

Abstract
Gliomas, the most common primary brain tumors in adults, constitute clinically, histologically, and molecularly a most heterogeneous type of cancer. Owing to this, accurate clinical prognosis for short-term vs. long-term survival for patients with grade II or III glioma is currently nonexistent. A rigorous, multi-method bioinformatic approach was used to identify the top most differentially expressed genes as captured by mRNA sequencing of tumor tissue. Mathematical modeling was employed to develop the model, and three different and independent methods of validation were used to assess its performance. I present here a mathematical model that can identify with a high accuracy (sensitivity=92.9%, specificity=96.0%) those patients with glioma (grade II or III) who will experience short-term survival (≤ 1 year), as well as those with long-term survival (≥ 3 years), at the time of diagnosis and prior to surgery and adjuvant chemotherapy. The 5 gene input variables to the model are: FAM120AOS, PDLIM4, OCIAD2, PCDH15, and MXI1. MXI1, a transcriptional repressor, represents the top biomarker of survival and the most promising target for the development of a pharmacological treatment.
Keywords: Glioma, cancer genomics, survival, computational biology, mathematical modeling, systems biology, RNA-Sequencing
Introduction
Gliomas constitute approximately 80% of all malignant primary brain tumors in adults [1]. According to their histology and morphological features, gliomas are classified by the World Health Organization (WHO) mainly into astrocytomas, oligodendrogliomas, and oligoastrocytomas [2]. In addition to this lineage-based classification, gliomas are graded by WHO into four grades (I-IV), ranging from the most benign with low proliferative potential (grade I) to the most aggressive with very high proliferative activity (grade IV), such as glioblastomas. However general, the WHO grading system is used as a predictor of biological progression and clinical outcome; and in the clinic, it is influencing the choice of treatment [2]. Excluding the two extreme grades (I and IV), however, accurate clinical prognosis for short-term vs. long-term survival for patients with grade II or grade III glioma is currently nonexistent. This has to do with, inter alia, the following two observations: 1) Gliomas are histologically and molecularly very heterogeneous, and, therefore, they result in widely different clinical outcomes in terms of survival - so much so, that even among the same type of gliomas, there is wide variability in terms of survival [1,3]. 2) The WHO grading system is in effect a malignancy scale based on tumor histology [2], with only four broad classifications, and with a large overlap between the two middle classifications, i.e. grade II and grade III. For instance, patients with grade II astrocytoma have a five-year survival rate of ~50%; whereas patients with grade III astrocytoma have a five-year survival rate of ~30% [1,3]. Furthermore, as can be seen in Table S1, which contains clinical information about all 89 subjects used in this study, out of the 75 subjects (Subjects No 1-75) that were long-term survivors (survival ≥ 3 yrs), 40 had grade II glioma and 35 had grade III glioma. This clearly, therefore, underscores the need for an accurate prognostic method for clinical outcome in terms of survival that can be used in the clinic to guide the choice of treatment for patients with grade II or grade III glioma. Other studies have accentuated the same clinical need, as well [4-6]. The hypothesis of this study is that there exists a significant difference in the tumor gene expression that determines short-term versus long-term survival in patients with either grade II (G2) glioma or grade III (G3) glioma. In order to enhance this genetic difference, and in order to place greater emphasis on the detection of the specific genetic difference that is characteristic of those particularly aggressive tumors that result in very poor clinical outcome, i.e. very short survival, I defined the boundaries of the two groups as follows: 1) short-term survivors (STS) with survival ≤ 1 year and 2) long-term survivors (LTS) with survival ≥ 3 years. Using mRNA sequencing of tumor tissue from 14 short-term survivors and 75 long-term survivors, and by employing a rigorous, multi-method bioinformatic approach that I have developed and presented previously [7-10], I identified 29 genes that were the most significant in terms of differential expression between short-term and long-term survivors. Furthermore, using a general methodology that I have developed and presented previously [7,11,12], I generated a function that - based on the input of 5 genes from the 29 most significant genes - was able to identify/classify accurately all but one of the short-term survivors [sensitivity=(13/14)=0.929] and all but three of the long-term survivors [specificiy=(72/75)=0.960]. This model was designed to detect/identify those patients with either G2 or G3 glioma who - owing to the aggressive genetic make-up of the tumor tissue, and contrary to expectations according to the WHO grading system - will turn out to have a very poor clinical outcome (survival ≤ 1 year); in fact, their clinical outcome will be the same as, if not worse than, that of patients with the most aggressive type of glioma (grade IV), such as glioblastoma (survival ~14 months). The ability to identify those patients at the time of the diagnosis and to select more appropriate, intensive treatment(s) - that constitutes the clinical utility of this model.
Methods
Data acquisition
I downloaded the normalized data for 89 subjects with glioma (G2 or G3) [14 short-term survivors and 75 long-term survivors] generated from mRNA sequencing of tumor tissue (using the Illumina HiSeq 2000 sequencer) from The Cancer Genome Atlas (TCGA) of the National Cancer Institute under the category LGG (accessed on 2013-09-06).
Clinical study design
Hypothesis
The hypothesis of this study is that there exists a significant difference in the expression of a number of exome genes of the tumor tissue, and that that significant genetic difference is responsible for the discrimination between short-term survival and long-term survival in patients with either G2 or G3 glioma.
Inclusion/exclusion criteria
As was stated above, I defined the boundaries of the two groups as follows: 1) short-term survivors with survival ≤ 1 year and 2) long-term survivors with survival ≥ 3 years. The purpose of this was a) to enhance the tumor genetic contrast between the two groups by imposing a two-year gap between their survival boundaries, and b) to place greater emphasis on the detection of the specific genetic difference that is characteristic of those particularly aggressive tumors that result in very poor clinical outcome, i.e. very short survival.
All subjects selected for this study had supratentorial G2 or G3 glioma; all had one of the following types of glioma: astrocytoma, oligodendroglioma, or oligoastrocytoma; and all had surgery and adjuvant chemotherapy (mainly temozolomide).
None of the subjects selected for this study had a prior diagnosis of brain cancer, and none of them received radiation therapy.
Table S1 contains clinical and demographical information about all 89 subjects selected for this study.
Statistical methods
Control genes
Using 18 control genes, I first assessed the quality of the data by examining the expression of all 89 subjects [14 short-term survivors (STS) and 75 long-term survivors (LTS)] with respect to those 18 control genes. There was no statistically significant differential expression between the two groups with respect to any of those 18 genes. Table S2 shows those results in great detail. The significance tests used here are the same as those used in the main differential expression analysis, and they are, therefore, fully described next.
Differential expression analysis
In order to assess statistical significance, I used the following three different and independent methods.
1) ROC curve analysis. Using a methodology that I have developed and introduced previously [7-14], I performed ROC curve analysis on all gene variables in order to assess their discriminating power with respect to the two groups (STS vs. LTS); and with respect to this method, I set statistical significance at ROC AUC ≥ 0.930 (or ROC AUC ≤ -0.930 for those variables where the mean expression value of the STS group is less than that of the LTS group).
2) Fold Change. For all gene variables, I defined fold change (FC) as the mean expression value of the STS subjects over the mean expression value of the LTS subjects, and I set statistical significance at FC ≥ 1.50 (or FC ≤ -1.50 for those variables where the mean expression value of the STS group is less than that of the LTS group).
3) P-value. I used the independent t-Test (TT) for parametric gene variables (both normality and homogeneity of variance conditions were met); the Aspin-Welch unequal-variance test (AW) for gene variables that met the normality condition but not the homogeneity of variance condition; and the Mann-Whitney U test (MW) for the non-parametric gene variables. Taking into account that there are 20,531 gene variables (these are all the exome genes that were quantified by the mRNA sequencing of all tumor tissue samples using the Illumina HiSeq 2000 sequencer), and using the Bonferroni correction, I set the significance level for the entire study at α=2.43 x 10-6. Therefore, in order for any variable to be deemed significant according to the P-value method, the following condition must be met: P < α. Regarding the Mann-Whitney U test (MW), since none of the non-parametric variables had any sets of ties (a subject from one group having the same expression value as a subject from the other group), I used the exact probability for all MW tests.
Incorporating the three aforementioned independent methods of statistical significance assessment, and in order to minimize the number of both false positives and false negatives [15-18], I set the overall significance criterion as follows: in order for any variable to be included in the final list of the most significant variables, it would have to meet the significance criteria of each one of those three different and independent methods. Therefore, the overall criterion of significance required: 1) ROC AUC ≥ 0.930 (or ROC AUC ≤ -0.930), 2) FC ≥ 1.50 (or FC ≤ -1.50), and 3) P < 2.43 x 10-6.
Twenty nine genes fulfilled the overall criterion of significance and made up the final list of the most significantly differentially expressed genes between the two groups. Table 1 shows in great detail those results.
Table 1.
Top 29 most significantly differentially expressed genes
| No | Gene ID | DE (STS) | ROC AUC | FC | P | ML | SDL | MS | SDS | Notes |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | ABI1 | ↓ | -0.9505 | -1.920 | 3.17E-10 | 2653.474 | 843.333 | 1381.926 | 395.935 | MW |
| 2 | ADO | ↓ | -0.9438 | -1.627 | 1.15E-12 | 1259.006 | 195.237 | 773.854 | 228.252 | TT |
| 3 | AP1S3 | ↑ | 0.9400 | 3.973 | 1.49E-09 | 15.175 | 11.468 | 60.287 | 40.091 | MW |
| 4 | ARNTL2 | ↑ | 0.9352 | 3.529 | 2.83E-09 | 31.718 | 32.163 | 111.939 | 63.775 | MW |
| 5 | ASCC1 | ↓ | -0.9314 | -1.920 | 4.64E-09 | 1140.867 | 277.625 | 594.266 | 216.801 | MW |
| 6 | CMYA5 | ↑ | 0.9486 | 8.946 | 4.26E-10 | 90.587 | 237.137 | 810.392 | 681.291 | MW |
| 7 | CTBP2 | ↓ | -0.9333 | -1.605 | 8.89E-09 | 1326.318 | 280.781 | 826.362 | 196.115 | TT |
| 8 | DIAPH1 | ↑ | 0.9305 | 1.672 | 5.23E-09 | 1027.339 | 366.561 | 1717.753 | 413.980 | MW |
| 9 | EIF4EBP2 | ↓ | -0.9314 | -1.587 | 1.17E-08 | 4246.702 | 893.414 | 2676.768 | 595.612 | TT |
| 10 | EMP3 | ↑ | 0.9362 | 9.438 | 2.50E-09 | 121.852 | 250.739 | 1149.994 | 733.065 | MW |
| 11 | ETV7 | ↑ | 0.9410 | 5.667 | 1.30E-09 | 7.867 | 10.086 | 44.585 | 43.793 | MW |
| 12 | FABP5 | ↑ | 0.9305 | 9.416 | 5.23E-09 | 25.399 | 32.416 | 239.143 | 208.037 | MW |
| 13 | FAM120AOS | ↑ | 0.9571 | 1.504 | 1.06E-10 | 640.018 | 107.859 | 962.671 | 166.590 | MW |
| 14 | FBXO17 | ↑ | 0.9371 | 6.376 | 2.20E-09 | 73.694 | 74.914 | 469.885 | 254.136 | MW |
| 15 | GJD3 | ↑ | 0.9333 | 5.973 | 3.63E-09 | 12.486 | 17.679 | 74.581 | 71.060 | MW |
| 16 | LOC254559 | ↓ | -0.9352 | -3.671 | 2.83E-09 | 1929.637 | 871.222 | 525.618 | 575.678 | MW |
| 17 | MAP1LC3C | ↑ | 0.9762 | 32.187 | 2.33E-12 | 1.569 | 1.397 | 50.486 | 52.122 | MW |
| 18 | MARCH5 | ↓ | -0.9457 | -1.539 | 6.56E-10 | 1054.041 | 162.799 | 684.873 | 141.365 | MW |
| 19 | MRPL43 | ↓ | -0.9324 | -1.686 | 4.11E-09 | 1103.024 | 268.844 | 654.234 | 190.321 | MW |
| 20 | MXI1 | ↓ | -0.9438 | -2.049 | 8.39E-09 | 2964.509 | 851.196 | 1446.766 | 589.826 | TT |
| 21 | OCIAD2 | ↑ | 0.9400 | 7.625 | 1.49E-09 | 87.062 | 81.982 | 663.873 | 617.919 | MW |
| 22 | PCDH15 | ↓ | -0.9438 | -6.073 | 4.17E-16 | 1262.292 | 740.195 | 207.866 | 206.486 | AW |
| 23 | PDLIM4 | ↑ | 0.9495 | 12.332 | 3.68E-10 | 69.461 | 139.253 | 856.613 | 1071.121 | MW |
| 24 | RAP2A | ↓ | -0.9305 | -2.317 | 1.16E-11 | 6054.014 | 2475.325 | 2612.444 | 996.043 | AW |
| 25 | RBM17 | ↓ | -0.9448 | -1.691 | 1.98E-08 | 2122.824 | 506.611 | 1255.584 | 303.376 | TT |
| 26 | SEPHS1 | ↓ | -0.9419 | -1.776 | 1.14E-09 | 1581.372 | 479.258 | 890.179 | 202.707 | MW |
| 27 | SLC12A7 | ↑ | 0.9324 | 2.265 | 4.11E-09 | 378.113 | 225.375 | 856.256 | 286.952 | MW |
| 28 | SLC27A3 | ↑ | 0.9362 | 3.583 | 2.50E-09 | 120.516 | 109.021 | 431.788 | 282.630 | MW |
| 29 | TMPRSS3 | ↑ | 0.9381 | 15.190 | 1.93E-09 | 5.256 | 7.993 | 79.834 | 85.619 | MW |
The top 29 most significantly differentially expressed genes between the STS and the LTS subjects in alphabetical order. The arrows indicate differential expression [over-expression (↑) or under-expression (↓)] of the STS subjects as compared with the LTS subjects. The ROC AUC value, the fold change (FC) value, the P-value, the mean expression value of the LTS subjects (ML) and their standard deviation (SDL), the mean expression value of the STS subjects (MS) and their standard deviation (SDS) are listed for each gene variable. (AW): The Aspin-Welch unequal-variance test was used for the calculation of the P-value for those variables. (MW): The Mann-Whitney U test was used for the calculation of the P-value for those variables. (TT): The independent t-Test for parametric variables was used for the calculation of the P-value for those variables. As can be seen, all of those 29 genes met the overall criterion of significance set for, and required by, this study: 1) ROC AUC ≥ 0.930 (or ROC AUC ≤ -0.930), 2) FC ≥ 1.50 (or FC ≤ -1.50), and 3) P < 2.43 x 10-6.
The model
Development of the model
Using a split of approximately 75% and 25%, I randomly partitioned all of the subjects of this study (89 in all) into two fixed sets: a) the training set comprising 66 subjects (10 STS and 56 LTS) and b) the validation set comprising 23 subjects (4 STS and 19 LTS). The training set was used only for the development of the model, whereas the validation set was used only for the validation of the model. This split into two fixed sets, whereby one is used only for training and the other only for validation, represents the simplest implementation of K-fold cross validation [19,20]. The aforementioned 29 most significant gene variables provided the pool for the input variables of the model.
The conditions for the development of a model (function) were as follows: 1) A function could have as its input variables any subset of the 29 most significant gene variables described above. 2) Only the 66 subjects (10 STS and 56 LTS) of the training set may be used for the development of a function. 3) The 23 subjects (4 STS and 19 LTS) of the validation set may be used as unknown (test) subjects for the validation of a function since they were different from, and independent of, the 66 subjects of the training set. 4) Pertaining to the development phase, a function was deemed promising only if it exhibited a sensitivity ≥ 0.90 and a specificity ≥ 0.90 in connection with the 66 subjects of the training set. 5) Pertaining to the validation phase, a function was deemed promising only if it exhibited a sensitivity ≥ 0.90 and a specificity ≥ 0.90 in connection with the 23 unknown subjects of the validation set.
I was able to generate the following function that met all of the aforementioned conditions:
F 1 = [[In[((X 1)2 + (X 2)2 + (X 3)5/2)/(X 4)·(X 5)1/4]]1/2]·(10)
where X1=FAM120AOS, X2=PDLIM4, X3=OCIAD2, X4=PCDH15, and X5=MXI1. The X1-X5 are the normalized RNA-Seq gene expression values of the above listed 5 genes, and they constitute the 5 input variables of the F1 function.
The cut-off score of the F1 was determined by taking into account the calculation of the optimal point on the ROC curve based on the 66 F1 scores of the 66 subjects used in the development phase [optimal point is defined as the point with the highest sensitivity and the lowest false positive rate (1-specificity)]. The cut-off score of the F1 was determined to be 24.72. If a subject’s F1 score is ≥ 24.72, then that subject is classified as STS; otherwise, if the F1 score is < 24.72, then that subject is classified as LTS.
The results of the F1 in the development phase are shown in Table S3 and Figure S1.
Validation of the model
The F1 was validated with the following three different and independent validation methods:
1) Fixed validation (test) set. As was explained above in the Development of the model, the data of all 89 subjects were randomly partitioned into two fixed sets: a) the training set comprising 66 subjects (10 STS and 56 LTS) and b) the validation set comprising 23 subjects (4 STS and 19 LTS). The training set was used only for the development of the model, whereas the validation set was used only for the validation of the model. The F1 was validated with the 23 unknown subjects (4 STS and 19 LTS) of the validation set. Table S4 shows those results.
2) Ten-fold cross validation. The data of all 89 subjects were randomly partitioned into a training set and into a test set 10 times, and each one of those 10 times (folds) the performance of the model was assessed [21]. All of the specific details and the confusion matrix, with the results for all 10 folds, are shown in Table S5.
3) Leave-one-out cross validation. Out of the 89 subjects, one subject was randomly selected and used as a test subject. During the second round, another subject was randomly selected and used as a test subject. This continued until all 89 subjects had been selected and served as test subjects [21]. Since there were totally 89 subjects, there were 89 rounds of cross validation. All of the specific details and the confusion matrix, with the results for all 89 rounds of cross validation, are shown in Table S5.
Supervised principal component analysis (PCA)
PCA is one of the most widely used statistical multivariate methods. In order to increase considerably its classification accuracy and attain the best possible PCA results in terms of classification accuracy, I employed the PCA methodology I have developed and presented elsewhere [9]. Briefly, using all 89 subjects, I performed supervised PCA employing the correlation matrix and only the 29 most significant gene variables described above. Table S6 shows those results.
Computer software
All analyses in this study were carried out with custom software written by JBN in MATLAB R2012b. All computer programs in connection with the model were also created by JBN using MATLAB R2012b.
Results
Top 29 most significant genes
As was stated in the Methods, having employed three different and independent methods of statistical significance, namely, ROC curve analysis, fold change, and P-value, I was able to identify 29 genes that were the most significant in terms of differential expression between the two groups (STS vs. LTS). Figure 1 depicts the heat map that resulted by plotting the expression of the 29 most significant genes for all 89 subjects (14 STS and 75 LTS). Figure 2 also depicts the differential expression of the 29 most significant genes between the two groups (14 STS and 75 LTS) in a filled-contour plot. The direction of the differential expression of those 29 genes, along with all statistical results, appears in Table 1. As can been seen by the relative expression intensities (Figures 1 and 2), there is a distinct overall separation between the two groups.
Figure 1.
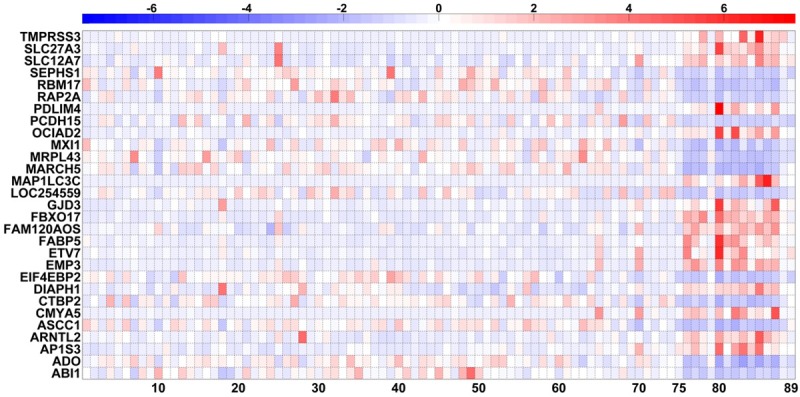
Heat map of the expression of the 29 most significant tumor tissue genes of all 89 subjects. Heat map of the tumor tissue gene expression, generated from mRNA sequencing, of 75 LTS subjects (columns # 1-75) (x-axis) and 14 STS subjects (columns # 76-89) (x-axis) with respect to the 29 most significant genes (rows # 1-29) (y-axis). The order of those 29 genes is alphabetical (the same as the one in Table 1). All 29 gene variables were standardized (mean=0 and SD=1). The intensity scale of the standardized expression values represents, therefore, the z scores; and it ranges from -7.5 [blue: low expression (7.5 SD below the mean)] to +7.5 [red: high expression (7.5 SD above the mean)], with 0 [white (mean=0)] representing the reference intensity value (mean expression value of all 89 subjects). As can be seen, based on the expression of those 29 most significant genes, there is a distinct overall separation between the LTS and the STS subjects.
Figure 2.
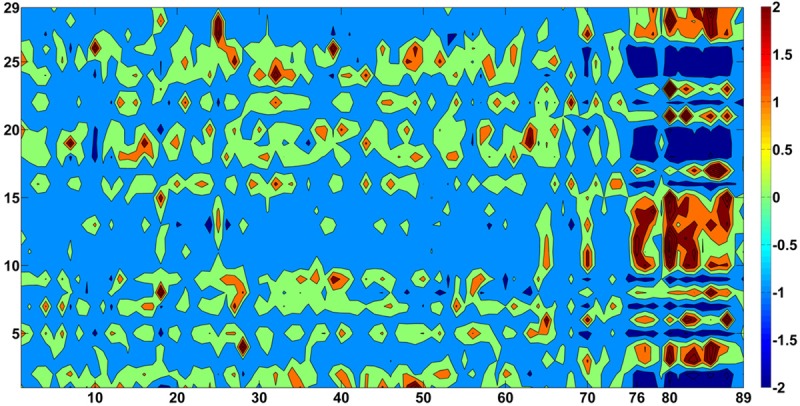
Filled-contour plot of the expression of the 29 most significant tumor tissue genes of all 89 subjects. Tumor tissue gene expression, generated from mRNA sequencing, of 75 LTS subjects (columns # 1-75) (x-axis) and 14 STS subjects (columns # 76-89) (x-axis) with respect to the 29 most significant genes (rows # 1-29) (y-axis). The order of those 29 genes is alphabetical (the same as the one in Table 1). All 29 gene variables were standardized (mean=0 and SD=1). The intensity scale of the standardized expression values represents, therefore, the z scores; and it ranges from -2 [dark blue: low expression (2 SD below the mean)] to +2 [dark red: high expression (2 SD above the mean)], with 0 [light green (mean=0)] representing the reference intensity value (mean expression value of all 89 subjects). As can be seen, based on the expression of those 29 most significant genes, there is a distinct overall separation between the LTS and the STS subjects.
Mathematical modeling of survival
As was explained in great detail in the Methods, I was able to generate the following function:
F 1 = [[In[((X 1)2 + (X 2)2 + (X 3)5/2)/(X 4)·(X 5)1/4]]1/2]·(10)
where X1=FAM120AOS, X2=PDLIM4, X3=OCIAD2, X4=PCDH15, and X5=MXI1. The X1-X5 are the normalized RNA-Seq gene expression values of the above listed 5 genes, and they constitute the 5 input variables of the F1 function. This F1 function (the model), based on the input of 5 genes from the 29 most significant genes, was able to identify/classify accurately all but one of the short-term survivors [sensitivity= (13/14)=0.929] and all but three of the long-term survivors [specificity=(72/75)=0.960]. Figure 3 and Table S7 show the results of the overall performance of the F1. Those results of the overall performance of the F1 were obtained by combining the results from the development and the validation phases. Figure 4 depicts the 3D space position of all 89 subjects used in this study (14 STS and 75 LTS) according to their F1 scores. Figure S1 and Table S3 show the performance results of the F1 in the development phase, i.e. in connection with the 66 subjects of the training set; whereas Table S4 shows the performance results of the F1 in the validation phase, i.e. in connection with the 23 unknown subjects of the validation set.
Figure 3.
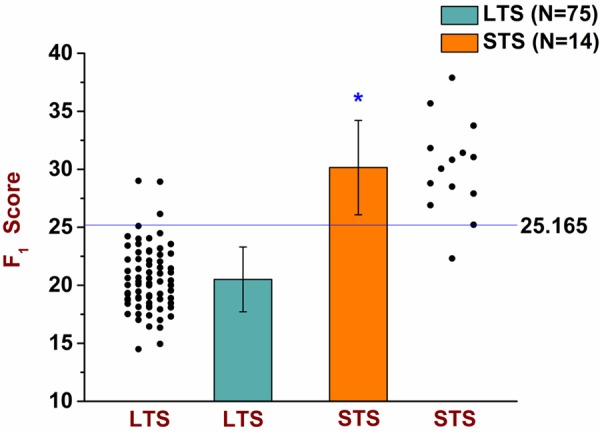
Overall results of the F1 function. The F1 uses 5 of the 29 most significant genes as its input variables. Using the expression value of those 5 genes for a particular subject, the F1 yields the F1 score of that subject; and, based on the determined cut-off score of 25.165, the F1 classifies that subject as a long-term survivor (LTS) if the F1 score is < 25.165 or as a short-term survivor (STS) if the F1 score is ≥ 25.165. The results of the overall performance of the F1 were obtained by combining the results from the development and the validation phases. As can be seen by its overall performance in this dot plot & bar graph, the F1 classified correctly all but one of the STS subjects [sensitivity=(13/14)=0.929] and all but three of the LTS subjects [specificity=(72/75)=0.960]. The mean F1 score of the LTS subjects was 20.511 (top of the green bar) and their standard deviation (whiskers above or below the top of the green bar) was 2.790. Using bootstrapping with a sample size of 100,000, the 99.99% confidence interval of the mean F1 score of the LTS subjects was: [19.150, 21.738]. The mean F1 score of the STS subjects was 30.157 (top of the orange bar) and their standard deviation (whiskers above or below the top of the orange bar) was 4.068. Using bootstrapping with a sample size of 100,000, the 99.99% confidence interval of the mean F1 score of the STS subjects was: [26.160, 34.108].The significance level was set at α=0.001 (two-tailed), and the probability of significance for the F1 was P=4.05 × 10-18 (independent t-Test with T-value=-10.986). The F1 is parametrically distributed with respect to both groups. The F1 scores of all 89 subjects are shown in Table S7.
Figure 4.
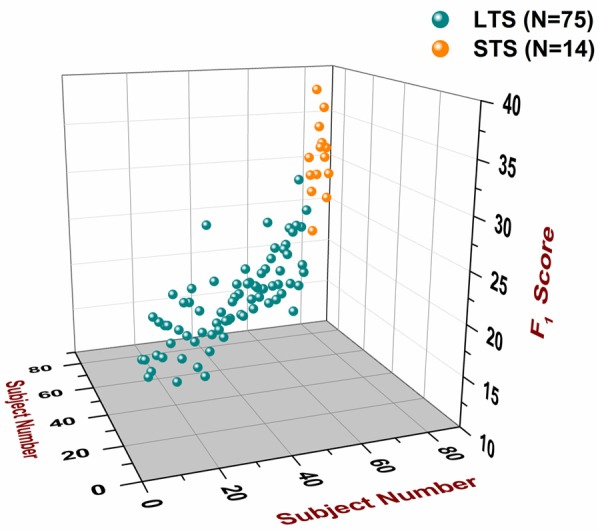
3D space position of all 89 subjects according to their F1 scores. The F1 scores of all 89 subjects [75 LTS (#1-75) and 14 STS (#76-89)] are plotted in the z-axis. The subject number (#1-89) is plotted in the x-axis and the y-axis. The order of the subjects is the same as the one that appears in Table S7. A plane parallel to the x-y plane that intersects the z-axis at the point 25.165, which is the cut-off score, represents the cut-off plane. Subjects that are classified as STS lie above the cut-off plane, whereas subjects that are classified as LTS lie below the cut-off plane.
As can be seen in Table S4, in the validation phase, and according to the cut-off score of 24.72 that was determined in the development phase, the F1 misclassified 3 unknown LTS subjects (Subjects No 25, 53, and 75) and one unknown STS subject (Subject No 79). If the results from the development and the validation phases are combined, and if a new ROC curve analysis is performed on all F1 scores of all 89 subjects (66 subjects from the development phase and 23 unknown subjects from the validation phase), then there exists a better cut-off score, i.e. one that yields a higher specificity, than the original cut-off score of 24.72 that was determined during the development phase (see “Development of the model” section in the Methods). According to the aforementioned new ROC curve analysis, that more optimal cut-off score is 25.165, such that if a subject’s F1 score is ≥ 25.165, then that subject is classified as STS; otherwise, if the F1 score is < 25.165, then that subject is classified as LTS. According to the new, more optimal cut-off score of 25.165, as can be seen in Table S4, subject no 75 (an unknown LTS subject with F1 score of 25.1102) is no longer misclassified by the F1. The new, more optimal cut-off score of 25.165 yields a higher specificity [(72/75)=0.960 as opposed to (71/75)=0.947], without affecting the sensitivity [(13/14)=0.929]. Therefore, according to this validation method [fixed validation (test) set], in total, there were 4 misclassifications out of 89 subjects, and that yielded a misclassification rate of 0.045 in connection with the F1.
In addition to the above validation method [fixed validation (test) set], the performance of the F1 was further assessed by two other different and independent methods:
1) Ten-fold cross validation. According to this validation method, in total, there were 5 misclassifications out of 89 subjects, and that yielded a misclassification rate of 0.056 and a mean-squared error of 0.056 in connection with the F1. All of the specific details and the confusion matrix, with the results for all 10 folds, are shown in Table S5.
2) Leave-one-out cross validation. According to this validation method, in total, there were 5 misclassifications out of 89 subjects, and that yielded a misclassification rate of 0.056 and a mean-squared error of 0.056 in connection with the F1. All of the specific details and the confusion matrix, with the results for all 89 rounds of cross validation, are shown in Table S5.
In order to compare the performance of the model (F1) with well-known and popular statistical multivariate methods, such as principal component analysis (PCA), I performed a supervised PCA, as was explained in the Methods, seeking, thus, to increase considerably the classification accuracy of PCA and attain the best possible PCA results in terms of classification accuracy. Given that the first principal component (PC1) has the highest classification accuracy of all principal components, and looking at the PC1 scores of all 89 subjects in Table S6, one can see that the supervised PCA misclassified 11 out of 89 subjects (misclassification rate=0.124); whereas the F1 misclassified 4 out of 89 subjects (misclassification rate=0.045).
Finally, it should be noted here that the F1 needs to be further validated with a larger, independent cohort.
As was mentioned earlier, the five genes that constitute the five input variables to the F1 are: FAM120AOS, PDLIM4, OCIAD2, PCDH15, and MXI1, and their respective function and properties will be discussed next.
Discussion
On the 5 gene input variables to the model
1) FAM120AOS. This gene encodes a protein whose function remains unknown. Nevertheless, FAM120AOS has been observed to play a role in medulloblastoma [22], in glioblastoma [23], and in the NCI-60 human tumor cell lines [24].
2) PDLIM4. This gene encodes a protein whose function is protein binding. PDLIM4 has been observed to be involved in medulloblastoma [22], in glioblastoma [23], and in breast and colorectal cancers [25].
3) OCIAD2. This gene, whose official full name is: ovarian carcinoma immunoreactive antigen domain containing 2, encodes a protein whose function remains unknown. OCIAD2 has been observed to be involved, among other, in glioblastoma [23], in lung adenocarcinoma [26], and in breast and colorectal cancers [25].
4) PCDH15. This gene is a member of the cadherin superfamily, whose members encode integral membrane proteins that mediate calcium-dependent cell-cell adhesion. As can be seen in Table 1, PCDH15 is significantly under-expressed in the STS subjects compared with the LTS subjects. That suggests that in the case of the STS subjects, the tumor cells have significantly lower cell-cell adhesion, or, to put it equivalently, they have significantly higher mobility, which facilitates greater spreading and metastasis, than the tumor cells of the LTS subjects. PCDH15 has been observed to be involved in pediatric low-grade glioma [27], in medulloblastoma [28], and in colon and rectal cancers [29].
5) MXI1. This gene encodes a protein whose function is to inhibit transcriptional activity. More on the biological function of this gene and its implications for this study will be discussed next. MXI1 has been observed to be involved in numerous types of cancer, including in glioblastoma [23,30], in oligodendroglioma [31], in colon and rectal cancers [29], in breast cancer [32], and in pancreatic cancer [33].
On the biological and clinical importance of MXI1
As was mentioned above, and based on current knowledge, MXI1 is a transcriptional repressor, and it has been theorized that one of its targets is MYC, a well-known oncogene. According to the results of this study, however, the expression of MYC in the STS subjects is not significantly different from that in the LTS subjects. That suggests that both the role and the range of MXI1 as a transcriptional repressor are much wider than previously thought. Insofar as the results of this study are concerned, MXI1 acts as a tumor suppressor. Other studies have also suggested different and more general mechanisms of action for the tumor-suppressing function of MXI1, such as arresting the cell cycle via binding to, and inhibiting, cyclin B1 (CCNB1) in the case of glioma cells [34]. I should point out here that the results of this study are in accord with the observations and the proposed mechanism of action for MXI1 of the aforementioned study, for there is an inversely proportional trend regarding the expression of cyclin B1 in connection with the expression of MXI1. More specifically, the STS subjects have a significantly lower expression of MXI1 and higher expression of cyclin B1 compared with the LTS subjects. Other studies have proposed even more different and much wider mechanisms of action regarding the function of MXI1 as a transcriptional repressor and inhibitor of cell proliferation, such as different isoforms of MXI1, with different properties and effects, binding to, and acting on, many different families of proteins to arrest the cell cycle in different ways [35].
As can be seen in Table 1, MXI1 is significantly under-expressed in the STS subjects compared with the LTS subjects. That suggests that in the tumor cells of the STS subjects, transcriptional repression is significantly lower than that in the tumor cells of the LTS subjects; and from that it follows that the tumor cells of the STS subjects experience significantly higher rates of cell proliferation than the tumor cells of the LTS subjects. This conclusion accords very well with the experimental observations (clinical outcome between STS and LTS subjects) and provides the theoretical (biological) foundation of the model. Finally, I should point out here that, based on the results of this study, MXI1 presents itself not only as the top biomarker for survival in patients with either G2 or G3 glioma, but also as the most promising target for the development of a drug treatment.
On the development of a possible treatment
The results in Table 1 show that the top 29 genes are the most significant discriminators between the STS and the LTS subjects, and that they can collectively account for the overall differential survival between the two groups. Those 29 most significant genes, therefore, represent a comprehensive list of targets wherefrom a pharmacological treatment may be developed. Such a treatment would be administered right after surgery, and prior to the administration of the adjuvant chemotherapy, in order to treat the post-surgery residual tumor load in the identified STS subjects by normalizing the expression of those 29 genes, i.e. by suppressing those genes that are over-expressed and by increasing the expression of those genes that are under-expressed. The goal of such a treatment would be to render the post-surgery residual tumor cells in the identified STS subjects sensitive to the adjuvant chemotherapy, resulting thus in long-term survival, just like in the case of the LTS subjects.
The results of this study in connection with the model demonstrated that the five genes that constitute the five input variables to the model, namely, FAM120AOS, PDLIM4, OCIAD2, PCDH15, and MXI1, can discriminate between the STS and LTS subjects with a high accuracy. Moreover, as was discussed above, all of those five genes have been observed to be involved in numerous types of cancer. Those five genes, therefore, represent a selective and prime list of targets wherefrom a pharmacological treatment may be developed. Furthermore, as was mentioned in the previous section, MXI1, based on its biological function and significance, is the top target in that selective and prime list.
On the clinical utility of the model
The clinical utility of the model presented here is defined by its ability to detect/identify those patients with either G2 or G3 glioma who - owing to the aggressive genetic make-up of the tumor tissue - will turn out to have a very poor clinical outcome (survival ≤ 1 year). Currently, the means to identify those patients at the time of the diagnosis and prior to surgery and adjuvant chemotherapy is nonexistent. This means that an appropriate treatment will not be timely available for those patients. For instance, at the time of the diagnosis, a patient with G2 (grade II) glioma who - on account of the genetic make-up of the tumor tissue - will turn out to be a short-term survivor (survival ≤ 1 year) will not receive the same intensive treatment as a patient with glioblastoma (grade IV glioma), even though the former will experience the same or, more likely, shorter survival than the latter. According to the WHO grading system, at the time of the diagnosis, the clinical outcome of the former patient (with grade II glioma) will be considered far better than that of the latter patient (with grade IV glioma).
Finally, the clinical utility of the model is further defined by the potential to develop a pharmacological treatment based on the aforementioned identified gene targets. If such a treatment became a reality, then the model could be used, at the time of the diagnosis, to identify the STS patients, i.e. the subset of patients for whom such a treatment would be appropriate and necessary.
Acknowledgements
This study was supported by Genomix, Inc. in terms of salary provided. The funders had no role in study design, data collection, and analysis, decision to publish, or preparation of the manuscript. The author would like to thank Sarah L. Nikas for helpful discussions.
Disclosure of conflict of interest
The author is a partner, and owns shares, of Genomix, Inc.
Supporting Information
References
- 1.Ostrom QT, Gittleman H, Farah P, Ondracek A, Chen Y, Wolinsky Y, Stroup NE, Kruchko C, Barnholtz-Sloan JS. CBTRUS Statistical Report: Primary Brain and Central Nervous System Tumors Diagnosed in the United States in 2006-2010. Neuro-Oncology. 2013;15:ii1–ii56. doi: 10.1093/neuonc/not151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, Scheithauer BW, Kleihues P. The 2007 WHO Classification of Tumours of the Central Nervous System. Acta Neuropathol. 2007;114:97–109. doi: 10.1007/s00401-007-0243-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goodenberger ML, Jenkins RB. Genetics of adult glioma. Cancer Genetics. 2012;205:613–621. doi: 10.1016/j.cancergen.2012.10.009. [DOI] [PubMed] [Google Scholar]
- 4.Gravendeel LAM, de Rooi JJ, Eilers PHC, van den Bent MJ, Sillevis Smitt PAE, French PJ. Gene expression profiles of gliomas in formalin-fixed paraffin-embedded material. Br J Cancer. 2012;106:538–545. doi: 10.1038/bjc.2011.547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Elsir T, Qu M, Berntsson SG, Orrego A, Olofsson T, Lindstrom MS, Nister M, von Deimling A, Hartmann C, Ribom D, Smits A. PROX1 is a predictor of survival for gliomas WHO grade II. Br J Cancer. 2011;104:1747–1754. doi: 10.1038/bjc.2011.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vitucci M, Hayes DN, Miller CR. Gene expression profiling of gliomas: merging genomic and histopathological classification for personalised therapy. Br J Cancer. 2011;104:545–553. doi: 10.1038/sj.bjc.6606031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nikas JB. Inflammation and Immune System Activation in Aging: A Mathematical Approach. Sci Rep. 2013;3:3254. doi: 10.1038/srep03254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Burns MB, Lackey L, Carpenter MA, Rathore A, Land AM, Leonard B, Refsland EW, Kotandeniya D, Tretyakova N, Nikas JB, Yee D, Temiz NA, Donohue DE, McDougle RM, Brown WL, Law EK, Harris RS. APOBEC3B is an enzymatic source of mutation in breast cancer. Nature. 2013;494:366–370. doi: 10.1038/nature11881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nikas JB, Low WC. ROC-supervised principal component analysis in connection with the diagnosis of diseases. Am J Transl Res. 2011;3:180–196. [PMC free article] [PubMed] [Google Scholar]
- 10.Nikas JB, Keene CD, Low WC. Comparison of Analytical Mathematical Approaches for Identifying Key Nuclear Magnetic Resonance Spectroscopy Biomarkers in the Diagnosis and Assessment of Clinical Change of Diseases. J Comp Neurol. 2010;518:4091–4112. doi: 10.1002/cne.22365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nikas JB, Low WC, Burgio PA. Prognosis of Treatment Response (Pathological Complete Response) in Breast Cancer. Biomarker Insights. 2012;7:59–70. doi: 10.4137/BMI.S9387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nikas JB, Boylan KLM, Skubitz APN, Low WC. Mathematical Prognostic Biomarker Models for Treatment Response and Survival in Epithelial Ovarian Cancer. Cancer Inform. 2011;10:233–247. doi: 10.4137/CIN.S8104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nikas JB, Low WC. Application of clustering analyses to the diagnosis of Huntington disease in mice and other diseases with well-defined group boundaries. Comput Methods Programs Biomed. 2011;104:e133–e147. doi: 10.1016/j.cmpb.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nikas JB, Low WC. Linear Discriminant Functions in Connection with the micro-RNA Diagnosis of Colon Cancer. Cancer Inform. 2012;11:1–14. doi: 10.4137/CIN.S8779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McCarthy DJ, Smyth GK. Testing significance relative to a fold-change threshold is a TREAT. Bioinformatics. 2009;25:765–771. doi: 10.1093/bioinformatics/btp053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Witten DM, Tibshirani R. A comparison of fold-change and the t-statistic for microarray data analysis. Stanford University, Department of Statistics. 2007 [Google Scholar]
- 17.Perneger TV. What’s wrong with Bonferroni adjustments. BMJ. 1998;316:1236–1238. doi: 10.1136/bmj.316.7139.1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Feise RJ. Do multiple outcome measures require p-value adjustment? BMC Med Res Methodol. 2002;2:8. doi: 10.1186/1471-2288-2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Amari S, Murata N, Muller KR, Finke M, Yang HH. Asymptotic Statistical Theory of Overtraining and Cross-Validation. IEEE Trans Neural Netw. 1997;8:985–996. doi: 10.1109/72.623200. [DOI] [PubMed] [Google Scholar]
- 20.Efron B, Tibshirani R. An Introduction to the Bootstrap. London: Chapman and Hall; 1993. [Google Scholar]
- 21.Hastie T, Tibshirani R, Friedman J. The Elements of Statistical Learning. New York: Springer; 2001. [Google Scholar]
- 22.Parsons DW, Li M, Zhang X, Jones S, Leary RJ, Lin JC, Boca SM, Carter H, Samayoa J, Bettegowda C, Gallia GL, Jallo GI, Binder ZA, Nikolsky Y, Hartigan J, Smith DR, Gerhard DS, Fults DW, VandenBerg S, Berger MS, Marie SK, Shinjo SM, Clara C, Phillips PC, Minturn JE, Biegel JA, Judkins AR, Resnick AC, Storm PB, Curran T, He Y, Rasheed BA, Friedman HS, Keir ST, McLendon R, Northcott PA, Taylor MD, Burger PC, Riggins GJ, Karchin R, Parmigiani G, Bigner DD, Yan H, Papadopoulos N, Vogelstein B, Kinzler KW, Velculescu VE. The Genetic Landscape of the Childhood Cancer Medulloblastoma. Science. 2011;331:435–439. doi: 10.1126/science.1198056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Siu IM, Gallia GL, Olivi A, McLendon R, Rasheed BA, Keir S, Nikolskaya T, Nikolsky Y, Busam DA, Tekleab H, Diaz LA Jr, Hartigan J, Smith DR, Strausberg RL, Marie SK, Shinjo SM, Yan H, Riggins GJ, Bigner DD, Karchin R, Papadopoulos N, Parmigiani G, Vogelstein B, Velculescu VE, Kinzler KW. An Integrated Genomic Analysis of Human Glioblastoma Multiforme. Science. 2008;321:1807–1812. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Abaan OD, Polley EC, Davis SR, Zhu YJ, Bilke S, Walker RL, Pineda M, Gindin Y, Jiang Y, Reinhold WC, Holbeck SL, Simon RM, Doroshow JH, Pommier Y, Meltzer PS. The Exomes of the NCI-60 Panel: A Genomic Resource for Cancer Biology and Systems Pharmacology. Cancer Res. 2013;73:4372–4382. doi: 10.1158/0008-5472.CAN-12-3342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sjoblom T, Jones S, Wood LD, Parsons DW, Lin J, Barber TD, Mandelker D, Leary RJ, Ptak J, Silliman N, Szabo S, Buckhaults P, Farrell C, Meeh P, Markowitz SD, Willis J, Dawson D, Willson JKV, Gazdar AF, Hartigan J, Wu L, Liu C, Parmigiani G, Park BH, Bachman KE, Papadopoulos N, Vogelstein B, Kinzler KW, Velculescu VE. The Consensus Coding Sequences of Human Breast and Colorectal Cancers. Science. 2006;314:268–274. doi: 10.1126/science.1133427. [DOI] [PubMed] [Google Scholar]
- 26.Imielinski M, Berger AH, Hammerman PS, Hernandez B, Pugh TJ, Hodis E, Cho J, Suh J, Capelletti M, Sivachenko A, Sougnez C, Auclair D, Lawrence MS, Stojanov P, Cibulskis K, Choi K, de Waal L, Sharifnia T, Brooks A, Greulich H, Banerji S, Zander T, Seidel D, Leenders F, Ansén S, Ludwig C, Engel-Riedel W, Stoelben E, Wolf J, Goparju C, Thompson K, Winckler W, Kwiatkowski D, Johnson BE, Jänne PA, Miller VA, Pao W, Travis WD, Pass HI, Gabriel SB, Lander ES, Thomas RK, Garraway LA, Getz G, Meyerson M. Mapping the Hallmarks of Lung Adenocarcinoma with Massively Parallel Sequencing. Cell. 2012;150:1107–1120. doi: 10.1016/j.cell.2012.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang J, Wu G, Miller CP, Tatevossian RG, Dalton JD, Tang B, Orisme W, Punchihewa C, Parker M, Qaddoumi I, Boop FA, Lu C, Kandoth C, Ding L, Lee R, Huether R, Chen X, Hedlund E, Nagahawatte P, Rusch M, Boggs K, Cheng J, Becksfort J, Ma J, Song G, Li Y, Wei L, Wang J, Shurtleff S, Easton J, Zhao D, Fulton RS, Fulton LL, Dooling DJ, Vadodaria B, Mulder HL, Tang C, Ochoa K, Mullighan CG, Gajjar A, Kriwacki R, Sheer D, Gilbertson RJ, Mardis ER, Wilson RK, Downing JR, Baker SJ, Ellison DW St. Jude Children’s Research Hospital-Washington University Pediatric Cancer Genome Project. Whole-genome sequencing identifies genetic alterations in pediatric low-grade gliomas. Nat Genet. 2013;45:602–612. doi: 10.1038/ng.2611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pugh TJ, Weeraratne SD, Archer TC, Pomeranz Krummel DA, Auclair D, Bochicchio J, Carneiro MO, Carter SL, Cibulskis K, Erlich RL, Greulich H, Lawrence MS, Lennon NJ, McKenna A, Meldrim J, Ramos AH, Ross MG, Russ C, Shefler E, Sivachenko A, Sogoloff B, Stojanov P, Tamayo P, Mesirov JP, Amani V, Teider N, Sengupta S, Francois JP, Northcott PA, Taylor MD, Yu F, Cr-abtree GR, Kautzman AG, Gabriel SB, Getz G, Jäger N, Jones DT, Lichter P, Pfister SM, Roberts TM, Meyerson M, Pomeroy SL, Cho YJ. Medulloblastoma exome sequencing uncovers subtype-specific somatic mutations. Nature. 2012;488:106–110. doi: 10.1038/nature11329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–337. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bettegowda C, Agrawal N, Jiao Y, Sausen M, Wood LD, Hruban RH, Rodriguez FJ, Cahill DP, McLendon R, Riggins G, Velculescu VE, Oba-Shinjo SM, Marie SKN, Vogelstein B, Bigner D, Yan H, Papadopoulos N, Kinzler KW. Mutations in CIC and FUBP1 Contribute to Human Oligodendroglioma. Science. 2011;333:1453–1455. doi: 10.1126/science.1210557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stephens PJ, Tarpey PS, Davies H, Van Loo P, Greenman C, Wedge DC, Nik-Zainal S, Martin S, Varela I, Bignell GR, Yates LR, Papaemmanuil E, Beare D, Butler A, Cheverton A, Gamble J, Hinton J, Jia M, Jayakumar A, Jones D, Latimer C, Lau KW, McLaren S, McBride DJ, Menzies A, Mudie L, Raine K, Rad R, Chapman MS, Teague J, Easton D, Langerød A Oslo Breast Cancer Consortium (OSBREAC) Lee MT, Shen CY, Tee BT, Huimin BW, Broeks A, Vargas AC, Turashvili G, Martens J, Fatima A, Miron P, Chin SF, Thomas G, Boyault S, Mariani O, Lakhani SR, van de Vijver M, van’t Veer L, Foekens J, Desmedt C, Sotiriou C, Tutt A, Caldas C, Reis-Filho JS, Aparicio SA, Salomon AV, Børresen-Dale AL, Richardson AL, Campbell PJ, Futreal PA, Stratton MR. The landscape of cancer genes and mutational processes in breast cancer. Nature. 2012;486:400–404. doi: 10.1038/nature11017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Kamiyama H, Jimeno A, Hong SM, Fu B, Lin MT, Calhoun ES, Kamiyama M, Walter K, Nikolskaya T, Nikolsky Y, Hartigan J, Smith DR, Hidalgo M, Leach SD, Klein AP, Jaffee EM, Goggins M, Maitra A, Iacobuzio-Donahue C, Eshleman JR, Kern SE, Hruban RH, Karchin R, Papadopoulos N, Parmigiani G, Vogelstein B, Velculescu VE, Kinzler KW. Core Signaling Pathways in Human Pancreatic Cancers Revealed by Global Genomic Analyses. Science. 2008;321:1801–1806. doi: 10.1126/science.1164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Manni I, Tunici P, Cirenei N, Albarosa R, Colombo BM, Roz L, Sacchi A, Piaggio G, Finocchiaro G. Mxi1 inhibits the proliferation of U87 glioma cells through down-regulation of cyclin B1 gene expression. British Journal of Cancer. 2002;86:477–484. doi: 10.1038/sj.bjc.6600065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dugast-Darzacq C, Pirity M, Blanck JK, Scherl A, Schreiber-Agus N. Mxi1-SRa: a novel Mxi1 isoform with enhanced transcriptional repression potential. Oncogene. 2004;23:8887–8899. doi: 10.1038/sj.onc.1208107. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.