

Abstract
Hyperforin is a pharmacologically active component of the medicinal plant Hypericum perforatum (St. John's wort), recommended as a treatment for a range of ailments including mild to moderate depression. Part of its action has been attributed to TRPC6 channel activation. We found that hyperforin induces TRPC6-independent H+ currents in HEK-293 cells, cortical microglia, chromaffin cells and lipid bilayers. The latter demonstrates that hyperforin itself acts as a protonophore. The protonophore activity of hyperforin causes cytosolic acidification, which strongly depends on the holding potential, and which fuels the plasma membrane sodium-proton exchanger. Thereby the free intracellular sodium concentration increases and the neurotransmitter uptake by Na+ cotransport is inhibited. Additionally, hyperforin depletes and reduces loading of large dense core vesicles in chromaffin cells, which requires a pH gradient in order to accumulate monoamines. In summary the pharmacological actions of the “herbal Prozac” hyperforin are essentially determined by its protonophore properties shown here.
Hypericum perforatum (St. John's wort) has been used for centuries in herbal treatment of bacterial and viral infections, respiratory diseases, skin wounds, peptic ulcera, inflammation and mild depression1. Hyperforin, isolated from its flowering parts, is the most studied natural component of this plant, and has been reported to induce apoptosis in tumor cells2 and to inhibit tumor cell growth3, cancer invasion and metastasis4, as well as angiogenesis5. In addition, hyperforin is used as “herbal Prozac” to treat mild to moderate depression6, reveals antibiotic7 and antimalarial8 activity, and induces hepatic drug metabolism by activating the cytochrome P450 system via high affinity binding to the steroid- and xenobiotic-sensing nuclear pregnan X receptor (PXR)9, making it a critical candidate in drug interaction.
The mechanisms of hyperforin actions are not yet understood, but may include inhibition of 5-lipoxygenase10, high affinity binding to the pregnane X receptor9, release of Ca2+ and/or Zn2+ from intracellular stores11,12, and affecting of presynaptic and vesicular uptake, storage and release of neurotransmitters such as serotonin, dopamine, norepinephrine, acetylcholine, GABA and glutamate13,14,15,16,17,18. Gobbi et al.14 proposed an impairment of the monoamine storage due to a reserpine-like inhibition of the vesicular monoamine transporter (VMAT). But, whereas reserpine potently targets VMATs in the SLC18 gene family19 for serotonin, epinephrine, norepinephrine and dopamine, hyperforin additionally affects acetylcholine18, substrate of the vesicular acetylcholine transporter (VAChT, also SLC18 gene family), and GABA and glutamate15,16, substrates of the vesicular inhibitory amino acid transporters (VIAATs, SLC32 gene family)20 and the vesicular glutamate transporters (VGluTs, SLC17 gene family)21. Storage of neurotransmitters heavily relies on a vesicular electrical and proton gradient, produced by a H+-ATPase, utilizing energy to pump H+ continuously into synaptic vesicles. The neurotransmitters can then be concentrated against a gradient in exchange of two protons leaving the vesicle19. It has been shown that acidification of isolated synaptic vesicles monitored by acridine orange fluorescence quenching was abolished in the presence of hyperforin16,22, and it was suggested that dissipation of the H+ gradient by a protonophore-like activity eliminates the driving force for neurotransmitter uptake into the vesicles16,22.
Not just vesicular, but also plasma membrane transport of neurotransmitters seems to be affected by hyperforin. In this respect, hyperforin was suggested to increase the intracellular sodium concentration, subsequently reducing the sodium gradient required for neurotransmitter (re)uptake from the extracellular space by the presynaptic neurotransmitter transporters23,24. Later it was found that hyperforin activates non-selective cation channels in human platelets and rat phaeochromocytoma (PC12) cells25, and in 2007 hyperforin was introduced as specific activator for the non-selective cation channel TRPC6 (transient receptor potential canonical 6)26. TRPC6 is expressed in human platelets27 and PC12 cells26,28, and their hyperforin-induced Na+ influx, might be due to TRPC6 activation. However, other effects of hyperforin are described which might also participate in its pharmacological actions. For example, hyperforin attenuates various voltage- and ligand-gated ionic conductances in isolated hippocampal neurons or cerebellar Purkinje neurons29,30,31,32, it changes the fluidity of membranes33 and collapses the mitochondrial membrane potential leading to the release of Zn2+ and Ca2+ into the cytosol12.
In the present study we used fluorescent imaging to monitor intracellular pH, cytosolic sodium changes and neurotransmitter release, and the whole-cell patch clamp technique to identify and to characterize the hyperforin-induced conductance in four independent systems, in HEK-293 cells, primary mouse microglia and chromaffin cells, and lipid bilayer membranes. Our data show that hyperforin itself acts as a protonophore and thereby mediates a significant proton conductance. This conductance does not require the presence of channel proteins like TRPC6 and its direction depends on the existing driving forces such as membrane potential and pH gradient. In line with these results, accumulation of neurotransmitters is abolished in primary mouse chromaffin cells in the presence of hyperforin.
Results
OAG-induced TRPC6 versus hyperforin-induced currents
Diacylglycerol (DAG), generated by Gq/11-coupled signaling pathways, or directly applied DAG analogs, are known to stimulate TRPC3, TRPC6, TRPC7 and mouse TRPC2 channels, independent from a DAG-mediated activation of protein kinase C (PKC). Using the whole-cell patch clamp technique we measured the development of inward and outward currents, extracted from voltage ramps at −80 and +80 mV, respectively, upon external application of 100 μM 1-oleoyl-2-acetyl-sn-glycerol (OAG), an analog of DAG, in HEK-293 cells stably expressing TRPC6 cDNA (HEK-TRPC6 cells; Fig. 1a, black trace). 100 μM flufenamic acid (FFA), also shown to activate TRPC6, revealed the same current as OAG in HEK-TRPC6 cells (Fig. 1a, blue trace). Figure 1b shows the corresponding current-voltage relationships (IVs) revealing the typical double-rectifying TRPC6 currents. External application of 1, 3 and 10 μM hyperforin dose-dependently activated a different current in HEK-TRPC6 cells (Fig. 1c). The corresponding IVs (Fig. 1d) did not represent the typical shape of the TRPC6-mediated current (see Fig. 1b). When applying hyperforin onto non-transfected HEK-293 cells, not expressing TRPC6 (HEK; Fig. 1e), or on primary mouse cortical microglial cells (Fig. 1g) we found the same current with the same IV (Fig. 1f,h) as seen in HEK-TRPC6 cells. Thus, the current induced by hyperforin was not mediated via TRPC6. The same current appeared using hyperforin supplied as dicyclohexylammonium (DCHA) salt (Sigma; Fig. 1a–f), or as free acid in methanol (Sigma or Cayman Chemical; Fig. 1g,h).
Figure 1. OAG-induced TRPC6 versus hyperforin-induced currents.

Inward and outward currents at −80 and +80 mV, respectively, from HEK-293 cells, stably expressing TRPC6 cDNA (HEK-TRPC6; a, c), non-transfected HEK (HEK) cells (e) and primary mouse cortical microglial cells (g). As indicated by the bars 100 μM OAG or 100 μM flufenamic acid (FFA; a), 1, 3 or 10 μM hyperforin supplied as dicyclohexylammonium (DCHA) salt (c, e), and 10 μM hyperforin supplied as free acid in methanol (MtOH; g) were applied. Currents were normalized to the cell size and basic currents before compound application were subtracted. The corresponding current-voltage relationships (IVs) are displayed in (b, d, f) and (h). Data represent means ± S.E.M. of the indicated number of experiments (cells).
Hyperforin-induced currents in microglial cells
The hyperforin-induced current depends on the dose of hyperforin, as shown in mouse microglial cells (Fig. 2a,b), with half-maximal concentrations (EC50) of 9.3 and 8.7 μM hyperforin for the inward (Fig. 2d) and outward current (Fig. 2e), respectively. In parallel the cellular capacitance increased with higher hyperforin concentrations as long as hyperforin was present, and decreased thereafter again (Fig. 2c). Current and capacitance did not reach a plateau during hyperforin application, but the rate of increase decelerated. Apparently, the capacitance changes are due to an incorporation of the lipophilic hyperforin into the lipid bilayer of the plasma membrane. Microglial cells isolated from TRPC3/TRPC6-deficient mice revealed the same hyperforin-mediated current (Fig. 2f) with the same IV (Fig. 2g) as microglial cells isolated from wild-type mice, and neither cells developed a specific current upon OAG application (Fig. 2h,i). RT-PCR of 50 sorted microglial cells from wild-type mice did not reveal any transcript for TRPC6 (Fig. 2j), proofing that the hyperforin-induced current, appearing in all so far tested cells, does not depend on the presence of TRPC6.
Figure 2. Hyperforin dose-dependently induces currents in microglial cells.

Inward and outward currents at −80 and +80 mV, respectively, from primary microglial cells isolated from wild-type (a, h, k) and TRPC3/TRPC6-deficient mice (f, h). Currents were normalized to the cell size and, except in (h), basic currents before compound application were subtracted. As indicated by the bars different concentrations of hyperforin (a, f), 100 μM OAG or bath solution (no OAG; h) were applied. (b, g) and (i) display the corresponding IVs. (c) Dose-dependent changes of the normalized capacitance of the cells in (a). Sigmoidal fits of the maximal hyperforin-induced currents in (a) reveal a half-maximal concentration (EC50) of 9.3 and 8.7 μM for inward (d) and outward currents (e), respectively. Microglial cells isolated from C3/C6-deficient (ko; f, g) and wild-type mice (a, b) reveal similar hyperforin-induced currents, and neither developed a specific current upon OAG application (h, i). (j) RT-PCR for TRPC6 (197 bp) and HPRT transcripts (160 bp) from 50 FACS-sorted microglial cells. Total RNA from brain served as control. (k) Hyperforin-induced currents in the presence (black) and absence of extracellular monovalent cations, replaced by NMDG+ (blue), Cl−, replaced by aspartate (red), and Ca2+ and Mg2+ (0Ca0Mg; no substitute; green). (l) Corresponding IVs normalized to the current amplitude at 100 mV. Data represent means ± S.E.M. of the indicated number of experiments (cells).
To test which ions are responsible for the hyperforin-induced current, we modified the ion composition of the external solution. But, neither external substitution of Na+ and K+ (replaced by the non-permeable cation NMDG+), or removal of divalent cations (nominal 0Ca0Mg), nor substitution of Cl− (replaced by the non-permeable anion aspartate) significantly changed the development of the hyperforin-induced current in microglial cells (Fig. 2k). The normalized IVs of the hyperforin-mediated currents in the presence and absence of Na+, Ca2+/Mg2+ and Cl− all reveal virtually the same shape and reversal potential, indicating that the hyperforin-induced current neither is driven by the typical monovalent and divalent cations, nor the typical anions (Fig. 2l).
Hyperforin mediates a proton conductance
We next changed the pH of the external solution, since the only remaining possibly permeable ions were protons. Applying hyperforin at more acidic or more basic external pH than 7.2 (intracellular always pH 7.2) either way resulted in a significant decrease of the current amplitude (Fig. 3a). The reversal potentials obtained from the IVs (Fig. 3b) significantly shifted towards more positive and more negative values upon extracellular acidification and alkalization, respectively (Fig. 3c). Since the development of the hyperforin-induced current at acidic or basic external conditions was significantly reduced, we activated the current at external pH 7.2 and changed the pH after the current had already developed (Fig. 3d). Changing to pH 8.9 resulted in an immediate decrease of the current amplitude towards similar values as seen for the development of the hyperforin-induced current in pH 8.9. In contrast, after an initial increase of inward current, switching to pH 5.4 only led to a small decrease of the current amplitude. However, as already seen in figures 3b and c, the reversal potentials of the hyperforin-induced currents shifted towards more positive values upon external acidification and more negative values upon external alkalization (Fig. 3e,f).
Figure 3. Hyperforin induces a proton-dependent current in microglial cells.

Inward and outward currents at −80 and +80 mV, respectively, from primary mouse microglial cells (a, d, h, j). As indicated by the bars either 10 μM hyperforin was applied at different external pH (pHx; a) or external pH was changed in the absence (h) or during 10 μM hyperforin (d) or 10 μM CCCP (j) application. Currents were normalized to the cell size and basic currents before compound application were subtracted. The corresponding IVs are displayed in (b, e, i) and (k). (c, f) and (l) show the change of the reversal potentials, during the experiments in (a, d) and (j), respectively. Note that the reversal potentials of the hyperforin- and CCCP-induced currents change with different external pH (see c, f and l), and that the IVs and the pH 5.4-dependent changes of the reversal potential are similar for hyperforin- and CCCP-induced currents (see e and k as well as f and l, respectively). In (g) all experimentally obtained values of reversal potentials of the hyperforin-induced currents (black dotes) are plotted versus the external pH (intracellular pH 7.2). The dotted red line in (g) depicts the H+ reversal potentials calculated via the Nernst equation for a proton current at intracellular pH 7.2 and external pH as indicated (see experimental procedures). Data represent means ± S.E.M. of the indicated number of experiments (cells).
Figure 3g shows the plot of all measured reversal potentials in dependence of the external pH (black dots; intracellular pH 7.2). The red dotted line depicts the theoretical H+ reversal potentials, calculated by the Nernst equation under the given intra- and extracellular pH, i.e. proton concentrations. The experimentally detected and calculated reversal potentials are similar, and the slight difference is most probably due to the substantial proton flux, which might change the H+ concentration close to the membrane, and thus the expected reversal potential. Changing the external pH from 5.4 to 8.9 without applying hyperforin did not evoke similar currents in microglial cells (Fig. 3h; see Fig. 3i for IVs). In summary, the results so far suggest that hyperforin mediates a proton conductance in HEK as well as microglial cells.
Carbonyl cyanide m-chlorophenylhydrazone (CCCP), a protonophore mainly used as mitochondrial un-coupler, activated currents in microglial cells (Fig. 3j) with similar IV (Fig. 3k; black trace), but significantly smaller amplitude than hyperforin. Upon external acidification to pH 5.4 the CCCP-mediated inward current increased (Fig. 3j,k), and the reversal potential changed exactly the same way as in hyperforin towards more positive values, as expected from the proton distribution (Fig. 3l; see red trace in Fig. 3f for comparison). The red trace in figure 3k shows the initial IV of the hyperforin-mediated current directly after switching to pH 5.4. The orange trace depicts the IV at the maximal current amplitude in pH 5.4, where most probably an additional mainly outward current contributed, which was not further investigated.
Assuming that hyperforin mediates a proton conductance the cytosol of intact cells is expected to become more acidic upon hyperforin application, since the negative membrane potential presents a driving force for protons into the cell. To proof this, we loaded intact HEK cells with the pH-sensitive fluorescent dye BCECF-AM, applied different concentrations of hyperforin, and measured the fluorescence ratio F490/F450 as indicator for intracellular pH changes. With increasing concentrations of hyperforin the BCECF fluorescence ratio dose-dependently decreased (Fig. 4a). The BCECF ratio revealed a constant rundown (see also Fig. 4c). Therefore, we calculated the hyperforin-induced relative changes of the BCECF ratio in respect to the control application of 1% DMSO, which did not significantly alter the signal. The sigmoidal fit of the relative changes of the BCECF ratio at different external hyperforin concentrations, extracted at 200 s in figure 4a, revealed an EC50 of 8.5 μM hyperforin (Fig. 4b), which is virtually the same as for the hyperforin-induced currents in microglial cells (see Fig. 2d,e). To validate the pH measurements, NH4Cl (20 mM) was extracellularly applied. NH3 enters the cells and binds H+ elevating the intracellular pH (alkalization) resulting in an increase of the BCECF ratio (Fig. 4c), proofing that a decrease, as seen for hyperforin, represents an intracellular acidification. The relatively small negative membrane potential of HEK cells (−50 mV34) represents a limited driving force for protons into the cell. Therefore, we performed BCECF fluorescence imaging experiments in combination with the whole-cell patch clamp technique, to control the membrane potential of the HEK cells and thus the driving force for H+ into and out of the cell. BCECF (free acid) was added into the patch pipette solution. After reaching a stable fluorescence ratio (Fig. 4f), the membrane potential (holding potential, Vh) was clamped to different values (Fig. 4d). Figures 4e, f, g, and h represent the normalized capacitance, intracellular pH changes (BCECF F490/F450), inward and outward currents at −80 mV and +80 mV, respectively, and the reversal potential before and during the application of 10 μM hyperforin. Only in the presence of hyperforin (indicated by the box), when the putative proton current appears (Fig. 4g), holding potentials of −80 mV and +80 mV mediate intracellular acidification and alkalization (Fig. 4d,f), respectively, as expected from the driving force for protons into the cell at −80 mV and out of the cell at +80 mV. As consequence the reversal potential shifts to more positive values as calculated by the Nernst equation for intracellular decrease (alkalization), and more negative values as calculated for intracellular increase (acidification) of the proton concentration (Fig. 4h). Figures 4i and j show the IVs extracted at the indicated time points (arrowheads in Fig. 4g), and thus the reversal potentials before (Fig. 4i) and during (Fig. 4j) 10 μM hyperforin application, respectively, at a holding potential of −80, 0 and +80 mV.
Figure 4. Hyperforin induces changes of the intracellular pH in HEK cells.

(a, c) Relative changes of the fluorescence ratio (F490/F450) of the pH-sensitive dye BCECF-AM in HEK cells. As indicated by the bars different concentrations of hyperforin or 1% DMSO (a), or 20 mM NH4Cl (c) were applied. External NH4Cl induces intracellular alkalization and thus increase of F490/F450 (c). The sigmoidal fit of the relative decrease of the BCECF ratio at different hyperforin concentrations, calculated at 200 s in respect to the control application of DMSO, reveal a half-maximal concentration (EC50) of 8.5 μM (b). Normalized capacitance (e), intracellular pH changes (F490/F450; f), inward and outward currents at −80 mV and +80 mV, respectively, normalized to the cell size (g), and reversal potential (h) before and during 10 μM hyperforin, measured in HEK cells with the free acid of BCECF in the patch pipette. (d) depicts the changes of the holding potential (Vh), providing the driving force for the currents. IVs, extracted at the indicated time points (see arrowheads in g) before and during 10 μM hyperforin, are displayed in (i) and (j), respectively. Data represent means ± S.E.M. of the indicated number of experiments (cells).
Hyperforin acts as a protonophore
All whole-cell patch clamp and pH imaging experiments so far clearly indicate that hyperforin mediates a proton conductance in microglia and HEK cells. To proof whether this conductance is due to proton channels in the cellular membranes of these cells, activated by hyperforin, or if hyperforin itself reveals protonophore activity, we performed patch clamp experiments on lipid bilayers35, devoid of any ion channel proteins. The tip of a patch pipette was dipped into the lipid film on top of the bath solution, and after establishment of a tightly sealed lipid bilayer we ran the same voltage protocol as for the whole-cell patch clamp experiments, and applied 10 μM hyperforin, 100 μM CCCP or 1% DMSO to the lipid bilayer at the tip of the patch pipette (Fig. 5a). A gigaohm-resistance and a small basic current (IV see Fig. 5b) revealed the successful formation of a tight lipid bilayer. Upon application of 10 μM hyperforin a significant current appeared, showing, that hyperforin itself is able to mediate an ion conductance. The IV of the hyperforin-mediated current was similar as in microglia and HEK cells (Fig. 5c). The small shift of the reversal potential towards more negative values (black trace Fig. 5c) most probably results from an accumulation of protons, and thus pH decrease, in the very small volume inside the tip of the patch pipette, driven by the negative potential inside the pipette at the beginning of the ramp. This is a similar situation as the intracellular acidification in whole-cell experiments during hyperforin application, driven by a negative holding potential, which also results in a shift of the reversal potential of the hyperforin-mediated current towards more negative values (see Fig. 4h,j). The protonophore CCCP (100 μM) resulted in a much smaller current than hyperforin, and 1% DMSO did not change the basic current at all (Fig. 5a,c). In summary, hyperforin itself reveals strong protonophore activity in lipid bilayers independent of any channel protein.
Figure 5. Hyperforin induces currents in lipid bilayers.

(a) Inward and outward currents at −80 and +80 mV, respectively, from tight lipid bilayers at the tip of a patch pipette. As indicated by the bars either 10 μM hyperforin, 100 μM CCCP or 1% DMSO were applied. (b) depicts the current-voltage relationship (IV) of the basic current measured from the tight lipid bilayer. In (c) the corresponding IVs from the experiments in (a) are displayed after subtraction of the basic current (see b). Note that 10 μM hyperforin induced a significant conductance in the lipid bilayer. The protonophore CCCP (100 μM) yields a significant but much smaller current, and 1% DMSO did not result in any current at all (blue IV trace on top of x-axis in c). Data represent means ± S.E.M. of the indicated number of lipid bilayer experiments.
Hyperforin induces proton currents and depletes and reduces loading of monoamine-containing vesicles in chromaffin cells
To evaluate the action of hyperforin on neurotransmitter storage and release we prepared primary mouse chromaffin cells, in which catecholamines are stored in large dense core vesicles (LDCVs) possessing the vesicular monoamine transporter 1 (VMAT1)19. They are excitable neuroendocrine cells, have the same neural crest precursor cells as postganglionic sympathetic neurons, and various aspects of neurotransmitter metabolism, storage and release have been extensively studied with these cells.
As expected, application of 10 μM hyperforin induced the same whole-cell current with the same IV in chromaffin cells (Fig. 6a,b) as seen in HEK cells, microglial cells and lipid bilayers. To visualize effects on vesicular monoamine storage and release, we incubated chromaffin cells from wild-type and TRPC6-deficient mice with the fluorescent “false” neurotransmitter FFN51136. Like endogenous monoamines, FFN511 is carried by the plasma membrane monoamine transporter (MAT) into the cytosol and accumulates in a pattern consistent with LDCVs (Fig. 6c). As shown in figure 6d, e, f and g the cellular FFN511-dependent fluorescence gradually decreased under control conditions (0.1% DMSO) indicating the basic rate of FFN511 destaining, which was similar in cells from wild-type and TRPC6-deficient mice, and which apparently depended on light-activated bleaching and spontaneous release of FFN511. However, upon application of 10 μM hyperforin or membrane depolarization (70 mM KCl) the destaining of the FFN511-dependent fluorescence was significantly accelerated. Similar results were obtained in the presence of the protonophore CCCP (10 μM; see Fig. 6e). Hyperforin-induced destaining, i.e. release of FFN511, was similar in cells isolated from wild-type or TRPC6-deficient mice (Fig. 6d,e,f). The initial increase of the FFN511-dependent fluorescence during hyperforin and CCCP application might be due to a dequenching effect after reduction of the massive accumulation of FFN511 in the monoamine vesicles or due to the hyperforin-mediated pH changes. However, finally hyperforin resulted in a decrease of the FFN511-dependent fluorescence. As control we used HEK cells, which do not contain transporters for neurotransmitters and accordingly did not accumulate FFN511 (Fig. 6h).
Figure 6. Hyperforin induces membrane currents and depletes and reduces loading of monoamine-containing vesicles in chromaffin cells.

(a) Inward and outward currents at −80 and +80 mV, respectively, from primary mouse chromaffin cells. The bar indicates application of 10 μM hyperforin. Currents were normalized to the cell size and basic currents before application were subtracted. (b) shows the corresponding IV. (c) Confocal picture of FFN511-dependent fluorescence in a chromaffin cell. (d, e) Relative changes of the FFN511-dependent fluorescence (F400, normalized to the value at start of application) in chromaffin cells from wild-type (d) and TRPC6-deficient mice (e). At the indicated time (arrow) 0.1% DMSO, 10 μM hyperforin or 70 mM KCl were applied in (d), and 0.1% DMSO, 10 μM hyperforin or 10 μM CCCP in (e). (f) depicts the statistical analysis of the remaining fluorescence (F400 in d and e) at 1400 s. The asterisks (* p < 0.05; ** p < 0.01) denote a significant difference to the corresponding control in 0.1% DMSO. (g) Representative pictures of FFN511-dependent fluorescence at the start and 1450 s after application of 0.1% DMSO (upper set) or 10 μM hyperforin (lower set) in chromaffin cells. The right panel shows the transmission image of the respective chromaffin cells. HEK cells do not reveal a significant fluorescence after FFN511 loading (h). (i) FFN511-dependent fluorescence (F400) in chromaffin cells after 15 min FFN511 incubation in the absence (0.1% DMSO control normalized to 100%) and presence of 10 μM hyperforin. Data represent means ± S.E.M. of the indicated number of experiments (cells) in (a) and (b), and of the indicated number (n) of experiments including x cells (n/x) in (d, e, f) and (i).
In addition to an accelerated FFN511 destaining, loading of FFN511 into monoamine vesicles of chromaffin cells was significantly reduced in the presence of 10 μM hyperforin (Fig. 6i). Cellular (synaptic) monoamine uptake is arranged by the plasma membrane monoamine-sodium symporter (MAT; Fig. 7c (1)), driven by the Na+ gradient and the negative membrane potential (EM). The hyperforin-induced current did not depend on extracellular sodium (see Fig. 2k,l), however, imaging experiments on HEK cells, loaded with the Na+-sensitive fluorescent dye SBFI, revealed a significant increase of SBFI ratio, i.e. increase in intracellular sodium, upon hyperforin application (Fig. 7a). Using NMDG+ instead of Na+ in the external solution proved that the increase in SBFI ratio was not due to hyperforin-mediated pH-dependent effects on SBFI. However, the hyperforin-induced decrease of BCECF fluorescence ratio (see Fig. 4a), i.e. intracellular acidification, was accelerated in HEK cells in the absence of extracellular Na+ (Fig. 7b), suggesting a sodium-dependent pH regulating mechanism via e.g. the plasma membrane sodium-proton exchanger (NHE), which transports protons out of the cell and, in exchange, Na ions along their concentration gradient into the cell. The protonophore action of hyperforin moves H+ into the cell (synapse) (Fig. 7c (3)) and out of the vesicles (Fig. 7c (4)) due to the negative EM and the vesicular H+ gradient, respectively, resulting in an intracellular acidification. Thus, along with the intracellular acidification, hyperforin leads to an increase of the intracellular Na+ concentration, probably due to stimulation of the NHE (Fig. 7c (5)), and thereby reduces the driving force for the cellular MAT-dependent monoamine uptake (Fig. 7c (1)). Vesicular monoamine uptake is arranged by the vesicular monoamine-proton antiporter (VMAT; Fig. 7c (2)), driven by the huge H+ gradient and the vesicular membrane potential, both established by the vesicular proton pump (H+-ATPase). The hyperforin-induced dissipation of the vesicular H+ gradient (see above) ruins the driving force for the vesicular VMAT-dependent monoamine uptake.
Figure 7. Model of hyperforin action on monoamine uptake.

(a, b) Relative changes of the SBFI fluorescence ratio (F340/F380), representing intracellular Na+ changes (a), and the BCECF fluorescence ratio (F490/F450), representing intracellular pH changes (b), in HEK cells during the application (see arrowhead) of 30 μM (a) and 10 μM (b) hyperforin in the presence and absence of Na+ (replaced by NMDG+). In (a) 0.3% DMSO was applied as control. In (b) control experiments without application of hyperforin are shown in faint colors. Data represent means ± S.E.M. of the indicated number (n) of experiments including x cells (n/x). (c) Mechanism of monoamine uptake (left side of the model): Cellular (synaptic) uptake of monoamines (MA+) is arranged by the plasma membrane monoamine-sodium symporter (MAT; (1)). The negative membrane potential and low intracellular Na+ concentration provide the driving force for cellular Na+ and MA+ uptake. Vesicular monoamine uptake is arranged by the vesicular monoamine-proton antiporter (VMAT; (2)), moving MA+ into the vesicle in exchange for two protons. The huge H+ gradient and the vesicular membrane potential, both established by the vesicular proton pump (H+-ATPase) drives H+ out of the vesicle and thereby promotes vesicular MA+ uptake. Effects of hyperforin on monoamine uptake (right side of the model): The protonophore action of hyperforin moves H+ into the cell (synapse) due to the negative membrane potential (3). In addition, hyperforin moves H+ out of the vesicles, driven by the huge H+ gradient and positive vesicular membrane potential (4). The dissipation of the vesicular H+ gradient ruins the driving force for the vesicular VMAT-dependent MA+ uptake (2). The cytosolic acidification, mediated by the hyperforin-dependent H+ influx and H+ release from vesicles, increases the activity of the plasma membrane sodium-proton exchanger (NHE; (5)), which is driven by the Na+ gradient and the negative membrane potential, and moves one H+ out of the cell in exchange of one Na+ into the cell. This results in an increase of cytosolic Na+ concentration, reducing the driving force for the MAT-dependent cellular MA+ uptake (1).
Taken together these data show that release of FFN511 was enhanced and accumulation of FFN511 was reduced in the presence of hyperforin, which, due to its protonophore activity, collapses the vesicular pH and increases intracellular Na+ by enhanced activation of the sodium-proton exchanger.
Discussion
The present study shows that hyperforin, the major active ingredient of St. John's wort extract, induces a prominent ion conductance in primary mouse cortical microglia and chromaffin cells, HEK-293 cells and lipid bilayers. This ion conductance is independent of the presence of TRPC6 channels, neither mediated by Na+, K+, Ca2+, and Mg2+ nor by Cl−, but significantly depends on the extracellular and intracellular pH. Performing whole-cell patch clamp and pH imaging experiments we could show that the hyperforin-induced current is carried by protons. The presence of the same hyperforin-mediated conductance in lipid bilayers like in the three cell types indicates that hyperforin does not require a channel protein for its proton conducting activity, but rather itself acts as a protonophore. The non-saturating current and capacitance increase (see Fig. 2c) indicates accumulation of the lipophilic hyperforin in the plasma membrane, which might explain the effects of even low doses of hyperforin during St. John's wort extract treatment37.
Compounds with protonophore activity are e.g. the mitochondrial un-couplers carbonyl cyanide p-trifluoromethoxyphenylhydrazone (FCCP), carbonyl cyanide m-chlorophenylhydrazone (CCCP) or 2,4-dinitrophenol (DNP). They all share an aromatic structure with an acid-dissociable group of weak acidic character (pKa: FCCP 6.2, CCCP 5.59, DNP 4.1), strong electron withdrawing moiety and bulky hydrophobic groups38. In contrast to the general protonophores the biycyclic polyprenylated acylphloroglucinol hyperforin, is not a typical aromatic, but a phenol-like compound8. It shares the same hydrophobic and weak acidic character (pKa 4.87) as known protonophores, and other phloroglucinols such as moronone (bis-geranylacylphloroglucinol)39, 2,4-diacetylphloroglucinol (DAPG)40, and the anti-cancer agent nemorosone41, have been shown to reveal protonophore and mitochondrial uncoupling activity.
Several models describe the H+ translocation by protonophores across lipid bilayers. The simplest model bases on a weak acidic molecule, which is absorbed in its anionic form at the solution/membrane interface on the positive side of the membrane and there binds a H+. Due to the interaction with the H+ the molecule becomes neutral and diffuses towards the negative side of the membrane. Here the H+ leaves the weak acid again, which then, in its anionic form, moves back electrophoretically to the more positive side of the membrane and the cycle restarts38. The necessary driving forces are provided by the membrane potential and the proton concentration on both sides of the membrane. Since hyperforin reveals a weak acidic character7 and seems to accumulate in the cellular membrane (see Fig. 2c), this model might also apply for the protonophore mode of action of hyperforin.
The uncoupling protein 1 (UCP1) which transports protons across the inner membrane of brown fat mitochondria is activated by long-chain fatty acids (LCFAs)42. It also mediates proton currents after reconstitution in planar lipid bilayers upon activation by LCFAs43. Recently, Fedorenko et al.44 suggested a mechanism where the LCFAs bind to UCP1, catch protons on one side of the membrane and guide them trough UCP1 to the other side of the membrane. The protons are then released and the LCFAs stay bound at the UCP1. The mechanism of the hyperforin-mediated proton conductance might be more related to the transport mechanism of other weak acids (see above) rather than UCP1. However, the current-voltage relationship of the LCFA-activated UCP1-mediated proton current in mitoplasts, isolated from mouse brown adipose tissue44, looks exactly the same as the IV of the hyperforin-induced currents in the present study. In addition, the reversal potentials of the currents shift in a very similar manner upon pH changes.
Chatterjee et al.29 reported about a dose-dependent hyperforin-activated inward current in isolated rat hippocampal neurons. The sigmoidal fit revealed an EC50 of 9.1 μM, very close to the EC50 for the hyperforin-induced inward and outward currents (9.3 μM and 8.7 μM, respectively) and the intracellular acidification (8.5 μM) measured in the present study. However, they claimed that some preliminary data on the voltage and ionic dependence of the hyperforin-activated inward current, which was not shown in their paper, suggested a predominant role of anions.
Our data clearly show that hyperforin itself acts as a strong protonophore mediating significant proton currents. These currents can occur at the plasma membrane, leading to H+ influx due to the negative membrane potential, and at intracellular organelles. Most organelles keep an acidic intraorganellar environment and protonophore activity leads to efflux of protons into the cytosol. Thus, either way, the cytosolic pH becomes more acidic. This intracellular acidification and/or the dissipation of the mitochondrial proton gradient with subsequent Ca2+ and Zn2+ release12 might directly contribute to the various effects of hyperforin, including its antiinflammatory, proapoptotic, antiproliferative and antibiotic action1.
St. John's wort has been shown to alleviate symptoms of mild to moderate depression6,45, and recent meta-analysis shows that St. John's wort extracts are also effective antidepressants for major depression46. The most probably major active ingredient of St. John's wort, hyperforin is a potent inhibitor of 5-HT, dopamine, norepinephrine, acetylcholine, GABA and glutamate uptake in synaptosomal preparations13,14,15,17,18, and part of its antidepressant actions has been attributed to changes in intracellular H+ and Na+ concentrations and of neurotransmitter storage in synaptic vesicles17,22,23,24,25,26. Cellular neurotransmitter uptake by presynaptic transporters relies on Na+ cotransport47, and conditions that decrease the Na+ gradient of the neuronal membrane, either by lowering extracellular or elevating intracellular Na+, significantly reduce the efficiency of these transporters. Singer et al.23 suggested already that hyperforin inhibits serotonin uptake by elevating the intracellular Na+ concentration in human platelets, but the mechanism how hyperforin increases the cytosolic Na+ was not known at that time. In 2007, Leuner et al.26 proposed that hyperforin mediates Na+ increase, and thus effects neurotransmitter re-uptake, by specifically activating the non-selective cation channel TRPC6. However, by cytosolic acidification, as shown in figure 4, hyperforin fuels the plasma membrane sodium-proton exchanger (NHE), thereby increases free intracellular Na+ concentration, as shown in figure 7a, which inhibits the neurotransmitter uptake, depending on Na+ cotransport. At the same time hyperforin dissipates the H+ gradient in neurotransmitter vesicles16,22 by its protonophore activity and thereby impairs vesicular uptake, storage and release of neurotransmitters (see Fig. 6). Our data indicate that hyperforin's protonophore activity is underlying both processes (see Fig. 7c) and explain why hyperforin in contrast to reserpine or the classical antidepressant monoamine re-uptake inhibitors is unique in that it exerts its effects not only on monoamines but also on other neurotransmitters including acetylcholine, GABA and glutamate.
Methods
Microglia preparation and culture
Microglial cells were obtained from the cerebral cortex of newborn mice (postnatal days 0–3) as previously described48. In brief, pups were decapitated and their brains were removed and collected under sterile conditions in Dulbecco's Modified Eagle Medium (DMEM, Invitrogen) supplemented with 1% penicillin/streptomycin (Sigma) and 1% L-alanyl-L-glutamine (GlutaMAX, Invitrogen). Cortices were dissected, the meninges removed, and triturated after enzymatic digestion with 0.25% trypsin/EGTA and washing in medium. The homogenate was then forced through a 40 μm filter and the resulting suspension was plated in 75 cm2 poly-L-lysine-coated culture flasks containing medium with 10% fetal bovine serum (FBS). The cells were kept in an incubator at 37°C and 5% CO2, and the medium was changed 3 days following preparation and once per week thereafter. After about 2–3 weeks in culture, the flasks showed a confluent layer of astrocytes with microglial cells growing on top. Microglial cells were harvested and isolated by shaking the flasks to release the loosely attached microglia into the supernatant. Microglia from the supernatant were washed, plated on poly-L-lysine-coated glass coverslips (1 cm in diameter) and kept in medium with 10% FBS. To allow the cells to adapt to the subculture conditions plated cells were used for patch clamp experiments 3–7 days after harvest.
FACS and RT-PCR of microglial cells
Microglial cells were harvested from mixed cultures (see above), selectively stained by a monoclonal mouse anti CD11b antibody conjugated to FITC (Miltenyi Biotec Inc., Auburn, CA, USA) and sorted as 50 cells per tube by fluorescence-activated cell sorting (FACS; MoFlo flow cytometer, Cytomation, Purdue, CO, USA). For each RT-PCR a number of 50 sorted microglial cells were used. RT-PCR was performed using a SuperScript™ One-Step RT-PCR with Platinum® Taq (Stratagene, La Jolla, California, USA). The following primer pairs were used: 5′-TCC AGG AAA TTG AGG ATG ATG CG-3′ and 5′-TTG GAA GCC TTG CTT TTG ACC C-3′ for Trpc6, and 5′-GTC AAC GGG GGA CAT AAA AGT TAT TGG-3′ and 5′-GCT TGC AAC CTT AAC CAT TTT GGG-3′ for Hprt (hypoxanthin-guanin-phosphoribosyltransferase). Total RNA from mouse brain served as positive control.
Chromaffin cell preparation and culture
Chromaffin cells were obtained from the adrenal glands of newborn mice (postnatal days 0–1). Pups were decapitated and their adrenal glands were dissected, collected in Locke's Medium (in mM: 154 NaCl, 5.6 KCl, 3.6 NaHCO3, 5.6 glucose, 5 HEPES, pH 7.3), and connective tissue was removed. After 45 min of enzymatic digestion with 10 units/ml papain in DMEM containing 0.02% L-cysteine, 1 mM CaCl2 and 0.5 mM EDTA the enzymatic reaction was stopped by DMEM supplemented with 10% FBS, 0.25% albumin and 0.25% trypsin-inhibitor (Type II-O, chicken egg). Then glands were washed in “enriched” DMEM (4.5 g/l glucose, 0.1% Pen/Strep, 1% Insulin-Transferrin-Selenium-X) and gently triturated with a 200 μl pipette tip until getting a cell suspension. Cells were then plated on glass cover slips (1 cm in diameter) and kept in “enriched” DMEM in the incubator at 10% CO2 and 37°C. Experiments were performed within 1–5 days after preparation.
Mice
TRPC6−/− and TRPC6−/−/TRPC3−/− mice have been described elsewhere49,50. Single TRPC6- and TRPC3-deficient mice were generated and kindly provided by the laboratory of Lutz Birnbaumer (NIEHS/NIHR, Research Triangle Park, NC 27709, USA). All animal care and experimental procedures were carried out in accordance with the approved guidelines. Guidelines and all experimental protocols were approved by the animal welfare committee of the University of Saarland School of Medicine.
HEK cell culture
HEK-293 cells (ATCC, CRL 1573) obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA) and HEK cells stably expressing the TRPC6 cDNA (kindly provided by Dr. M.X. Zhu, University of Texas Health Science Center, Houston) were cultured in MEM and DMEM, respectively, with 10% FBS. The cells were kept in an incubator at 37°C and 5% CO2, and the medium was changed every third day. Cells were plated on glass coverslips of 1 cm in diameter coated with poly-L-lysine.
Electrophysiological recordings
Membrane currents were recorded in the tight seal whole-cell patch clamp configuration using an EPC-9 amplifier (HEKA Electronics, Lambrecht, Germany). Patch pipettes were pulled from glass capillaries GB150T-8P (Science Products, Hofheim, Germany) at a vertical Puller (PC-10, Narishige, Tokyo, Japan) and had resistances between 2 and 4 MΩ when filled with standard internal solution (in mM: 120 Cs-glutamate, 8 NaCl, 1 MgCl2, 10 HEPES, 10 Cs-BAPTA, 3.1 CaCl2 (100 nM free Ca2+, calculated with WebMaxC (http://www.stanford.edu/~cpatton/webmaxcS.htm)), pH adjusted to 7.2 with CsOH). Standard external solution contained (in mM): 140 NaCl, 2.8 KCl, 2 MgCl2, 1 CaCl2, 10 HEPES, 10 glucose, pH adjusted to 7.2 with NaOH. For some experiments the pH was adjusted to 5.4 or 6.5 and 7.9 or 8.9 by adding HCl and NaOH, respectively. In monovalent cation-free solution Na+ and K+ were replaced by NMDG+ (N-methyl-d-glucamine), and for nominally divalent cation-free solution CaCl2 and MgCl2 were omitted. Chloride-free external solution comprised of (in mM): 140 Na-aspartate, 1 Ca-gluconate, 2 MgSO4, 10 HEPES and 10 glucose, pH adjusted to 7.2 with NaOH. Hyperforin, carbonyl cyanide m-chlorophenylhydrazone (CCCP), 1-oleoyl-2-acetyl-sn-glycerol (OAG) or flufenamic acid (FFA) were added to the standard external solution from a 100 mM or 50 mM stock solution in DMSO to reach final concentrations as indicated. OAG and FFA experiments on TRPC6-expressing HEK cells were performed at 200 μM external CaCl2. All modified solutions were directly applied onto the patch-clamped cell via a pressure-driven application pipette. In some experiments a second application pipette was used. Osmolarity of all solutions ranged between 290 and 310 mOsm. Voltage ramps of 50 ms duration spanning a voltage range from -100 to 100 mV were applied at 0.5 Hz from a holding potential (Vh) of 0 mV over a period of up to 500 s using the PatchMaster software (HEKA). All voltages were corrected for a 10 mV liquid junction potential. Currents were filtered at 2.9 kHz and digitized at 100 μs intervals. Capacitive currents and series resistance were determined and corrected before each voltage ramp using the automatic capacitance compensation of the EPC-9. Basic currents before an application were subtracted to get the net developing current. Inward and outward currents were extracted from each individual ramp current recording by measuring the current amplitudes at −80 and +80 mV, respectively, and plotted versus time. Current-voltage (IV) relationships were extracted at indicated time points. Currents were normalized to the initial size of the cell to obtain current densities (pA/pF). For some experiments changes of the cell size (normalized capacitance as measured and extracted from the automatic capacitance compensation of the EPC9) and the reversal potential of currents were plotted versus time.
pH imaging
Intracellular live cell pH imaging experiments were performed using a Polychrome II and photomultiplier (MEA1530SF-V2DN, SMT, Seefeld, Germany) -based imaging system from TILL Photonics (Martinsried, Germany) at a Zeiss Axiovert 135 M fluorescence microscope equipped with a Zeiss 40×/0.65 Achroplan objective. Control of the polychrome II and data acquisition was accomplished via the EPC-9 and PatchMaster software. Prior to the experiments cells were incubated in media supplemented with 2 μM of the pH-sensitive fluorescent dye BCECF-AM for 30 min in the dark at 37°C, and washed with standard external solution to remove excess BCECF-AM. The BCECF-loaded cells, growing on 1 cm glass coverslips, were transferred to a bath chamber containing standard external solution, and BCECF fluorescence of a single cell was monitored with the photomultiplier at >510 nm after excitation at 490 and 450 nm for 15 ms each at a rate of 0.5 Hz for up to 600 s. Hyperforin was added to the standard external solution from a 100 mM stock (DMSO) to reach final concentrations as indicated and directly applied onto single cells via a pressure-driven application pipette (see Fig. 4a–c). For some experiments 20 mM NH4Cl was added to the standard external solution and pressure applied to the measured cell. The ratio of the background-corrected BCECF fluorescence at 490 and 450 nm (F490/F450) was plotted versus time. For some experiments BCECF-dependent fluorescence was detected by a camera (SensiCam) -based imaging system (Polychrom V, TILL Photonics) at a Zeiss Axiovert 100 M fluorescence microscope via a Zeiss Fluar 20×/0.75 objective, and hyperforin was added into the bath as indicated (see Fig. 7b). Data acquisition and analysis were performed by the software TILLvisION (TILL Photonics).
Combined electrophysiological recordings and pH imaging
In order to determine the hyperforin-induced changes of the intracellular pH under defined membrane potentials, the photometric technique (see pH imaging) was used in combination with the whole-cell patch clamp technique (see electrophysiological recordings). The patch pipette contained standard internal solution supplemented with 100 μM of BCECF (free acid). BCECF was excited at 490 and 450 nm for 25 ms each at a rate of 0.5 Hz for up to 800 s. Hyperforin was applied to the cell via a pressure driven application pipette as described for the pH imaging and the patch clamp experiments. The holding potential between the voltage ramps was changed from 0 mV to −80 or +80 mV as indicated (see Fig. 4d–h). Cellular currents and fluorescence changes were analyzed as described above.
Sodium imaging
Intracellular live cell Na+ imaging experiments were performed using the same camera-based imaging system (Polychrom V, TILL Photonics) as for pH imaging (see above). Prior to the experiments cells were incubated in media supplemented with 2 μM of the Na+-sensitive fluorescent dye SBFI-AM for 30–45 min in the dark at 37°C, and washed with standard external solution to remove excess SBFI-AM. The SBFI-loaded cells, growing on 1 cm glass coverslips, were transferred to a bath chamber containing external solution, and SBFI fluorescence of single cells was monitored at >510 nm after excitation at 340 and 380 nm for 100 ms each at a rate of 0.5 Hz for up to 800 s. Hyperforin and DMSO were added into the bath as indicated. The ratio of the background-corrected SBFI fluorescence at 340 and 380 nm (F340/F380) was plotted versus time (see Fig. 7a). Data acquisition and analysis were performed by the software TILLvisION (TILL Photonics).
FFN511 imaging experiments
Mouse chromaffin and HEK-293 cells, plated on glass coverslips, were incubated at 37°C for 15 min in culture media containing 10 μM of the fluorescent false neurotransmitter FFN511 (trifluoroacetate salt hydrate). For some experiments 10 μM hyperforin or 0.1% DMSO (as control) was already present during incubation with FFN511. Cells were washed with standard external ringer solution (see electrophysiological recordings) and kept in the dark for another 15 min. During the experiment every 5 s FFN511 was excited at 400 nm for a period of 10 ms and the FFN511-dependent fluorescence (>510 nm) of single cells was detected using the same camera-based imaging system (Polychrom V, TILL Photonics) as for pH and Na+ imaging (see above). Hyperforin, CCCP, high KCl or DMSO were applied into the bath as indicated. Data acquisition and analysis were performed by the software TILLvisION (TILL Photonics).
Confocal microscopy
Mouse chromaffin cells were loaded the same way as for FFN511 imaging experiments (see above). In addition to its peak excitation at around 400 nm FFN511 also shows a substantial excitation at 488 nm. Thus, the 488 nm line of an argon laser was used to visualize the FFN511-dependent fluorescence (GFP filter settings) with a Zeiss LSM780 confocal microscope controlled by the software ZEN System 2012 (both Carl Zeiss, Oberkochen, Germany).
Lipid bilayer experiments
Lipid bilayers on the tip of patch pipettes were made from phospholipid monolayers on the surface of the bath solution (see Ref. 35). In brief, 10 mg lipids (Ionovation, Osnabrück, Germany) were dissolved in 100 μl n-decan, and 1 μl was added gently onto the surface of 1 ml bath solution in which a patch pipette was already immerged. Bath and pipette solution comprised of 150 mM KCl and 10 mM HEPES, and the pH was adjusted to 7.0 with KOH. Within about 10 minutes a lipid monolayer was formed on top of the bath solution. The patch pipette was gently moved out of the bath with a lipid monolayer forming at its tip. By immerging the patch pipette back into the lipid monolayer-covered bath solution a tight lipid bilayer (sometimes) forms at the tip of the patch pipette, indicated by a gigaohm resistance. Voltage ramps were applied and currents were measured and analyzed as described for the whole-cell patch clamp experiments above. 10 μM hyperforin, 100 μM CCCP or 1% DMSO were applied onto the lipid bilayer at the tip of the patch pipette.
Data Analysis
Initial analysis of electrophysiological and photomultiplier-based pH imaging experiments were performed with FitMaster (HEKA), and the camera-based FFN511, SBFI and pH imaging experiments were analyzed using TILLvisION (TILL Photonics). IGOR Pro (Wave Metrics, Lake Oswego, OR, USA), ImageJ (NIH, USA) and Adobe Illustrator were used for further analysis and for preparing the figures. Where applicable the data was averaged and given as means ± S.E.M for an indicated number of cells. Two tailed two-sample equal variance Student's t-tests (Microsoft Excel) were used to test for significant differences. The theoretical reversal potentials for proton currents were calculated via the Nernst equation 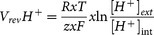 with R = 8.314 J/(mol*K), T = 297 K, F = 96485 C/mol, z = 1, and [H+]ext and [H+]int representing the external and internal proton concentrations, respectively.
with R = 8.314 J/(mol*K), T = 297 K, F = 96485 C/mol, z = 1, and [H+]ext and [H+]int representing the external and internal proton concentrations, respectively.
Chemicals
Hyperforin-dicyclohexylammonium (-DCHA) salt was obtained from Sigma, and hyperforin dissolved as free acid in methanol from Sigma and Biomol (Cayman Chemical). BCECF-AM and the free acid of BCECF were obtained from Molecular Probes (Eugene, OR, USA), and SBFI-AM from TEFLabs (Austin, TX, USA). The lipids for the lipid bilayer were kindly provided by Dr. Martin Jung, Medizinische Biochemie und Molekularbiologie, Homburg. FFN511 and all other chemicals were obtained from Sigma.
Author Contributions
T.S.S. performed and analyzed the patch clamp experiments. A.B. designed the experiments, performed and analyzed further patch clamp and all imaging experiments, and wrote the manuscript together with V.F. T.B. did the sorting and the PCR of the mouse microglial cells and contributed to the pH imaging experiments.
Acknowledgments
We thank Dr. Petra Weissgerber and the members of the Homburg SPF-animal facility for providing animals, Heidi Löhr, Ute Soltek, and Sandra Plant for expert technical assistance, Dr. Stephan Philipp for cell sorting, Martin Simon-Thomas for confocal microscopy, Dr. Dieter Bruns and Dr. Ralf Mohrmann for support in matters of chromaffin cells, Armin Weber and Andreas Helfer for mass spectrometry, and Dr. J.W. Deitmer, Dr. Holger Becker and Dr. Peter Wollenberg for helpful discussion. Supported by the Deutsche Forschungsgemeinschaft (SFB894 - A.B., V.F.; GK1326 - A.B., T.B., T.S.S., V.F.), the Homburg Forschungsförderungsprogramm HOMFOR (A.B.), and the Forschungskommission der Universität des Saarlandes (A.B., V.F.).
References
- Medina M. A., Martinez-Poveda B., Amores-Sanchez M. I. & Quesada A. R. Hyperforin: more than an antidepressant bioactive compound? Life Sci. 79, 105–111, 10.1016/j.lfs.2005.12.027 (2006). [DOI] [PubMed] [Google Scholar]
- Hostanska K., Reichling J., Bommer S., Weber M. & Saller R. Hyperforin a constituent of St John's wort (Hypericum perforatum L.) extract induces apoptosis by triggering activation of caspases and with hypericin synergistically exerts cytotoxicity towards human malignant cell lines. Eur. J. Pharm. Biopharm. 56, 121–132 (2003). [DOI] [PubMed] [Google Scholar]
- Schempp C. M. et al. Inhibition of tumour cell growth by hyperforin, a novel anticancer drug from St. John's wort that acts by induction of apoptosis. Oncogene 21, 1242–1250, 10.1038/sj.onc.1205190 (2002). [DOI] [PubMed] [Google Scholar]
- Dona M. et al. Hyperforin inhibits cancer invasion and metastasis. Cancer Res. 64, 6225–6232, 10.1158/0008-5472.CAN-04-0280 (2004). [DOI] [PubMed] [Google Scholar]
- Martinez-Poveda B., Quesada A. R. & Medina M. A. Hyperforin, a bio-active compound of St. John's Wort, is a new inhibitor of angiogenesis targeting several key steps of the process. Int. J. Cancer 117, 775–780, 10.1002/ijc.21246 (2005). [DOI] [PubMed] [Google Scholar]
- Di Carlo G., Borrelli F., Ernst E. & Izzo A. A. St John's wort: Prozac from the plant kingdom. Trends Pharmacol. Sci. 22, 292–297 (2001). [DOI] [PubMed] [Google Scholar]
- Gurevich A. I., Dobrynin V. N., Kolosov M. N., Popravko S. A. & Riabova I. D. [Antibiotic hyperforin from Hypericum perforatum L]. Antibiotiki 16, 510–513 (1971). [PubMed] [Google Scholar]
- Verotta L., Appendino G., Bombardelli E. & Brun R. In vitro antimalarial activity of hyperforin, a prenylated acylphloroglucinol. A structure-activity study. Bioorg. Med. Chem. Lett. 17, 1544–1548, 10.1016/j.bmcl.2006.12.100 (2007). [DOI] [PubMed] [Google Scholar]
- Moore L. B. et al. St. John's wort induces hepatic drug metabolism through activation of the pregnane X receptor. Proc. Natl. Acad. Sci. U S A 97, 7500–7502, 10.1073/pnas.130155097 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albert D. et al. Hyperforin is a dual inhibitor of cyclooxygenase-1 and 5-lipoxygenase. Biochem. Pharmacol. 64, 1767–1775 (2002). [DOI] [PubMed] [Google Scholar]
- Koch E. & Chatterjee S. S. Hyperforin stimulates intracellular calcium mobilisation and enhances extracellular acidification in DDT1-MF2 smooth muscle cells. Pharmacopsychiatry 34 Suppl 1S70–73 (2001). [DOI] [PubMed] [Google Scholar]
- Tu P., Gibon J. & Bouron A. The TRPC6 channel activator hyperforin induces the release of zinc and calcium from mitochondria. J. Neurochem. 112, 204–213, 10.1111/j.1471-4159.2009.06446.x (2010). [DOI] [PubMed] [Google Scholar]
- Chatterjee S. S., Bhattacharya S. K., Wonnemann M., Singer A. & Muller W. E. Hyperforin as a possible antidepressant component of hypericum extracts. Life Sci. 63, 499–510 (1998). [DOI] [PubMed] [Google Scholar]
- Gobbi M. et al. Hypericum perforatum L. extract does not inhibit 5-HT transporter in rat brain cortex. Naunyn Schmiedebergs Arch. Pharmacol. 360, 262–269 (1999). [DOI] [PubMed] [Google Scholar]
- Wonnemann M., Singer A. & Muller W. E. Inhibition of synaptosomal uptake of 3H-L-glutamate and 3H-GABA by hyperforin, a major constituent of St. John's Wort: the role of amiloride sensitive sodium conductive pathways. Neuropsychopharmacology 23, 188–197, 10.1016/S0893-133X(00)00102-0 (2000). [DOI] [PubMed] [Google Scholar]
- Chatterjee S. S., Biber A. & Weibezahn C. Stimulation of glutamate, aspartate and gamma-aminobutyric acid release from synaptosomes by hyperforin. Pharmacopsychiatry 34 Suppl 1S11–19 (2001). [DOI] [PubMed] [Google Scholar]
- Roz N., Mazur Y., Hirshfeld A. & Rehavi M. Inhibition of vesicular uptake of monoamines by hyperforin. Life Sci. 71, 2227–2237 (2002). [DOI] [PubMed] [Google Scholar]
- Buchholzer M. L., Dvorak C., Chatterjee S. S. & Klein J. Dual modulation of striatal acetylcholine release by hyperforin, a constituent of St. John's wort. J. Pharmacol. Exp. Ther. 301, 714–719 (2002). [DOI] [PubMed] [Google Scholar]
- Eiden L. E., Schafer M. K., Weihe E. & Schutz B. The vesicular amine transporter family (SLC18): amine/proton antiporters required for vesicular accumulation and regulated exocytotic secretion of monoamines and acetylcholine. Pflugers Arch. 447, 636–640, 10.1007/s00424-003-1100-5 (2004). [DOI] [PubMed] [Google Scholar]
- Gasnier B. The SLC32 transporter, a key protein for the synaptic release of inhibitory amino acids. Pflugers Arch 447, 756–759, 10.1007/s00424-003-1091-2 (2004). [DOI] [PubMed] [Google Scholar]
- Reimer R. J. & Edwards R. H. Organic anion transport is the primary function of the SLC17/type I phosphate transporter family. Pflugers Arch. 447, 629–635, 10.1007/s00424-003-1087-y (2004). [DOI] [PubMed] [Google Scholar]
- Roz N. & Rehavi M. Hyperforin inhibits vesicular uptake of monoamines by dissipating pH gradient across synaptic vesicle membrane. Life Sci. 73, 461–470 (2003). [DOI] [PubMed] [Google Scholar]
- Singer A., Wonnemann M. & Muller W. E. Hyperforin, a major antidepressant constituent of St. John's Wort, inhibits serotonin uptake by elevating free intracellular Na+1. J. Pharmacol. Exp. Ther. 290, 1363–1368 (1999). [PubMed] [Google Scholar]
- Muller W. E., Singer A. & Wonnemann M. Hyperforin–antidepressant activity by a novel mechanism of action. Pharmacopsychiatry 34 Suppl 1S98–102 (2001). [DOI] [PubMed] [Google Scholar]
- Treiber K., Singer A., Henke B. & Muller W. E. Hyperforin activates nonselective cation channels (NSCCs). Br. J. Pharmacol. 145, 75–83, 10.1038/sj.bjp.0706155 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leuner K. et al. Hyperforin–a key constituent of St. John's wort specifically activates TRPC6 channels. FASEB J. 21, 4101–4111, 10.1096/fj.07-8110com (2007). [DOI] [PubMed] [Google Scholar]
- Hassock S. R., Zhu M. X., Trost C., Flockerzi V. & Authi K. S. Expression and role of TRPC proteins in human platelets: evidence that TRPC6 forms the store-independent calcium entry channel. Blood 100, 2801–2811, 10.1182/blood-2002-03-0723 (2002). [DOI] [PubMed] [Google Scholar]
- Kumar S. et al. Mechanisms controlling neurite outgrowth in a pheochromocytoma cell line: the role of TRPC channels. J. Cell. Physiol. 227, 1408–1419, 10.1002/jcp.22855 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee S. et al. Hyperforin attenuates various ionic conductance mechanisms in the isolated hippocampal neurons of rat. Life Sci. 65, 2395–2405 (1999). [DOI] [PubMed] [Google Scholar]
- Fisunov A. et al. Hyperforin modulates gating of P-type Ca2+ current in cerebellar Purkinje neurons. Pflugers Arch. 440, 427–434 (2000). [DOI] [PubMed] [Google Scholar]
- Krishtal O. et al. Modulation of ion channels in rat neurons by the constituents of Hypericum perforatum. Pharmacopsychiatry 34 Suppl 1S74–82 (2001). [DOI] [PubMed] [Google Scholar]
- Kumar V. et al. NMDA receptor-antagonistic properties of hyperforin, a constituent of St. John's Wort. J. Pharmacol. Sci. 102, 47–54 (2006). [DOI] [PubMed] [Google Scholar]
- Eckert G. P. & Muller W. E. Effects of hyperforin on the fluidity of brain membranes. Pharmacopsychiatry 34 Suppl 1S22–25 (2001). [DOI] [PubMed] [Google Scholar]
- Babai N. et al. Anion-sensitive regions of L-type CaV1.2 calcium channels expressed in HEK293 cells. PLoS One 5, e8602, 10.1371/journal.pone.0008602 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coronado R. & Latorre R. Phospholipid bilayers made from monolayers on patch-clamp pipettes. Biophys. J. 43, 231–236, 10.1016/S0006-3495(83)84343-4 (1983). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gubernator N. G. et al. Fluorescent false neurotransmitters visualize dopamine release from individual presynaptic terminals. Science 324, 1441–1444, 10.1126/science.1172278 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cervo L. et al. Role of hyperforin in the antidepressant-like activity of Hypericum perforatum extracts. Psychopharmacology (Berl) 164, 423–428, 10.1007/s00213-002-1229-5 (2002). [DOI] [PubMed] [Google Scholar]
- Terada H. The interaction of highly active uncouplers with mitochondria. Biochim. Biophys. Acta. 639, 225–242 (1981). [DOI] [PubMed] [Google Scholar]
- Datta S. et al. Glycolysis inhibitor screening identifies the bis-geranylacylphloroglucinol protonophore moronone from Moronobea coccinea. J. Nat. Prod. 75, 2216–2222, 10.1021/np300711e (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troppens D. M., Dmitriev R. I., Papkovsky D. B., O'Gara F. & Morrissey J. P. Genome-wide investigation of cellular targets and mode of action of the antifungal bacterial metabolite 2,4-diacetylphloroglucinol in Saccharomyces cerevisiae. FEMS Yeast Res. 13, 322–334, 10.1111/1567-1364.12037 (2013). [DOI] [PubMed] [Google Scholar]
- Pardo-Andreu G. L. et al. The anti-cancer agent nemorosone is a new potent protonophoric mitochondrial uncoupler. Mitochondrion 11, 255–263, 10.1016/j.mito.2010.10.008 (2011). [DOI] [PubMed] [Google Scholar]
- Nicholls D. G. The physiological regulation of uncoupling proteins. Biochim. Biophys. Acta 1757, 459–466, 10.1016/j.bbabio.2006.02.005 (2006). [DOI] [PubMed] [Google Scholar]
- Urbankova E., Voltchenko A., Pohl P., Jezek P. & Pohl E. E. Transport kinetics of uncoupling proteins. Analysis of UCP1 reconstituted in planar lipid bilayers. J. Biol. Chem. 278, 32497–32500, 10.1074/jbc.M303721200 (2003). [DOI] [PubMed] [Google Scholar]
- Fedorenko A., Lishko P. V. & Kirichok Y. Mechanism of fatty-acid-dependent UCP1 uncoupling in brown fat mitochondria. Cell 151, 400–413, 10.1016/j.cell.2012.09.010 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiskey E., Werneke U. & Taylor D. A systematic review and meta-analysis of Hypericum perforatum in depression: a comprehensive clinical review. Int. Clin. Psychopharmacol 16, 239–252 (2001). [DOI] [PubMed] [Google Scholar]
- Linde K., Berner M. M. & Kriston L. St John's wort for major depression. Cochrane Database Syst. Rev. CD000448, 10.1002/14651858.CD000448.pub3 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres G. E., Gainetdinov R. R. & Caron M. G. Plasma membrane monoamine transporters: structure, regulation and function. Nat. Rev. Neurosci. 4, 13–25, 10.1038/nrn1008 (2003). [DOI] [PubMed] [Google Scholar]
- Giulian D. & Baker T. J. Peptides released by ameboid microglia regulate astroglial proliferation. J. Cell Biol. 101, 2411–2415 (1985). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietrich A. et al. Increased vascular smooth muscle contractility in TRPC6-/- mice. Mol. Cell. Biol. 25, 6980–6989, 10.1128/MCB.25.16.6980-6989.2005 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper M. T. et al. Transient receptor potential channels function as a coincidence signal detector mediating phosphatidylserine exposure. Sci. Signal 6, ra50, 10.1126/scisignal.2003701 (2013). [DOI] [PubMed] [Google Scholar]