

Abstract
The purpose of this study was to investigate whether caveolin-3 (Cav3) regulates localization of β2-adrenergic receptor (β2AR) and its cAMP signaling in healthy or failing cardiomyocytes. We co-expressed wildtype Cav3 or its dominant-negative mutant (Cav3DN) together with the Förster resonance energy transfer (FRET)-based cAMP sensor Epac2-camps in adult rat ventricular myocytes (ARVMs). FRET and scanning ion conductance microscopy were used to locally stimulate β2AR and to measure cytosolic cAMP. Cav3 overexpression increased the number of caveolae and decreased the magnitude of β2AR-cAMP signal. Conversely, Cav3DN expression resulted in an increased β2AR-cAMP response without altering the whole-cell L-type calcium current. Following local stimulation of Cav3DN-expressing ARVMs, β2AR response could only be generated in T-tubules. However, the normally compartmentalized β2AR-cAMP signal became diffuse, similar to the situation observed in heart failure. Finally, overexpression of Cav3 in failing myocytes led to partial β2AR redistribution back into the T-tubules. In conclusion, Cav3 plays a crucial role for the localization of β2AR and compartmentation of β2AR-cAMP signaling to the T-tubules of healthy ARVMs, and overexpression of Cav3 in failing myocytes can partially restore the disrupted localization of these receptors.
Keywords: beta-adrenergic receptors, cardiomyocytes, T-tubules, FRET, SICM
1. INTRODUCTION
The beta-adrenergic receptors (βAR) [1] are the main G-protein coupled receptors which mediate the functional effects of catecholamines in the heart. There are two main βAR subtypes (β1and β2AR) which are expressed in human and rodent cardiomyocytes [1–3]. Selective acute or chronic stimulation of β1 or β2AR elicits different cellular responses with respect to contractility, calcium cycling, hypertrophy and apoptosis. Whilst chronic β1AR stimulation usually promotes hypertrophy and apoptosis, β2AR elicits rather protective effects unless overexpressed at very high levels [4–7].
Highly localized activation of signaling pathways within different subcellular microdomains may be the key reason for these differences. β1AR has been shown to act exclusively by increasing cAMP levels and stimulating CAMKII via the stimulatory G-proteins (Gs), while β2AR can also activate inhibitory G-proteins (Gi), thereby inhibiting cAMP production, activating anti-apoptotic pathways and altering target protein phosphorylation through actions on phosphatases [5, 8, 9]. Previously, we used Förster resonance energy transfer (FRET)-based imaging to investigate subtype-specific cAMP signaling by β1AR and β2AR. Using a transgenically expressed FRET sensor in mouse cardiomyocytes, we demonstrated that β1-ARmediated cAMP signals diffused over long distances throughout the cell, whereas β2AR-receptor signals were locally confined [10].
Such differences in spatial cAMP dynamics might result from differential localization of both receptor subtypes in caveolae micro-domains, whilst β1AR has been found in both caveolar and non-caveolar membrane fractions of neonatal and adult cardiomyocytes; β2AR was shown to exclusively localize in caveolae [11–15]. Caveolae are invaginations of the plasma membrane enriched in cholesterol, glycophospholipids, and glycosylphosphatidylinositol-anchored proteins [16–20]. Caveolin, the main protein component of caveolae, recruits components of various signaling pathways, including Gi proteins [21], endothelial nitric oxide synthase [22, 23] and several protein kinases.
Neonatal cardiomyocytes lack transverse tubules (T-tubules), but have an increased caveolae density. Caveolae may be developmental precursors of T-tubules and share some of their functions [24]. T-tubular development in striated muscle depends on cholesterol and caveolin-3 (Cav3), the principal protein component of cardiac caveolae [17]. Several studies using transgenic mice overexpressing Cav3 revealed increased number of sarcolemmal muscle cell caveolae [25]. Overexpression of Cav3 in Duchene muscular dystrophy skeletal myofibers increased caveolae number [26]. In contrast, Cav3 knockout mice showed complete loss of cardiomyocyte caveolae, with associated T-tubular disorganization and dramatic cardiomyopathy [27, 28]. Experiments in adult rat ventricular myocytes (ARVMs) indicated that cholesterol depletion disrupted β2AR coupling to inhibitory G-proteins, and acute chemical caveolae disruption using methyl-β-cyclodextrin (MβCD) led to an increase in β2AR mediated cAMP and cell contractility [29]. This agrees with experimental evidence placing the β2AR in a functional relationship with the L-type Ca2+ channel, both of which are predominantly localized in the T-tubules and caveolae [30, 31].
However, no studies have addressed subtype-specific βAR signaling and its regulation by caveolae in failing cardiomyocytes. Recently, we and others have shown that during the development of heart failure (HF) after myocardial infarction, a dramatic remodelling of intracellular T-tubular network occurs which is characterized by its disorganisation and dilation at earlier stages [32], as well as loss of cell surface T-tubular openings in severe chronic HF [33]. Importantly, the latter condition leads to a redistribution of β2AR from T-tubules to detubulated membrane areas. This triggers a loss of β2AR-cAMP signal confinement, the diffusion of which is normally restricted to this microdomain in healthy cells, as revealed by a novel scanning ion conductance microscopy (SICM)/FRET-based imaging technique [34].
In this study, we tested the hypothesis that Cav3 regulates T-tubular localization of β2AR and its signalling to cAMP which are altered in HF. Using SICM/FRET and adenovirus-mediated expression of Cav3 or the dominant-negative mutant of Cav3 (Cav3DN), we found that Cav3 selectively modulates the spatial compartmentation of β2AR-cAMP responses in the T-tubular compartment which is altered in HF. Cav3 overexpression in failing cardiomyocytes reversed the pathological redistribution of β2AR-cAMP signalling.
2. MATERIALS AND METHODS
2.1 Heart Failure Model and Cell isolation
All animal surgical procedures and perioperative management were carried out in accordance with the Guide for the Care and Use of Laboratory Animals published by the U.S. National Institutes of Health under assurance number A5634-01. All animal surgical procedures and perioperative management conformed to the UK Animals (Scientific Procedures) Act 1986. Adult male Sprague-Dawley rats (250–300 g) underwent proximal coronary ligation to induce myocardial infarction as described [33]. Anaesthesia was induced by administration of 5% isoflurane for induction and reduced to 2% isoflurane once intubated and ventilated. It was ensured that pain reflexes were absent prior to an incision being made by testing pedal and palpebral reflexes. Preoperatively, buprenorphine was administered subcutaneously at a dose of 0.05 mg/kg to ensure adequate analgesia in addition to anaesthesia. Further doses of buprenorphine were administered as required post-operatively if there was any sign of distress. In addition, enrofloxacin (5 mg/kg) and 0.9 % saline (10 ml/kg) were administered pre-operatively. Cardiac failure was assessed via biometric and echocardiographic means. Heart weight corrected to tibia length provided a measure of hypertrophy. Echocardiography was performed under anaesthesia (2 % isoflurane) in the week of sacrifice. B-Mode echocardiographic images were acquired in the parasternal long axis at 70 Hz using a Visualsonics Vevo 770. This chronic HF model associated with massive hypertrophy and dilation has been previously well characterized by histological and functional analysis [33]. Isolated failing cardiomyocytes have been shown to have lower β1AR and unchanged β2AR densities [34].
Hearts were harvested for cell isolation 16 weeks post myocardial infarction, when a chronic HF phenotype has developed. Rats were sacrificed by cervical dislocation following brief exposure to 5% isoflurane until righting reflex was lost. Myocytes from healthy or failing hearts were isolated by the Langendorff perfusion method [34], plated on laminin coated coverslips and infected for 48h with Epac2-camps, Cav3 or Cav3DN adenoviral vectors [35]. For control experiments shown in Figures 3 and 4, cells were isolated from age-matched sham-operated control animals.
Figure 3. Cav3 modulates cAMP levels and its spatial distribution after β2AR stimulation.

A) Expression of the dominant negative Cav3DN construct results in an increase in intracellular cAMP stimulated by β2AR. Cells were transduced with either LacZ (control) or Cav3DN together with the Epac2-camps sensor adenovirus (all at MOI 500) and stimulated as described in Figure 2C and D. Data are means ± SE (n=12–13 cells). * - Difference from control is significant at P<0.05.
B) SICM/FRET analysis of β2AR distribution in Cav3DN expressing cells. β2AR was locally stimulated with the scanning pipette in either single T-tubules or in cell crests located between the Z-lines. The experiment was performed exactly as previously described [34]. β2AR-cAMP signals can be detected only upon local receptor stimulation in the T-tubules, suggesting that β2AR localization is unaltered in Cav3DN expressing cardiomyocytes. Data are presented as means±SE (n=5–8 cells). ** - Difference is significant at P<0.01.
C) Local β2AR stimulation in single T-tubules (white arrow denotes the position of the SICM pipette) of control (LacZ expressing) cardiomyocytes results in a highly localized cAMP signal which is measurable only in the subcellular region adjacent to the pipette. Data in the graph show YFP/CFP ratios in different color-labeled regions across the cell cytosol. Representative experiment, n=>6 cells
D) Local β2AR stimulation in single T-tubules of Cav3DN expressing cardiomyocytes results in a far-reaching cAMP gradient which diffuses across the entire cell. Data are presented as in C. Representative experiment, n=>8 cells.
E) Local T-tubular β2AR stimulation in failing cardiomyocytes leads to a similar far-reaching cAMP gradient. Representative experiment, n=10 cells.
Figure 4. Cav3 overexpression in failing cardiomyocytes partially restores the T-tubular localization of β2AR.

A) Cav3 overexpression for 48 h (MOI 500) does not lead to a significant improvement of the cell surface morphology. Representative SICM image of a failing cardiomyocyte overexpressing Cav3 (n=20 cells).
B) Quantification of the Z-groove index in these cells compared to healthy (Control) and failing (HF) cardiomyocytes without Cav3 overexpression (LacZ transfected, MOI 500). Shown are means ± SE (n=12–16 cells). ** - Difference is significant at P<0.01. NS - no significant difference.
C) SICM/FRET analysis of β2AR localization in Cav3 expressing cells, isolated from rats 16 weeks post-MI. FRET responses are significantly higher when agonist is applied to the t-tubules compared to the cellular crests. Graphs show Mean ± SE (n=15–19 cells) *P<0.05. This suggests some restoration of the normal healthy character of β2AR has been achieved by Cav3 overexpression; this is in contrast to cells derived from the same animal without Cav3 overexpression.
E) A representative trace of the β2AR response from a heart failure cell with Cav3 overexpressed.
2.2 Electron Microscopy
Cardiomyocytes were fixed with 2.5 % glutaraldehyde for 2 to 4h and then centrifuged at 500g for 5 min and the pellet was left overnight. The pellet was washed three times in cacodylate buffer and fixed in 1% osmium-tetroxide, followed by a 5–10 min washing with pure water. A small amount (25 to 50 μl) of liquid 2% agar at 45°C was added to the pellet. Drops were left to solidify on polythene, providing agar blocks with evenly distributed cells. The blocks were dehydrated through a series of graded alcohols, propylene oxide, and embedded in araldite. For low power examination by light microscopy before EM examination, 1μm thick sections were cut and stained with 1% toluidine blue in 1 % borax. For transmission electron microscopy, ultra-thin sections were stained with uranyl acetate and lead citrate. The ultrastructural features of cardiac myocytes, especially the membrane area were examined.
2.3 Western blot analysis
ARVMs were transfected with the control (LacZ), Cav3 or Cav3DN adenoviruses (all at MOI 500). 48h later cell were washed once and homogenized in the lysis buffer containing: 300mM sucrose, 150mM NaCl, 1mM EGTA, 2mM CaCl2, 1% Triton-100, 10 mM HEPES, pH=7.4. 5 μg protein samples were separated on a 15 % SDS-polyacrylamide gel and blotted onto nitrocellulose membrane (Millipore Corp., Bedford, MA, USA). Membranes were blocked for 1h at room temperature with 5 % non-fat milk in PBS containing 0.05 % Tween-20 and incubated with primary monoclonal Cav3 antibodies (1:5000, Santa Cruz Biotech, USA) overnight at 4°C. After washing, the blots were probed with a 1:5000 dilution of horseradish peroxidase (HRP)-conjugated anti-mouse IgG (Sigma, St Louis, MO, USA) and visualized using the ECL kit (Amersham Biosciences).
2.4 Radioligand binding studies
Radioligand binding studies were performed as previously described [36]. Briefly, isolated cell membranes were incubated for 1h at 30°C with 60–100 pM 125Icyanopindolol (125I-CYP) (PerkinElmer Life Sciences, Dreieich, Germany) and increasing concentrations of ICI118, 551. Competition binding curves were fitted and analyzed with the Prism software (GraphPad, San Diego, CA).
2.5 FRET imaging of cAMP in living cardiac myocytes
FRET was performed in cells infected for 48h with Epac2-camps adenovirus [37]. Cells were washed once and measured at room temperature in the buffer containing 144mM NaCl, 5.4mM KCl, 1mM MgCl2, 2mM CaCl2, and 10mM HEPES, pH=7.3. The imaging system was build around the Nikon TE2000 microscope equipped with halogen lamp illuminator pillar, EX436/20 excitation filter combined with DM455 dichroic mirror. Cell fluorescence was split into YFP and CFP channels using the DualView (Optical Insights, equipped with 535/40 and 480/30 emission filters) and monitored by the ORCA-ER CCD camera (Hamamatsu Photonics, Welwyn Garden City, UK). Cell images were analyzed using SimplePCI software (Hamamatsu). FRET ratios were corrected for the bleed through of CFP into the YFP channel and analyzed using the Origin software (OriginLab Corporation, Northhampton, MA).
2.6 SICM and SICM/FRET experiments
SICM/FRET measurements were performed exactly as previously described [34]. These included scanning of the living cell surface, local ligand application into single T-tubules via the scanning pipette and sub-cellular analysis of the induced cAMP signal by FRET microscopy. For high-resolution SICM, resistance of the pipette tip was around 200MΩ. To calculate the Z-groove index, we measured the maximum length of Z-grooves observed on single SICM images and divided this value by the total estimated Z-groove length as previously described [33].
2.7 Electrophysiological recordings from adult cardiac myocytes
Macroscopic currents were recorded using the whole-cell patch clamp technique with the external recording solution containing 1mM CaCl2, 0.5mM MgCl2, 5mM HEPES, 140mM choline chloride, 5mM CsCl, 5.5mM glucose, pH 7.4 with CsOH, ~305mOsm, and the internal pipette solution containing 130mM Cs-methanesulphonate, 11mM EGTA, 10mM HEPES, 2mM MgCl2, 5mM Mg-ATP, 0.3mM Na-GTP, pH 7.2 with CsOH, ~290mOsm/kg.
2.8 Statistical analysis
Data were analyzed using Origin Pro 8.6 software (OriginLab Corporation, Northhampton, MA) and presented as means ± SE from the indicated number of independent experiments, animals or cells isolated from several rats per condition, as indicated in the figure legends. Differences were tested using one-way ANOVA with Bonferroni post-test and considered significant at p<0.05.
2.9 Computer simulations
The β-adrenergic signaling formulation of Heijman et al. [39] was modified to represent one dimensional spatial distribution along the long axis of a simulated ventricular myocyte. A brief summary of aspects of the Heijman et al. model that are not directly germane to the simulations shown here can be found in the Supplementary Text. Crucial aspects are listed here together with modifications for the present study. The model includes distinct caveolar, extra-caveolar, and cytosolic signaling domains (cav, ecav, and cyt, respectively) and separate representations for β1 and β2ARs and their associated G-proteins. Here, we distinguish T-tubular from non-T-tubular, or “crest” sarcolemma (named based on topological appearance in SICM scans). In both membrane types, β2ARs reside primarily in cav domains (85%, with 15% in ecav in T-tubule; 100% cav in crest). The cyt domain is without β2ARs.
The 100 μm, 38e-6 μL (volume) cell was divided into 120 evenly spaced, equal volume segments. Segments alternated between T-tubule and crest type: 6 of each within 10 μm, as in SICM scans. Diffusion of cAMP and other molecules (i.e. PKA, phosphodiesterases, and PKI) between ecav and cyt domains in adjacent segments was at a rate of 1.0e-7 μL/s. The specific choice of 1.0e-7 μL/s was similar to the inter-domain diffusion rates in the original formulation by Heijman et al. and was supported by the empirical observation that either a ten-fold increase (to 1.0e-6 μL/s) or a ten-fold decrease (1.0e-8 μL/s) prevented cAMP concentration gradients from forming in disease cases where a diffusion gradient was expected (e.g Cav3DN, heart failure).
Cav domains are discrete and isolated from one another. Therefore, there was no direct inter-segmental diffusion of cAMP emanating from cav domains. However, indirect inter-segmental diffusion between adjacent T-tubule and crest segments could occur via ecav and cyt domains, which were represented as connected and contiguous. That is, ecav was connected to same-segment cav and cyt domains and to the neighboring segment ecav domain; cyt was connected to same-segment cav and ecav domains and to the neighboring segment cyt domain.
3. RESULTS
3.1 Caveolin-3 regulates the number of caveolae in cardiomyocytes
To analyze the effect of the functional Cav3 protein on the amount of caveolae in adult cardiomyocytes, we manipulated the function of endogenous Cav3 in two ways. First, we overexpressed the c-myc-tagged human Cav3 via the previously described adenoviral vector which has been shown to protect NRCM from hypertrophy [35]. Transduction of ARVMs for 48 h with this Cav3 adenovirus resulted in a 3-fold increase caveolae number, as revealed by electron microscopy of longitudinal cardiomyocyte sections (Figure 1A, B). We quantified the degree of Cav3 overexpression in ARVMs by Western blot analysis with a monoclonal Cav3 antibody, which detects both endogenous and the c-myc tagged Cav3 bands, and observed a 3.8 ± 0.1 fold increase (mean ± SE, n=3) in the amount of Cav3. In living Cav3 overexpressing ARVMs, cell-surface caveolae could be detected by high-resolution SICM (Figure 1D). Second, we used an adenovirus which encodes an untagged mutant of Cav3 lacking 3 amino acids (threonine, phenylalanine and threonine) in the caveolin scaffolding domain and behaves in a dominant-negative fashion (Cav3DN) [40], causing limb-girdle muscular dystrophy in humans [35]. Cav3DN could also be detected in Cav3 western blot, at a 2.9 ± 0.4 fold (mean ± SE, n=3) higher level than the endogenous Cav3 (Figure 1C). In contrast to wildtype Cav3, expression of the Cav3DN mutant led to a moderate but significant decrease in caveolae number (see Figure 1A, B).
Figure 1. Cav3 overexpression increases caveolae number, while the expression of the dominant-negative Cav3 mutant slightly decreases it.

A) Representative electron microscopy image of longitudinal cardiomyocyte sections showing caveolae (marked with black arrows) in control cells (infected with LacZ adenovirus) as well as in cells overexpressing wildtype Cav3 or Cav3DN mutant (all at MOI 500).
B) Quantification of the number of caveolae per μm cross-section of the membrane from the electron microscopy images. Data are means ± SE, n=4–5 per condition. *, ** - Differences are significant at P<0.05 or 0.01, respectively.
C) Western blots analysis of the Cav3 overexpression 48 h after transduction with the control (LacZ), Cav3 or Cav3DN adenoviruses (all at MOI 500).
D) High resolution SICM image of an ARVM overexpressing Cav3 (MOI 500) which shows clearly visible caveolae.
3.2 Caveolin-3 regulates βAR-cAMP signaling in a subtype-specific manner
We next investigated whether Cav3 might have an effect on βAR subtype-specific cAMP signaling. To test whether Cav3 overexpression had an effect on the total and relative numbers of β1 and β2ARs, we prepared cell membranes from LacZ or Cav3 transfected ARVMs and used them in total radioligand binding and competition displacement experiments. There was no significant difference in total amounts of βAR receptors (~0.5 fmol/μg membrane protein) and in the relative fractions of β1 and β2ARs as measured by radioligand displacement with ICI 118551 (Supplementary Figure 1A). To elucidate whether functional activity of β1 or β2AR is affected by Cav3 overexpression, we performed FRET-based imaging of cAMP levels in living ARVMs. 48 hours after co-transfection with adenoviruses for the Epac2-camps FRET biosensor and Cav3, we monitored intracellular cAMP levels upon selective stimulation of ARVM with either β1 or β2AR ligands (Figure 2). We stimulated entire cardiomyocytes (bath application) with saturating concentrations of the βAR agonist isoproterenol (ISO, 100 nM) in presence of selective antagonists of either β2AR (ICI118551, 50 nM) or β1AR (CGP20712A, 100 nM). Subsequently, the adenylyl cyclase activator forskolin, 10 μM, was applied to determine the maximal cAMP response. In control cardiomyocytes, cAMP signals from β2AR were smaller than β1AR responses, consistent with our previous results in adult mouse and rat cardiomyocytes [10, 34]. Selective stimulation of β1AR led to similar amounts of cAMP produced in case of control or Cav3 expressing cells (Figure 2A, B). In sharp contrast, the cAMP signals upon selective β2AR stimulation were ~2-fold smaller when Cav3 was overexpressed, without any change in the FRET-responses to the direct adenylyl cyclase activation (Figure 2C, D).
Figure 2. FRET-based cAMP measurements upon selective stimulation of β1 or β2AR in cardiomyocytes overexpressing Cav3.

A) Cav3 overexpression does not affect β1AR responses. ARVMs were infected for 48 h with Epac2-camps cAMP sensor adenovirus in combination with LacZ or the Cav3 virus (all at MOI 500). cAMP levels are shown as % change of the YFP/CFP ratio. A decrease in this ratio corresponds to an increase in intracellular cAMP. Cells were first treated with 100 nM isoproterenol (ISO) plus 50 nM of the β2AR blocker ICI118551 (ICI) and subsequently fully stimulated with 10μM forskolin. Quantification of the FRET responses from β1AR is presented in B (means ± SE, n=5–14cells).
C) Cav3 overexpression decreases cAMP production after β2AR stimulation. Cells were first treated with 100nM ISO plus 100nM of the β1AR blocker CGP20712A (CGP) and subsequently fully stimulated with 10μM forskolin. Quantification of the FRET responses from β2AR is shown in D (means ± SE, n=7–12 cells). * - Differences between the control and Cav3 groups are statistically significant at P<0.05.
3.3 Cav3DN increases the levels and intracellular diffusion of β2AR-cAMP
We hypothesized that endogenous Cav3 restricts local cAMP gradients generated by β2AR stimulation to the T-tubular compartment. To test this, we studied whether the expression of the dominant negative Cav3 mutant affects the spatio-temporal characteristics of the β2AR-cAMP responses. In Cav3DN expressing cells, selective whole-cell β2AR stimulation led to significantly higher total levels of intracellular cAMP (~2-fold increase in the FRET signal amplitude) (Figure 3A). To study whether this might have any effect on β2AR localization and the sub-cellular cAMP dynamics, we performed SICM/FRET experiments in Cav3DN expressing cells. Previously, using this technique we showed that in healthy ARVMs, functional β2ARs are restricted to T-tubules, while in failing cardiomyocytes or in cells after chemical cholesterol depletion using MβCD, β2AR was redistributed from the T-tubules to non-tubular membrane areas where this receptor induced far-reaching cAMP signals [34]. Here we took advantage of Cav3DN as a more specific tool to address the role of Cav3 in β2AR localization and signaling. In contrast to our previous MβCD results, Cav3DN expressing cells did not show any β2AR redistribution (see Figure 3B). However, the spatial distribution of the β2AR-cAMP signals after local stimulation of single T-tubules was dramatically changed. In contrast to control cells where these signals are stringently localized (Figure 3C), Cav3DN expression led to far-reaching cAMP signals (Figure 3D) similar to those which can be observed in failing cardiomyocytes isolated from rats 16 weeks after myocardial infarction (Figure 3E). These results suggest that Cav3 plays a crucial role in the confinement of β2AR-cAMP signaling to the T-tubular compartment. Similar to the previously published results with MβCD [41], the density of the whole-cell L-type Ca2+ channel currents were not affected by Cav3DN (Supplementary Figure 2), suggesting that the basal activity of these channels is not altered by the disruption of the T-tubular cAMP micro-domain.
3.4 Overexpression of Cav3 partially restores the disrupted β2AR localization in heart failure
Finally, we asked whether Cav3 overexpression might improve the abnormal localization of β2AR and β2AR-cAMP signals found in failing cardiomyocytes [34]. We isolated cells from the same rat chronic HF model used in this former study (Supplementary Figure 3), and overexpressed Cav3 together with Epac2-camps for 48 h to analyse the membrane structure and receptor localization by SICM/FRET. Cav3 overexpression for this short period of time did not result in any significant increase in the membrane curvature or the appearance of the T-tubular openings, as judged based on overall membrane topography and on the Z-groove index (Figure 4A, B). However, cells analyzed by SICM/FRET showed redistribution of β2AR back into the remaining T-tubules (Figure 4C) in comparison to heart failure cells without Cav3 overexpression. This suggests that Cav3 is important for T-tubular localization of β2AR, and its overexpression can restore the disrupted β2AR distribution in the failing cardiomyocytes. However, Cav3 overexpression also reduces the amplitude of β2AR responses as demonstrated by the bath application experiments described above. (Figure 4D-representative trace).
3.5 Mechanisms by which Cav3 regulates localization of the β2ARs response
We developed a theoretical framework so that a hypothetical explanation for Cav3 interactions with β2ARs under the various conditions considered here and in our previous related work [34] could be proposed. The conditions considered include control, Cav3 overexpression (Cav3), expression of dominant negative mutant Cav3 (Cav3DN), application of MβCD, heart failure, and failure with Cav3 overexpression (failure+Cav3). The framework is illustrated in Figure 5.
Figure 5.
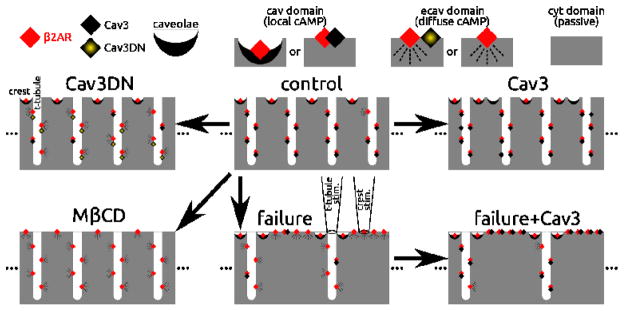
Illustration of the relationships represented in the computational model. Model components included β2AR (red diamonds), Cav3 protein (black diamonds), mutant Cav3 protein (Cav3DN, yellow shaded diamonds), and fully formed caveolae (black crescents). cAMP emanating from discrete cav domains remained local, while that from ecav domains diffused freely. The cyt domain was passive, lacking β2ARs. In control (top middle), nearly all β2ARs were in cav domains. In the crest all β2ARs were caveolae associated. In T-tubules, 85% of β2ARs were cav3 associated (lone β2ARs were rare, 15%). Cav3 overexpression (top right) associated lone β2ARs with Cav3 in the T-tubules. Cav3DN (top left) replaced 75% of β2AR:Cav3 associations with β2AR:Cav3DN. MβCD (bottom left) removed all caveolae and Cav3, leaving only lone β2ARs. Heart failure (bottom middle) involved disassociation of β2AR:Cav3 pairs (75%) and T-tubule loss — represented by relocation of previously T-tubular β2AR:Cav3 pairs and lone β2ARs to the crest. Overexpression of Cav3 in the case of failure (bottom right) restored cav functionality to all lone β2ARs at the crest, and to 75% of lone β2ARs in remaining T-tubules. As depicted above the heart failure case, pipette stimulation at a T-tubule site captured many β2ARs down the entire length of a T-tubule; in contrast, at the flat crest surface, far fewer β2ARs could be captured beneath the pipette and stimulated (1000-fold fewer than at a T-tubule).
The working components of the model, which associate with β2ARs, include Cav3, Cav3DN, and fully formed caveolae; β2ARs can also be unassociated (lone). Pairings of β2AR with caveolae or Cav3 were considered to be discrete “cav” signaling domains, where cAMP was local due to spatial isolation (see Methods section 2.9) and where relatively high phosphodiesterase (PDE) activity partially muted signals (see Supplementary Text). Lone β2ARs or β2ARs paired with dysfunctional Cav3DN constituted ecav signaling domains. cAMP generated by ecav β2ARs was readily diffusible because ecav elements were contiguous (not discrete like cav, c.f. Methods 2.9) and because PDE activity was low enough to permit cAMP accumulation. Ecav lacks PDE type 3, which is found in cav, and ecav has a volume that is twice that of cav, causing relative dilution of PDE activity (see Supplementary Text). The cytosol was devoid of β2ARs. Passive diffusion of cAMP in cyt occurred, but signals were greatly diminished by the fact that cyt volume was roughly ten-fold larger than sub-membrane volume (cav+ecav).
Our electron micrographs showed fully formed, flask-shaped caveolae in crest regions, but they were not observed in T-tubules. Recently, Wong et al. presented a similar finding on the basis of a highly novel fluorescent microscopy technique coupled with electron microscopy (their Supplementary Figure S4 [38]). Cav3 protein is apparently abundant in T-tubules but it is not clear whether fully stabilized caveolae domains can be formed in this region (e.g. [38]). Thus, to represent the control case, β2ARs were placed in caveolae in the crest and were Cav3-associated in T-tubules (exception: 15% of T-tubular β2ARs were lone; Figure 5, top middle).
Cav3 overexpression (Figure 5, top right) paired the few lone T-tubular β2ARs with Cav3 and added nonfunctional Cav3 and caveolae lacking β2ARs. We hypothesized that Cav3DN (Figure 5, top left) affected T-tubular β2AR:Cav3 pairs, replacing 75% of native Cav3 with mutant Cav3DN, and changing their designation from cav to ecav in the model. Importantly, we hypothesized that β2AR:caveolae pairs at the crest were immune to Cav3DN replacement. This concept is supported further below. We hypothesized that formation of β2AR:caveolae and β2AR:Cav3 complexes requires cholesterol. Therefore, MβCD was represented by strictly lone β2ARs in crest and T-tubules (Figure 5, bottom left). Failure was represented by Cav3 loss (affecting 75% of β2AR:Cav3 pairs in intact T-tubules) and also loss of T-tubules (half of them, as indicated by Z-groove index reduction shown here and previously [34]). Lost T-tubules purged their contents to the crest, dissociating Cav3 from 90% of β2ARs in the process (Figure 5, bottom middle). Finally, overexpressing Cav3 in heart failure was represented as pairing lone β2ARs with Cav3 (1000% and 300% increase in β2ARs:Cav3 pairings in crest and T-tubules, respectively, Figure 5, bottom right).
The central assumption of this theoretical framework is summarized as follows (supported below). β2AR associations with caveolae, found in the crest, are stable in the face of Cav3DN expression, while associations with Cav3 protein(s) in the T-tubules are not. This assumption can alternatively be viewed as causing limited pools of Cav3 to prioritize the crest over T-tubule when it comes to the localization of cAMP by forming stronger cAMP compartments. Other assumptions (e.g. the % changes above), though not incidental, were of quantitative but not qualitative consequence and needed to be made in order to realize a computational model to test the validity of the framework in Figure 5.
Figure 6 presents simulation results. Plotted are sub-membrane cAMP concentration landscapes ([cAMP] in μM) computed in space and time along a longitudinal line scan (100 microns) following β2AR stimulation at either a distal T-tubular site (panel A) or at a distal crest site (panel B). We chose to measure [cAMP] at the membrane where the majority of important signaling targets reside (rationale outlined further in Supplementary Text and Supplementary Figure 4). Pipette placement over a T-tubule stimulates many β2ARs, all along the entire depth of the structure, while stimulation at crest sites affects only the few β2ARs immediately beneath the pipette (e.g. pipette cartoon in Figure 5, bottom middle). Simulations accounted for this with a 1000-fold T-tubule to crest β2AR capture ratio (results were not different using a lower estimate of 100-fold).
Figure 6.

Simulations based on the relationships proposed in Figure 5 qualitatively reproduced cAMP experimental measurements. Shown are surface maps of cAMP concentration at the membrane ([cAMP], from 0.5 to 4 μM) along the cell length (100 microns) during the time just prior to and minutes following β2AR stimulation. Stimulation was at distal T-tubule (panel A, top) or distal crest sites (panel B, bottom) — indicated in space and time by black rectangles labeled “β2AR stim.”. The protocol was designed to mimic experiments shown in Figures 3C, D, E and 4D. Results for control, Cav3 overexpression, Cav3DN, MβCD, heart failure, and heart failure with Cav3 overexpression are shown. Cases are arranged as in Figure 5. A) Following Ttubular stimulation, cAMP was robustly generated at the stimulus site in all cases. However, it did not diffuse in control, Cav3, and failure+Cav3 cases (flat [cAMP] landscapes, “no diffusion” straight arrow). Pathological diffusion was exhibited by Cav3DN, MβCD, and heart failure (contoured [cAMP] landscapes, “diffusion” curved arrow). B) Crest stimulation did not always generate cAMP: e.g. the non-disease cases of control and Cav3, but also Cav3DN and failure+Cav3 cases. cAMP was generated following crest stimulation for MβCD and failure cases, and it diffused along the cell. Color scales are the same for all cases. Z-axes scales are the same for each case.
T-tubular stimulation resulted in sharp local spikes in [cAMP] at the stimulation site in all cases. However, diffuse cAMP (i.e. smooth concentration gradient from the stimulus site) occurred only in Cav3DN, failure, and MβCD cases, but not in cases of control, Cav3 overexpression, or heart failure with Cav3 overexpression. This matches experiments shown here and in our previous work: Cav3DN – Figure 3D, heart failure – Figure 3E, MβCD – Supplementary Figure S8C of Nikolaev et al. [34], control – Figure 3C, and failure+Cav3 – Figure 4D. The cAMP signal was reduced by Cav3 overexpression (integrated total cAMP was ~70% of control). This is in agreement with Figure 2D. Cav3DN simulations showed an increase in overall cAMP relative to control (73% increase in integrated total cAMP). Although the amplitude is not different, more cAMP response is present due to propagation throughout the cell. This is in agreement with Figure 3A,
Simulation results showed that a response to crest stimulation was only registered for the MβCD and heart failure cases. This is in accord with experiments (no response: control – Figure 2G of Nikolaev et al. [34], Cav3DN – Figure 3B, and failure+Cav3 – Figure 4C. response: MβCD – Supplementary Figure S8B of Nikolaev et al. [34], and failure – Figure 4C). Importantly, when we relaxed the central assumption that Cav3DN does not disrupt β2AR associations with caveolae in the crest, there was a robust crest response in Cav3DN simulations, in violation of Figure 3B experimental results. Without this central assumption, the Cav3DN case was nearly identical to the heart failure case. Though crest caveolae were not degraded in failure, T-tubule loss caused redistribution of newly lone β2ARs to the crest. This distinguished the failure response from the Cav3DN response.
Simulations predicted baseline cAMP elevation in pathological states (i.e. Cav3DN, failure and MβCD). This was caused by the greater proportion of β2ARs in the ecav domains, where PDE activity was relatively low. FRET experiments cannot support or refute this prediction.
4. DISCUSSION
The β2AR has been shown to associate with caveolin and caveolae in neonatal and adult ventricular cardiomyocytes. However, functional consequences of the receptor-caveolin interactions remained poorly defined, especially in failing cardiomyocytes. In this work, we analyzed the role of Cav3 in β2AR localization and in β2AR-cAMP compartmentation in healthy and failing ARVMs and showed that β2AR localization and its compartmentalized signaling are directly regulated by Cav3.
Overexpression of Cav3 in healthy ARVMs led to an increase in caveolae number (see Figure 1) and to a subtype-specific decrease of β2AR-cAMP production which was not associated with altered receptor densities or with changes in the β1/β2AR expression ratio (see Figure 2, Supplementary Figure 1). This suggests that Cav3 restricts cAMP accumulation or its diffusion from β2AR into the cytosol. Conversely, adenoviral gene transfer of the dominant-negative mutant Cav3DN led to an increase of the total β2AR-cAMP levels (see Figure 3A). Interestingly, Cav3 had no effect on the amplitude of the β1AR-stimulated cAMP response. Recently, it was demonstrated that β1AR-mediated cAMP production in the cytosolic compartment of ARVM assessed by the Epac2-camps sensor was unchanged after cholesterol depletion by MβCD. However, it was increased in the compartment associated with caveolae and the type II cAMP-dependent protein kinase, suggesting that there is a population of β1AR specifically coupled to caveolae which cannot be functionally detected by our cytosolic cAMP sensor Epac2-camps [13].
Previously, Calaghan and White demonstrated that β1AR-dependent modulation of cell shortening and Ca2+ transient was not potentiated by caveolar disruption, while β2AR stimulation significantly increased phospholamban phosphorylation, cell shortening and Ca2+ transients after MβCD treatment [29]. Therefore, β2AR association with caveolae is required for the localized signaling and relative dampening of the positive inotropic and lusitropic response mediated by this receptor. Using a specific Cav3 inhibitory peptide, the same group showed that selective disruption of Cav3 results in increased β2AR-stimulated phospholamban phosphorylation. This was not only due to an increase in cAMP levels but also due to enhanced inhibitory modulation of phosphatases at the sarcoplasmic reticulum which can be explained by changes in receptor-cAMP compartmentation [42]. However, the role of Cav3 in β2AR localization has not been studied before due to the lack of appropriate techniques. To directly visualize the functional interactions of β2AR with the sub-cellular pools of cAMP, we used the recently developed SICM/FRET method which can correlate the localization of the functional receptor with the cAMP microdomains. In our previous study, we used MβCD to disrupt cardiomyocyte caveolae and found β2AR responses to be present both in the T-tubular and in non-tubular areas of the cell surface. Stimulating β2AR in either T-tubules or crests of MβCD-treated cardiomyocytes generated far-reaching cAMP signals. This contrasted with the normal cell in which β2AR signal is exclusively present in T-tubules and is stringently confined [34]. Here we disrupted caveolae more precisely by expressing a dominant-negative Cav3DN mutant. We found that the β2AR signal does not migrate from T-tubules to the non-tubular cell surface areas, suggesting unaltered receptor localization. However, upon stimulation of the β2AR in the T-tubules, the resulting cAMP signal becomes diffuse, similar to its dynamics in failing cardiomyocytes (see Figure 3). This strongly suggests that Cav3 may be responsible for the compartmentation of the β2AR-cAMP response to the T-tubules of healthy cardiomyocytes.
Since the β2AR signaling and its regulation by caveolae in failing cardiomyocytes has not been systematically analyzed before, we studied how Cav3 overexpression might affect the altered β2AR localization and cAMP dynamics in HF. It is widely accepted that β2AR plays a cardio-protective role in the healthy heart [43]. Whilst transgenic overexpression of Cav3 in mouse cardiomyocytes induces endogenous cardiac protection from ischemia/reperfusion injury via increased activation of survival kinases, this effect is absent in Cav3 knockout mice [44]. The previously reported down-regulation of Cav3 in various animal HF models [45–48], and the adverse cardiac phenotype and T-tubular abnormalities in Cav3 knockout mice [27, 28] support the idea of Cav3 overexpression as a beneficial treatment option for HF. We tested this possibility in cells isolated from the chronic rat HF model where we overexpressed Cav3 for 48h (see Figure 4). This short treatment did not lead to any measurable improvement of cell surface morphology, but it was associated with redistribution of β2AR’s functional effects back into the T-tubules, suggesting that Cav3 might potentially restore the β2AR localization and its confined signaling to cAMP.
That the computational model was able to reproduce essential experimental observations is support for our hypothesis and its underlying assumptions. That is, β2AR associations with Cav3 molecules in T-tubules are more readily disrupted than are β2AR associations with fully formed caveolae. The exact reasons for this should be the subject of future work. We speculate that binding among the 14–16 Cav3 molecules in the formation of a caveolae [17] is cooperative, and that this could be the stabilizing factor. The mechanism by which caveolae or Cav3 association can confine cAMP response resulting from β2AR stimulation is likely to be indirect. We speculate that individual β2AR:Cav3 and β2AR:caveolae are diffusively separated from the general milieu. Moreover, we speculate that relatively high phosphodiesterase activity in the cav domain (included in the Heijman et al. model, see Supplementary Text for more detail), can mute cAMP levels and thereby limit its reach.
Implications of this study extend beyond Cav3 mutations and heart failure. A recent publication showed that a diet high in palmitate disrupted Cav3 in ventricular myocytes [49]. High dietary palmitate may lead to cellular behavior similar to Cav3DN case, where local cAMP responses to sympathetic β2AR stimulation were lost. This may have consequences for individuals’ cardiovascular physiology. Finally, although the rat cardiomyocyte is the model presented in this paper the authors are aware of the limitations of this species with regard to the necessity of culture. The t-tubular network is observed to degrade during the course of the culturing process. Equally, adenovirus appears to be the only method of gene delivery robust enough for use with this cell type. The physical and molecular changes caused by adenoviral entry and t-tubular disruption as well as the effects of unloading in culture may reduce the relevance of data from studies using this cell to failing cells in vivo.
Future studies should analyze β2AR localization and cAMP signaling by SICM/FRET in cardiomyocyte-specific Cav3 transgenic and knockout mice where the differences in membrane morphology might be even more pronounced due to constitutive expression or deletion of Cav3. Transgenic mice are in existence, which constitutively express FRET sensors similar to the one employed by this study [34]. Crossing these mice with Cav3 transgenics may provide an avenue to studying the role of Cav3 in modulating β2AR function in freshly isolated cells. Finally, measurements in failing human myocytes with Cav3 re-expression could be performed when technically possible, as this would also require culturing. These studies might pave the way towards a new gene therapy option for HF patients.
5. CONCLUSIONS
In healthy adult rat cardiomyocytes, Cav3 regulates β2AR-cAMP signaling, playing a crucial role for the compartmentation of cAMP signals to the T-tubular compartment. Disruption of Cav3 function with a dominant-negative mutant leads to far-reaching β2AR-cAMP signals similar to those observed in heart failure. In failing cardiomyocytes, Cav3 overexpression can partially restore the disrupted localization of these receptors.
Supplementary Material
HIGHLIGHTS.
Caveolin-3 (Cav-3) controls cardiomyocyte β2AR signaling via effects on cAMP responses
Cav-3 is necessary for proper compartmentation of β2AR-cAMP responses
Loss of Cav-3 function disrupts β2AR-cAMP’s spatial compartmentation specifically at the T-tubules
Overexpression of Cav-3 in failing myocytes can restore T-tubular β2AR localization and spatial compartmentation
Acknowledgments
We thank Peter O’Gara for cardiomyocyte isolation, Christian Dees for the radioligand binding studies and Karina Zimmermann for Western blot analysis. This work was supported by grants from the Wellcome Trust (WTN090594 to JG and 092852 to MS), the British Heart Foundation (NH/10/3/28574 to JG and SEH, and FS/11/67/28954 to ARL) and the Deutsche Forschungsgemeinschaft (SFB 1002, TP A01 to VON).
Abbreviations
- β-AR
β-adrenergic receptor
- ARVM
adult rat ventricular myocytes
- Cav3
caveolin-3
- Cav3DN
caveolin-3 dominant negative mutant
- FRET
fluorescence resonance energy transfer
- HF
heart failure
- MβCD
methyl-β-cyclodextrin
- SICM
scanning ion conductance microscopy
Footnotes
DISCLOSURES: NONE DECLARED
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Brodde OE, Michel MC. Adrenergic and muscarinic receptors in the human heart. Pharmacol Rev. 1999;51:651–90. [PubMed] [Google Scholar]
- 2.Xiang Y, Kobilka BK. Myocyte adrenoceptor signaling pathways. Science. 2003;300:1530–2. doi: 10.1126/science.1079206. [DOI] [PubMed] [Google Scholar]
- 3.Lohse MJ, Engelhardt S, Eschenhagen T. What is the role of beta-adrenergic signaling in heart failure? Circ Res. 2003;93:896–906. doi: 10.1161/01.RES.0000102042.83024.CA. [DOI] [PubMed] [Google Scholar]
- 4.Schäfer M, Frischkopf K, Taimor G, Piper HM, Schlüter K-D. Hypertrophic effect of selective β1-adrenoceptor stimulation on ventricular cardiomyocytes from adult rat. American Journal of Physiology - Cell Physiology. 2000;279:C495–C503. doi: 10.1152/ajpcell.2000.279.2.C495. [DOI] [PubMed] [Google Scholar]
- 5.Zhu W-Z, Zheng M, Koch WJ, Lefkowitz RJ, Kobilka BK, Xiao R-P. Dual modulation of cell survival and cell death by β2-adrenergic signaling in adult mouse cardiac myocytes. Proceedings of the National Academy of Sciences. 2001;98:1607–12. doi: 10.1073/pnas.98.4.1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Engelhardt S, Hein L, Wiesmann F, Lohse MJ. Progressive hypertrophy and heart failure in beta1-adrenergic receptor transgenic mice. Proc Natl Acad Sci U S A. 1999;96:7059–64. doi: 10.1073/pnas.96.12.7059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dorn GW, 2nd, Tepe NM, Lorenz JN, Koch WJ, Liggett SB. Low- and high-level transgenic expression of beta2-adrenergic receptors differentially affect cardiac hypertrophy and function in Galphaq-overexpressing mice. Proc Natl Acad Sci U S A. 1999;96:6400–5. doi: 10.1073/pnas.96.11.6400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xiao R-P. {beta}-Adrenergic Signaling in the Heart: Dual Coupling of the {beta}2-Adrenergic Receptor to Gs and Gi Proteins. Science Signaling. 2001;2001:re15. doi: 10.1126/stke.2001.104.re15. [DOI] [PubMed] [Google Scholar]
- 9.Xiao RP, Ji X, Lakatta EG. Functional coupling of the beta 2-adrenoceptor to a pertussis toxin-sensitive G protein in cardiac myocytes. Molecular Pharmacology. 1995;47:322–9. [PubMed] [Google Scholar]
- 10.Nikolaev VO, Bünemann M, Schmitteckert E, Lohse MJ, Engelhardt S. Cyclic AMP Imaging in Adult Cardiac Myocytes Reveals Far-Reaching β1-Adrenergic but Locally Confined β2-Adrenergic Receptor–Mediated Signaling. Circulation Research. 2006;99:1084–91. doi: 10.1161/01.RES.0000250046.69918.d5. [DOI] [PubMed] [Google Scholar]
- 11.Head BP, Patel HH, Roth DM, Lai NC, Niesman IR, Farquhar MG, et al. G-protein- coupled Receptor Signaling Components Localize in Both Sarcolemmal and Intracellular Caveolin-3-associated Microdomains in Adult Cardiac Myocytes. Journal of Biological Chemistry. 2005;280:31036–44. doi: 10.1074/jbc.M502540200. [DOI] [PubMed] [Google Scholar]
- 12.Xiang Y, Rybin VO, Steinberg SF, Kobilka B. Caveolar Localization Dictates Physiologic Signaling of β2-Adrenoceptors in Neonatal Cardiac Myocytes. Journal of Biological Chemistry. 2002;277:34280–6. doi: 10.1074/jbc.M201644200. [DOI] [PubMed] [Google Scholar]
- 13.Agarwal SR, MacDougall DA, Tyser R, Pugh SD, Calaghan SC, Harvey RD. Effects of cholesterol depletion on compartmentalized cAMP responses in adult cardiac myocytes. Journal of Molecular and Cellular Cardiology. 2011;50:500–9. doi: 10.1016/j.yjmcc.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ostrom RS, Violin JD, Coleman S, Insel PA. Selective Enhancement of β-Adrenergic Receptor Signaling by Overexpression of Adenylyl Cyclase Type 6: Colocalization of Receptor and Adenylyl Cyclase in Caveolae of Cardiac Myocytes. Molecular Pharmacology. 2000;57:1075–9. [PubMed] [Google Scholar]
- 15.Rybin VO, Xu X, Lisanti MP, Steinberg SF. Differential Targeting of β-Adrenergic Receptor Subtypes and Adenylyl Cyclase to Cardiomyocyte Caveolae. Journal of Biological Chemistry. 2000;275:41447–57. doi: 10.1074/jbc.M006951200. [DOI] [PubMed] [Google Scholar]
- 16.Cohen AW, Hnasko R, Schubert W, Lisanti MP. Role of Caveolae and Caveolins in Health and Disease. Physiological Reviews. 2004;84:1341–79. doi: 10.1152/physrev.00046.2003. [DOI] [PubMed] [Google Scholar]
- 17.Razani B, Woodman SE, Lisanti MP. Caveolae: From Cell Biology to Animal Physiology. Pharmacological Reviews. 2002;54:431–67. doi: 10.1124/pr.54.3.431. [DOI] [PubMed] [Google Scholar]
- 18.Shaul PW, Anderson RGW. Role of plasmalemmal caveolae in signal transduction. American Journal of Physiology - Lung Cellular and Molecular Physiology. 1998;275:L843–L51. doi: 10.1152/ajplung.1998.275.5.L843. [DOI] [PubMed] [Google Scholar]
- 19.Harvey RD, Calaghan SC. Caveolae create local signalling domains through their distinct protein content, lipid profile and morphology. J Mol Cell Cardiol. 2012;52:366–75. doi: 10.1016/j.yjmcc.2011.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gratton JP, Bernatchez P, Sessa WC. Caveolae and caveolins in the cardiovascular system. Circ Res. 2004;94:1408–17. doi: 10.1161/01.RES.0000129178.56294.17. [DOI] [PubMed] [Google Scholar]
- 21.Lisanti MP, Scherer PE, Vidugiriene J, Tang Z, Hermanowski-Vosatka A, Tu YH, et al. Characterization of caveolin-rich membrane domains isolated from an endothelial-rich source: implications for human disease. The Journal of Cell Biology. 1994;126:111–26. doi: 10.1083/jcb.126.1.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Feron O, Balligand JL. Caveolins and the regulation of endothelial nitric oxide synthase in the heart. Cardiovasc Res. 2006;69:788–97. doi: 10.1016/j.cardiores.2005.12.014. [DOI] [PubMed] [Google Scholar]
- 23.Barouch LA, Harrison RW, Skaf MW, Rosas GO, Cappola TP, Kobeissi ZA, et al. Nitric oxide regulates the heart by spatial confinement of nitric oxide synthase isoforms. Nature. 2002;416:337–9. doi: 10.1038/416337a. [DOI] [PubMed] [Google Scholar]
- 24.Parton RG, Way M, Zorzi N, Stang E. Caveolin-3 Associates with Developing T-tubules during Muscle Differentiation. The Journal of Cell Biology. 1997;136:137–54. doi: 10.1083/jcb.136.1.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Galbiati F, Volonte D, Chu JB, Li M, Fine SW, Fu M, et al. Transgenic overexpression of caveolin-3 in skeletal muscle fibers induces a Duchenne-like muscular dystrophy phenotype. Proc Natl Acad Sci U S A. 2000;97:9689–94. doi: 10.1073/pnas.160249097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Repetto S, Bado M, Broda P, Lucania G, Masetti E, Sotgia F, et al. Increased Number of Caveolae and Caveolin-3 Overexpression in Duchenne Muscular Dystrophy. Biochemical and Biophysical Research Communications. 1999;261:547–50. doi: 10.1006/bbrc.1999.1055. [DOI] [PubMed] [Google Scholar]
- 27.Woodman SE, Park DS, Cohen AW, Cheung MWC, Chandra M, Shirani J, et al. Caveolin-3 Knock-out Mice Develop a Progressive Cardiomyopathy and Show Hyperactivation of the p42/44 MAPK Cascade. Journal of Biological Chemistry. 2002;277:38988–97. doi: 10.1074/jbc.M205511200. [DOI] [PubMed] [Google Scholar]
- 28.Galbiati F, Engelman JA, Volonte D, Zhang XL, Minetti C, Li M, et al. Caveolin-3 Null Mice Show a Loss of Caveolae, Changes in the Microdomain Distribution of the Dystrophin-Glycoprotein Complex, and T-tubule Abnormalities. Journal of Biological Chemistry. 2001;276:21425–33. doi: 10.1074/jbc.M100828200. [DOI] [PubMed] [Google Scholar]
- 29.Calaghan S, White E. Caveolae modulate excitation–contraction coupling and β2-adrenergic signalling in adult rat ventricular myocytes. Cardiovascular Research. 2006;69:816–24. doi: 10.1016/j.cardiores.2005.10.006. [DOI] [PubMed] [Google Scholar]
- 30.Balijepalli RC, Foell JD, Hall DD, Hell JW, Kamp TJ. Localization of cardiac L-type Ca2+ channels to a caveolar macromolecular signaling complex is required for β2-adrenergic regulation. Proceedings of the National Academy of Sciences. 2006;103:7500–5. doi: 10.1073/pnas.0503465103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen-Izu Y, Xiao R-P, Izu LT, Cheng H, Kuschel M, Spurgeon H, et al. Gi-Dependent Localization of β2-Adrenergic Receptor Signaling to L-Type Ca2+ Channels. Biophysical Journal. 2000;79:2547–56. doi: 10.1016/S0006-3495(00)76495-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wagner E, Lauterbach MA, Kohl T, Westphal V, Williams GS, Steinbrecher JH, et al. Stimulated emission depletion live-cell super-resolution imaging shows proliferative remodeling of T-tubule membrane structures after myocardial infarction. Circ Res. 2012;111:402–14. doi: 10.1161/CIRCRESAHA.112.274530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lyon AR, MacLeod KT, Zhang Y, Garcia E, Kanda GK, Lab MJ, et al. Loss of T-tubules and other changes to surface topography in ventricular myocytes from failing human and rat heart. Proc Natl Acad Sci U S A. 2009;106:6854–9. doi: 10.1073/pnas.0809777106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nikolaev VO, Moshkov A, Lyon AR, Miragoli M, Novak P, Paur H, et al. Beta2-adrenergic receptor redistribution in heart failure changes cAMP compartmentation. Science. 2010;327:1653–7. doi: 10.1126/science.1185988. [DOI] [PubMed] [Google Scholar]
- 35.Koga A, Oka N, Kikuchi T, Miyazaki H, Kato S, Imaizumi T. Adenovirus-Mediated Overexpression of Caveolin-3 Inhibits Rat Cardiomyocyte Hypertrophy. Hypertension. 2003;42:213–9. doi: 10.1161/01.HYP.0000082926.08268.5D. [DOI] [PubMed] [Google Scholar]
- 36.Hannawacker A, Krasel C, Lohse MJ. Mutation of Asn293 to Asp in Transmembrane Helix VI Abolishes Agonist-Induced but Not Constitutive Activity of the β2-Adrenergic Receptor. Molecular Pharmacology. 2002;62:1431–7. doi: 10.1124/mol.62.6.1431. [DOI] [PubMed] [Google Scholar]
- 37.Nikolaev VO, Gambaryan S, Engelhardt S, Walter U, Lohse MJ. Real-time Monitoring of the PDE2 Activity of Live Cells. Journal of Biological Chemistry. 2005;280:1716–9. doi: 10.1074/jbc.C400505200. [DOI] [PubMed] [Google Scholar]
- 38.Wong J, Baddeley D, Bushong EA, Yu Z, Ellisman MH, Hoshijima M, et al. Nanoscale Distribution of Ryanodine Receptors and Caveolin-3 in Mouse Ventricular Myocytes: Dilation of T-Tubules near Junctions. Biophys J. 2013;104:L22–4. doi: 10.1016/j.bpj.2013.02.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Heijman J, Volders PG, Westra RL, Rudy Y. Local control of beta-adrenergic stimulation: Effects on ventricular myocyte electrophysiology and Ca(2+)-transient. J Mol Cell Cardiol. 2011;50:863–71. doi: 10.1016/j.yjmcc.2011.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Galbiati F, Volonté D, Minetti C, Chu JB, Lisanti MP. Phenotypic Behavior of Caveolin-3 Mutations That Cause Autosomal Dominant Limb Girdle Muscular Dystrophy (LGMD-1C) Journal of Biological Chemistry. 1999;274:25632–41. doi: 10.1074/jbc.274.36.25632. [DOI] [PubMed] [Google Scholar]
- 41.Agarwal SR, MacDougall DA, Tyser R, Pugh SD, Calaghan SC, Harvey RD. Effects of cholesterol depletion on compartmentalized cAMP responses in adult cardiac myocytes. J Mol Cell Cardiol. 2011;50:500–9. doi: 10.1016/j.yjmcc.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Macdougall DA, Agarwal SR, Stopford EA, Chu H, Collins JA, Longster AL, et al. Caveolae compartmentalise beta2-adrenoceptor signals by curtailing cAMP production and maintaining phosphatase activity in the sarcoplasmic reticulum of the adult ventricular myocyte. J Mol Cell Cardiol. 2012;52:388–400. doi: 10.1016/j.yjmcc.2011.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Communal C, Singh K, Sawyer DB, Colucci WS. Opposing effects of beta(1)- and beta(2)-adrenergic receptors on cardiac myocyte apoptosis : role of a pertussis toxin-sensitive G protein. Circulation. 1999;100:2210–2. doi: 10.1161/01.cir.100.22.2210. [DOI] [PubMed] [Google Scholar]
- 44.Tsutsumi YM, Horikawa YT, Jennings MM, Kidd MW, Niesman IR, Yokoyama U, et al. Cardiac-Specific Overexpression of Caveolin-3 Induces Endogenous Cardiac Protection by Mimicking Ischemic Preconditioning. Circulation. 2008;118:1979–88. doi: 10.1161/CIRCULATIONAHA.108.788331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ratajczak P, Damy T, Heymes C, Oliviero P, Marotte F, Robidel E, et al. Caveolin-1 and -3 dissociations from caveolae to cytosol in the heart during aging and after myocardial infarction in rat. Cardiovasc Res. 2003;57:358–69. doi: 10.1016/s0008-6363(02)00660-0. [DOI] [PubMed] [Google Scholar]
- 46.Oka N, Asai K, Kudej RK, Edwards JG, Toya Y, Schwencke C, et al. Downregulation of caveolin by chronic beta-adrenergic receptor stimulation in mice. Am J Physiol. 1997;273:C1957–62. doi: 10.1152/ajpcell.1997.273.6.C1957. [DOI] [PubMed] [Google Scholar]
- 47.Piech A, Massart PE, Dessy C, Feron O, Havaux X, Morel N, et al. Decreased expression of myocardial eNOS and caveolin in dogs with hypertrophic cardiomyopathy. Am J Physiol Heart Circ Physiol. 2002;282:H219–31. doi: 10.1152/ajpheart.2002.282.1.H219. [DOI] [PubMed] [Google Scholar]
- 48.Fujita T, Toya Y, Iwatsubo K, Onda T, Kimura K, Umemura S, et al. Accumulation of molecules involved in alpha1-adrenergic signal within caveolae: caveolin expression and the development of cardiac hypertrophy. Cardiovasc Res. 2001;51:709–16. doi: 10.1016/s0008-6363(01)00348-0. [DOI] [PubMed] [Google Scholar]
- 49.Knowles CJ, Cebova M, Pinz IM. Palmitate diet-induced loss of cardiac caveolin-3: a novel mechanism for lipid-induced contractile dysfunction. PLoS One. 2013;8:e61369. doi: 10.1371/journal.pone.0061369. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.