

Graphical abstract
Keywords: Leishmania infantum, Tyrosine aminotransferase, Infectivity, KMTB
Highlights
-
•
The tyrosine aminotransferase from Leishmania infantum has a cytoplasmic distribution and is able to use the oxoacid ketomethiobutyrate, as a co-substrate.
-
•
L. infantum tyrosine aminotransferase is over-expressed in infective and nitric oxide resistant parasites.
-
•
The structural differences with the mammalian TAT, together with cellular distribution, expression pattern and activity, support that LiTAT is a drug target candidate.
-
•
The structure-based model of the pharmacophore of LiTAT with specific substrate ketomethiobutyrate has been generated.
Abstract
Leishmania infantum is the etiological agent of zoonotic visceral leishmaniasis in the Mediterranean basin. The disease is fatal without treatment, which has been based on antimonial pentavalents for more than 60 years. Due to resistances, relapses and toxicity to current treatment, the development of new drugs is required. The structure of the L. infantum tyrosine aminotransferase (LiTAT) has been recently solved showing important differences with the mammalian orthologue. The characterization of LiTAT is reported herein. This enzyme is cytoplasmic and is over-expressed in the more infective stages and nitric oxide resistant parasites. Unlike the mammalian TAT, LiTAT is able to use ketomethiobutyrate as co-substrate. The pharmacophore model of LiTAT with this specific co-substrate is described herein. This may allow the identification of new inhibitors present in the databases. All the data obtained support that LiTAT is a good target candidate for the development of new anti-leishmanial drugs.
1. Introduction
Leishmaniases are a group of parasitic diseases caused by species of the genus Leishmania with an estimated annual incidence of almost 1.8 million cases (WHO, 2012). Leishmania infantum (synonym to L. chagasi, in the New World) (Kinetoplastida: Trypanosomatidae) is the etiological agent of zoonotic visceral leishmaniasis, which is fatal without treatment. In the Mediterranean basin, domestic dogs are the main reservoir of the parasite and an increase in co-infection with HIV has been registered (Pasquau et al., 2005). For over 60 years, pentavalent antimonials have been the most important drugs for treatment of leishmaniases and are still widely used. However, resistances against them have increased over the years. Other drugs like amphotericin B, its lipid formulations and miltefosine are effective against leishmaniasis, but their high cost, toxicity and appearance of resistances limit their usefulness (Haldar et al., 2011). Therefore there is a need for finding new targets involved in pathogenic mechanisms and infectivity of the parasite.
The dimorphic life cycle of the parasite alternates between a mobile extracellular promastigote form in the phlebotomine insect vector and an immobile intracellular amastigote form in the mammalian host (Handman, 2001). Once inside the gut of the vector, promastigotes undergo a process of differentiation increasing their infectivity prior to be inoculated into the mammalian host completing the biological cycle. This process can be mimicked in culture, where significant differences in gene expression profile between logarithmic and stationary phases have been described (Alcolea et al., 2010a). A minor subpopulation (about 1%) within the stationary phase does not agglutinate with peanut agglutinin (PNA−) and is more infective. Gene expression profiling carried out comparing these parasite subpopulations showed a certain group of genes up-regulated in the more infective PNA− subpopulation (Alcolea et al., 2009). One of these genes was the L. infantum tyrosine aminotransferase (LiTAT) gene, which may play a key function in the parasite. In the genus Leishmania, metabolism of amino acids play an important role throughout differentiation from promastigotes to amastigotes as well as inside the gut of the haematophagous vector, where the main nutrients available are amino acids and lipids (Rosenzweig et al., 2008). TAT deficiency leads to metabolic disease in humans (Natt et al., 1987), whereas consequence of TAT alterations in Leishmania are not well understood. Aromatic amino acid catabolism consists of two steps in trypanosomatids. The first one is reversible transamination of aromatic amino acid to its corresponding oxoacid (al-Hemidan and al-Hazzaa, 1995) and is catalyzed by TAT. This broad specificity enzyme transfers the amino group from the aromatic amino acids to an incoming oxoacid that accepts it. The deaminated amino acid is then reduced by a dehydrogenase and the resulting product is supposed to be excreted. This role has been related in other pathogenic trypanosomatids with pathogenesis. As an example, depletion of certain amino acids has been correlated with pathology of sleeping sickness, a disease caused by Trypanosoma brucei (Seed et al., 1983). The lack of certain amino acids in the host, such as tryptophan, would lead to the shortage of essential metabolites for the host (Tizard et al., 1978; Seed et al., 1983; Hall et al., 1985; Leelayoova et al., 1992). Moreover, the toxicity of end-products such as phenylpyruvate has been related to the pathogeny of the disease (Stibbs and Seed, 1973). These end-products of the aromatic amino acid oxidation process were identified and detected in high amounts into the supernatant of T. cruzi epimastigotes axenic culture (Montemartini et al., 1994a).
Broad substrate specificity aminotransferases have been associated to the methionine recycling pathway in trypanosomatids such as T. brucei and Crithidia fasciculata, where this protein catalyzes the last step of this pathway, the conversion of ketomethiobutyrate (KMTB) to methionine via transamination (Berger et al., 1996, 2001). Methionine regeneration has been targeted as an important pathway for anti-malarial and anti-T. brucei drug therapy based on 5′-deoxy-5′-(methylthio) adenosine analogues (Sufrin et al., 1995; Goldberg et al., 1998). Additionally, as methionine is an essential amino acid involved in a number of critical processes in fast growing cells, this pathway has been previously targeted for the development of antiproliferative compounds (Quash et al., 2004).
LiTAT belongs to the Iγ subfamily and is involved in tyrosine metabolism. At present, just a few TAT structures have been solved. The first structure released corresponds to the trypanosomatid T. cruzi (TcTAT) (PDB code: 1BW0) (Blankenfeldt et al., 1999), followed by the human (PDB code: 3DYD) and the mouse TAT (PDB code: 3PDX) (Mehere et al., 2010). Recently, the structure of LiTAT has been solved to 2.3 Å (PDB code: 4IX8) (Moreno et al., 2014). On the basis of this background, the aim of the work is the characterization of the protein regarding its cellular localization and expression at the different stages as well as the excretion of the final product p-hydroxyphenyllactate (pHPL) to the culture medium. Additionally we have studied structural differences found not only with the mammalian enzyme but also with the other trypanosomatid orthologue described so far. Structural comparison and subsequent docking assays have led us to generate a preliminary pharmacophore using a specific substrate of LiTAT which will help to identify inhibitors currently present in the databases. These data support that this enzyme may be a good candidate for novel structure-based development of drugs affecting the parasite but not the mammalian host.
2. Material and methods
2.1. Promastigote and axenic amastigote cultures
L. infantum isolate M/CAN/ES/98/10445 (zymodeme MON-1) was cultured in RPMI 1640 supplemented with l-glutamine (Thermo Fisher, Massachusetts, USA), 10% heat inactivated foetal bovine serum (HIFBS) (Sigma) and 100 μg/ml streptomycin – 100 IU/ml penicillin (Cambrex, Karlskoga, Sweden) (complete medium) at 27 °C and a starting density of 2 × 106 promastigotes/ml. L. chagasi resistant (LBM/LVC/SE/30) and sensitive (LBM/LVC/SE/19) strains to nitric oxide (NO) were kindly provided by R. Almeida (Universidade Federal de Sergipe, Brazil). Experiments were performed on three replicates and promastigotes were recovered in early-logarithmic (day 2) and stationary phase (day 6). The agglutination procedure was carried out as previously described (Alcolea et al., 2009). Briefly, stationary phase promastigotes were recovered and harvested at 2000 g. Then, they were resuspended in complete medium and incubated with 50 μg/ml PNA at a cell density of 2 × 108 (Sigma–Aldrich, Buchs, Switzerland) at room temperature for 30 min. 108 promastigotes of each subpopulation (PNA+ and PNA−) were centrifuged and washed twice with 100 mM PBS pH 7.4. Axenically grown amastigote forms of L. infantum were obtained at 37 °C with 5% CO2 by weekly passages in Medium 199 (Sigma), 10% HIFBS (Sigma), 10 mM PBS pH 5.5 and 100 μg/ml streptomycin – 100 IU/ml penicillin (Cambrex) from a inoculum of 5 × 106 stationary promastigotes/ml. After that, 108 axenic amastigotes obtained at a starting cell density of 5 × 106 were centrifuged on day 4 and washed twice with 100 mM PBS pH 7.4.
2.2. Identification of p-HPL
The extraction procedure was carried out as previously described (Montemartini et al., 1994a) slightly modified. The negative control used was sterile complete medium. A total volume of 230 ml of cell-free culture medium was used in each extraction (including the negative control). To summarize, pHPL was separated by liquid chromatography on an Agilent 1200 HPLC system equipped with a C18/5 μm 25 × 0.45 cm column and a diode array detector. A discontinuous gradient of acetonitrile (10–30% ACN 6 min; 30% ACN 6–12 min; 30–100% ACN 12–19 min) was used at 1 ml/min flow rate. Commercially available pHPL (Sigma) was used as standard. Subsequently, the eluted peak was derivatized by trimethylsilylation and analyzed in a gas chromatography coupled to mass spectrometry (GC–MS) Agilent 7980A-5975C system equipped with an injector split-splitless Inlet (T = 320 °C; P = 12.5 psi) and an HP5 MS column 30 m × 250 μm × 0.25 μm (polysiloxane). The mass spectrometer was set to register components in a mass range between 40 and 500 g/mol. The sample was diluted five times in water and the final concentration of pHPL in the culture medium was obtained by integrating the area under the peak and interpolating it to four different concentrations of pHPL standard (0.5, 0.1, 0.05 and 0.01 mM).
2.3. Determination of p-hydroxyphenylpyruvate dehydrogenase activity in promastigotes
The determination of p-hydroxyphenylpyruvate (pHPP) reduction catalyzed by pHPP dehydrogenase was determined following a previous report with some modifications (Montemartini et al., 1994b) using commercial pHPP (SIGMA). The decrease of absorbance at 340 nm was measured spectrophotometrically at 27 °C in a Cary4000 system in the first five minutes of the reaction. Early-logarithmic, late-logarithmic and stationary phase promastigotes were lysed mildly in Tris–HCL 50 mM pH 7.4, EDTA 2 mM and Triton X-100 0.2%. Protein extracts were quantified by the Bradford method (Bradford, 1976) and 200 μg of total protein extracts were used in each reaction. The final value is the mean of three replicates in two different cultures sampled in every phase.
2.4. Determination of recombinant LiTAT activity
LiTAT activity was assayed with KMTB by the method of Diamondstone (Diamondstone, 1966) without the addition of diethylthiocarbamate. One unit of enzyme activity is defined as the amount of LiTAT catalyzing the formation of 1 μmol of p-hydroxyphenylpyruvate. Four replicates were performed.
2.5. Western blot analysis
Cell extracts of L. infantum promastigotes were obtained as described previously (Alcolea et al., 2011). A specific polyclonal antibody against LiTAT was obtained with 2 mg of purified native recombinant LiTAT as described (Moreno et al., 2014) in a New Zealand rabbit. For this purpose it was administered in four weekly subcutaneous shots together with the Freund’s Adjuvant. P. Michels (University of Edinburgh, UK) kindly provided anti-L. mexicana glycosomal glyceraldehyde 3-phosphate dehydrogenase (gGAPDH) rabbit serum and a dilution of 1:10000 was used (Gualdron-Lopez et al., 2013). Western blotting of the protein extracts was carried out as described (Gonzalez-Aseguinolaza et al., 1997) using a 1:3000 dilution of primary antibody anti-LiTAT and 1:2000 dilution of HRP-conjugated anti-rabbit IgG secondary antibody (Dako, Denmark). In all cases, 10 μg of total protein were loaded per well. L. infantum and L. chagasi protein extracts were quantified as described in Section 2.3.
2.6. Subcellular localization by immunofluorescence
L. infantum early-logarithmic promastigotes (day 2) were laid on the slides (∼2 × 105 each) and dried at room temperature. The cells were fixed for 5 min at room temperature in 4% paraformaldehyde. Then they were washed in PBS and permeabilized with PBS-0.5% Triton X-100. Blocking was carried out with 5% skimmed milk in PBS-0.1% Tween 20 at 37 °C for 30 min in a wet chamber. The coverslips were incubated with a 1:1000 dilution of the LiTAT polyclonal antibody in blocking buffer for 1 h. After washing, the slides were incubated with a 1:200 dilution of goat anti-rabbit IgG conjugated with Alexa Fluor 488 (Life Technologies, USA) in blocking buffer in darkness for 1 h. During the last 10 min of incubation with the secondary antibody, coverslips were treated with DAPI (Life Technologies) at a final concentration of 0.2 μg/ml in darkness. After washing, the coverslips were mounted on the slides with Mowiol (Merck-Millipore, Germany) and observed in a confocal microscope Leica TCS SP5, with a magnification of 630×.
2.7. Subcellular localization by digitonin fractionation extractions
For digitonin treatment fresh early-log cells were harvested, washed once in PBS, and resuspended in 20 mM Tris–HCL (pH 8.0), 1 mM EDTA, 1 mM DTT containing a protease inhibitor cocktail (Roche) at a cell density of 5 × 107 cells/ml. Aliquots of 100 μl were made and supplemented with increasing (0–10 mg/mg of total protein extracts) concentrations of digitonin (Roche) and incubated at 26 °C for 10 min. Cells were then centrifuged at 20,800g for 2 min. Supernatants were immediately removed and resolved by 10% SDS–PAGE. Protein extracts quantification and Western blotting incubations were done as described above.
2.8. Molecular dynamics simulations
Complexes of LiTAT with the transition states containing tyrosine bonded to pyridoxal phosphate in its quinonoid resonance form, pyridoxamine (PMP) and PMP with KMTB bonded (all of them are described as transition states involved in transamination reaction) were obtained by molecular docking using the software Discovery Studio 4.0 (Accelrys Software Inc., San Diego, CA.) The molecules for docking were prepared by using Smi2Depict tool to generate 2D images from SMILES (Chen et al., 2007). Input site sphere of interaction without PLP ligand (coordinate in a three-dimensional system which can represent a single point or the head of a vector whose tail is located at the origin: X,Y,Z, Radius): 64.8412 Å, 15.5681 Å, 9.92923 Å, 18.7782 Å. Simulations with the transition states were conducted with LiTAT using CDocker (Wu et al., 2003) as docking algorithm to predict binding positions, conformations and orientations of the transition states and the ligand KMTB. Default parameters were used for all the simulations. The root mean square deviation (RMSD) values derived from the docked transition states were obtained using PLP bonded to LiTAT structure as the model to compare. The features that match the interactions between KMTB and the protein binding site with PMP have been used to build the pharmacophore model. Each sphere defines the location tolerance of a particular feature point. The default radius tolerance for all features is 1.6 Å.
3. Results
3.1. The end-product of tyrosine catabolism is produced in high amounts and excreted into the culture medium by promastigotes
The ether-soluble fraction extracted from three replicates of stationary-phase promastigote culture allowed to detect different peaks corresponding to excreted products from the parasite. The peak depicted in Fig. 1A corresponds to p-hydroxyphenyllactic (pHPL) standard, which elutes at 11.4 min. The supernatant of the L. infantum culture was run and the chromatogram differs greatly from the negative control (Fig. 1B and C). Among all peaks, the one eluting at 10.7 min was isolated from the rest and then analyzed by GC. The mass spectrum of the peak eluting at 13.68 min (Fig. 1D) was identical to the mass spectrum of the standard (Fig. 1E and F), which is indicative of the presence of pHPL in the supernatant of L. infantum promastigotes. The final concentration of pHPL in the culture medium is 3.39 mM. The activity of pHPP dehydrogenase measured in different promastigote growth phases is similar between early and mid-logarithmic stage, however the activity is increased by two times at the stationary phase (Table 1).
Fig. 1.
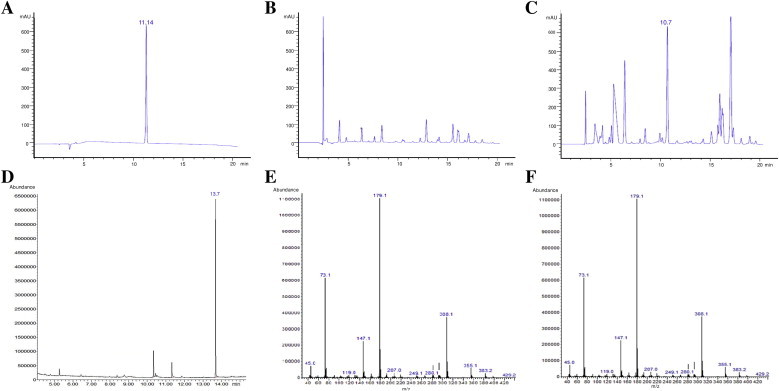
pHPL is excreted in high amounts by L. infantum promastigotes. Figures A, B and C correspond to HPLC chromatograms. (A) 20 μl of a solution containing 0.5 mM pHPL as standard was run. The peak corresponding to pHPL eluted at 11.14 min. (B) A total volume of 230 ml of complete medium was used to extract the ether soluble fraction as control in order to asses that there are no peaks at the same elution time than the standard in the chromatogram. 20 μl was loaded in the run. (C) A total volume of 230 ml of supernatant obtained from a stationary-phase promastigote culture was used to extract the ether-soluble fraction and analyzed by HPLC. The peak which its retention time is similar to the retention time of the standard was eluted and sent for further analysis by GC–MS. (D) Gas chromatogram of the peak eluted in Fig. 1C. The major peak with an elution time of 13.7 min was isolated and analyzed by mass spectrometry. (E and F) The mass spectrum of the peak (E) was identical to the mass spectrum of the pHPL standard (F).
Table 1.
p-hydroxyphenyl pyruvate dehydrogenase activity in different promastigote growth phases.
| Culture phase | Specific activity (U/mg) |
|---|---|
| Early-logarithmic | 8.16 × 10−3 ± 0.21 × 10−3 |
| Mid-logarithmic | 8.54 × 10−3 ± 0.43 × 10−3 |
| Stationary | 17.12 × 10−3 ± 0.38 × 10−3 |
3.2. LiTAT is over-expressed in more infective promastigotes and in resistant strains to nitric oxide
LiTAT protein expression was evaluated by Western blotting of L. infantum cell extracts obtained from samples taken at different growth phases. The protein was mainly expressed in promastigotes at the proliferative early-logarithmic stage whereas it was almost absent at the stationary phase (Fig. 2A). However, over-expression was observed in the more infective PNA− subpopulation compared to the PNA+ subpopulation within the stationary phase (Fig. 2B). The protein was also over-expressed in amastigote-like forms of L. infantum (Fig. 2A). Furthermore, in early-logarithmic stage promastigotes a nitric oxide resistant strain of L. chagasi TAT displayed a over-expression compared to a NO sensitive strain (Fig. 2C).
Fig. 2.
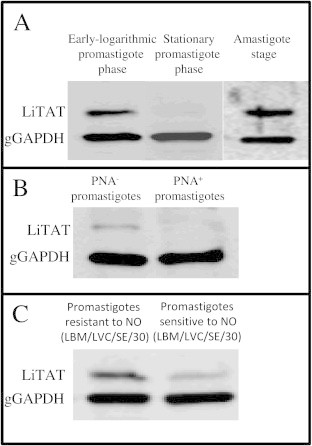
Expression profile of LiTAT. In all cases a 1:3000 dilution and 1:10000 of specific anti-LiTAT and anti-gGAPDH polyclonal antibody respectively was used. 10 μg of total protein extracts were loaded per well. (A) L. infantum early-logarithmic and stationary phase promastigotes and axenic amastigotes protein extracts. (B) L. infantum stationary PNA+ and PNA− protein extracts. (C) L. chagasi early-logarithmic stage of resistant and sensitive strains.
3.3. Subcellular localization of TAT
LiTAT was detected by indirect immunofluorescence (IFI) in L. infantum promastigotes at different growth phases. The distribution pattern of the protein is mainly cytoplasmic though not homogenously distributed. By contrast, we were unable to detect fluorescence in the stationary phase promastigotes (Fig. 3A). In order to assess cytoplasmic subcellular localization, L. infantum promastigotes were incubated with increasing digitonin concentrations to extract soluble and insoluble proteins fractions, which were then subjected to Western blot analysis. LiTAT was extracted to the supernatant at low concentrations of digitonin (Fig. 3B). The gGAPDH used as a compartmentalized protein control was not released completely even at 10 mg digitonin per mg of total protein. According to these results, LiTAT is mainly cytoplasmic.
Fig. 3.
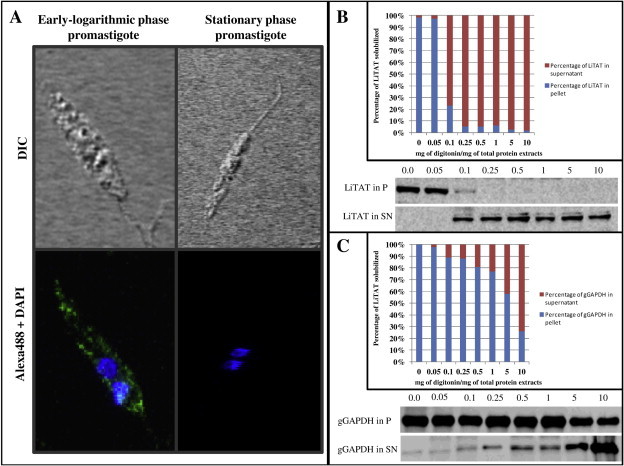
Subcellular localization of LiTAT. (A) Confocal images were obtained using L. infantum early-logarithmic promastigotes and the green fluorescence corresponds to the Alexa Fluor 488 coupled to the primary antibodies specifically raised against the recombinant TAT. The blue fluorescence corresponds to the nuclear (N) and kinetoplast (K) DNA, stained with DAPI. LiTAT is distributed non-homogenously throughout the entire cytoplasm in early-logarithmic phase promastigotes. In stationary phase promastigotes any green fluorescence could be detected. (B) 10 μg of total protein extracts of selected fractions resulting from the digitonin gradient were subjected to Western blot analysis and probed with specific polyclonal antibody anti-LiTAT and anti-L. mexicana gGAPDH. Digitonin gradient assay allowed suggesting that the protein is cytoplasmic since the compartmentalized control protein (gGAPDH) is released to the supernatant with higher concentration of detergent than LiTAT as depicted in the graphics. (For interpretation of the references to colours in this figure legend, the reader is referred to the web version of this paper.)
3.4. Enzymatic activity and in silico studies
First, we determined the enzymatic activity of LiTAT protein purified as reported (Moreno et al., 2014) with tyrosine as substrate and KMTB as co-substrate (AEE = 33.4 ± 2 U/mg of purified protein). The scheme of the KMTB transamination using tyrosine as amino donor is shown in Fig. 4A. In order to identify the structural features involved in the recognition of KMTB, we performed molecular docking simulations. The formation of the intermediate state pyridoxamine phosphate (PMP) is preceded by a quinonoid intermediate which is rapidly hydrolyzed to PMP (Kirsch et al., 1984). We first determined the disposition of the quinonoid intermediate with tyrosine with the lowest free energy value, depicted in Fig. 4B. The RMSD compared to the PLP in the 2.3 Å-solved structure of LiTAT, for that position is 2.53 Å. After that, the position adopted by PMP within the active center was simulated yielding 106 poses for the PMP transit state. These poses were compared with those of the quinonoid transit state, being the most similar not the lowest in free energy value but the second one (RMSD is 3.6 and 1.19 Å, respectively), which is showed in Fig. 4C. The distance between the secondary amine of PMP transferred to the acceptor (the oxoacid group of KMTB) is 3.89 Å. The docked KMTB molecule allowed generating the pharmacophore group of LiTAT with KMTB (Fig. 5A). The residues involved in the pharmacophore and the distances between them are depicted in Fig. 5B.
Fig. 4.
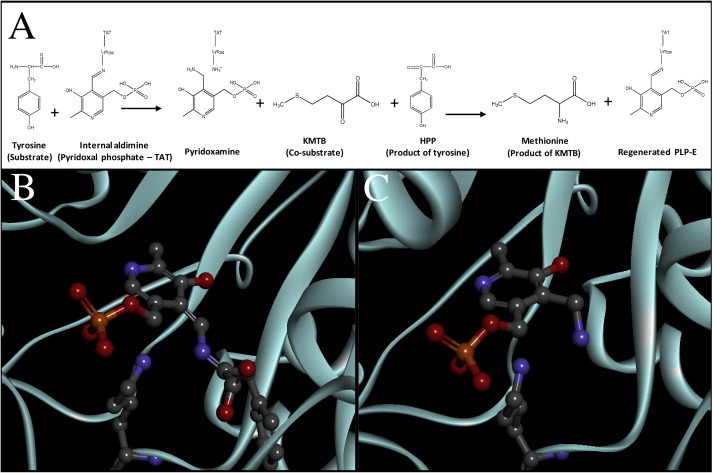
Molecular docking simulation with transition states. (A) Scheme reaction of transamination catalyzed by LiTAT using tyrosine as amino donor and KMTB as amino acceptor. (B) LiTAT with quinonoid form transit state complex. (C) LiTAT-PMP transition state complex. LiTAT protein is shown as a cyan cartoon. Lys286, which covalently bonds PLP, and ligands are shown in balls and sticks and residues from LiTAT which bond KMTB molecule are shown by thin sticks colored by elements. Carbons, oxygens, nitrogens and phosphates are shown by gray, red, blue and orange respectively. (For interpretation of the references to colours in this figure legend, the reader is referred to the web version of this paper.)
Fig. 5.
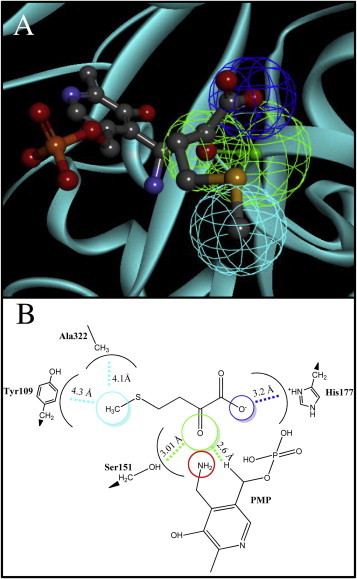
The pharmacophore model was developed using the X-ray L. infantum tyrosine aminotransferase structure. Amino acceptor group of KMTB is directed towards amine group of PMP transit state (surrounded by red circle). Carbons, oxygens, nitrogens, sulfurs and phosphates are shown by gray, red, blue, yellow and orange respectively. Each sphere indicates the location tolerance of a particular feature point. Hydrogen bonds, hydrophobic and electrostatic interactions are shown by green, dark blue and cyan respectively. (For interpretation of the references to colours in this figure legend, the reader is referred to the web version of this paper.)
4. Discussion
Despite visceral leishmaniasis is the most severe form of the disease and is fatal without treatment, the current therapy for leishmaniasis is limited to a few drugs. The oldest and most used in therapy against leishmaniasis is pentavalent antimonials. Nevertheless, the increase in resistances, relapses and patients who did not respond to treatment (Ouakad et al., 2011) has caused the substitution of antimonials by other drugs like the oral formulation of miltefosine (Thakur et al., 2009). However, resistances to this treatment have been also reported (Cojean et al., 2012). Therefore, there is a need for finding new leishmanicidal compounds. The identification of targets involved in metabolic pathways present in Leishmania but absent in the host is the first step to find new drug targets. Differentiation of promastigotes transmitted by the sand fly to the mammalian host to amastigotes surviving inside phagocytes is a crucial process in the life cycle of the parasite. In the gut of the vector, one of the most available sources of energy are amino acids, which are normally oxidized and get into the TCA cycle, obtaining NAD(P)+ (Rosenzweig et al., 2008). The availability of nutrients is lower in the phagolysosome of the host cell (McConville et al., 2007), where amastigotes scavenge essential amino acids released by the proteolysis of host proteins (Antoine et al., 1998). Therefore, the amino acid transporters systems, which are essential, are over-expressed in the amastigote stage, suggesting that catabolism of amino acids is highly activated (Alcolea et al., 2010b; Landfear, 2011). Amino acid metabolism may be a good option in searching for targets due to the differences between mammals and trypanosomatids as it has been previously proposed (Nowicki and Cazzulo, 2008). Consequently, catabolism of amino acids might be relevant for infectivity and survival within the host macrophage. Excretion of the end-products of the aromatic amino acid catabolism was also reported in T. cruzi but it is remarkable that certain aromatic products are not excreted in L. mexicana (Montemartini et al., 1994a). It was proposed that the symptomatology of the African trypanosomiasis is partially due to excretion of the end-products of aromatic amino acid catabolic pathways by the parasite, highlighting the tryptophan catabolic pathway (Seed et al., 1983). In this work, we have demonstrated that there is pHPP dehydrogenase activity and that the aromatic 2-hydroxyacid corresponding to tyrosine is being excreted in high amounts in L. infantum and this can be related to the infectivity of the parasite (Fig. 1). In fact, an increase in the LiTAT transcript in the PNA− subpopulation was described suggesting that this increase may be explained by the excretion of the end-product pHPL (Alcolea et al., 2009). In fact, it is probably related with virulence in T. brucei (Nowicki and Cazzulo, 2008). The over-expression of LiTAT has been confirmed at the protein level both in the more infective PNA− promastigote subpopulation (Fig. 2B) and in a resistant strain of L. chagasi to nitric oxide, (Fig. 2C) which is one of the main mechanisms used by the host macrophage against the parasite. LiTAT is also strongly over-expressed in the proliferating early logarithmic phase, as well as in the amastigote stage. Conversely, the protein almost completely disappears in the stationary phase (Fig. 2A). The expression of LiTAT in more infective promastigotes suggests that the enzyme has a relevant function during the process of promastigote pre-adaptation to the new host.
LiTAT in early-logarithmic promastigotes is almost entirely distributed along the cytoplasm in spite of appearing as non-homogenously distributed according to the IFI assays. The subcellular fractionation experiment supports that the protein is cytoplasmic as in L. major (Marciano et al., 2009) (Fig. 3). This subcellular localization pattern can be advantageous in order to improve the access of a putative inhibitor to the target once it crosses the plasma membrane, which is an important requirement to consider the protein as a good target (Myler, 2008).
The differences between LiTAT and the mammalian orthologues both in sequence identity (Jensen and Gu, 1996) and in the structure of the active center reflect their different substrate specificity and activity (Mehere et al., 2010; Moreno et al., 2014). This is probably critical for the recognition of substrates or analogues. Unlike in trypanosomatid TATs, the specificity of the human is restricted to the substrate pair tyrosine/α-ketoglutarate (Sivaraman and Kirsch, 2006). Although both LiTAT and TcTAT are able to use pyruvate as amino acceptor, LiTAT does not use α-ketoglutarate as the TcTAT and the mammalian TAT do because of the absence of two important residues in the N-terminal domain that are essential for the recognition of the dicarboxylic oxoacid (Sobrado et al., 2003; Moreno et al., 2014;). In trypanosomatids such as T. brucei and C. fasciculata, the final reaction of the methionine regeneration pathway was identified to be catalyzed by a broad substrate aminotransferases (Berger et al., 1996). In Leishmania, TAT has also been proposed to be responsible for catalyzing this final step based on the ability of the enzyme to use KMTB as substrate (Marciano et al., 2009).This, agrees with our data (see Section 3.4.). Therefore we aimed to define the active sites that stabilize KMTB prior to its transamination by molecular dynamic simulations. As a result, we have predicted the bonds formed for recognition of the transit state involving KMTB and PMP pair, and we have generated a pharmacophore model that will allow the identification of novel inhibitors in the treatment against leishmaniases. In the case of LiTAT, most of the residues determined involved in the pharmacophore are clearly different to the correspondent ones described for the mammalian enzyme. With the model generated, KMTB is forming a hydrogen bond with PMP and there are electrostatic interactions with His177 and a hydrophobic interaction with Ala322. Interestingly, these residues are not present in the mammalian orthologues suggesting that they may play an important role in the selectivity of the substrates in LiTAT.
5. Conclusions
The final product of the tyrosine catabolic pathway, pHPL, is excreted by L. infantum promastigotes. LiTAT is a cytoplasmic protein over-expressed in the more infective L. infantum and in resistant to nitric oxide L. chagasi promastigotes, as well as in axenic amastigotes and, as a difference with the mammalian TAT, is able to transaminate KMTB. The pharmacophore model developed in this study may be useful for the screening of new ligands against LiTAT. For these reasons, this enzyme can be considered as a candidate for the development of new drugs against leishmaniasis.
Conflict of interest
The authors declared that there is no conflict of interest.
Acknowledgments
The authors thank the whole SSGCID team. This research was funded under Federal Contract No. HHSN272201200025C from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services. The project was also funded by grant AGL 2010-21806-C02-01 of the Spanish Ministry of Economy and Competitiveness and by a contract No. 050204100014 by Fundación Ramón Areces. We would like to thank Paul Michels for lending us the anti L. mexicana gGAPDH antibody and Alicia Prieto for its assistance with HPLC and GC–MS experiments. The author M.A. Moreno thanks also the National Research Council for the grant 2012EST JAE Predoc.
Contributor Information
Miguel Angel Moreno, Email: mamoreno@cib.csic.es.
Ana Alonso, Email: amalonso@cib.csic.es.
Pedro Jose Alcolea, Email: pjalcolea@cib.csic.es.
Mario García de Lacoba, Email: mario@cib.csic.es.
Peter John Myler, Email: peter.myler@sbri.org.
Vicente Larraga, Email: vlarraga@cib.csic.es.
References
- al-Hemidan A.I., al-Hazzaa S.A. Richner–Hanhart syndrome (tyrosinemia type II). Case report and literature review. Ophthalmic Genet. 1995;16:21–26. doi: 10.3109/13816819509057850. [DOI] [PubMed] [Google Scholar]
- Alcolea P.J., Alonso A., Gomez M.J., Moreno I., Dominguez M., Parro V., Larraga V. Transcriptomics throughout the life cycle of Leishmania infantum: high down-regulation rate in the amastigote stage. Int. J. Parasitol. 2010;40:1497–1516. doi: 10.1016/j.ijpara.2010.05.013. [DOI] [PubMed] [Google Scholar]
- Alcolea P.J., Alonso A., Gomez M.J., Sanchez-Gorostiaga A., Moreno-Paz M., Gonzalez-Pastor E., Torano A., Parro V., Larraga V. Temperature increase prevails over acidification in gene expression modulation of amastigote differentiation in Leishmania infantum. BMC Genomics. 2010;11:31. doi: 10.1186/1471-2164-11-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alcolea P.J., Alonso A., Larraga V. Proteome profiling of Leishmania infantum promastigotes. J. Eukaryot. Microbiol. 2011 doi: 10.1111/j.1550-7408.2011.00549.x. [DOI] [PubMed] [Google Scholar]
- Alcolea P.J., Alonso A., Sanchez-Gorostiaga A., Moreno-Paz M., Gomez M.J., Ramos I., Parro V., Larraga V. Genome-wide analysis reveals increased levels of transcripts related with infectivity in peanut lectin non-agglutinated promastigotes of Leishmania infantum. Genomics. 2009;93:551–564. doi: 10.1016/j.ygeno.2009.01.007. [DOI] [PubMed] [Google Scholar]
- Antoine J.C., Prina E., Lang T., Courret N. The biogenesis and properties of the parasitophorous vacuoles that harbour Leishmania in murine macrophages. Trends Microbiol. 1998;6:392–401. doi: 10.1016/s0966-842x(98)01324-9. [DOI] [PubMed] [Google Scholar]
- Berger B.J., Dai W.W., Wang H., Stark R.E., Cerami A. Aromatic amino acid transamination and methionine recycling in trypanosomatids. Proc. Natl. Acad. Sci. U.S.A. 1996;93:4126–4130. doi: 10.1073/pnas.93.9.4126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger L.C., Wilson J., Wood P., Berger B.J. Methionine regeneration and aspartate aminotransferase in parasitic protozoa. J. Bacteriol. 2001;183:4421–4434. doi: 10.1128/JB.183.15.4421-4434.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blankenfeldt W., Nowicki C., Montemartini-Kalisz M., Kalisz H.M., Hecht H.J. Crystal structure of Trypanosoma cruzi tyrosine aminotransferase: substrate specificity is influenced by cofactor binding mode. Protein Sci. 1999;8:2406–2417. doi: 10.1110/ps.8.11.2406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Cojean S., Houze S., Haouchine D., Huteau F., Lariven S., Hubert V., Michard F., Bories C., Pratlong F., Le Bras J., Loiseau P.M., Matheron S. Leishmania resistance to miltefosine associated with genetic marker. Emerg. Infect. Dis. 2012;18:704–706. doi: 10.3201/eid1804.110841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J.H., Linstead E., Swamidass S.J., Wang D., Baldi P. ChemDB update – Full-text search and virtual chemical space. Bioinformatics. 2007;23:2348–2351. doi: 10.1093/bioinformatics/btm341. [DOI] [PubMed] [Google Scholar]
- Diamondstone T.I. Assay of tyrosine transaminase activity by conversion of p-hydroxyphenylpyruvate to p-hydroxybenzaldehyde. Anal. Biochem. 1966;16:395–401. [Google Scholar]
- Goldberg B., Rattendi D., Lloyd D., Sufrin J.R., Bacchi C.J. Effects of intermediates of methionine metabolism and nucleoside analogs on S-adenosylmethionine transport by Trypanosoma brucei brucei and a drug-resistant Trypanosoma brucei rhodesiense. Biochem. Pharmacol. 1998;56:95–103. doi: 10.1016/s0006-2952(98)00118-x. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Aseguinolaza G., Almazan F., Rodriguez J.F., Marquet A., Larraga V. Cloning of the gp63 surface protease of Leishmania infantum. Differential post-translational modifications correlated with different infective forms. Biochim. Biophys. Acta. 1997;1361:92–102. [PubMed] [Google Scholar]
- Gualdron-Lopez M., Chevalier N., Van Der Smissen P., Courtoy P.J., Rigden D.J., Michels P.A. Ubiquitination of the glycosomal matrix protein receptor PEX5 in Trypanosoma brucei by PEX4 displays novel features. Biochim. Biophys. Acta. 2013;1833:3076–3092. doi: 10.1016/j.bbamcr.2013.08.008. [DOI] [PubMed] [Google Scholar]
- Haldar A.K., Sen P., Roy S. Use of antimony in the treatment of leishmaniasis: current status and future directions. Mol. Biol. Int. 2011;2011:571242. doi: 10.4061/2011/571242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall J.E., Seed J.R., Sechelski J.B. Multiple alpha-keto aciduria in Microtus montanus chronically infected with Trypanosoma brucei gambiense. Comp. Biochem. Physiol. B. 1985;82:73–78. doi: 10.1016/0305-0491(85)90130-0. [DOI] [PubMed] [Google Scholar]
- Handman E. Leishmaniasis: current status of vaccine development. Clin. Microbiol. Rev. 2001;14:229–243. doi: 10.1128/CMR.14.2.229-243.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen R.A., Gu W. Evolutionary recruitment of biochemically specialized subdivisions of Family I within the protein superfamily of aminotransferases. J. Bacteriol. 1996;178:2161–2171. doi: 10.1128/jb.178.8.2161-2171.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirsch J.F., Eichele G., Ford G.C., Vincent M.G., Jansonius J.N., Gehring H., Christen P. Mechanism of action of aspartate aminotransferase proposed on the basis of its spatial structure. J. Mol. Biol. 1984;174:497–525. doi: 10.1016/0022-2836(84)90333-4. [DOI] [PubMed] [Google Scholar]
- Landfear S.M. Nutrient transport and pathogenesis in selected parasitic protozoa. Eukaryot Cell. 2011;10:483–493. doi: 10.1128/EC.00287-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leelayoova S., Marbury D., Rainey P.M., Mackenzie N.E., Hall J.E. In vitro tryptophan catabolism by Leishmania donovani donovani promastigotes. J. Protozool. 1992;39:350–358. doi: 10.1111/j.1550-7408.1992.tb01329.x. [DOI] [PubMed] [Google Scholar]
- Marciano D., Maugeri D.A., Cazzulo J.J., Nowicki C. Functional characterization of stage-specific aminotransferases from trypanosomatids. Mol. Biochem. Parasitol. 2009;166:172–182. doi: 10.1016/j.molbiopara.2009.04.001. [DOI] [PubMed] [Google Scholar]
- McConville M.J., de Souza D., Saunders E., Likic V.A., Naderer T. Living in a phagolysosome; metabolism of Leishmania amastigotes. Trends Parasitol. 2007;23:368–375. doi: 10.1016/j.pt.2007.06.009. [DOI] [PubMed] [Google Scholar]
- Mehere P., Han Q., Lemkul J.A., Vavricka C.J., Robinson H., Bevan D.R., Li J. Tyrosine aminotransferase: biochemical and structural properties and molecular dynamics simulations. Protein Cell. 2010;1:1023–1032. doi: 10.1007/s13238-010-0128-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montemartini M., Santome J.A., Cazzulo J.J., Nowicki C. Production of aromatic alpha-hydroxyacids by epimastigotes of Trypanosoma cruzi, and its possible role in NADH reoxidation. FEMS Microbiol. Lett. 1994;118:89–92. doi: 10.1111/j.1574-6968.1994.tb06808.x. [DOI] [PubMed] [Google Scholar]
- Montemartini M., Santome J.A., Cazzulo J.J., Nowicki C. Purification and partial structural and kinetic characterization of an aromatic L-alpha-hydroxy acid dehydrogenase from epimastigotes of Trypanosoma cruzi. Mol. Biochem. Parasitol. 1994;68:15–23. doi: 10.1016/0166-6851(94)00145-6. [DOI] [PubMed] [Google Scholar]
- Moreno M.A., Abramov A., Abendroth J., Alonso A., Zhang S., Alcolea P.J., Edwards T., Lorimer D., Myler P.J., Larraga V. Structure of tyrosine aminotransferase from Leishmania infantum. Acta Crystallogr. F Struct. Biol. Commun. 2014;70:583–587. doi: 10.1107/S2053230X14007845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myler P.J. Searching the Tritryp genomes for drug targets. Adv. Exp. Med. Biol. 2008;625:133–140. doi: 10.1007/978-0-387-77570-8_11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natt E., Westphal E.M., Toth-Fejel S.E., Magenis R.E., Buist N.R., Rettenmeier R., Scherer G. Inherited and de novo deletion of the tyrosine aminotransferase gene locus at 16q22.1––q22.3 in a patient with tyrosinemia type II. Hum. Genet. 1987;77:352–358. doi: 10.1007/BF00291426. [DOI] [PubMed] [Google Scholar]
- Nowicki C., Cazzulo J.J. Aromatic amino acid catabolism in trypanosomatids. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 2008;151:381–390. doi: 10.1016/j.cbpa.2007.03.010. [DOI] [PubMed] [Google Scholar]
- Ouakad M., Vanaerschot M., Rijal S., Sundar S., Speybroeck N., Kestens L., Boel L., De Doncker S., Maes I., Decuypere S., Dujardin J.C. Increased metacyclogenesis of antimony-resistant Leishmania donovani clinical lines. Parasitology. 2011;138:1392–1399. doi: 10.1017/S0031182011001120. [DOI] [PubMed] [Google Scholar]
- Pasquau F., Ena J., Sanchez R., Cuadrado J.M., Amador C., Flores J., Benito C., Redondo C., Lacruz J., Abril V., Onofre J. Leishmaniasis as an opportunistic infection in HIV-infected patients: determinants of relapse and mortality in a collaborative study of 228 episodes in a Mediterranean region. Eur. J. Clin. Microbiol. Infect. Dis. 2005;24:411–418. doi: 10.1007/s10096-005-1342-6. [DOI] [PubMed] [Google Scholar]
- Quash G., Roch A.M., Charlot C., Chantepie J., Thomas V., Hamedi-Sangsari F., Vila J. 4-methylthio 2-oxobutanoate transaminase: a specific target for antiproliferative agents. Bull. Cancer. 2004;91:E61–79. [PubMed] [Google Scholar]
- Rosenzweig D., Smith D., Opperdoes F., Stern S., Olafson R.W., Zilberstein D. Retooling Leishmania metabolism: from sand fly gut to human macrophage. FASEB J. 2008;22:590–602. doi: 10.1096/fj.07-9254com. [DOI] [PubMed] [Google Scholar]
- Seed J.R., Hall J.E., Price C.C. A physiological mechanism to explain pathogenesis in African trypanosomiasis. Contrib. Microbiol. Immunol. 1983;7:83–94. [PubMed] [Google Scholar]
- Sivaraman S., Kirsch J.F. The narrow substrate specificity of human tyrosine aminotransferase – The enzyme deficient in tyrosinemia type II. FEBS J. 2006;273:1920–1929. doi: 10.1111/j.1742-4658.2006.05202.x. [DOI] [PubMed] [Google Scholar]
- Sobrado V.R., Montemartini-Kalisz M., Kalisz H.M., De La Fuente M.C., Hecht H.J., Nowicki C. Involvement of conserved asparagine and arginine residues from the N-terminal region in the catalytic mechanism of rat liver and Trypanosoma cruzi tyrosine aminotransferases. Protein Sci. 2003;12:1039–1050. doi: 10.1110/ps.0229403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stibbs H.H., Seed J.R. Chromatographic evidence for the synthesis of possible sleep-mediators in Trypanosoma brucei gambiense. Experientia. 1973;29:1563–1565. doi: 10.1007/BF01943919. [DOI] [PubMed] [Google Scholar]
- Sufrin J.R., Meshnick S.R., Spiess A.J., Garofalo-Hannan J., Pan X.Q., Bacchi C.J. Methionine recycling pathways and antimalarial drug design. Antimicrob. Agents Chemother. 1995;39:2511–2515. doi: 10.1128/aac.39.11.2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakur C.P., Meenakshi Thakur A.K., Thakur S. Newer strategies for the kala-azar elimination programme in India. Indian J. Med. Res. 2009;129:102–104. [PubMed] [Google Scholar]
- Tizard I., Nielsen K.H., Seed J.R., Hall J.E. Biologically active products from African Trypanosomes. Microbiol. Rev. 1978;42:664–681. doi: 10.1128/mr.42.4.664-681.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G., Robertson D.H., Brooks C.L., 3rd, Vieth M. Detailed analysis of grid-based molecular docking: a case study of CDOCKER-A CHARMm-based MD docking algorithm. J. Comput. Chem. 2003;24:1549–1562. doi: 10.1002/jcc.10306. [DOI] [PubMed] [Google Scholar]