

Rare copy number variations (CNVs) contribute to genetic risk for many developmental and neuropsychiatric conditions. The growing uptake of chromosomal microarray analysis in clinical practice1, 2 is predicated on the assumption that there exist valid protocols to guide the interpretation of very rare or private CNVs. Many protocols currently in use can be strongly influenced by the results of parental studies.1, 3, 4 We used a simple Bayesian model to demonstrate that in the case of inherited variants, the observed parental phenotype will be heavily and predictably influenced by the decreased reproductive fitness associated with disease expression. This means that transmitting parents with high penetrance variants will nonetheless often be ‘unaffected'.
Biases inherent in parental studies
In any individual most genetic variants, including rare variants, are inherited.5 Pathogenicity of very rare or private inherited CNVs is often inferred in part from the parental phenotype. Disease concordance between parent and offspring may be used to conclude that a shared CNV likely had a causal role, despite the fact that the a priori probability of both the parent and a child sharing that CNV (given that the parent had the CNV) was 50% in most cases. Disease discordance in the form of an ‘unaffected' or ‘healthy' parent, on the other hand, may be used to argue that a shared variant is likely benign. However, by definition, transmitting parents have been able to find a partner and to reproduce (and were available and willing to have genetic testing). This will bias downward the observed prevalence in transmitting parents of major phenotypes associated with reduced reproductive fitness (Figure 1), such as major developmental and neuropsychiatric conditions.6
Figure 1.

Observed penetrance in transmitting parents for major phenotypes associated with reduced reproductive fitness. See text, Equation (1), and Supplementary Tables S1 and S2 for details.
A simple Bayesian model
One can quantify the potential impact of reproductive fitness differences associated with expression of a disease (X) on the observed prevalence of X in transmitting parents with a rare autosomal dominant-acting CNV (g). Let P be the penetrance of variant g for disease X and f be the relative reproductive fitness of individuals with g and with X (compared with individuals with g and without X), where 0<P, f<1. Using standard Bayesian analysis methods (Supplementary Table S1), the probability of disease X in a randomly selected transmitting parent is:
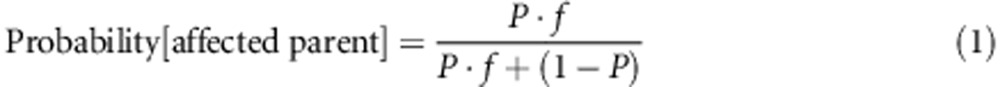 |
In those cases where P<1/(1+f) (eg, where a variant has penetrance P=0.5 for disease X but where those affected with X have on average only half of the number of offspring of those without X), only a minority of transmitting parents would be expected to be affected with X (Figure 1). Thus, while the observation in clinical practice that a particular very rare or novel CNV has been inherited from an unaffected parent may demonstrate that penetrance is incomplete, it is less informative with respect to quantifying penetrance.
A practical illustration: 22q11.2 deletion syndrome and schizophrenia
As a practical illustration of the general principle highlighted by Equation (1), recent data6, 7 can be used to explain historical observations regarding the prevalence of schizophrenia in transmitting parents with 22q11.2 deletion syndrome (22q11.2DS; OMIM #188400/#192430). Take genetic variant g to be a typical 22q11.2 deletion and disease X to be schizophrenia. The penetrance P for schizophrenia in 22q11.2DS is ∼0.25 (25%).7 Individuals with 22q11.2DS and schizophrenia have approximately one tenth as many children as do individuals with 22q11.2DS and without schizophrenia, and thus the relative fitness f associated with schizophrenia in 22q11.2DS is ∼0.10 (10%).6 The observed penetrance of schizophrenia in transmitting parents with 22q11.2DS would therefore be on the order of 0.03 (3%) (Figure 1; Supplementary Table S2). Early in the study of 22q11.2DS, most adults reported in the literature and being seen in clinical practice were transmitting parents.8 As expected per the above calculation, only a small minority was reported to have a serious psychotic illness.8, 9 Thus, this ascertainment bias effectively masked the true extent of the association of schizophrenia with 22q11.2DS for some time. Most adults with other genomic disorders thus far reported in the literature are transmitting parents;10, 11 this will similarly confound accurate genotype–phenotype correlations across the life span.
Other implications for clinical and research practice
The above observations may seem self-evident, and yet there is some evidence to suggest that this ‘transmitting parent' bias has affected decision-making in clinical practice. For example, in a recent large-scale study of prenatal microarray testing,12 a maternally inherited 10q11.2 duplication (chr10:46,482,304–51,558,563; hg18) was deemed of uncertain significance and ultimately not reported to the family, while a comparable de novo 10q11.2 duplication (chr10:49,033,015–51,005,023 and 51,407,449–52,135,488) was reported. A corollary to this caveat of parental studies is that imposing a major distinction between de novo variants and those that might have appeared in the immediate preceding generation may sometimes be unnecessarily artificial.
There are also implications for designing family-based gene hunting studies in complex diseases. A disease-concordant sibpair design has had a major perceived disadvantage with respect to the discovery of associated autosomal dominant mutations: any variant shared by both affected siblings would typically also have to be present in an unaffected parent. The above discussion and calculations suggest that such a study design may nonetheless have the ability to discover high penetrance variants. Expression in siblings may be a better way of gauging penetrance than expression in parents, albeit with the potential for different biases like stoppage rules.13
Conclusions
In summary, simple probabilistic reasoning challenges a convenient narrative: that de novo variants and variants inherited from a similarly affected parent are likely associated with the proband's phenotype, whereas those inherited from an unaffected parent are likely benign. We do not advocate abandoning existing protocols and conventions; rather, we simply urge caution in mistaking reasonable (and generally useful) heuristics for absolute tenets. Just as de novo variants are not necessarily causal,14 very rare or private variants inherited from apparently unaffected parents are not necessarily benign. The interpretation of rare variants in complex disease is not yet driven by an understanding of the underlying etiopathogenesis. In the future, current approaches to rare CNV interpretation may be superseded by population-based estimates of disease-specific penetrance (for recurrent variants) and additional functional approaches to variant classification.
Studying contemporary reproductive fitness in genomic disorders will have positive repercussions for genetic counseling and understanding the evolutionary biology of these structural rearrangements.6 As for 22q11.2DS,6 reproductive fitness and transmission patterns of pathogenic CNVs will change over time. Regardless, the purpose of Equation (1) is to model a general principle that is immediately relevant to clinical practice, rather than to perform specific calculations in the (rare) cases where the necessary data would be available. Finally, although we have focused on clinical adjudication of rare CNVs, many of the observations and results are generalizable to rare sequence variants and to discovery-based science. Use of whole exome and whole-genome sequencing in clinical practice provides further impetus to optimize methods of interpreting rare variants.
Acknowledgments
I thank Drs Anne S Bassett and Danielle A Baribeau for helpful discussions. This work was supported by MD/PhD Studentships from the Canadian Institutes of Health Research (CIHR) and McLaughlin Centre.
The author declares no conflict of interest.
Footnotes
Supplementary Information accompanies this paper on European Journal of Human Genetics website (http://www.nature.com/ejhg)
Supplementary Material
References
- Miller DT, Adam MP, Aradhya S, et al. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet. 2010;86:749–764. doi: 10.1016/j.ajhg.2010.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coughlin CR, 2nd, Scharer GH, Shaikh TH. Clinical impact of copy number variation analysis using high-resolution microarray technologies: advantages, limitations and concerns. Genome Med. 2012;4:80. doi: 10.1186/gm381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearney HM, Thorland EC, Brown KK, Quintero-Rivera F, South ST. American College of Medical Genetics standards and guidelines for interpretation and reporting of postnatal constitutional copy number variants. Genet Med. 2011;13:680–685. doi: 10.1097/GIM.0b013e3182217a3a. [DOI] [PubMed] [Google Scholar]
- Hanemaaijer NM, Sikkema-Raddatz B, van der Vries G, et al. Practical guidelines for interpreting copy number gains detected by high-resolution array in routine diagnostics. Eur J Hum Genet. 2012;20:161–165. doi: 10.1038/ejhg.2011.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C, Scherer SW. The clinical context of copy number variation in the human genome. Expert Rev Mol Med. 2010;12:e8. doi: 10.1017/S1462399410001390. [DOI] [PubMed] [Google Scholar]
- Costain G, Chow EW, Silversides CK, Bassett AS. Sex differences in reproductive fitness contribute to preferential maternal transmission of 22q11.2 deletions. J Med Genet. 2011;48:819–824. doi: 10.1136/jmedgenet-2011-100440. [DOI] [PubMed] [Google Scholar]
- Fung WL, McEvilly R, Fong J, Silversides CK, Chow EWC, Bassett AS. Elevated prevalence of generalized anxiety disorder in adults with 22q11.2 deletion syndrome. Am J Psychiatry. 2010;167:998. doi: 10.1176/appi.ajp.2010.09101463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen E, Chow EW, Weksberg R, Bassett AS. Phenotype of adults with the 22q11 deletion syndrome: A review. Am J Med Genet. 1999;86:359–365. doi: 10.1002/(sici)1096-8628(19991008)86:4<359::aid-ajmg10>3.0.co;2-v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald-McGinn DM, Tonnesen MK, Laufer-Cahana A, et al. Phenotype of the 22q11.2 deletion in individuals identified through an affected relative: cast a wide FISHing net! Genet Med. 2001;3:23–29. doi: 10.1097/00125817-200101000-00006. [DOI] [PubMed] [Google Scholar]
- Dolcetti A, Silversides CK, Marshall CR, et al. 1q21.1 Microduplication expression in adults. Genet Med. 2013;15:282–289. doi: 10.1038/gim.2012.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper GM, Coe BP, Girirajan S, et al. A copy number variation morbidity map of developmental delay. Nat Genet. 2011;43:838–846. doi: 10.1038/ng.909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wapner RJ, Martin CL, Levy B, et al. Chromosomal microarray versus karyotyping for prenatal diagnosis. N Engl J Med. 2012;367:2175–2184. doi: 10.1056/NEJMoa1203382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones MB, Szatmari P. Stoppage rules and genetic studies of autism. J Autism Dev Disord. 1988;18:31–40. doi: 10.1007/BF02211816. [DOI] [PubMed] [Google Scholar]
- Vermeesch JR, Balikova I, Schrander-Stumpel C, Fryns JP, Devriendt K. The causality of de novo copy number variants is overestimated. Eur J Hum Genet. 2011;19:1112–1113. doi: 10.1038/ejhg.2011.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.