotting for the expression of apoptosis-related proteins.
GT1b-related gene expression in A549 lung cancer cells
We used cDNA microarrays to analyze GT1b-related gene expression in lung cancer cells. The microarray data were filtered and combined based on gene symbols, and network analyses were performed using the Database for Annotation, Visualization, and Integrated Discovery (DAVID) tools (http://david.abcc.ncifcrf.gov/). Lists of genes that were at least two-fold up- or downregulated in GT1b-treated cells were uploaded to DAVID for Gene Ontology analysis. The upregulated genes included those involved in the regulation of signal transduction (including p53-mediated signaling), cytoskeleton-dependent intracellular transport, and cell projection morphogenesis (Figure 3A). The downregulated genes included those involved in the negative regulation of cell proliferation, migration, and the regulation of apoptosis (Figure 3B). We next identified genes that were regulated by GT1b treatment and that play roles in apoptosis using the GeneCards database (http://www.genecards.org/). We then independently evaluated the association between the apoptosis-associated genes and those whose expression changed more than two-fold in GT1b-treated cells. The results are illustrated schematically as Venn diagrams in Figure 3C, and the gene lists are provided in Figure 3D.
Figure 3.
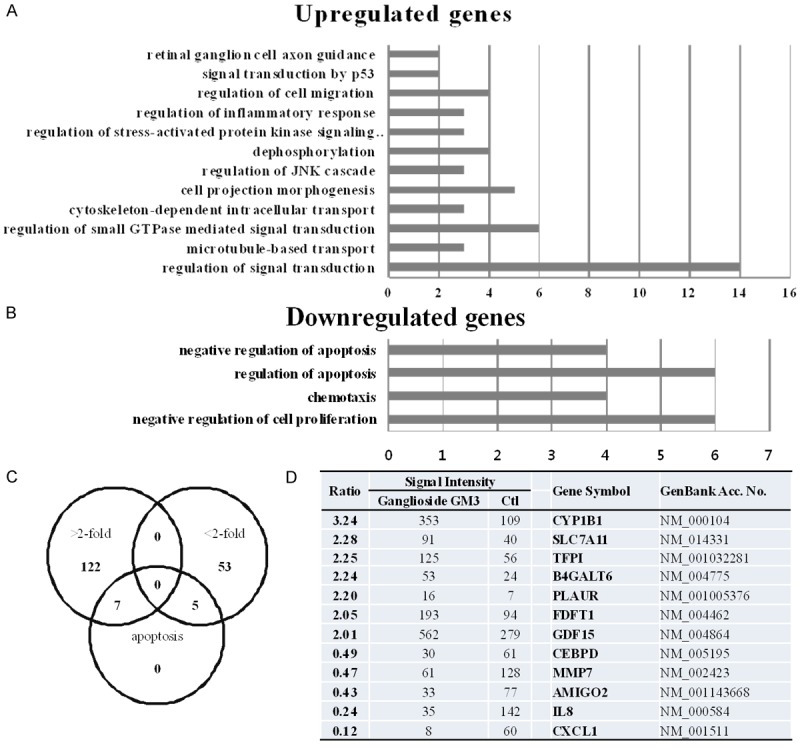
Microarray and Gene Ontology analyses and the apoptotic genes whose expression was altered by GT1b. Gene Ontology analysis revealed differentially expressed genes (> 2-fold, A, or >-fold-regulated genes, B) in A549 human lung cancer cells in response to GT1b. Three gene lists (> 2-fold increase, > 2-fold decrease, and apoptosis-related genes) are shown, which are also intersected individually using Venn diagrams (C). The apoptosis-related genes are shown in (D). The relative expression ratio was calculated by dividing the test channel intensity (GT1b) by the control channel intensity.
Gene ontology analysis
We next used Gene Ontology analysis to explore the function of the GT1b-related proteins. The 187 proteins that were up- or downregulated by GT1b were queried against the IPA program, resulting in a distinct interconnected network of 36 proteins (Figure 4). There were quantitative alterations in gene expression in GT1b-treated lung cancer cells compared with control cells. Among these, uPAR was identified as the center of the GT1b-related protein network in lung cancer cells. uPAR expression was increased in GT1b-treated lung cancer cells.
Figure 4.
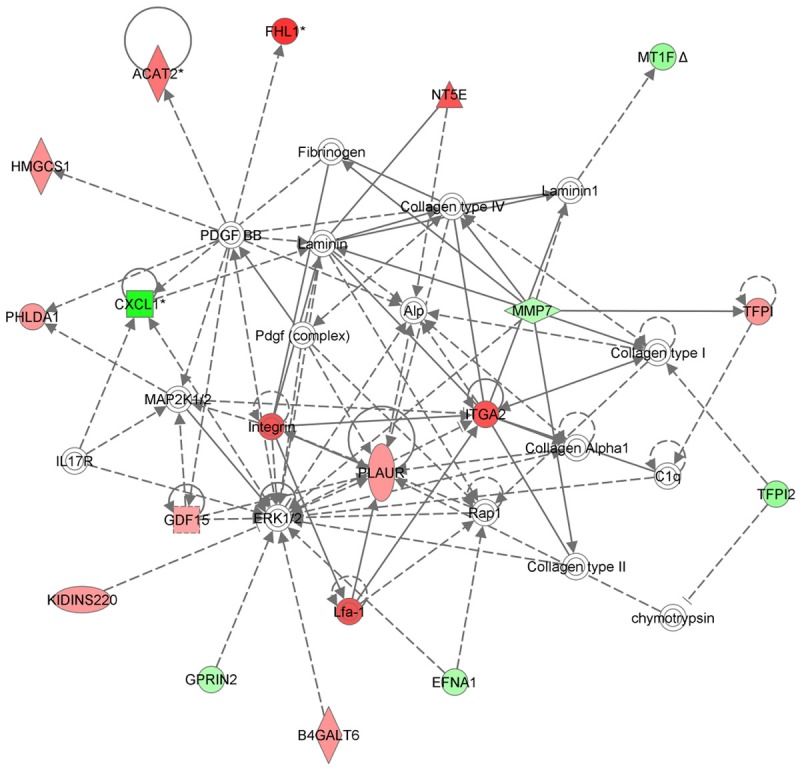
Network analysis based on Gene Ontology data. Proteins whose expression was changed at least two-fold in response to GT1b were queried in IPA, which yielded a distinct interconnected network of 36 proteins.
The caveolin-1-dependent effects of GT1b on uPAR signaling pathway
To investigate the caveolin-dependent uPAR-α5β1 integrin-ERK signaling pathway, the present study was undertaken to identify the function of caveolin-1 as a membrane adaptor or a scaffold to mediate uPAR-dependent activation of α5β1 integrin and ERK. uPAR and α5β1 integrin and its ability to bind FN, FAK, ERK were monitored. A549 lung adenocarcinoma cells expressed caveolin-1, uPAR, α5β1 integrin, FAK, and ERK. When GT1b was applied to A549 cells, expression of uPAR was increased, whereas caveolin-1, FN, α5β1 integrin, and FAK, pERK were decreased, indicating that uPAR, complexed with caveolin and signaling molecules, enters the cluster and then binds to α5β1 integrin with FN (Figure 5), and caveolin-1 siRNA also decreased the abundance of caveolin-1, FN, FAK1 and p-ERK (Figure 6B). These data demonstrate that GT1b downregulated caveolin-1 in uPAR-expressing A549 cells to disrupt uPAR/integrin complexes, resulting in downregulations of FN-α5β1-integrin-ERK signaling. Thus, these findings reveal that GT1b is involved in caveolin-dependent uPAR-α5β1 integrin-ERK signaling.
Figure 5.
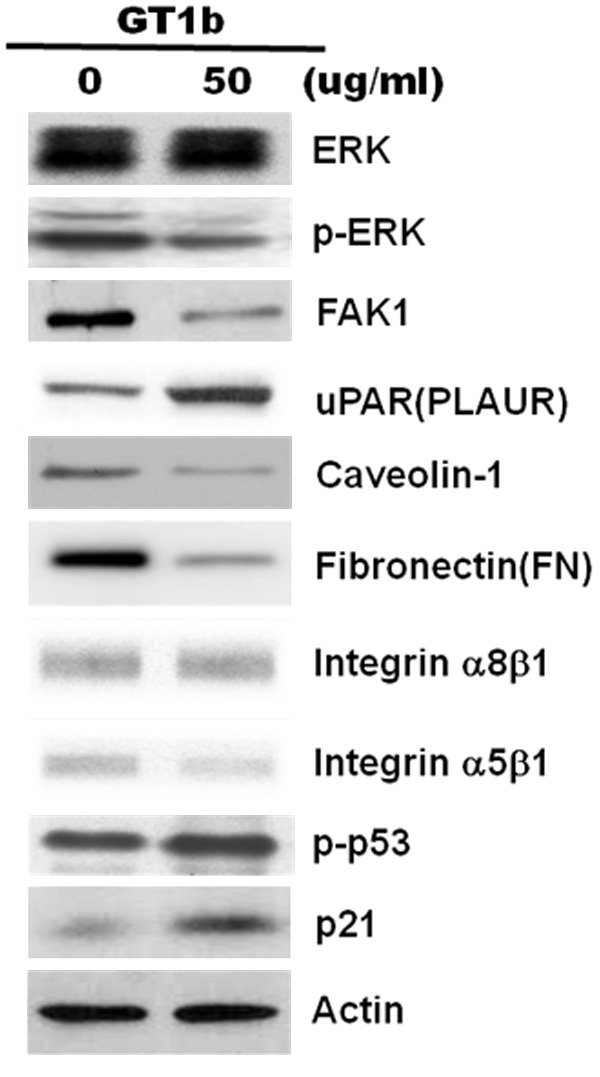
GT1b inhibits uPAR-α5β1 integrin-ERK signaling. A549 cells were incubated with 50 μg/mL GT1b for 24 h, and the expression of ERK, phospho-ERK, FAK1, uPAR, caveolin-1, fibronectin, integrin-α8β1, integrin-α5β1, p21, and p53 was analyzed using western blotting. Actin was used as an internal standard.
Figure 6.
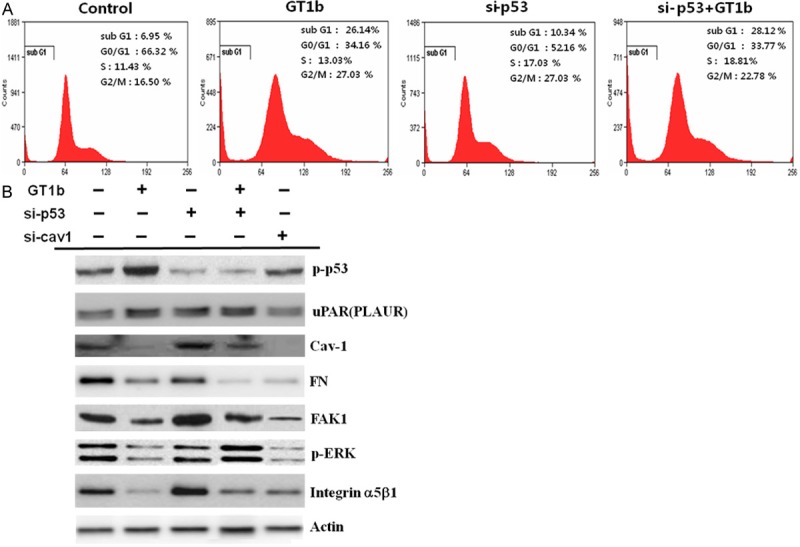
GT1b induces proapoptotic signaling through caveolin-1-dependent and reentry uPAR signaling pathways. A. Control and p53 siRNAs were transfected into A549 cells, which were then treated with GT1b for 24 h. The data are representative of three independent experiments. B. Western blotting for the levels of phospho-p53, uPAR, caveolin-1, FAK1 and pERK. Silencing p53 upregulated uPAR levels compared with control. GT1b inhibited the interaction between uPAR and α5β1 integrin, which upregulated uPAR and downregulated α5β1 integrin; the expression of uPAR and p53 was also increased. Caveolin-1, FN, α5β1 integrin, FAK1, and pERK in A549 cells were downregulated by si-caveolin-1. Actin was used as an internal standard.
The caveolin-1-independent uPAR-FAK-ERK signaling pathway
To investigate the caveolin-1-independent uPAR-FAK-ERK signaling pathway, the present study was undertaken to identify the caveolin-1-independent reentry uPAR-mediated downstream signaling pathway. GT1b inhibited caveolin-1, FAK, and p-ERK expression. In addition, the expression of FN was downregulated by GT1b. Downregulating the adhesion of α5β1-integrin to FN inhibited the interaction between uPAR and α5β1-integrin through downregulation of caveolin-1, resulting in inhibition of uPAR-α5β1 integrin-ERK signaling pathway. This suggests that GT1b activates uPAR, which inhibits α5β1 integrin to promote the development of lung cancer. GT1b increased the levels of p-p53 and uPAR. To investigate the effect of p53 expression on uPAR signaling, we tried to clarify whether or not p53 mediates GT1b-induced early apoptosis and growth arrest in A549 lung cancer cells. C ontrol scrambled siRNA and p53 siRNA were transfected into A549 cells, which were then treated with GT1b for 24 h. Cells were stained with PI for cell cycle, and the left peak (sub-G1) in the resulting analyses indicated early apoptotic cells. GT1b and p53 siRNA plus GT1b inhibited cell growth. The proportion of early apoptotic cells was increased from 6.95% in the control treatment to 26.14% in the GT1b treatment. The number of cells in the sub-G1 phase was increased to 28.12% in p53 siRNA cells treated with GT1b compare to 10.34% in p53 siRNA cells (Figure 6A). When p53 siRNA-transfected A549 cells were treated with GT1b, FAK and p-ERK levels were recovered, whereas there was no significantly change in caveolin-1 expression (Figure 6B), suggesting that uPAR activated rentry FAK-ERK signaling pathway through downregulation of the uPAR complexed molecule, caveolin-1, by GT1b. These findings reveal that GT1b is involved in caveolin-1-independent reentry uPAR-FAK-ERK signaling through upregulation of p53. Thus, these results suggest that GT1b induces proapoptotic signaling through dual regulation of uPAR-ERK signaling by modulating caveolin and p53 in A549 lung cancer cells.
Discussion
GT1b promotes cell proliferation, migration, and invasion by affecting cell signaling at the membrane [8], and also induces apoptosis in various cancers [2,8,10]. However, the mechanisms underlying the role of p53 and caveolin-1 in the induction of apoptosis remain unclear. This study showed that pretreatment with 50 μg/mL GT1b reduced cell viability and strongly inhibited the growth of A549 cells (Figure 1A). However, treatment with GT1b had little effect on the morphology of A549 cells compared with control treatment (Figure 1B). The results of annexin V-PI staining using FACS demonstrated that GT1b could induce pro-apoptosis. GT1b transformed cells from the normal state (untreated group: 80.0% normal, 16.0% early apoptotic, and 3% late apoptotic) to the apoptotic state (18.42% normal, 52.0% early apoptotic, and 5.0% late apoptotic), but there were no changes treated with GD2 (Figure 2A). Therefore, these results suggest that GT1b exhibits anti-lung cancer activity and promotes pro-apoptosis compared with GD2. The anti-apoptotic proteins Bcl-2 and Bcl-XL and the pro-apoptotic protein Bax regulate critical steps in cancer cell death. Figure 2B shows that GT1b-treated lung cancer cells had increased Bax and decreased Bcl-2 and Bcl-XL expression. The ratio of Bax/Bcl-2 expression is a key factor in the regulation of apoptosis [11] and as a late apoptosis marker caspase 8 had not changed. Thus, GT1b clearly induced early apoptosis in lung cancer cells.
To analyze GT1b-related gene expression in lung cancer cells, we used a cDNA microarray approach. The clustered microarray data identified groups of genes that were regulated differently in control and GT1b-treated cells. The genes whose expression changed by at least two-fold are shown in Figure 3A and 3B. Three groups of genes (> 2-fold increased, > 2-fold decreased, and apoptosis-related genes) are shown. Among these, 122 genes were increased by > 2-fold, and 53 were decreased by > 2-fold with apoptosis related genes individually using Venn diagrams (Figure 3C). The apoptosis-related genes are listed in Figure 3D. To explore the major GT1b-related proteins identified using Gene Ontology analyses, we used IPA to query 187 proteins that were up- or downregulated by GT1b. These analyses resulted in a distinct interconnected network of 35 proteins (Figure 4). Among these, uPAR was the center of the GT1b-related protein network.
uPAR interacts with integrins, and is a critical regulator of cancer cell proliferation and angiogenesis. In this study, we investigated the potential function of uPAR in lung cancer. uPAR expression was increased in GT1b-treated lung cancer cells. Protein-protein network analysis suggested that uPAR interacts with multiple proteins. Although it is clear that uPAR plays a key role in lung cancer [12], the mechanism is unclear. The accumulation of uPAR has been associated with lung cancer therapy. uPAR ligands such as FN and vitronectin are also crucial for this process. In addition, uPAR-modulated signal transduction requires α5β1 integrins [13]. In various cells, uPA binding to uPAR stimulates integrins, through as yet unknown partner molecules, which triggers FAK1, and activates myosin light-chain kinase [14]. Inhibiting uPAR induces apoptosis [15]. However, our data suggest that even though GT1b activates uPAR, it also inhibits α5β1 integrin and activates p53. The role of p53 in the regulation of uPAR-α5β1 integrin signaling during apoptosis has not yet been fully characterized. Based on the results of this study, we hypothesize that p53 interferes with uPAR signaling.
p53 plays a central role in many cellular responses by triggering cell cycle arrest or apoptosis [16]. In addition, mutant p53 drives cell invasion by promoting integrin recycling [17], and can also regulate uPAR expression [6]. Although previous studies have reported only inhibitory effects of GT1b on uPAR-α5β1 integrin signaling, in the current study, uPAR was increased, whereas caveolin-1, FN, FAK, ERK, and α5β1 integrin were decreased in GT1b-treated A549 lung cancer cells (Figure 5). The downregulation of caveolin and α5β1 integrin implies caveolin-dependent effects of GT1b on the uPAR-α5β1 integrin signaling pathway. In contrast, the expression of α5β1 integrin, FAK, and ERK was downregulated, indicating dissociation of the interaction between uPAR and α5β1 integrin.
In an effort to verify the role of p53 in GT1b-induced early apoptosis, we attempted to determine whether p53 is required for the induction of proapoptosis. We transfected cells with p53 siRNA and then treated them with GT1b. Inhibiting p53 in control cells had effect on the cell cycle, whereas the proportion of proapoptotic cells was shifted from 6.95% to 26.14% in A549 cells treated with GT1b alone, and was further increased to 28.12% in p53 siRNA cells treated with GT1b compare to 10.34% in p53 siRNA (Figure 6A). In addition, treating p53 siRNA cells with GT1b increased uPAR and decreased caveolin-1, FAK, and ERK expression (Figure 6B). These results are consistent with a previous study demonstrating that uPAR induces apoptosis [18,19]. Interestingly, this study suggests that GT1b could inhibit integrin-α5β1 but activate p53, even in the absence of the uPAR/α5β1-integrin interaction. Inhibiting the interaction between uPAR and α5β1-integrin activated p53. In view of previous observations that caveolin-1 stimulates uPAR-dependent integrin activation and FN by uPAR [20,21], and the association of activated uPAR with α5β1-integrin requires the presence of caveolin [8,21]. we investigated whether si-caveolin-1 affects u-PAR interaction with integrins. We found that the si-caveolin-1 decreased the association of FN with a5b1-integrin, FAK, and pERK (Figure 6B). Thus, the caveolin-dependent effects of GT1b on the uPAR-α5β1 integrin-ERK signaling pathway suggest that GT1b regulates uPAR-α5β1 integrin-ERK signaling through downregulation of caveolin. Although previous studies have reported only inhibitory effects of GT1b on uPAR-α5β1 integrin signaling, in the present study conducted in A549 lung cancer cells, α5β1 integrin was inhibited, whereas uPAR increased. When p53-silenced cells were treated with GT1b, FAK and p-ERK expression increased, whereas there was little change in caveolin protein expression, suggesting the involvement of a caveolin-independent reentry uPAR-FAK-ERK signaling pathway in these effects.
The ability of GT1b to inhibit uPAR signaling in a caveolin-independent manner suggests the existence of unique signaling circuits in these cells. uPAR activated FAK and ERK phosphorylation through GT1b, which led to the activation of the classic mitogenic pathway, whereas upregulation of p53 by GT1b inactivated FAK and ERK. These results suggest that GT1b induces early apoptotic signaling through c aveolin-1-dependent and caveolin-1-independent reentry uPAR signaling pathways by modulating caveolin-1 and p53 in A549 lung cancer cells.
Acknowledgements
This work was supported by a grant to Y.H.C. from the Korea Basic Science Institute (D34400), and by the Creative Fusion Research Program through the Creative Allied Project funded by the Korea Research Council of Fundamental Science and Technology (CAP-12-1).
Disclosure of conflict of interest
None.
References
- 1.Sun P, Wang XQ, Lopatka K, Bangash S, Paller AS. Ganglioside loss promotes survival primarily by activating integrin-linked kinase/Akt without phosphoinositide 3-OH kinase signaling. J Invest Dermatol. 2002;119:107–117. doi: 10.1046/j.1523-1747.2002.01802.x. [DOI] [PubMed] [Google Scholar]
- 2.Wang XQ, Sun P, Paller AS. Ganglioside modulation regulates epithelial cell adhesion and spreading via ganglioside-specific effects on signaling. J Biol Chem. 2002;277:40410–40419. doi: 10.1074/jbc.M207117200. [DOI] [PubMed] [Google Scholar]
- 3.Hildenbrand R, Gandhari M, Stroebel P, Marx A, Allgayer H, Arens N. The urokinase-system--role of cell proliferation and apoptosis. Histol Histopathol. 2008;23:227–236. doi: 10.14670/HH-23.227. [DOI] [PubMed] [Google Scholar]
- 4.Mekkawy AH, Pourgholami MH, Morris DL. Involvement of Urokinase-Type Plasminogen Activator System in Cancer: An Overview. Med Res Rev. 2014;34:918–56. doi: 10.1002/med.21308. [DOI] [PubMed] [Google Scholar]
- 5.Blasi F, Carmeliet P. uPAR: a versatile signalling orchestrator. Nat Rev Mol Cell Biol. 2002;3:932–943. doi: 10.1038/nrm977. [DOI] [PubMed] [Google Scholar]
- 6.Shetty S, Velusamy T, Idell S, Shetty P, Mazar AP, Bhandary YP, Shetty RS. Regulation of urokinase receptor expression by p53: novel role in stabilization of uPAR mRNA. Mol Cell Biol. 2007;27:5607–5618. doi: 10.1128/MCB.00080-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ossowski L, Aguirre-Ghiso JA. Urokinase receptor and integrin partnership: coordination of signaling for cell adhesion, migration and growth. Curr Opin Cell Biol. 2000;12:613–620. doi: 10.1016/s0955-0674(00)00140-x. [DOI] [PubMed] [Google Scholar]
- 8.Wang XQ, Sun P, Paller AS. Gangliosides inhibit urokinase-type plasminogen activator (uPA)-dependent squamous carcinoma cell migration by preventing uPA receptor/alphabeta integrin/epidermal growth factor receptor interactions. J Invest Dermatol. 2005;124:839–848. doi: 10.1111/j.0022-202X.2005.23669.x. [DOI] [PubMed] [Google Scholar]
- 9.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 2009. CA Cancer J Clin. 2009;59:225–249. doi: 10.3322/caac.20006. [DOI] [PubMed] [Google Scholar]
- 10.Chung TW, Choi HJ, Kim SJ, Kwak CH, Song KH, Jin UH, Chang YC, Chang HW, Lee YC, Ha KT, Kim CH. The ganglioside GT1b is associated with cisplatin-induced apoptosis in human colon cancer cells. PLoS One. 2014;9:e92786. doi: 10.1371/journal.pone.0092786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zinkel S, Gross A, Yang E. BCL2 family in DNA damage and cell cycle control. Cell Death Differ. 2006;13:1351–1359. doi: 10.1038/sj.cdd.4401987. [DOI] [PubMed] [Google Scholar]
- 12.Rao JS, Gujrati M, Chetty C. Tumor-associated soluble uPAR-directed endothelial cell motility and tumor angiogenesis. Oncogenesis. 2013;2:e53. doi: 10.1038/oncsis.2013.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Monaghan-Benson E, McKeown-Longo PJ. Urokinase-type plasminogen activator receptor regulates a novel pathway of fibronectin matrix assembly requiring Src-dependent transactivation of epidermal growth factor receptor. J Biol Chem. 2006;281:9450–9459. doi: 10.1074/jbc.M501901200. [DOI] [PubMed] [Google Scholar]
- 14.Koshelnick Y, Ehart M, Hufnagl P, Heinrich PC, Binder BR. Urokinase receptor is associated with the components of the JAK1/STAT1 signaling pathway and leads to activation of this pathway upon receptor clustering in the human kidney epithelial tumor cell line TCL-598. J Biol Chem. 1997;272:28563–28567. doi: 10.1074/jbc.272.45.28563. [DOI] [PubMed] [Google Scholar]
- 15.Aguirre-Ghiso JA, Liu D, Mignatti A, Kovalski K, Ossowski L. Urokinase receptor and fibronectin regulate the ERK(MAPK) to p38(MAPK) activity ratios that determine carcinoma cell proliferation or dormancy in vivo. Mol Biol Cell. 2001;12:863–879. doi: 10.1091/mbc.12.4.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Marchetti A, Cecchinelli B, D’Angelo M, D’Orazi G, Crescenzi M, Sacchi A, Soddu S. p53 can inhibit cell proliferation through caspase-mediated cleavage of ERK2/MAPK. Cell Death Differ. 2004;11:596–607. doi: 10.1038/sj.cdd.4401368. [DOI] [PubMed] [Google Scholar]
- 17.Muller PA, Caswell PT, Doyle B, Iwanicki MP, Tan EH, Karim S, Lukashchuk N, Gillespie DA, Ludwig RL, Gosselin P, Cromer A, Brugge JS, Sansom OJ, Norman JC, Vousden KH. Mutant p53 drives invasion by promoting integrin recycling. Cell. 2009;139:1327–1341. doi: 10.1016/j.cell.2009.11.026. [DOI] [PubMed] [Google Scholar]
- 18.Stepanova V, Jerke U, Sagach V, Lindschau C, Dietz R, Haller H, Dumler I. Urokinase-dependent human vascular smooth muscle cell adhesion requires selective vitronectin phosphorylation by ectoprotein kinase CK2. J Biol Chem. 2002;277:10265–10272. doi: 10.1074/jbc.M109057200. [DOI] [PubMed] [Google Scholar]
- 19.Besch R, Berking C, Kammerbauer C, Degitz K. Inhibition of urokinase-type plasminogen activator receptor induces apoptosis in melanoma cells by activation of p53. Cell Death Differ. 2007;14:818–829. doi: 10.1038/sj.cdd.4402065. [DOI] [PubMed] [Google Scholar]
- 20.Wei Y, Yang X, Liu Q, Wilkins JA, Chapman HA. A role for caveolin and the urokinase receptor in integrin-mediated adhesion and signaling. J Cell Biol. 1999;144:1285–1294. doi: 10.1083/jcb.144.6.1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bist A, Fielding CJ, Fielding PE. p53 regulates caveolin gene transcription, cell cholesterol, and growth by a novel mechanism. Biochemistry. 2000;39:1966–1972. doi: 10.1021/bi991721h. [DOI] [PubMed] [Google Scholar]