

Abstract
The intestinal epithelium is a single-cell layer that constitutes the largest and most important barrier against the external environment. It acts as a selectively permeable barrier permitting the absorption of nutrients, electrolytes and water, while maintaining an effective defense against intraluminal toxins, antigens and enteric flora. The epithelium maintains its selective barrier function through the formation of complex protein-protein networks that mechanically link adjacent cells and seal the intercellular space. The protein networks connecting epithelial cells form three adhesive complexes: desmosomes, adherens junctions and tight junctions. These complexes consist of transmembrane proteins that interact extracellularly with adjacent cells and intracellularly with adaptor proteins that link to the cytoskeleton. Over the past decade, there has been increasing recognition of an association between disrupted intestinal barrier function and the development of autoimmune and inflammatory diseases. In this review, we summarize the evolving understanding of the molecular composition and regulation of intestinal barrier function. We discuss the interactions between innate and adaptive immunity and intestinal epithelial barrier function, as well as the impact of exogenous factors on intestinal barrier function. Finally, we summarize clinical and experimental evidence demonstrating intestinal epithelial barrier dysfunction as a major factor contributing to the predisposition to inflammatory diseases including food allergy, inflammatory bowel diseases and celiac disease.
Introduction
The intestinal epithelium is a single layer of cells lining the gut lumen and has two critical functions. Firstly, it acts as a barrier to prevent the passage of harmful intraluminal entities including foreign antigens, microorganisms and their toxins 1, 2. Its second function is to act as a selective filter allowing the translocation of essential dietary nutrients, electrolytes and water from the intestinal lumen into the circulation 1, 3–5. The intestinal epithelium mediates selective permeability via two major routes: transepithelial/transcellular and paracellular pathways 6 (Figure 1). Transcellular permeability is generally associated with solute transport through the epithelial cells and predominantly regulated by selective transporters for amino acids, electrolytes, short chain fatty acids and sugars 3–5. Paracellular permeability is associated with transport in the space between epithelial cells, and is regulated by intercellular complexes localized at the apical-lateral membrane junction and along the lateral membrane 7. Contact between intestinal epithelial cells includes three components that can be identified at the ultrastructural level: desmosomes, adherens junctions (AJs) and tight junctions (TJs) (Figure 2) 8. The adhesive junctional complexes consist of transmembrane proteins that link adjacent cells to the actin cytoskeleton via cytoplasmic scaffolding proteins. The AJs and desmosomes are thought to be more important in the mechanical linkage of adjacent cells 9–11. The TJs, on the other hand, are the apical-most junctional complex and responsible for sealing the intercellular space and regulating selective paracellular ionic solute transport 6, 12–14. The AJ and TJ complexes are also important in the regulation of cellular proliferation, polarization and differentiation 6, 11–16.
Figure 1. Pathways of epithelial permeability.

Transcellular permeability is associated with solute or water movement through intestinal epithelial cells. Paracellular permeability is associated with movement in the intercellular space between epithelial cells and is regulated by tight junctions localized at the junction of the apical-lateral membranes.
Figure 2. Overview of intestinal epithelial junctional complexes.
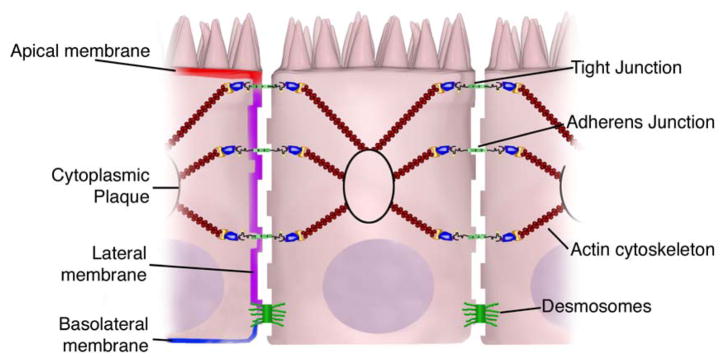
The intestinal epithelium consists of a single layer of polarized epithelial cells. Adjacent cells are connected by 3 main junctional complexes: desmosomes, adherens junctions and tight junctions. Desmosomes are localized dense plaques that are connected to keratin filaments. Adherens and tight junctions both consist of transcellular proteins connected intracellularly via adaptor proteins to the actin cytoskeleton. The collection of proteins in the junctional complexes form “cytoplasmic plaques”.
Structural Components of Junctional Complexes
Adherens junctions (AJs)
AJs (also known as zonula adherens) are protein complexes on the lateral membrane that occur at points of cell-cell contact (Figure 2). They are formed by interactions between transmembrane proteins, intracellular adaptor proteins and the cytoskeleton. The major AJs are formed by cadherin-catenin interactions. Epithelial (E)-cadherins (calcium-dependent adhesion molecules) are Type-I single transmembrane spanning glycoproteins that possess an intracellular C-terminus and extracellular N-terminus. The extracellular domain forms homotypical interactions with cadherins of neighboring cells to promote cell-cell adhesion. The intracellular domain contains a catenin-binding domain that interacts with members of the armadillo repeat superfamily, β-, γ- and p120-catenin 11, 17–21. The catenins link the AJ to the cytoskeletal network via direct binding to the C-terminal domain of F-actin or indirectly through interactions with other adaptor proteins such as afadin 22–26. Cadherin-catenin complexes are important not only for linking adjacent cells, but also for maintaining cell polarity, regulating epithelial migration and proliferation and the formation of other adhesive complexes such as desmosomes 21, 27, 19. In support of this, downregulation of E-cadherin in the intestinal epithelium weakens cell-cell adhesion and has been linked with perturbed intestinal epithelial proliferation and migration 28, 29.
Nectin-afadin interactions form another important AJ complex 21, 30, 31. Nectins (nectin-1-4) are immunoglobulin-like proteins that undergo homophilic and heterophilic interactions with nectins on adjacent cells 32. Nectins can interact with the cytoskeleton via afadin, an F-actin binding protein, or alternatively via interactions with other F- or α-actin binding proteins including ponsin/SH3P12, vinculin and afadin dil domain-interacting protein 33–37.
Tight junctions (TJs)
TJs are the apical-most adhesive junctional complexes in mammalian epithelial cells that form a continuous belt-like ring around epithelial cells at the border between the apical and lateral membrane regions (Figure 2) 8. TJs are dynamic, multi-protein complexes that function as a selective/semipermeable paracellular barrier, which facilitates the passage of ions and solutes through the intercellular space, while preventing the translocation of luminal antigens, microorganisms and their toxins. The evolution of TJ biology emerged in the 1960’s with the development of electron microscopy. Analysis of epithelial cells revealed a series of apparent fusions, where the space between adjacent epithelial cells was eliminated 6, 38, 39. These so-called “kissing points” are morphologically different from AJs and desmosomes, where adjacent cell membranes remain 15–20nm apart 6. Since these initial observations, TJs have been found to consist of four unique families of transmembrane proteins: occludin, claudins, junctional adhesion molecules (JAMs) and tricellulin.
The extracellular domains of transmembrane TJ proteins in adjacent cells anastomose to form the TJ seal. These interactions include those involving proteins in the same membrane (in cis) and those involving proteins in adjacent cells (in trans). In addition, TJ proteins can form either homophilic (with the same protein) or heterophilic (between non-identical TJ proteins) interactions. Similar to the AJs, the intracellular domains interact with various scaffolding proteins, adaptor proteins and signaling complexes to regulate cytoskeletal attachment, cell polarity, cell signaling and vesicle trafficking (Figure 3). The intracellular regions of AJs possess PDZ-binding domains, which recruit and interact with PDZ domain containing proteins. The PDZ domain (Post synaptic density-95/Drosophila disc large/Zonula occludens-1 protein) is a common structural domain of 80–90 amino acids that functions to anchor transmembrane proteins to the cytoskeleton. The intracellular domains can also interact with non-PDZ-binding domain containing proteins such as cingulin, which can interact with junctional membrane proteins, the actin cytoskeleton and signaling proteins 40. The complex network of network of intracellular protein interactions is also known as the “cytoplasmic plaque”.
Figure 3. Tight Junctions.

TJs are localized to the apical-lateral membrane junction. They consist of integral transmembrane proteins (occludin, claudins and junctional adhesion molecules (JAMs)) that interact in the paracellular space with proteins on adjacent cells. Interactions can be homophilic (eg claudin-1/claudin-1) or heterophilic (claudin-1/claudin-3). The intracellular domains of transmembrane proteins interact with PDZ-domain-containing adaptor proteins that mechanically link the TJ complex to the actin cytoskeleton. TJ proteins are regulated by phosphorylation by kinases, phosphatases and other signaling molecules.
Tight junction formation in the gastrointestinal tract
The intestinal epithelium forms the largest and most important barrier between our internal and external environments. The barrier is maintained by the expression of AJs and TJs, including cadherins, claudins, occludin and JAM proteins, which seal together adjacent cells and provide cytoskeletal anchorage (Figure 3) 41. Expression of junctional proteins in the intestine is highly regulated and dependent on the intestinal compartment (small or large intestine), villus/crypt localization and cell membrane specificity (apical, lateral or basolateral). The complex pattern of TJ expression in the intestine is related to the specific functions of a particular intestinal region and localization. Expression of AJ and TJ proteins are also regulated by phosphorylation (Table 1). Phosphorylation can either promote TJ formation and barrier function, or alternatively promote TJ protein redistribution and complex destabilization 42, 43.
Table 1.
Transgenic or knockout mice and effects on intestinal barrier function.
| Protein | Transgenic Or knockout | Function | Phenotype | reference |
|---|---|---|---|---|
| Occludin | Gene deletion | TJ protein | No change in TJs or permeability | 56 and 57 |
| Claudin-1 | Gene deletion | TJ protein | Die within 1 day of birth | 72 |
| Claudin-6 | Transgenic | TJ protein | Epidermis disrupted TJ formation and increased epithelial permeability | 73 |
| JAM-A | Gene deletion | TJ protein | Increased intestinal permeability Elevated claudin-10 and 15 expression Increased susceptibilty to DSS colitis |
94 |
| IL-9 | Intestinal transgenic | cytokine | Elevated intestinal mast cell Increased intestinal permeability Increased susceptibility to oral antigen sensitization and anaphylaxis |
206 |
| IL-10 | Gene deletion | cytokine | Increased permeability; Spontaneously develop chronic colitis similar to CD | 107, 108 and 111 |
| STAT-6 | Gene deletion | Signaling molecule | Protected against IL-4- and IL-13-induced intestinal epithelial barrier dysfunction | 106 |
| mMCP-1 | Gene deletion | Mast cell protease | Protected against T. spiralis infestation-induced intestinal epithelial barrier dysfunction | 126 |
| MLCK | Transgenic | Signaling molecule | Increased intestinal permeability Increased onset and severity of immune-related colitis. |
219 |
i. Occludin
The first TJ-specific integral membrane protein identified was occludin 44. Occludin is expressed predominately at TJs in epithelial and endothelial cells, but also by astrocytes, neurons and dendritic cells 44,45–47.Occludin (60–82 kDa) is a tetraspanning integral membrane protein with two extracellular loops, a short cytoplasmic N-terminus and a long cytoplasmic C-terminus 11, 13. Functional analysis indicates that the extracellular loops and transmembrane domains of occludin regulate selective paracellular permeability. Intracellularly, the C-terminus interacts with the PDZ-domain containing protein ZO-1, which is required to link occludin to the actin cytoskeleton (Figure 3) 48, 49.
Several occludin isoforms have been identified and are thought to be a result of alternative mRNA splicing 50, 51. Notably, several splice variants demonstrate altered subcellular distribution and interaction with other TJ molecules 50, 51. Analysis of the splice variants revealed that the cytoplasmic C-terminal domain is essential for the intracellular trafficking of occludin to the lateral cell membrane, and that the fourth transmembrane domain is critical for targeting occludin to the TJ and for ZO-1 interactions 51.
The function of occludin is not fully delineated; however, in vitro and in vivo data suggest a role for occludin in the regulation of paracellular permeability 52, 53. Notably, the major allergen of the house dust mite, Der p 1, has been found to proteolyticly cleave occludin leading to the disruption of the TJ complex and increased paracellular permeability 54. Furthermore hydrocortisone treatment of bovine retinal endothelial cells increased occludin expression two-fold and enhanced monolayer barrier properties 55. Although occludin is an important constituent of TJs, TJ formation and paracellular permeability barrier function are not dependent on occludin. Experimental analyses of occludin−/− mice demonstrated equivalent numbers and organization of TJs and similar paracellular ion conductance as wild-type mice 56. Furthermore, epithelial transport and barrier function were normal in occludin−/− mice 57. In addition to regulating paracellular permeability, there is evidence suggesting occludin is involved in cellular adhesion 58. Expression of occludin in occludin−/− rat fibroblasts conferred cell–cell adhesion that was abrogated by synthetic peptides corresponding to the first extracellular loop of occludin, underscoring the importance of this region of occludin in cell adhesion 59.
In vitro analysis suggests that occludin localization to the TJ complex is regulated by phosphorylation. Several potential phosphorylation sites at tyrosine, serine, and threonine residues of occludin have been identified and regulation of occludin phosphorylation is proposed to occur by kinases, including the non-receptor tyrosine kinase c-Yes and protein kinase C (PKC), and phosphatases including the serine/threonine protein phosphatase 2A 60, 61 (Figure 3). PKCη, a novel protein kinase predominantly expressed in the intestinal epithelium, has been shown to directly phosphorylate occludin at threonine residues (T403 and T404). Blockade of PKCη-mediated occludin phosphorylation disrupted junctional distribution of occludin and ZO-1 and compromised epithelial barrier function62. These data suggest that occludin phosphorylation regulates occludin-ZO-1 interactions and the maintenance of intact TJ complexes and paracellular barrier function.
ii. Claudins
Claudins are 20–27kDa integral membrane proteins with four hydrophobic transmembrane domains, two extracellular loops and N- and C-terminal cytoplasmic domains 7, 63–65. The extracellular loops are critical for homophilic and/or heterophilic TJ protein-protein interactions and the formation of ion-selective channels 7. The intracellular C-terminal domain is involved in anchoring claudin to the cytoskeleton via interactions with PDZ-binding domain proteins, including ZO-1, -2 and -3 66–68 (Figure 3). Currently, 24 distinct claudin family gene members have been identified in humans with a number of orthologues expressed in other species 6, 69. They exhibit distinct tissue-, cell- and developmental stage-specific expression patterns 88–93
Claudin-claudin interactions between adjacent cells can be either homophilic or heterophilic 70, 71. Homophilic interactions have been demonstrated for claudins 1, 2, 3, 5, 6, 9, 11, 14 and 19. On the other hand, heterophilic interactions are more restricted and primarily have been observed with claudin-3, which can interact with claudins-1, -2 and -5 70. Notably, there is specificity in heterophilic trans-interactions. For example, transfection of fibroblasts with claudins-1, -2 and -3 led to claudin-3 interactions with both claudin-1 and -2; however no interactions between claudin-1 and -2 were observed 71. These selective interactions are thought to explain the diversity in TJ formations and provide a molecular basis for tissue-specific heterogeneity of barrier function 65.
Recent studies, with claudin-deficient mice also provide corroborative data supporting a role for claudins in the regulation of barrier function. Claudin-1−/− mice die within one day of birth due to significant transepidermal water loss 72. In addition, transgenic overexpression of claudin-6 in the epidermis disrupted tight junction formation and increased epithelial permeability 73. Notably, experimental data suggests that claudins can have differential effects on paracellular permeability. For example, introduction of claudin-2 into MDCK I cells that express claudin-1 and -4 induces a decrease in transepithelial resistance (TER); whereas transfection of claudin-3 had no effect suggesting that claudin-2 markedly decreased claudin-1/claudin-4 based TJ strand tightness74. In support of these data recent experimental evidence suggests that claudins can form size- and charge-specific paracellular channels. Transfection of claudin-8 into MDCK II cells which lacks endogenous claudin-8 significantly reduced paracellular movement of mono- and divalent cations without affecting anion and uncharged solute movement 75. Experimental analyses suggest that the first extracellular loop of claudins play an important role in determining charge selectivity. Interchanging of the first or both extracellular domains of claudin-4 on claudin-2 profoundly decreased the ion conductance of Na+ relative to Cl− 76. Furthermore, substitution of a negatively-charged lysine to a positively charged aspartic acid (K65D) within the loop of claudin-15 caused an increase in Na+ permeability, while mutation in the same region of three positively charged amino acids to negatively charged aspartic acid, arginine and aspartic acid (E46K, D55R and E64K) switched the ion selectivity of claudin-15 from Na+ to Cl− channel 77. Pore density and size may also influence paracellular movement of charged and non charged solutes 78.
Claudins also play a role in epithelial cell invasion and motility. Overexpression of claudins-3 and -4 in human ovarian epithelial cells, which lack endogenous expression of these proteins, was associated with increased epithelial cell survival and enhanced invasion and motility 79. Consistent with this observation, siRNA-mediated knockdown of claudins-3 and -4 in ovarian cancer cell lines reduced invasion 79. The effects of claudin-3 appear to be linked to altered matrix metalloprotease-2 activity, which suggests that claudin-induced invasion may be mediated by metalloprotease proteins.
As with occludin, claudin localization to the TJ complex and its function are regulated by post-translational phosphorylation and via interactions with PDZ-binding domains. The intracellular C-terminal domain of claudin possesses multiple regulatory sites including potential serine and theronine phosphorylation sites and PDZ-binding domains 7. Phosphorylation of claudins-3 and -4 in ovarian cancer cells is linked to the regulation of paracellular permeability 80, 81. For example, patients with pseudohypoaldosteronism type II (PHA II; or chloride shunt syndrome) present with hyperkalemic metabolic acidosis, hypertension and dysregulated paracellular ion transport 82. The molecular basis is linked to a loss-of-function mutation in the serine-threonine kinases, WNK1 and WNK4, which regulate epithelial chloride cotransporters. This leads to an increase in the phosphorylation of claudins-1-4 and an increase in paracellular permeability 82. A number of signaling pathways have been implicated in the phosphorylation of claudins including PKC, Rho GTPases, mitogen-activated protein kinases (MAPKs) and phosphatases 83. For example, MAPK phosphorylation of claudin-1 is required for claudin-1-mediated barrier function 84. Furthermore, claudins-1, -2, -7, -8, -16 and -17 possess putative PKC phosphorylation sites 83
All claudins, except claudin-12, end in the dipeptide sequence YV, which has been shown to interact with PDZ-binding domain proteins include ZO-1, -2 and -3, multi-PDZ domain protein 1 and PALS1-associated TJ protein 63, 65, 67,85 (Figure 3). Many of these scaffolding proteins contain multiple PDZ domains, which facilitates the formation of dense localized protein complexes, also known as “cytoplasmic plaques”. Furthermore, the scaffolding proteins can interact with signaling molecules, including heterodimeric GTP binding proteins (Rab13 and Gα12), transcriptional factors and RNA-processing factors, to link TJ complexes to the actin-cytoskeleton and regulate aspects of epithelial polarization, differentiation and barrier function (For review see 6, 21, 38, 86, 87).
iii. Junctional adhesion molecules (JAMs)
JAMs are integral membrane proteins that belong to the immunoglobulin superfamily and have two immunoglobulin folds (VH- and C2-type) in the extracellular domain 88, 89. JAMs are expressed by multiple cell types, including epithelial, endothelial and immune cells 90, 91. They are subdivided based on the expression of Type I or II PDZ-binding motifs in the intracellular C-terminus, which suggests that the two types interact with unique scaffolding and cytoplasmic proteins 88. JAM-A, -B and -C (or JAM1-3) have Type II binding motifs, while the atypical JAMs, including JAM-4, coxsackie and adenovirus receptor (CAR) and endothelial selective adhesion molecule contain Type I PDZ-binding domains 21, 88. Similar to other TJ proteins, these JAM-PDZ interactions provide anchorage to the actin cytoskeleton (Figure 3).
The extracellular region of JAMs bind to multiple ligands through homophilic and heterophilic interactions, which are proposed to regulate the cellular functions and paracellular permeability of JAMs 88, 89. Homophilic JAM-A or -B interactions regulate the formation of functional TJs and cell-cell border formation 92, 93, while heterophilic JAM interactions play a role in leukocyte-endothelial cell adhesion 89.
Recent studies demonstrate the importance of JAM-A in the formation and assembly of TJs in intestinal epithelial cells. siRNA downregulation of JAM-A in SK-C015 epithelial cells induced an increase in permeability 94. Consistent with this, JAM-A−/− mice had increased mucosal permeability as indicated by enhanced dextran flux and decreased TER 94. However, these mice also had an increase in claudin-10 and -15 expression, which are thought to form selective pores in the TJ complex, enhancing paracellular permeability 77, 95. Interestingly, JAM-A−/− mice have increased susceptibility to chemical-induced colitis. Dextran sodium sulfate administration to JAM-A−/− mice induced more severe colonic injury as compared to WT control animals 94. These studies suggest altered intestinal permeability as a susceptibility factor to intestinal disease.
Dysregulation of TJ formation and intestinal barrier function
i. Cytokine-mediated
In vitro and in vivo animal studies have demonstrated that intestinal permeability is regulated by multiple factors including exogenous factors, epithelial apoptosis, cytokines and immune cells (Figure 4). Immune-induced intestinal barrier dysfunction is thought to be critical in the predisposition to and exacerbation of numerous autoimmune and inflammatory conditions, including IBD, food allergy, celiac disease and diabetes 96. For example, interferon-γ (IFNγ) and tumor necrosis factor-α (TNFα), which are central mediators of intestinal inflammatory diseases including IBD, induce intestinal epithelial barrier function 97–99. Incubation of intestinal epithelial cell monolayers (Caco2 and T84) with IFNγ and TNFα promoted the reorganization of several TJ proteins: ZO-1, JAM-A, occludin, claudins-1 and -4 and decreased epithelial barrier function 100. The mechanism of action of these cytokines appears to be primarily mediated via myosin light chain kinase (MLCK)-mediated phosphorylation of myosin light chain (MLC), which promotes TJ disruption 100. In support of this, inhibition of TNFα- and IFNγ-induced MLC phosphorylation restored barrier function 100. . Alternatively, TNFα and IFNγ can disrupt TJs and increase intestinal permeability via dysregulation of claudin and occludin expression 101.
Figure 4. Immune regulation of intestinal barrier function.

T-cell-derived IFNγ and TNFα inhibit MLCK-mediated phosphorylation of myosin light chain leading to TJ junction disruption and intestinal barrier dysfunction. IFNγ can also promote the redistribution of TJ proteins, JAM-A, occludin, claudin-1 and claudin-4 from the apical TJ border by a micropinocytosis process. TNFα and IFNγ may alternatively disrupt TJ stability and increase intestinal permeability via dysregulation of occludin expression. IL-4 and IL-13-induced increase in intestinal permeability is mediated via induction of epithelial apoptosis and expression of the pore-forming tight junction protein claudin-2. IL-4 and not IL-13 regulates ion conductance via downregulation of epithelial CFTR Cl− channel expression. Intraepithelial (iIEL)γδ+ lymphocytes (TCR-Vγ7+) iIEL cells are important in serine phosphorylation of occludin and TJ stabilization. Mast cell mediators including cytokine TNFα, mast cell protease 1 (mcpt-1) and lipid mediators including histamine, PAF and prostaglandins promote increased Cl− conductance and increase intestinal permeability. Mcpt1 degrades the TJ protein occludin altering barrier function Eosinophil derived MBP down regulates tight junction protein occludin-1 expression in colonic epithelial cells.
Experimental and clinical data supports a role for Th2 cytokines in the regulation of intestinal barrier function (Figure 4). Stimulation of colonic epithelial cells (T84 and HT-29/B6) with IL-4 or -13 induced an increase in intestinal permeability 102–104. Notably, altered barrier function was associated with the induction of epithelial apoptosis and expression of the pore-forming TJ claudin-2. In vitro data suggests that the effects of IL-4 and -13 on barrier function is primarily mediated by phosphoinositide 3-kinases 103, 105. Blockade of phosphoinositide 3-kinase, but not STAT-6 activation blocked IL-4/IL-13-induced barrier dysfunction 103. However, studies in STAT-6−/− mice identified a role for STAT-6 signalling in IL-4 and -13-mediated intestinal epithelial barrier dysfunction 106. Moreover, IL-4- and IL-13-induced altered permeability, glucose absorption and chloride secretion was attenuated in STAT-6 deficient mice compared to WT mice 106.
The anti-inflammatory cytokine IL-10 has also been shown to regulate intestinal barrier function 107, 108. Stimulation of ileal segments from Sprague-Dawley rats with IL-10 enhanced intestinal electroneutral sodium and chloride absorption and inhibited stimulated chloride secretion 109. In addition, treatment of T84 epithelial cell monolayers with IL-10 blocked IFNγ-induced epithelial permeability 110. These results suggest that IL-10 plays a protective role in intestinal barrier function. In support of this, mice deficient in IL-10 have increased small intestinal permeability 107. Notably, IL-10−/− mice spontaneously develop chronic intestinal inflammation, which is manifested by symptoms commonly associated with Crohn’s Disease (CD), including weight loss, mucosal hyperplasia and chronic enterocolitis 111. These data suggest that increased permeability may predispose IL-10−/− mice to intestinal inflammation and colitis. Consistent with this hypothesis, increased permeability in IL-10−/− mice was observed prior to disease onset 107.
Mechanistic studies to delineate IL-10-mediated intestinal permeability have implicated the zonulin pathway and TNFα. Remarkably, inhibition of the zonulin receptor in IL-10−/− mice led to decreased intestinal permeability, reduced colonic TNFα secretion ex vivo and abrogated the spontaneous development of colitis 108. These findings further support a role for increased intestinal permeability in the development of intestinal inflammation and disease and a possible role for zonulin. The zonulin/zonulin receptor pathway is thought to regulate TJ formation via PKC-dependent actin reorganization 112. Whether decreased intestinal barrier function in IL-10−/− mice is primarily due to an inherent defect in the zonulin/zonulin receptor pathway or alternatively, a consequence of increased expression of cytokines such as IFNγ and TNFα remains to be delineated.
ii. Immune cells
T-cells
Anti-CD3-induced CD4+ T-cell activation in mice promotes an increase in transcellular and paracellular intestinal permeability, and the release of proinflammatory cytokines such as IFNγ and TNFα 113, 114 (Figure 4). Furthermore, injection of mice with TNFα provokes a breakdown in intestinal barrier function, diarrhea and PKCα-dependent inhibition of Na+/H+ exchange 115. T-cells regulate transcellular permeability through the downregulation of Na+/K+-ATPase, and disruption of Na+ absorption, Na+-glucose cotransport and inducible Cl− secretion 113, 114. Whereas, dysregulation of the paracellular permeability pathway, is mediated via MLCK-dependent TJ disruption 114.
Gamma/delta-positive intestinal intraepithelial lymphocytes (iIELγδ+), which are closely associated with the basolateral side of intestinal epithelial cells, have also been implicated in intestinal barrier maintenance 116. In response to enteric parasitic infestation, mice deficient in iIELγδ+ T-cells have abnormal claudin-3, occludin and ZO-1 localization, decreased occludin phosphorylation and abnormal epithelial TJ formation 116. Notably, the alterations in intestinal barrier function could be attributed to a single subset of iIELγδ+ lymphocytes: T-cells expressing Vγ7+ encoded T-cell receptors. Reconstitution of mice deficient in iIELγδ+ T-cells with Vγ7+ iIELs restored epithelial barrier function 116.
Mast cells
Mast cells are present in all compartments of the gastrointestinal (GI) tract 117. Upon activation, they release a powerful array of inflammatory mediators including histamine, 5-hydroxytryptamine (5-HT), neutral proteases (tryptases, chymases and carboxypeptidase A), prostaglandins, leukotrienes, platelet activating factor and several cytokines including TNFα, IL-3, -4, -5, -6 and GM-CSF 118–120. Employing models of food allergy or helminthic infestation (Nippostrongylus brasiliensis or Trichinalla spiralis), investigators have demonstrated mast cell involvement in intestinal barrier function 121 (Figure 4). Intraluminal challenge of egg albumin-sensitized rats induced a 15-fold increase in uptake of 51Cr-labeled EDTA compared to rats treated with unrelated protein 122. The antigen-induced decreased barrier function was associated with mast cell degranulation and an increase in the short-circuit current, a measure of net ion transport 122. The importance of mast cells was demonstrated by the absence of changes in barrier function in mast cell-deficient mice sensitized and challenged with egg albumin, which, was restored by bone marrow reconstitution 123, 124. Furthermore, several mast cell mediators have been shown to modulate intestinal epithelial ion transport. Pretreatment of egg albumin-sensitized rats with histamine-H1 or 5-HT2 receptor antagonists significantly reduced oral antigen-induced short-circuit current alterations 123, 125.
Experimental analyses employing models of parasitic infestation have identified a role for mast cell-derived proteases in intestinal barrier function 126. Murine infestation with the enteric nematode, T. spiralis, induced intestinal mastocytosis, occludin degradation and increased intestinal permeability 126. The alterations in barrier function were demonstrated to be mast cell-dependent as depletion of mast cells with a neutralizing anti-c-kit antibody ablated intestinal epithelial barrier dysfunction 126. Similarly, mice deficient in the murine mast cell protease 1 (mMCP-1) were also resistant to T. spiralis infestation-induced intestinal epithelial barrier dysfunction. Mast cell/MCP-1 regulation of intestinal permeability during T. Spiralis infection was linked to occludin degradation 126.
Eosinophils
Increased eosinophils and eosinophil granular proteins, including major basic protein, eosinophil peroxidase and eosinophilic cationic protein are often associated with IBD and altered barrier function 127–130. In vitro coculture of T84 intestinal epithelial cells with eosinophils or eosinophil-derived major basic protein decreased TER and increased permeability. Altered intestinal barrier function was associated with the downregulation of occludin 131.
Exogenous Regulation of Intestinal Barrier Function
Alcohol
Chronic alcohol consumption has been shown to be associated with increased intestinal permeability, inhibition of vitamin and nutrient transport and a reduction in sodium and water absorption 132, 133. Experimental analyses suggest involvement of the byproduct of ethanol metabolism, acetaldehyde and nitric oxide (NO) in alcohol-mediated barrier dysfunction. High levels of acetaldehyde have been detected in the intestine of rats following ethanol administration. Increased levels of acetaldehyde was associated with increased intestinal permeability and endotoxin translocation 134. Furthermore, incubation of Caco2 cells with acetaldehyde increased monolayer permeability and this increase was associated with elevated tyrosine phosphorylation of ZO-1, E-cadherin, and β-catenin 42. Exposure of Caco2 monolayers to ethanol also promotes inducible nitric oxide synthase expression, stimulating increased NO production and increased monolayer permeability 135. NO-induced changes were associated with an increase in unstable, non-polymerized tubulin and extensive damage to the microtubule cytoskeleton.
Experimental studies in rodents have also demonstrated that acute administration of alcohol induces mucosal damage in the upper small intestine including villus ulceration, submucosal blebbing and hemorrhagic erosions and intestinal barrier dysfunction 133, 136, 137. It is postulated that alcohol-induced intestinal permeability facilitates enhanced translocation of endotoxin to distant organs leading to inflammation and tissue damage 136, 138, 139. Intragastric administration of endotoxin in the presence of alcohol to rodents lead to significantly higher plasma endotoxin levels than animals fed endotoxin alone 139, 136. Similar lesions have been found in healthy volunteers and active alcoholics following acute alcohol consumption 139, 140 and plasma endotoxin levels in alcoholics were found to be 5-fold greater than healthy controls 141. While not fully understood, evidence suggests the mechanism underlying alcohol-induced barrier dysfunction is related to the influx of inflammatory cells and release of various mediators, including cytokines, reactive oxygen species, leukotrienes and histamine 142, 143.
Non-steroidal anti-inflammatory drugs (NSAIDs)
NSAID use is associated with a high incidence of GI side effects, and there is substantial evidence indicating that chronic use can alter intestinal barrier function and cause significant GI damage, including ulcers, perforation, hemorrhage and an exacerbation of IBD 144–151. Both acute and chronic ingestion of NSAIDs by healthy volunteers and patients promotes altered intestinal barrier dysfunction and hypermotility 146, 152, 153. In vitro studies with MKN28, a gastric epithelial cell line have demonstrated that aspirin-induced increase in permeability was accompanied by a significant decrease in the expression of claudin-7, but not claudins-3, -4, ZO-1 or occludin 154
NSAID-induced GI injury was initially found to be a consequence of cyclooxygenase inhibition and decreased prostaglandin synthesis; however, it has become evident that intestinal damage is a multi-stage process 145, 152,155. Experimental analyses have identified a contribution from neutrophils, microcirculatory disturbances, oxygen free radicals and bile acids in NSAID-induced GI damage 145, 156, 157. NSAIDs increase intestinal nitric oxide synthase expression and activity, leading to increased levels of NO, promoting increased intestinal permeability 158. NSAIDs can also uncouple mitochondrial oxidative phosphorylation, which impairs the mitochondrial energy production necessary for TJ complex integrity leading to increased intestinal inflammation and permeability 159. Finally, a recent study demonstrated that aspirin induced an increase in gastric epithelial cell permeability that was mediated by activation of p38 MAPK and a decrease in claudin-7, and treatment with a p38 MAPK inhibitor attenuated this response 154.
Pathogens
The intestine is home, both permanently and transiently, to an extraordinarily complex microflora that provides an abundant source of potentially pathogenic organisms, toxins and antigens. The dynamic and complex interactions between enteric pathogens and the intestinal epithelium often leads to disturbances in the intestinal barrier, altered fluid and electrolyte transport and the induction of an inflammatory response 160. Enteric pathogens can disrupt the intestinal barrier either directly, by binding to cell surface molecules and inducing changes in TJ protein expression. Alternatively, pathogens generate toxins and proteases, which can promote cell damage and apoptosis, alter epithelial ion transport and disrupt TJs and the cytoskeleton. Herein, we will provide examples and brief descriptions of several mechanisms by which pathogens disrupt barrier function.
Vibrio cholera
V cholera is a major enteric pathogen that alters intestinal barrier function through the disruption of TJs, dysregulation of intestinal ion and fluid transport and the initiation of inflammatory cascades. A major toxin produced by V cholera is the cytotoxin, hemagglutinin protease (HA/P), a zinc-binding metalloprotease that degrades TJ proteins and decreases barrier function 161, 162. Studies of mutant toxin-attenuated strains of V cholera have identified HA/P as the principle toxin responsible for alterations of TJs and decreased TER in cultured MDCK and T84 cells 161, 162. In vitro studies demonstrate that HA/P cleaves the extracellular domain of occludin. This disrupts intracellular occludin-ZO-1 interactions and destabilizes the TJ complex and cytoskeletal anchorage, resulting in increased paracellular permeability 163.
Another toxin elaborated by V cholera is zonula occludens toxin (Zot), an enterotoxin that reversibly increases intestinal epithelial permeability, disrupts the actin cytoskeleton and induces fragmentation of ZO-1 and occludin 164, 165, 166. Zot binds to the zonulin receptor on the apical side of intestinal epithelial cells and activates phospholipase C leading to PKCα-dependent polymerization of the actin cytoskeleton 165,167. Actin polymerization is thought to promote cytoskeletal reorganization and the destabilization of TJ complexes. Consistent with this hypothesis, pretreating intestinal epithelial monolayers with PKCα inhibitors prevented Zot-induced changes in actin polymerization and permeability 165. A human homologue for Zot, zonulin, has been identified and found to bind to the same receptor and regulate intestinal permeability 168. Zonulin is believed to regulate TJ function and its dysregulation has been has been implicated in several inflammatory diseases associated with intestinal barrier dysfunction including IBD, Type I Diabetes and celiac disease (see clinical review).
Enteropathogenic E. coli (EPEC)
EPEC is a diarrhea-causing bacteria that disrupts TJ proteins by adhering directly to the surface of epithelial cells. They form attaching and effacing lesions, characterized by the localized destruction of the adjacent epithelial microvilli and the formation of a pedestal-like structure from the accumulation of cytoskeletal proteins, such as actin, beneath the site of attachment 169. EPEC uses a syringe-like type III secretion system to trigger TJ disruption and alterations in intestinal epithelial ion secretion 170–172. Infection of intestinal T84 monolayers with EPEC increased epithelial permeability and this was associated with destabilization and dissociation of ZO-1, occludin and claudin-1 TJ complex from the lateral membrane 171–173. The molecular mechanisms associated with EPEC-mediated TJ alterations are still unclear; however, studies utilizing pharmacological agents that inhibit MLCK, have implicated involvement of the MLCK pathway in the process 174.
Clostridium perfringens
While many bacterial products have been demonstrated to alter TJs, the enterotoxin of Clostridium perfringens (CPE), which is a common cause of food poisoning, directly interacts with and utilizes TJs as receptors 175, 176. CPE binds to the extracellular loop of claudins-3 and -4 on the cell surface of enterocytes forming small protein complexes in the plasma membrane 176. These complexes promote oligomerization and the formation of larger plasma membrane complexes, which have been associated with increased plasma membrane permeability 177, 178. CPE also interacts with occludin to promote its removal from the TJ and redistribution into the cytoplasm 177. The redistribution of claudins and occludin induce destabilization of the TJ complex leading to altered intestinal paracellular permeability. For example, exposure of MDCK monolayers to CPE induced a reversible decrease in TER and increase in permeability 179. Finally, the large CPE and TJ-containing complexes are believed to insert into the plasma membrane to form a functional pore that induces Ca2+ influx that triggers host epithelial cell death by apoptosis or oncosis 180, 181.
Clinical Review
Introduction
A breakdown or impairment of the epithelial barrier has been implicated as a critical determinant in the predisposition to intestinal inflammation and a number of gastrointestinal (GI) diseases including inflammatory bowel disease (IBD) and food allergy 160, 182–187 (Table 2). While altered intestinal barrier function (increased intestinal epithelial permeability) can be a consequence of disease exacerbation, clinical evidence suggests that it may be a primary etiologic factor predisposing to disease development. For example, healthy first-degree relatives of IBD and celiac patients have increased intestinal permeability. Furthermore, altered intestinal permeability persisted in asymptomatic celiac disease patients treated with a gluten-free diet 188, 189 and is predictive of clinical relapse in patients with clinically inactive IBD 190, 191. In this section, we summarize the current clinical data relevant to intestinal epithelial barrier function in chronic disease susceptibility and describe the potential implications of these studies in disease pathogenesis.
Table 2.
Diseases associated with altered TJ protein expression and Intestinal epithelial barrier function.
| Disease State | TJ proteins | Inflammatory cell/Cytokine | Proposed Mechanism | references |
|---|---|---|---|---|
| Food Allergy | Not defined | Mast cells | Mast cell-mediated degradation of TJ proteins | 206 and 207 |
| IBD | ↓ Claudin-3, -4, -5 and -8 ↑ Claudin-2 |
CD4+ T-cells IL-10, IFNγ and TNFα and MLCK |
IFNγ and TNFα-mediated activation of MLCK leading to TJ disruption and dysregulation of claudin and occludin expression. | 97, 98, 108, 219, 227 and 228 |
| Celiac Disease | Occludin and ZO-1 | Zonulin | Gliadin-induced Zonulin secretion by intestinal epithelial cells. Zonulin-induced downregulation of occludin/ZO-1. |
235 and 236 |
| Diabetes | Not defined | Zonulin | Increased zonulin secretion | 246 and 247 |
| Stress | Occludin and ZO-1 | Mast cells and corticotrophin-releasing hormone | Destabilization and redistribution of occludin/ZO-1 | 250–254 |
Food Allergy
Food allergies are adverse, immune-mediated reactions to ingested food proteins/antigens. It is hypothesized that intestinal barrier dysfunction may contribute to both antigen sensitization and also the IgE/mast cell-mediated anaphylactic effector phase of disease. The development of food allergies is dependent on the exposure of the food antigen to the mucosal immune system, which leads to antigen sensitization and the production of dietary antigen-specific CD4+ Th2 cells and IgE. It is hypothesized that altered intestinal barrier function permits increased dietary antigen transport across the intestinal barrier and exposure of dietary antigens to the mucosal immune system leading to the development of the dietary antigen-specific response. Consistent with this hypothesis, intestinal permeability in infants with food allergy as assessed by lactulose/mannitol ratio in the urine, was significantly increased compared to healthy young children 192, 193. To determine whether the altered intestinal barrier function was a consequence of a recent adverse allergic reaction to dietary antigen, lactulose/mannitol ratio was examined in food allergic patients who had been on an allergen-free diet for at least six months. Intestinal permeability remained elevated in these individuals indicating that increased intestinal permeability persisted in the absence of food antigen stimulation 192.
Additional data supporting a role for increased intestinal permeability in the development of food antigen sensitization and food allergies is provided by recent clinical studies that demonstrate an association between increased intestinal permeability and the development of new-onset food allergies in patients following liver and heart transplantation. Patients treated with the immunosuppressant tacrolimus (FK506) have been shown to have increased intestinal permeability and elevated levels of food antigen-specific IgE 194–196. Notably, some of these patients developed new-onset food allergies 197, 198. The development of food allergies in immunosuppressed post-transplant patients was originally thought to be a consequence of the passive transfer of food antigen-specific IgE or lymphocytes from food-allergic donors to previously non-allergic recipients 199, 200. However, studies have reported the development of food allergies in patients where the donor had no history of food allergy 201, 202. Interestingly, in vitro and in vivo experiments with rats have demonstrated that tacrolimus induces a dose-dependent increase in intestinal permeability 203 suggesting that tacrolimus-induced altered intestinal barrier function may be a possible explanation for the new-onset food allergies in immunosuppressed post-transplant patients.
Notably, tacrolimus has been shown to uncouple mitochondrial oxidative phosphorylation, leading to impaired mitochondrial energy production and a significant decrease in cellular ATP 203, 204. Importantly, formation of the intestinal barrier and the maintenance of intercellular junctional complexes are energy-dependent processes and decreased cellular ATP is responsible for inducing a breakdown in TJ complexes and barrier dysfunction 44. Consistent with this rats treated with tacrolimus were shown to have a dose-dependent increase in intestinal permeability that correlated with decreased intracellular ATP levels and CO2 release 203. Similarly, liver transplant patients treated with tacrolimus were found to have reduced mitochondrial energy production associated with increased intestinal permeability and an increase in serum endotoxin levels 198.
The immunosuppressive activity of tacrolimus is through the inhibition of calcineurin, which is critical for IL-2 induced T-cell activation 205, Inhibition of IL-2, has been shown to promote T-helper 2 immune response 233. Th2 cells secrete IL-4, IL-5 and IL-13, which promote IgE-mediated allergic inflammation and set the stage for food antigen sensitization and the induction of food allergies. There are most likely several mechanisms involved in the pathogenesis of food allergies in tacrolimus-immunosuppressed patients and increased intestinal permeability appears to be an important mediator to facilitate presentation of food antigens to the immune system and oral antigen sensitization.
We have recently provided experimental evidence supporting a role for altered intestinal permeability in oral antigen sensitization and the development of food allergies in mice 206. We generated a transgenic mouse that overexpresses the cytokine interleukin-9 specifically in the enterocytes of the small intestine (iIL-9Tg). A consequence of transgenic overexpression of IL-9 was a pronounced intestinal mastocytosis and altered intestinal permeability 206. Repeated oral administration of OVA to iIL-9Tg BALB/c mice and not WT mice promoted the development of antigen-specific IgE, CD4+ IL-4+ T-cells and symptoms of a food allergic response in the absence of prior systemic sensitization or the use of adjuvant. Pharmacological mast cell depletion in iIL-9Tg mice was found to restore intestinal permeability to levels comparable to WT mice. Remarkably, reconstitution of barrier function and decreased intestinal permeability in iIL-9Tg mice prevented orally-induced antigen sensitization 206. These findings suggest that increased intestinal permeability facilitates enhanced antigen uptake and the oral induction of food antigen sensitization.
Intestinal barrier dysfunction is also thought to contribute to the severity of food allergen-induced clinical symptoms. Oral challenge of food allergic individuals with food allergen induced an increase in lactulose/mannitol ratio in the urine 192, 207. The level of intestinal barrier dysfunction positively correlated with the severity of clinical symptoms 192. Notably, treatment of the food allergic group with sodium cromoglycate a mast cell stabilizer prior to ingestion of food allergen, significantly reduced lactulose permeability compared to food allergen-challenged individuals not receiving sodium cromoglycate indicating a role for mast cells in dietary antigen-induced intestinal epithelial barrier dysfunction 207.
Consistent with clinical observations animal models of GI anaphylaxis and food allergy have also demonstrated increased intestinal permeability following oral antigen challenge 206, 208, 209. Intraluminal challenge of egg-sensitized rats with egg albumin induced a 15-fold increase in uptake of 51cr-labelled EDTA as compared to rats treated with unrelated protein 122. Studies utilizing mast cell-deficient animals or pharmacological agents to deplete mast cells have provided corroborative evidence demonstrating that mast cells are critical for altered intestinal barrier function during food allergic reactions 206, 209, 123, 124. Increased permeability following antigen challenge has been shown to initially be the result of increased antigen uptake and translocation by the transcellular route, as evidenced by an increase in HRP-containing endosomes within minutes of HRP challenge in rats that had been sensitized 210. The second phase, which occurs after sensitization and is mast cell-dependent, was associated with a disruption in the TJs and an increase in paracellular permeability 210. Collectively, these studies suggest a role for altered intestinal barrier function in food allergy. Furthermore, these studies suggest a role for mast cells in the regulation of intestinal barrier dysfunction in food allergy.
Inflammatory Bowel Disease (IBD)
The IBDs, Crohn’s disease (CD) and ulcerative colitis, are chronic, relapsing-remitting inflammatory diseases. An emerging model of the pathogenesis of IBD suggests there are three essential factors: 1) a breakdown in intestinal barrier function; 2) exposure of luminal contents to immune cells in the lamina propria; and 3) an exaggerated immune response 211. However, it is currently unclear which factor is responsible for initiating this self-perpetuating cycle, leading to disease exacerbation. There is a growing body of data to suggest that increased intestinal permeability is a primary etiologic factor contributing to IBD pathogenesis. CD patients with clinically active disease have increased intestinal permeability and in patients with inactive disease, increased intestinal permeability is predictive of clinical relapse 190, 191. In addition to patients with IBD, increased intestinal permeability occurs in 10–25% of their healthy first-degree relatives, indicating that increased intestinal permeability likely preceded the onset of clinical disease 212–214. Furthermore, studies have found that a subset of patients who are at high risk for CD have either increased intestinal permeability at baseline or an exaggerated increase in permeability in response to stimulation 183, 215. Notably, a case study on the long-term follow-up of a healthy 13-year-old girl who had elevated intestinal permeability and a parent with CD patient reported that she subsequently developed CD 8 years later 216.
There are extensive experimental studies employing models of experimental colitis that demonstrate a link between altered intestinal barrier function and IBD. Mouse models predisposed to developing IBD-like symptoms, including IL-10−/−, mdr1a−/− and SAMP/Yit mice have established that increased intestinal permeability precedes disease development 107, 217, 218. For example, IL-10−/− mice spontaneously develop a CD-like colitis by 12 weeks of age 111. These mice also had increased intestinal permeability, which was present prior to the onset of disease 107. Notably, when IL-10−/− mice were treated with a zonulin peptide inhibitor, intestinal permeability was reduced and development of colitis was significantly attenuated 108. While intestinal permeability is a key player in the development of IBD, recent data suggests that increased intestinal permeability alone is not sufficient to predispose to the development of IBD. Transgenic mice that constitutively express active myosin light chain kinase MLCK in the intestinal epithelia had significant intestinal barrier dysfunction and increased intestinal permeability 219. It was found that the decrease in barrier function did not predispose the mice to spontaneous development of colitis; however, it did accelerate the onset and severity of immune-mediated colitis in the MLCK transgenic mice 219.
A significant advance in IBD research came with the discovery of the nucleotide-binding oligomerization domain 2 (NOD2)/caspase-recruitment domain 15 (CARD15) gene as a CD genetic susceptibility loci 220, 221. Mutations in the CARD15 gene have been identified in patients with CD and their healthy first-degree relatives 183. Notably, 40% of relatives with one mutation and 75% of relatives with two mutations had increased intestinal permeability compared to controls.
Multiple molecular mechanisms for increased intestinal permeability in IBD have been reported, including altered TJ protein expression and/or distribution and increased epithelial apoptosis 222. Initial studies reported a downregulation in occludin expression in IBD patients with no change in claudin-1 expression in IBD patients 223; however, the role occludin may play in barrier dysfunction has been questioned since occludin-deficient mice have normal intestinal barrier function 224. There are a number of studies demonstrating significant barrier dysfunction is associated with the disruption of occludin. This seeming discrepancy is most likely a consequence of the dynamic nature of TJs, whereby TJ complexes can form “normally” in the absence of occludin, as in occludin-deficient mice; however, once occludin is intimately associated with the complex, its disruption can alter the barrier function of the TJ complex. Clinical studies have also reported an upregulation of the barrier-reducing TJ protein claudin-2, in particular in the crypt epithelium, as well as decreased expression and redistribution of the sealing TJ proteins claudin-3, -4, -5 and -8 in IBD 105, 224 (Table 2). However, these TJ modifications might be a consequence of disease pathogenesis, rather than a cause, as they were not altered in patients with inactive CD 224. Additionally, the breakdown in the protective barrier in IBD leads to an inflammatory infiltrate and enhanced production of cytokines and other mediators that can further contribute to altered barrier function. Increased levels of IFNγ and TNFα have been demonstrated in the intestinal mucosa of IBD patients, and both cytokines have been shown to alter intestinal epithelial barrier function in vitro 97, 98, 225, 226. Notably, treating CD patients with monoclonal anti-TNF antibodies downregulated the inflammatory response and restored intestinal barrier function leading to a decrease in intestinal permeability 227, 228.
Celiac Disease
Celiac disease is an immune-mediated enteropathy triggered by an inappropriate T cell-mediated response to ingested gluten and its component gliadin. Clinical and experimental studies suggest that altered intestinal barrier function may play an inciting role in the development of celiac disease by allowing gliadin to cross the intestinal barrier and activate the immune system. Patients with celiac disease have enhanced intestinal permeability and altered TJ morphology; and these disruptions persisted in asymptomatic patients who were on a gluten-free diet 188, 189, 229, 230. There is data demonstrating that the increased intestinal permeability exists prior to disease onset and suggests that permeability may play an inciting role in the development of celiac disease. For example, a significant proportion of healthy first degree relatives of celiac patients also have increased intestinal permeability 231. Consistent with these observations inbred Irish Setter dogs, which spontaneously develop a gluten-sensitive enteropathy similar to human celiac disease, have increased intestinal permeability 232. Notably, the increase in permeability was present prior to gluten exposure and disease onset, and when animals reared on a gluten-free diet were first exposed to gluten, they immediately developed disease. These findings suggest that altered barrier function is a predetermining factor in celiac disease susceptibility 233.
The environmental trigger for celiac disease, gluten and its toxic component gliadin, have been well studied and both have been shown to directly stimulate zonulin production and induce an increase in intestinal permeability 188, 189, 229, 230. Under physiological circumstances the intestinal epithelium is, for the most part, impermeable to gluten and gliadin; however, patients with celiac disease have been found to have compromised TJ integrity and enhanced paracellular permeability, which could allow for gliadin to cross the intestinal barrier and activate the immune system. Gliadin regulates intestinal barrier function in part by the upregulation of zonulin and gliadin has been found to bind to the CXCR3 receptor on intestinal epithelial and initiate a MyD88-dependent release of zonulin 234. Incubating human intestinal epithelial monolayers or biopsies from celiac patients with gliadin stimulated zonulin secretion and an increase in epithelial permeability 235. Clinical studies have also demonstrated a positive correlation between increased intestinal permeability and intestinal zonulin levels in patients with active celiac disease 236. Furthermore, antagonism of zonulin prevented gliadin-induced permeability changes in intestinal biopsies from patients with celiac disease 235.
Mechanistically, zonulin binds to the zonulin receptor on intestinal epithelial cells and induces a PKC-mediated rearrangement of the cytoskeleton, downregulation of ZO-1 and occludin and disruption of TJ complex integrity increasing epithelial permeability 235, 237. In line with the in vitro and ex vivo studies demonstrating that zonulin alters ZO-1 expression, additional studies have demonstrated decreased expression of ZO-1 and redistribution of F-actin in patients with celiac disease 238. Remarkably, when patients eliminated gluten from their diet, the celiac disease went into remission, normal intestinal permeability was reinstated and the abnormalities in ZO-1 and F-actin expression were reversed 239. In addition, celiac disease also clearly has a genetic component as evidenced by 95% of celiac patients being HLA-DQ2 positive 240. However, a role for this in barrier function is yet to be defined.
Type I Diabetes
It is hypothesized that a combination of predisposing genetics, dysregulated intestinal barrier function and aberrant immune responses play an inciting role in type I diabetes. Increased intestinal permeability has been reported in patients with type I diabetes at disease onset and is believed to facilitate increased exposure to antigens that can trigger autoimmune destruction of the insulin-producing, pancreatic beta cells 241–243. Additionally, ultra-structural examination of duodenum from diabetic patients revealed altered TJ structure and an increase in the paracellular space between epithelial cells as compared to healthy controls 244 (Table 2). Studies using the diabetes-prone BioBreeding rat (BBDP), an inbred line that spontaneously develops autoimmune diabetes when weaned onto a normal diet, supports a role for altered intestinal barrier function in the pathogenesis of type I diabetes. BBDP rats have been found to have increased intestinal permeability associated with decreased expression of the TJ protein claudin-1 prior to the onset of insulitis and clinical diabetes 96, 245. In support of this, a recent study examined barrier function in diabetics at various stages of disease progression. Intestinal permeability was increased in all diabetic groups; however, pre-diabetics had the greatest increase, suggesting that increased intestinal permeability precedes the onset of clinical diabetes 96, 245.
Similar to celiac disease and IBD, increased intestinal zonulin production is a potential mechanism leading to enhanced intestinal permeability in Type I diabetes. Experimental studies with pre-diabetic BBDP rats demonstrated increased intestinal zonulin secretion, which coincided with altered barrier function and preceded the development of autoantibodies 246. Notably, treatment of the rats with a zonulin receptor antagonist reconstituted normal barrier function and abrogated disease development 246. A recent clinical study examining type I diabetics and their first-degree relatives found significantly higher serum zonulin levels in diabetic patients, which correlated with the degree of intestinal barrier dysfunction 247. Elevated serum zonulin was also found in 70% of pre-diabetic relatives, who were classified by the presence of positive autoantibodies in the absence of clinical disease 247. Taken together, these studies suggest that an abnormal upregulation of zonulin can induce an increase in paracellular intestinal permeability that could facilitate the development of autoimmune diabetes.
Stress-Induced Barrier Dysfunction and Disease Exacerbation
Psychological and physical stress can induce a variety of changes in normal GI function, including changes in gut motility and permeability as well as alterations in ion, fluid and mucus secretion and absorption 248. Animal models of acute and chronic stress demonstrate that stress induces changes in intestinal barrier function. Administration of acute, cold-restraint stress induced an increase in transcellular and paracellular intestinal permeability in rats 249. Electron microscopic examination revealed an increase in the number and size of HRP-containing endosomes in enterocytes from stressed rats as compared to controls; additionally, HRP was found within the paracellular spaces of epithelial cells in the intestine from stressed rats but not control rats 249. The increased intestinal permeability induced by immobilization stress has also been associated with a temporary redistribution of TJ proteins, including occludin and ZO-1 (Table 2) 250. Experimental analyses have demonstrated the stress-induced permeability changes are mediated by mast cells, cholinergic and adrenergic nerves and corticotropin-releasing hormone 251. Studies have demonstrated that stress-induced permeability and ion secretion changes are attenuated in mast cell-deficient animals or following mast cell depletion or stabilization 252–254.
Psychological stress has been shown to influence the clinical course of chronic intestinal disorders including IBD and irritable bowel syndrome 255–257. Long-term stress has been associated with an increased risk and number of relapses in patients with UC 255. Additionally, studies using animal models of colitis have found that stress induces a worsening of colitis, enhances disease reactivation, reduces colonic mucus production and increases colonic permeability 258, 259. Furthermore, stress has been linked to the onset and exacerbation of irritable bowel syndrome and functional gastrointestinal disorders 260. The increased intestinal permeability induced by stress is believed to play an important role in disease progression and relapse. Blocking stress-induced barrier changes may represent a novel therapy to circumvent stress-induced IBD and irritable bowel syndrome relapse.
Summary
Dysregulation of the intestinal barrier has been associated with chronic immune diseases including food allergy, IBD and celiac disease. Whether or not intestinal epithelial barrier function is a primary etiologic factor in the predisposition to disease development remains unclear; however clinical and experimental evidence supports a role for intestinal epithelial barrier dysfunction in disease pathogenesis. Recent experimental studies have identified a role for a number of exogenous factors including bacterial pathogens and components of innate and adaptive immunity in the regulation of intestinal barrier function. Understanding the interactions between innate and adaptive immunity and intestinal barrier function will provide important insight into the pathogenesis of inflammatory and autoimmune diseases. Furthermore, delineation of the molecular pathways involved in the regulation of intestinal barrier function will have important clinical implications both to the treatment and prevention of chronic inflammatory disease as well as development of therapeutic agents targeted at modulating intestinal barrier function that could be useful for immunotherapy and as well as drug and vaccine absorption.
What do we know?
The intestinal epithelial barrier is maintained by complex protein-protein networks that form desmosomes, adherens junctions and tight junctions (TJs).
Alterations of TJ protein formation and distribution and/or destabilization of the TJ complexes leads to intestinal epithelial barrier dysfunction.
Intestinal barrier function is modulated by:
The immune system, including the Th1-cytokine IFNγ, Th2-cytokines IL-4 and IL-13, TNFα, T-cells, mast cells and eosinophils
Ingestion of alcohol or NSAIDs
Enteric pathogens directly and through the elaboration of toxins and proteases.
Altered intestinal barrier function and increased intestinal permeability is associated with:
Food allergies
Inflammatory bowel disease
Celiac Disease
Type I Diabetes
What is still unknown?
Molecular pathways involved in the regulation of homeostatic intestinal barrier function.
Molecular components of the tight junctional complex within the intestinal epithelium.
The contribution of individual TJ proteins to barrier function and formation and stabilization of TJ complexes.
Molecular mechanisms of inflammation-induced intestinal barrier dysfunction.
Key inflammatory cells involved in the regulation of intestinal barrier dysfunction in chronic inflammatory diseases including food allergy, IBD and celiac disease.
Whether intestinal barrier dysfunction is a primary etiologic factor predisposing to chronic inflammatory disease development.
Molecular basis of mast cell-mediated intestinal barrier dysfunction in food allergy.
Acknowledgments
Dr. Hogan’s laboratory is indebted to the following grants and/or organizations that have partly financed the work presented in this review: NIAID R01 R01 (AI 073553), Crohn’s Colitis Foundation of America Career Development Award; American Heart Association Grant-in-Aid. The authors thank numerous instrumental colleagues who have contributed to the ideas formulated in this review including Drs. Marc Rothenberg, Fred Finkelman, Rick Strait, Paul Foster, Pablo Abonia, Glenn Furuta, James Lee, Elizabeth Forbes and the dedicated laboratory workers, Richard Ahrens, Heather Osterfel, Drs David Wu and Muthuvel Aramagum.
Grant Support: This work was supported in part by Crohn’s Colitis Foundation of America Career Development Award; American Heart Association Grant-in-Aid and NIH grants R01 (AI 073553), F30 (DK082113) and T32 (GM063483).
Abbreviations
- AJs
adherens junctions
- TJs
tight junctions
- JAMs
junctional adhesion molecules
- PDZ
Post synaptic density-95/Drosophila disc large/Zonula occludens-1 protein
- PKC
protein kinase C
- TER
transepithelial resistance
- MAPK
mitogen-activated protein kinases
- IFNγ
interferon-gamma
- TNFα
tumor necrosis factor alpha
- MLCK
myosin light chain kinase
- IL
interleukin
- IBD
inflammatory bowel disease
- CD
Crohn’s disease
- iIEL
intestinal intraepithelial lymphocytes
- NO
nitric oxide
- iNOS
inducible nitric oxide synthase
- GI
gastrointestinal
- mMCP-1
murine mast cell protease-1
- NSAIDs
non-steroidal anti-inflammatory drugs
- HA/P
hemagglutinin protease
- Zot
zonula occludens toxin
- EPEC
enteropathogenic E. coli
- CPE
Clostridium perfringens
- HRP
horseradish peroxidase
- iIL-9 Tg
intestinal interleukin-9 transgenic mice
- mdr1a−/−
multi-drug resistance gene deficient mice
- NOD2/CARD15
nucleotide-binding oligomerization domain 2/caspase-recruitment domain 15
- CXCR3
CXC chemokine receptor-3
- BBDP
BioBreeding diabetes-prone rat
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Blikslager AT, Moeser AJ, Gookin JL, Jones SL, POdle J. Restoration of barrier function in injured intestinal mucosa. Physiol Rev. 2007:87. doi: 10.1152/physrev.00012.2006. [DOI] [PubMed] [Google Scholar]
- 2.Podolsky DK. Mucosal immunity and inflammation. V. Innate mechanisms of mucosal defense and repair: the best offense is a good defense. Am J Physiol. 1999;277:G495–9. doi: 10.1152/ajpgi.1999.277.3.G495. [DOI] [PubMed] [Google Scholar]
- 3.Kunzelmann K, Mall M. Electrolyte transport in the mammalian colon: Mechanisms and Implications for disease. Physiol Rev. 2002;82:245–89. doi: 10.1152/physrev.00026.2001. [DOI] [PubMed] [Google Scholar]
- 4.Broer S. Amino acid transport across mammalian intestinal and renal epithelia. Physiol Rev. 2008;88:249–86. doi: 10.1152/physrev.00018.2006. [DOI] [PubMed] [Google Scholar]
- 5.Ferraris RP, Diamond J. Regulation of intestinal sugar transport. Physiol Rev. 1997;77:257–302. doi: 10.1152/physrev.1997.77.1.257. [DOI] [PubMed] [Google Scholar]
- 6.Tsukita S, Furuse M, Itoh M. Multifunctional strands in tight junctions. Nat Rev Mol Cell Biol. 2001;2:285–93. doi: 10.1038/35067088. [DOI] [PubMed] [Google Scholar]
- 7.Van Itallie CM, Anderson JM. Claudins and epithelial paracellular transport. Annu Rev Physiol. 2006;68:403–29. doi: 10.1146/annurev.physiol.68.040104.131404. [DOI] [PubMed] [Google Scholar]
- 8.Farquher MG, Palade GE. Junctional complexes in various epithelia. J Cell Biol. 1963;17:375–412. doi: 10.1083/jcb.17.2.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schneeberger EE, Lynch RD. Structure, function, and regulation of cellular tight junctions. Am J Physiol. 1992:L647–L661. doi: 10.1152/ajplung.1992.262.6.L647. [DOI] [PubMed] [Google Scholar]
- 10.Gumbiner B. Breaking through the tight junction barrier. J Cell Biol. 1993;123:1631–3. doi: 10.1083/jcb.123.6.1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hartsock A, Nelson WJ. Adherens and tight junctions: structure, function and connections to the actin cytoskeleton. Biochim Biophys Acta. 2008;1778:660–9. doi: 10.1016/j.bbamem.2007.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Forster C. Tight junctions and the modulation of barrier function in disease. Histochem Cell Biol. 2008;130:55–70. doi: 10.1007/s00418-008-0424-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harhaj NS, Antonetti DA. Regulation of tight junction and loss of barrier function in pathophysiology. Inter J Biochem Cell Biol. 2004:36. doi: 10.1016/j.biocel.2003.08.007. [DOI] [PubMed] [Google Scholar]
- 14.Tsukita S, Furuse M. The structure and function of claudins, cell adhesion molecules at tight junctions. Ann N Y Acad Sci. 2000;915:129–35. doi: 10.1111/j.1749-6632.2000.tb05235.x. [DOI] [PubMed] [Google Scholar]
- 15.Cereijido M, Contreras RG, Shoshani L. Cell adhesion, polarity and epithelia in the dawn of metazoans. Physiol Rev. 2004;84:1229–62. doi: 10.1152/physrev.00001.2004. [DOI] [PubMed] [Google Scholar]
- 16.Weis WI, Nelson WJ. Re-solving the cadherin-catenin-actin conundrum. J Biol Chem. 2006;281:35593–7. doi: 10.1074/jbc.R600027200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Halbleib JM, Nelson WJ. Cadherins in development: cell adhesion, sorting, and tissue morphogenesis. Genes Dev. 2006;20:3199–214. doi: 10.1101/gad.1486806. [DOI] [PubMed] [Google Scholar]
- 18.Perez-Moreno M, Fuchs E. Catenins: keeping cells from getting their signals crossed. Dev Cell. 2006;11:601–12. doi: 10.1016/j.devcel.2006.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Perez-Moreno M, Jamora C, Fuchs E. Sticky business: orchestrating cellular signals at adherens junctions. Cell. 2003;112:535–48. doi: 10.1016/s0092-8674(03)00108-9. [DOI] [PubMed] [Google Scholar]
- 20.Gumbiner BM. Regulation of cadherin-mediated adhesion in morphogenesis. Nat Rev Mol Cell Biol. 2005;6:622–34. doi: 10.1038/nrm1699. [DOI] [PubMed] [Google Scholar]
- 21.Ebnet K. Organization of multiprotein complexes at cell-cell junctions. Histochem Cell Biol. 2008;130:1–20. doi: 10.1007/s00418-008-0418-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vasioukhin V, Bauer C, Yin M, Fuchs E. Directed actin polymerization is the driving force for epithelial cell-cell adhesion. Cell. 2000;100:209–19. doi: 10.1016/s0092-8674(00)81559-7. [DOI] [PubMed] [Google Scholar]
- 23.Vasioukhin V, Bowers E, Bauer C, Degenstein L, Fuchs E. Desmoplakin is essential in epidermal sheet formation. Nat Cell Biol. 2001;3:1076–85. doi: 10.1038/ncb1201-1076. [DOI] [PubMed] [Google Scholar]
- 24.Ikeda W, Nakanishi H, Miyoshi J, Mandai K, Ishizaki H, Tanaka M, et al. Afadin: A key molecule essential for structural organization of cell-cell junctions of polarized epithelia during embryogenesis. J Cell Biol. 1999;146:1117–32. doi: 10.1083/jcb.146.5.1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pokutta S, Weis WI. The cytoplasmic face of cell contact sites. Curr Opin Struct Biol. 2002;12:255–62. doi: 10.1016/s0959-440x(02)00318-4. [DOI] [PubMed] [Google Scholar]
- 26.Pokutta S, Weis WI. Structure and mechanism of cadherins and catenins in cell-cell contacts. Annu Rev Cell Dev Biol. 2007;23:237–61. doi: 10.1146/annurev.cellbio.22.010305.104241. [DOI] [PubMed] [Google Scholar]
- 27.Reynolds AB, Roczniak-Ferguson A. Emerging roles for p120-catenin in cell adhesion and cancer. Oncogene. 2004;23:7947–56. doi: 10.1038/sj.onc.1208161. [DOI] [PubMed] [Google Scholar]
- 28.Hermiston ML, Gordon JI. In vivo analysis of cadherin function in the mouse intestinal epithelium: essential roles in adhesion, maintenance of differentiation, and regulation of programmed cell death. J Cell Biol. 1995;129:489–506. doi: 10.1083/jcb.129.2.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Herminston ML, Gordon JI. Inflammaotry bowel disease and adenomas in mice expressing a dominant negative N-cadherin. Science. 1995;270:1203–7. doi: 10.1126/science.270.5239.1203. [DOI] [PubMed] [Google Scholar]
- 30.Miyoshi J, Takai Y. Nectin and nectin-like molecules: biology and pathology. Am J Nephrol. 2007;27:590–604. doi: 10.1159/000108103. [DOI] [PubMed] [Google Scholar]
- 31.Takai Y, Ikeda W, Ogita H, Rikitake Y. The immunoglobulin-like cell adhesion molecule nectin and its associated protein afadin. Annu Rev Cell Dev Biol. 2008;24:309–42. doi: 10.1146/annurev.cellbio.24.110707.175339. [DOI] [PubMed] [Google Scholar]
- 32.Sakisaka T, Ikeda W, Ogita H, Fujita N, Takai Y. The roles of nectins in cell adhesions: cooperation with other cell adhesion molecules and growth factor receptors. Curr Opin Cell Biol. 2007;19:593–602. doi: 10.1016/j.ceb.2007.09.007. [DOI] [PubMed] [Google Scholar]
- 33.Pokutta S, Drees F, Takai Y, Nelson WJ, Weis WI. Biochemical and structural definition of the l-afadin- and actin-binding sites of alpha-catenin. J Biol Chem. 2002;277:18868–74. doi: 10.1074/jbc.M201463200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tachibana K, Nakanishi H, Mandai K, Ozaki K, Ikeda W, Yamamoto Y, et al. Two cell adhesion molecules, nectin and cadherin, interact through their cytoplasmic domain-associated proteins. J Cell Biol. 2000;150:1161–76. doi: 10.1083/jcb.150.5.1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mandai K, Nakanishi H, Satoh A, Takahashi K, Satoh K, Nishioka H, et al. Ponsin/SH3P12: an l-afadin- and vinculin-binding protein localized at cell-cell and cell-matrix adherens junctions. J Cell Biol. 1999;144:1001–17. doi: 10.1083/jcb.144.5.1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Asada M, Irie K, Morimoto K, Yamada A, Ikeda W, Takeuchi M, et al. ADIP, a novel Afadin- and alpha-actinin-binding protein localized at cell-cell adherens junctions. J Biol Chem. 2003;278:4103–11. doi: 10.1074/jbc.M209832200. [DOI] [PubMed] [Google Scholar]
- 37.Ooshio T, Irie K, Morimoto K, Fukuhara A, Imai T, Takai Y. Involvement of LMO7 in the association of two cell-cell adhesion molecules, nectin and E-cadherin, through afadin and alpha-actinin in epithelial cells. J Biol Chem. 2004;279:31365–73. doi: 10.1074/jbc.M401957200. [DOI] [PubMed] [Google Scholar]
- 38.Gonzalez-Mariscal L, Betanzos A, Nava P, Jaramillo BE. Tight junction proteins. Prog Biophys Mol Biol. 2003;81:1–44. doi: 10.1016/s0079-6107(02)00037-8. [DOI] [PubMed] [Google Scholar]
- 39.Staehelin LA. Further observations on the fine structure of freeze-cleaved tight junctions. J Cell Sci. 1973;13:763–86. doi: 10.1242/jcs.13.3.763. [DOI] [PubMed] [Google Scholar]
- 40.Matter K, Balda MS. Epithelial tight junctions, gene expression and nucleojunctional interplay. J Cell Sci. 2007;120:1505–11. doi: 10.1242/jcs.005975. [DOI] [PubMed] [Google Scholar]
- 41.Laukoetter MG, Bruewer M, Nusrat A. Regulation of the intestinal epithelial barrier by the apical junctional complex. Curr Opin Gastroenterol. 2006;22:85–9. doi: 10.1097/01.mog.0000203864.48255.4f. [DOI] [PubMed] [Google Scholar]
- 42.Atkinson KJ, Rao RK. Role of protein tyrosine phosphorylation in acetaldehyde-induced disruption of epithelial tight junctions. Am J Physiol Gastrointest Liver Physiol. 2001;280:G1280–8. doi: 10.1152/ajpgi.2001.280.6.G1280. [DOI] [PubMed] [Google Scholar]
- 43.Seth A, Basuroy S, Sheth P, Rao RK. L-Glutamine ameliorates acetaldehyde-induced increase in paracellular permeability in Caco-2 cell monolayer. Am J Physiol Gastrointest Liver Physiol. 2004;287:G510–7. doi: 10.1152/ajpgi.00058.2004. [DOI] [PubMed] [Google Scholar]
- 44.Furuse M, Hirase T, Itoh M, Nagafuchi A, Yonemura S, Tsukita S, et al. Occludin: a novel integral membrane protein localizing at tight junctions. J Cell Biol. 1993;123:1777–88. doi: 10.1083/jcb.123.6.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bauer H, Stelzhammer W, Fuchs R, Weiger TM, Danninger C, Probst G, et al. Astrocytes and neurons express the tight junction-specific protein occludin in vitro. Exp Cell Res. 1999;250:434–8. doi: 10.1006/excr.1999.4558. [DOI] [PubMed] [Google Scholar]
- 46.Barber AJ, Antonetti DA, Gardner TW. Altered expression of retinal occludin and glial fibrillary acidic protein in experimental diabetes. The Penn State Retina Research Group. Invest Ophthalmol Vis Sci. 2000;41:3561–8. [PubMed] [Google Scholar]
- 47.Rescigno M, Urbano M, Valzasina B, Francolini M, Rotta G, Bonasio R, et al. Dendritic cells express tight junction proteins and penetrate gut epithelial monolayers to sample bacteria. Nat Immunol. 2001;2:361–7. doi: 10.1038/86373. [DOI] [PubMed] [Google Scholar]
- 48.Furuse M, Itoh M, Hirase T, Nagafuchi A, Yonemura S, Tsukita S, et al. Direct association of occludin with ZO-1 and its possible involvement in the localization of occludin at tight junctions. J Cell Biol. 1994;127:1617–26. doi: 10.1083/jcb.127.6.1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mitic LL, Van Itallie CM, Anderson JM. Molecular physiology and pathophysiology of tight junctions I. Tight junction structure and function: lessons from mutant animals and proteins. Am J Physiol Gastrointest Liver Physiol. 2000;279:G250–4. doi: 10.1152/ajpgi.2000.279.2.G250. [DOI] [PubMed] [Google Scholar]
- 50.Muresan Z, Paul DL, Goodenough DA. Occludin 1B, a variant of the tight junction protein occludin. Mol Biol Cell. 2000;11:627–34. doi: 10.1091/mbc.11.2.627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mankertz J, Waller JS, Hillenbrand B, Tavalali S, Florian P, Schoneberg T, et al. Gene expression of the tight junction protein occludin includes differential splicing and alternative promoter usage. Biochem Biophys Res Commun. 2002;298:657–66. doi: 10.1016/s0006-291x(02)02487-7. [DOI] [PubMed] [Google Scholar]
- 52.Balda MS, Whitney JA, Flores C, Gonzalez S, Cereijido M, Matter K. Functional dissociation of paracellular permeability and transepithelial electrical resistance and disruption of the apical-basolateral intramembrane diffusion barrier by expression of a mutant tight junction membrane protein. J Cell Biol. 1996;134:1031–49. doi: 10.1083/jcb.134.4.1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McCarthy KM, Skare IB, Stankewich MC, Furuse M, Tsukita S, Rogers RA, et al. Occludin is a functional component of the tight junction. J Cell Sci. 1996;109( Pt 9):2287–98. doi: 10.1242/jcs.109.9.2287. [DOI] [PubMed] [Google Scholar]
- 54.Wan H, Winton HL, Soeller C, Tovey ER, Gruenert DC, Thompson PJ, et al. Der p 1 facilitates transepithelial allergen delivery by disruption of tight junctions. J Clin Invest. 1999;104:123–33. doi: 10.1172/JCI5844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Antonetti DA, Wolpert EB, DeMaio L, Harhaj NS, Scaduto RC., Jr Hydrocortisone decreases retinal endothelial cell water and solute flux coincident with increased content and decreased phosphorylation of occludin. J Neurochem. 2002;80:667–77. doi: 10.1046/j.0022-3042.2001.00740.x. [DOI] [PubMed] [Google Scholar]
- 56.Saitou M, Fujimoto K, Doi Y, Itoh M, Fujimoto T, Furuse M, et al. Occludin-deficient embryonic stem cells can differentiate into polarized epithelial cells bearing tight junctions. J Cell Biol. 1998;141:397–408. doi: 10.1083/jcb.141.2.397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schulzke JD, Gitter AH, Mankertz J, Spiegel S, Seidler U, Amasheh S, et al. Epithelial transport and barrier function in occludin-deficient mice. Biochim Biophys Acta. 2005;1669:34–42. doi: 10.1016/j.bbamem.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 58.Furuse M, Fujimoto K, Sato N, Hirase T, Tsukita S, Tsukita S. Overexpression of occludin, a tight junction-associated integral membrane protein, induces the formation of intracellular multilamellar bodies bearing tight junction-like structures. J Cell Sci. 1996;109( Pt 2):429–35. doi: 10.1242/jcs.109.2.429. [DOI] [PubMed] [Google Scholar]
- 59.Van Itallie CM, Anderson JM. Occludin confers adhesiveness when expressed in fibroblasts. J Cell Sci. 1997;110( Pt 9):1113–21. doi: 10.1242/jcs.110.9.1113. [DOI] [PubMed] [Google Scholar]
- 60.Chen ML, Pothoulakis C, LaMont JT. Protein kinase C signaling regulates ZO-1 translocation and increased paracellular flux of T84 colonocytes exposed to Clostridium difficile toxin A. J Biol Chem. 2002;277:4247–54. doi: 10.1074/jbc.M109254200. [DOI] [PubMed] [Google Scholar]
- 61.Nunbhakdi-Craig V, Machleidt T, Ogris E, Bellotto D, White CL, 3rd, Sontag E. Protein phosphatase 2A associates with and regulates atypical PKC and the epithelial tight junction complex. J Cell Biol. 2002;158:967–78. doi: 10.1083/jcb.200206114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Suzuki T, Elias BC, Seth A, Shen L, Turner JR, Giorgianni F, et al. PKC eta regulates occludin phosphorylation and epithelial tight junction integrity. Proc Natl Acad Sci U S A. 2009;106:61–6. doi: 10.1073/pnas.0802741106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Harhaj NS, Antonetti DA. Regulation of tight junctions and loss of barrier function in pathophysiology. Int J Biochem Cell Biol. 2004;36:1206–37. doi: 10.1016/j.biocel.2003.08.007. [DOI] [PubMed] [Google Scholar]
- 64.Schneeberger EE, Lynch RD. The tight junction: a multifunctional complex. Am J Physiol Cell Physiol. 2004;286:C1213–28. doi: 10.1152/ajpcell.00558.2003. [DOI] [PubMed] [Google Scholar]
- 65.Turksen K, Troy TC. Barriers built on claudins. J Cell Sci. 2004;117:2435–47. doi: 10.1242/jcs.01235. [DOI] [PubMed] [Google Scholar]
- 66.Morita K, Furuse M, Fujimoto K, Tsukita S. Claudin multigene family encoding four-transmembrane domain protein components of tight junction strands. Proc Natl Acad Sci U S A. 1999;96:511–6. doi: 10.1073/pnas.96.2.511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Itoh M, Furuse M, Morita K, Kubota K, Saitou M, Tsukita S. Direct binding of three tight junction-associated MAGUKs, ZO-1, ZO-2, and ZO-3, with the COOH termini of claudins. J Cell Biol. 1999;147:1351–63. doi: 10.1083/jcb.147.6.1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.McCarthy KM, Francis SA, McCormack JM, Lai J, Rogers RA, Skare IB, et al. Inducible expression of claudin-1-myc but not occludin-VSV-G results in aberrant tight junction strand formation in MDCK cells. J Cell Sci. 2000;113(Pt 19):3387–98. doi: 10.1242/jcs.113.19.3387. [DOI] [PubMed] [Google Scholar]
- 69.Furuse M, Fujita K, Hiiragi T, Fujimoto K, Tsukita S. Claudin-1 and -2: novel integral membrane proteins localizing at tight junctions with no sequence similarity to occludin. J Cell Biol. 1998;141:1539–50. doi: 10.1083/jcb.141.7.1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Krause G, Winkler L, Mueller SL, Haseloff RF, Piontek J, Blasig IE. Structure and function of claudins. Biochim Biophys Acta. 2008;1778:631–45. doi: 10.1016/j.bbamem.2007.10.018. [DOI] [PubMed] [Google Scholar]
- 71.Furuse M, Sasaki H, Tsukita S. Manner of interaction of heterogeneous claudin species within and between tight junction strands. J Cell Biol. 1999;147:891–903. doi: 10.1083/jcb.147.4.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Furuse M, Hata M, Furuse K, Yoshida Y, Haratake A, Sugitani Y, et al. Claudin-based tight junctions are crucial for the mammalian epidermal barrier: a lesson from claudin-1-deficient mice. J Cell Biol. 2002;156:1099–111. doi: 10.1083/jcb.200110122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Turksen K, Troy TC. Permeability barrier dysfunction in transgenic mice overexpressing claudin 6. Development. 2002;129:1775–84. doi: 10.1242/dev.129.7.1775. [DOI] [PubMed] [Google Scholar]
- 74.Furuse M, Furuse K, Sasaki H, Tsukita S. Conversion of zonulae occludentes from tight to leaky strand type by introducing claudin-2 into Madin-Darby canine kidney I cells. J Cell Biol. 2001;153:263–72. doi: 10.1083/jcb.153.2.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yu AS, Enck AH, Lencer WI, Schneeberger EE. Claudin-8 expression in Madin-Darby canine kidney cells augments the paracellular barrier to cation permeation. J Biol Chem. 2003;278:17350–9. doi: 10.1074/jbc.M213286200. [DOI] [PubMed] [Google Scholar]
- 76.Colegio OR, Van Itallie C, Rahner C, Anderson JM. Claudin extracellular domains determine paracellular charge selectivity and resistance but not tight junction fibril architecture. Am J Physiol Cell Physiol. 2003;284:C1346–54. doi: 10.1152/ajpcell.00547.2002. [DOI] [PubMed] [Google Scholar]
- 77.Colegio OR, Van Itallie CM, McCrea HJ, Rahner C, Anderson JM. Claudins create charge-selective channels in the paracellular pathway between epithelial cells. Am J Physiol Cell Physiol. 2002;283:C142–7. doi: 10.1152/ajpcell.00038.2002. [DOI] [PubMed] [Google Scholar]
- 78.Van Itallie CM, Holmes J, Bridges A, Gookin JL, Coccaro MR, Proctor W, et al. The density of small tight junction pores varies among cell types and is increased by expression of claudin-2. J Cell Sci. 2008;121:298–305. doi: 10.1242/jcs.021485. [DOI] [PubMed] [Google Scholar]
- 79.Agarwal R, D’Souza T, Morin PJ. Claudin-3 and claudin-4 expression in ovarian epithelial cells enhances invasion and is associated with increased matrix metalloproteinase-2 activity. Cancer Res. 2005;65:7378–85. doi: 10.1158/0008-5472.CAN-05-1036. [DOI] [PubMed] [Google Scholar]
- 80.D’Souza T, Agarwal R, Morin PJ. Phosphorylation of claudin-3 at threonine 192 by cAMP-dependent protein kinase regulates tight junction barrier function in ovarian cancer cells. J Biol Chem. 2005;280:26233–40. doi: 10.1074/jbc.M502003200. [DOI] [PubMed] [Google Scholar]
- 81.D’Souza T, Indig FE, Morin PJ. Phosphorylation of claudin-4 by PKCepsilon regulates tight junction barrier function in ovarian cancer cells. Exp Cell Res. 2007;313:3364–75. doi: 10.1016/j.yexcr.2007.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yamauchi K, Rai T, Kobayashi K, Sohara E, Suzuki T, Itoh T, et al. Disease-causing mutant WNK4 increases paracellular chloride permeability and phosphorylates claudins. Proc Natl Acad Sci U S A. 2004;101:4690–4. doi: 10.1073/pnas.0306924101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gonzalez-Mariscal L, Tapia R, Chamorro D. Crosstalk of tight junction components with signaling pathways. Biochim Biophys Acta. 2008;1778:729–56. doi: 10.1016/j.bbamem.2007.08.018. [DOI] [PubMed] [Google Scholar]
- 84.Fujibe M, Chiba H, Kojima T, Soma T, Wada T, Yamashita T, et al. Thr203 of claudin-1, a putative phosphorylation site for MAP kinase, is required to promote the barrier function of tight junctions. Exp Cell Res. 2004;295:36–47. doi: 10.1016/j.yexcr.2003.12.014. [DOI] [PubMed] [Google Scholar]
- 85.Roh MH, Liu CJ, Laurinec S, Margolis B. The carboxyl terminus of zona occludens-3 binds and recruits a mammalian homologue of discs lost to tight junctions. J Biol Chem. 2002;277:27501–9. doi: 10.1074/jbc.M201177200. [DOI] [PubMed] [Google Scholar]
- 86.Aijaz S, Balda MS, Matter K. Tight junctions: molecular architecture and function. Int Rev Cytol. 2006;248:261–98. doi: 10.1016/S0074-7696(06)48005-0. [DOI] [PubMed] [Google Scholar]
- 87.Macara IG. Parsing the polarity code. Nat Rev Mol Cell Biol. 2004;5:220–31. doi: 10.1038/nrm1332. [DOI] [PubMed] [Google Scholar]
- 88.Ebnet K, Suzuki A, Ohno S, Vestweber D. Junctional adhesion molecules (JAMs): more molecules with dual functions? J Cell Sci. 2004;117:19–29. doi: 10.1242/jcs.00930. [DOI] [PubMed] [Google Scholar]
- 89.Bazzoni G. The JAM family of junctional adhesion molecules. Curr Opin Cell Biol. 2003;15:525–30. doi: 10.1016/s0955-0674(03)00104-2. [DOI] [PubMed] [Google Scholar]
- 90.Williams LA, Martin-Padura I, Dejana E, Hogg N, Simmons DL. Identification and characterisation of human Junctional Adhesion Molecule (JAM) Mol Immunol. 1999;36:1175–88. doi: 10.1016/s0161-5890(99)00122-4. [DOI] [PubMed] [Google Scholar]
- 91.Liu Y, Nusrat A, Schnell FJ, Reaves TA, Walsh S, Pochet M, et al. Human junction adhesion molecule regulates tight junction resealing in epithelia. J Cell Sci. 2000;113( Pt 13):2363–74. doi: 10.1242/jcs.113.13.2363. [DOI] [PubMed] [Google Scholar]
- 92.Bazzoni G, Martinez-Estrada OM, Mueller F, Nelboeck P, Schmid G, Bartfai T, et al. Homophilic interaction of junctional adhesion molecule. J Biol Chem. 2000;275:30970–6. doi: 10.1074/jbc.M003946200. [DOI] [PubMed] [Google Scholar]
- 93.Babinska A, Kedees MH, Athar H, Sobocki T, Sobocka MB, Ahmed T, et al. Two regions of the human platelet F11-receptor (F11R) are critical for platelet aggregation, potentiation and adhesion. Thromb Haemost. 2002;87:712–21. [PubMed] [Google Scholar]
- 94.Laukoetter MG, Nava P, Lee WY, Severson EA, Capaldo CT, Babbin BA, et al. JAM-A regulates permeability and inflammation in the intestine in vivo. J Exp Med. 2007;204:3067–76. doi: 10.1084/jem.20071416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Van Itallie CM, Rogan S, Yu A, Vidal LS, Holmes J, Anderson JM. Two splice variants of claudin-10 in the kidney create paracellular pores with different ion selectivities. Am J Physiol Renal Physiol. 2006;291:F1288–99. doi: 10.1152/ajprenal.00138.2006. [DOI] [PubMed] [Google Scholar]
- 96.Meddings JB, Jarand J, Urbanski SJ, Hardin J, Gall DG. Increased gastrointestinal permeability is an early lesion in the spontaneously diabetic BB rat. Am J Physiol. 1999;276:G951–7. doi: 10.1152/ajpgi.1999.276.4.G951. [DOI] [PubMed] [Google Scholar]
- 97.MacDonald TT, Hutchings P, Choy MY, Murch S, Cooke A. Tumour necrosis factor-alpha and interferon-gamma production measured at the single cell level in normal and inflamed human intestine. Clin Exp Immunol. 1990;81:301–5. doi: 10.1111/j.1365-2249.1990.tb03334.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Fais S, Capobianchi MR, Silvestri M, Mercuri F, Pallone F, Dianzani F. Interferon expression in Crohn’s disease patients: increased interferon-gamma and -alpha mRNA in the intestinal lamina propria mononuclear cells. J Interferon Res. 1994;14:235–8. doi: 10.1089/jir.1994.14.235. [DOI] [PubMed] [Google Scholar]
- 99.Ye D, Ma I, Ma TY. Molecular mechanism of tumor necrosis factor-alpha modulation of intestinal epithelial tight junction barrier. Am J Physiol Gastrointest Liver Physiol. 2006;290:G496–504. doi: 10.1152/ajpgi.00318.2005. [DOI] [PubMed] [Google Scholar]
- 100.Zolotarevsky Y, Hecht G, Koutsouris A, Gonzalez DE, Quan C, Tom J, et al. A membrane-permeant peptide that inhibits MLC kinase restores barrier function in in vitro models of intestinal disease. Gastroenterology. 2002;123:163–72. doi: 10.1053/gast.2002.34235. [DOI] [PubMed] [Google Scholar]
- 101.Mankertz J, Tavalali S, Schmitz H, Mankertz A, Riecken EO, Fromm M, et al. Expression from the human occludin promoter is affected by tumor necrosis factor alpha and interferon gamma. J Cell Sci. 2000;113( Pt 11):2085–90. doi: 10.1242/jcs.113.11.2085. [DOI] [PubMed] [Google Scholar]
- 102.Zund G, Madara JL, Dzus AL, Awtrey CS, Colgan SP. Interleukin-4 and interleukin-13 differentially regulate epithelial chloride secretion. J Biol Chem. 1996;271:7460–4. doi: 10.1074/jbc.271.13.7460. [DOI] [PubMed] [Google Scholar]
- 103.Ceponis PJ, Botelho F, Richards CD, McKay DM. Interleukins 4 and 13 increase intestinal epithelial permeability by a phosphatidylinositol 3-kinase pathway. Lack of evidence for STAT 6 involvement. J Biol Chem. 2000;275:29132–7. doi: 10.1074/jbc.M003516200. [DOI] [PubMed] [Google Scholar]
- 104.Berin MC, Yang PC, Ciok L, Waserman S, Perdue MH. Role for IL-4 in macromolecular transport across human intestinal epithelium. Am J Physiol. 1999;276:C1046–52. doi: 10.1152/ajpcell.1999.276.5.C1046. [DOI] [PubMed] [Google Scholar]
- 105.Prasad S, Mingrino R, Kaukinen K, Hayes KL, Powell RM, MacDonald TT, et al. Inflammatory processes have differential effects on claudins 2, 3 and 4 in colonic epithelial cells. Lab Invest. 2005;85:1139–62. doi: 10.1038/labinvest.3700316. [DOI] [PubMed] [Google Scholar]
- 106.Madden KB, Whitman L, Sullivan C, Gause WC, Urban JF, Jr, Katona IM, et al. Role of STAT6 and mast cells in IL-4- and IL-13-induced alterations in murine intestinal epithelial cell function. J Immunol. 2002;169:4417–22. doi: 10.4049/jimmunol.169.8.4417. [DOI] [PubMed] [Google Scholar]
- 107.Madsen KL, Malfair D, Gray D, Doyle JS, Jewell LD, Fedorak RN. Interleukin-10 gene-deficient mice develop a primary intestinal permeability defect in response to enteric microflora. Inflamm Bowel Dis. 1999;5:262–70. doi: 10.1097/00054725-199911000-00004. [DOI] [PubMed] [Google Scholar]
- 108.Arrieta MC, Madsen K, Doyle J, Meddings J. Reducing small intestinal permeability attenuates colitis in the IL10 gene-deficient mouse. Gut. 2009;58:41–8. doi: 10.1136/gut.2008.150888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Madsen KL, Tavernini MM, Mosmann TR, Fedorak RN. Interleukin 10 modulates ion transport in rat small intestine. Gastroenterology. 1996;111:936–44. doi: 10.1016/s0016-5085(96)70061-6. [DOI] [PubMed] [Google Scholar]
- 110.Madsen KL, Lewis SA, Tavernini MM, Hibbard J, Fedorak RN. Interleukin 10 prevents cytokine-induced disruption of T84 monolayer barrier integrity and limits chloride secretion. Gastroenterology. 1997;113:151–9. doi: 10.1016/s0016-5085(97)70090-8. [DOI] [PubMed] [Google Scholar]
- 111.Kuhn R, Lohler J, Rennick D, Rajewsky K, Muller W. Interleukin-10-deficient mice develop chronic enterocolitis [see comments] Cell. 1993;75:263–74. doi: 10.1016/0092-8674(93)80068-p. [DOI] [PubMed] [Google Scholar]
- 112.Fasano A, Shea-Donohue T. Mechanisms of Disease: The role of intestinal barrier function in the pathogenesis of gastrointestinal diseases. Nature Clin Pract Gastroenterol Hepatol. 2005:2. doi: 10.1038/ncpgasthep0259. [DOI] [PubMed] [Google Scholar]
- 113.Musch MW, Clarke LL, Mamah D, Gawenis LR, Zhang Z, Ellsworth W, et al. T cell activation causes diarrhea by increasing intestinal permeability and inhibiting epithelial Na+/K+-ATPase. J Clin Invest. 2002;110:1739–47. doi: 10.1172/JCI15695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Clayburgh DR, Barrett TA, Tang Y, Meddings JB, Van Eldik LJ, Watterson DM, et al. Epithelial myosin light chain kinase-dependent barrier dysfunction mediates T cell activation-induced diarrhea in vivo. J Clin Invest. 2005;115:2702–15. doi: 10.1172/JCI24970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Clayburgh DR, Musch MW, Leitges M, Fu YX, Turner JR. Coordinated epithelial NHE3 inhibition and barrier dysfunction are required for TNF-mediated diarrhea in vivo. J Clin Invest. 2006;116:2682–94. doi: 10.1172/JCI29218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Dalton JE, Cruickshank SM, Egan CE, Mears R, Newton DJ, Andrew EM, et al. Intraepithelial gammadelta+ lymphocytes maintain the integrity of intestinal epithelial tight junctions in response to infection. Gastroenterology. 2006;131:818–29. doi: 10.1053/j.gastro.2006.06.003. [DOI] [PubMed] [Google Scholar]
- 117.Yu LC, Perdue MH. Role of mast cells in intestinal mucosal function: studies in models of hypersensitivity and stress. Immunol Rev. 2001;179:61–73. doi: 10.1034/j.1600-065x.2001.790107.x. [DOI] [PubMed] [Google Scholar]
- 118.Galli SJ, Grimbaldeston M, Tsai M. Immunomodulatory mast cells: negative, as well as positive, regulators of immunity. Nat Rev Immunol. 2008;8:478–86. doi: 10.1038/nri2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Bischoff SC, Kramer S. Human mast cells, bacteria, and intestinal immunity. Immunol Rev. 2007;217:329–37. doi: 10.1111/j.1600-065X.2007.00523.x. [DOI] [PubMed] [Google Scholar]
- 120.Bischoff SC. Role of mast cells in allergic and non-allergic immune responses: comparison of human and murine data. Nat Rev Immunol. 2007;7:93–104. doi: 10.1038/nri2018. [DOI] [PubMed] [Google Scholar]
- 121.Pennock JL, Grencis RK. The mast cell and gut nematodes: damage and defence. Chem Immunol Allergy. 2006;90:128–40. doi: 10.1159/000088885. [DOI] [PubMed] [Google Scholar]
- 122.Crowe SE, Soda K, Stanisz AM, Perdue MH. Intestinal permeability in allergic rats: nerve involvement in antigen-induced changes. Am J Physiol. 1993;264:G617–23. doi: 10.1152/ajpgi.1993.264.4.G617. [DOI] [PubMed] [Google Scholar]
- 123.Perdue MH, Masson S, Wershil BK, Galli SJ. Role of mast cells in ion transport abnormalities associated with intestinal anaphylaxis. Correction of the diminished secretory response in genetically mast cell-deficient W/Wv mice by bone marrow transplantation. J Clin Invest. 1991;87:687–93. doi: 10.1172/JCI115047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Berin MC, Kiliaan AJ, Yang PC, Groot JA, Taminiau JA, Perdue MH. Rapid transepithelial antigen transport in rat jejunum: impact of sensitization and the hypersensitivity reaction. Gastroenterology. 1997;113:856–64. doi: 10.1016/s0016-5085(97)70180-x. [DOI] [PubMed] [Google Scholar]
- 125.Crowe SE, Sestini P, Perdue MH. Allergic reactions of rat jejunal mucosa. Ion transport responses to luminal antigen and inflammatory mediators. Gastroenterology. 1990;99:74–82. doi: 10.1016/0016-5085(90)91232-u. [DOI] [PubMed] [Google Scholar]
- 126.McDermott JR, Bartram RE, Knight PA, Miller HR, Garrod DR, Grencis RK. Mast cells disrupt epithelial barrier function during enteric nematode infection. Proc Natl Acad Sci U S A. 2003;100:7761–6. doi: 10.1073/pnas.1231488100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Jeziorska M, Haboubi N, Schofield P, Woolley DE. Distribution and activation of eosinophils in inflammatory bowel disease using an improved immunohistochemical technique. J Pathol. 2001;194:484–92. doi: 10.1002/path.904. [DOI] [PubMed] [Google Scholar]
- 128.Levy AM, Gleich GJ, Sandborn WJ, Tremaine WJ, Steiner BL, Phillips SF. Increased eosinophil granule proteins in gut lavage fluid from patients with inflammatory bowel disease. Mayo Clin Proc. 1997;72:117–23. doi: 10.4065/72.2.117. [DOI] [PubMed] [Google Scholar]
- 129.Carvalho AT, Elia CC, de Souza HS, Elias PR, Pontes EL, Lukashok HP, et al. Immunohistochemical study of intestinal eosinophils in inflammatory bowel disease. J Clin Gastroenterol. 2003;36:120–5. doi: 10.1097/00004836-200302000-00006. [DOI] [PubMed] [Google Scholar]
- 130.Laukoetter MG, Nava P, Nusrat A. Role of the intestinal barrier in inflammatory bowel disease. World J Gastroenterol. 2008;14:401–7. doi: 10.3748/wjg.14.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Furuta GT, Nieuwenhuis EE, Karhausen J, Gleich G, Blumberg RS, Lee JJ, et al. Eosinophils alter colonic epithelial barrier function: role for major basic protein. Am J Physiol Gastrointest Liver Physiol. 2005;289:G890–7. doi: 10.1152/ajpgi.00015.2005. [DOI] [PubMed] [Google Scholar]
- 132.Bode JC. Alcohol and the gastrointestinal tract. Ergeb Inn Med Kinderheilkd. 1980;45:1–75. doi: 10.1007/978-3-642-67632-1_1. [DOI] [PubMed] [Google Scholar]
- 133.Beck IT, Dinda PK. Acute exposure of small intestine to ethanol: effects on morphology and function. Dig Dis Sci. 1981;26:817–38. doi: 10.1007/BF01309614. [DOI] [PubMed] [Google Scholar]
- 134.Ferrier L, Berard F, Debrauwer L, Chabo C, Langella P, Bueno L, et al. Impairment of the intestinal barrier by ethanol involves enteric microflora and mast cell activation in rodents. Am J Pathol. 2006;168:1148–54. doi: 10.2353/ajpath.2006.050617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Banan A, Fields JZ, Decker H, Zhang Y, Keshavarzian A. Nitric oxide and its metabolites mediate ethanol-induced microtubule disruption and intestinal barrier dysfunction. J Pharmacol Exp Ther. 2000;294:997–1008. [PubMed] [Google Scholar]
- 136.Lambert JC, Zhou Z, Wang L, Song Z, McClain CJ, Kang YJ. Prevention of alterations in intestinal permeability is involved in zinc inhibition of acute ethanol-induced liver damage in mice. J Pharmacol Exp Ther. 2003;305:880–6. doi: 10.1124/jpet.102.047852. [DOI] [PubMed] [Google Scholar]
- 137.Barona E, Pirola RC, Leiber CS. Small intestinal damage and changes in cell population produced by ethanol ingestion in the rat. Gastroenterology. 1974;66:226–34. [PubMed] [Google Scholar]
- 138.Shibayama Y, Asaka S, Nakata K. Endotoxin hepatotoxicity augmented by ethanol. Exp Mol Pathol. 1991;55:196–202. doi: 10.1016/0014-4800(91)90053-z. [DOI] [PubMed] [Google Scholar]
- 139.Tamai H, Horie Y, Kato S, Yokoyama H, Ishii H. Long-term ethanol feeding enhances susceptibility of the liver to orally administered lipopolysaccharides in rats. Alcohol Clin Exp Res. 2002;26:75S–80S. doi: 10.1097/01.ALC.0000026981.32386.FD. [DOI] [PubMed] [Google Scholar]
- 140.Gottfried EB, Korsten MA, Lieber CS. Alcohol-induced gastric and duodenal lesions in man. Am J Gastroenterol. 1978;70:587–92. [PubMed] [Google Scholar]
- 141.Parlesak A, Schafer C, Schutz T, Bode JC, Bode C. Increased intestinal permeability to macromolecules and endotoxemia in patients with chronic alcohol abuse in different stages of alcohol-induced liver disease. J Hepatol. 2000;32:742–7. doi: 10.1016/s0168-8278(00)80242-1. [DOI] [PubMed] [Google Scholar]
- 142.Dinda PK, Leddin DJ, Beck IT. Histamine is involved in ethanol-induced jejunal microvascular injury in rabbits. Gastroenterology. 1988;95:1227–33. doi: 10.1016/0016-5085(88)90355-1. [DOI] [PubMed] [Google Scholar]
- 143.Dinda PK, Kossev P, Beck IT, Buell MG. Role of xanthine oxidase-derived oxidants and leukocytes in ethanol-induced jejunal mucosal injury. Dig Dis Sci. 1996;41:2461–70. doi: 10.1007/BF02100144. [DOI] [PubMed] [Google Scholar]
- 144.Robert A. An intestinal disease produced experimentally by a prostaglandin deficiency. Gastroenterology. 1975;69:1045–7. [PubMed] [Google Scholar]
- 145.Whittle BJ. Temporal relationship between cyclooxygenase inhibition, as measured by prostacyclin biosynthesis, and the gastrointestinal damage induced by indomethacin in the rat. Gastroenterology. 1981;80:94–8. [PubMed] [Google Scholar]
- 146.Bjarnason I, Zanelli G, Smith T, Prouse P, Williams P, Smethurst P, et al. Nonsteroidal antiinflammatory drug-induced intestinal inflammation in humans. Gastroenterology. 1987;93:480–9. doi: 10.1016/0016-5085(87)90909-7. [DOI] [PubMed] [Google Scholar]
- 147.Kaufmann HJ, Taubin HL. Nonsteroidal anti-inflammatory drugs activate quiescent inflammatory bowel disease. Ann Intern Med. 1987;107:513–6. doi: 10.7326/0003-4819-107-4-513. [DOI] [PubMed] [Google Scholar]
- 148.Fries JF, Miller SR, Spitz PW, Williams CA, Hubert HB, Bloch DA. Toward an epidemiology of gastropathy associated with nonsteroidal antiinflammatory drug use. Gastroenterology. 1989;96:647–55. doi: 10.1016/s0016-5085(89)80061-7. [DOI] [PubMed] [Google Scholar]
- 149.Lanas A, Sekar MC, Hirschowitz BI. Objective evidence of aspirin use in both ulcer and nonulcer upper and lower gastrointestinal bleeding. Gastroenterology. 1992;103:862–9. doi: 10.1016/0016-5085(92)90018-t. [DOI] [PubMed] [Google Scholar]
- 150.Langman MJ, Weil J, Wainwright P, Lawson DH, Rawlins MD, Logan RF, et al. Risks of bleeding peptic ulcer associated with individual non-steroidal anti-inflammatory drugs. Lancet. 1994;343:1075–8. doi: 10.1016/s0140-6736(94)90185-6. [DOI] [PubMed] [Google Scholar]
- 151.Lanas A, Serrano P, Bajador E, Esteva F, Benito R, Sainz R. Evidence of aspirin use in both upper and lower gastrointestinal perforation. Gastroenterology. 1997;112:683–9. doi: 10.1053/gast.1997.v112.pm9041228. [DOI] [PubMed] [Google Scholar]
- 152.Sigthorsson G, Jacob M, Wrigglesworth J, Somasundaram S, Tavares I, Foster R, et al. Comparison of indomethacin and nimesulide, a selective cyclooxygenase-2 inhibitor, on key pathophysiologic steps in the pathogenesis of nonsteroidal anti-inflammatory drug enteropathy in the rat. Scand J Gastroenterol. 1998;33:728–35. doi: 10.1080/00365529850171675. [DOI] [PubMed] [Google Scholar]
- 153.Sigthorsson G, Tibble J, Hayllar J, Menzies I, Macpherson A, Moots R, et al. Intestinal permeability and inflammation in patients on NSAIDs. Gut. 1998;43:506–11. doi: 10.1136/gut.43.4.506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Oshima T, Miwa H, Joh T. Aspirin induces gastric epithelial barrier dysfunction by activating p38 MAPK via claudin-7. Am J Physiol Cell Physiol. 2008;295:C800–6. doi: 10.1152/ajpcell.00157.2008. [DOI] [PubMed] [Google Scholar]
- 155.Meyer RA, McGinley D, Posalaky Z. The effects of 16,16-dimethyl prostaglandin E2 + aspirin on the canine gastric mucosal barrier. Virchows Arch A Pathol Anat Histopathol. 1987;412:119–25. doi: 10.1007/BF00716183. [DOI] [PubMed] [Google Scholar]
- 156.Wallace JL, Keenan CM, Granger DN. Gastric ulceration induced by nonsteroidal anti-inflammatory drugs is a neutrophil-dependent process. Am J Physiol. 1990;259:G462–7. doi: 10.1152/ajpgi.1990.259.3.G462. [DOI] [PubMed] [Google Scholar]
- 157.Takeuchi K, Ueshima K, Hironaka Y, Fujioka Y, Matsumoto J, Okabe S. Oxygen free radicals and lipid peroxidation in the pathogenesis of gastric mucosal lesions induced by indomethacin in rats. Relation to gastric hypermotility. Digestion. 1991;49:175–84. doi: 10.1159/000200718. [DOI] [PubMed] [Google Scholar]
- 158.Tanaka A, Kunikata T, Mizoguchi H, Kato S, Takeuchi K. Dual action of nitric oxide in pathogenesis of indomethacin-induced small intestinal ulceration in rats. J Physiol Pharmacol. 1999;50:405–17. [PubMed] [Google Scholar]
- 159.Somasundaram S, Sigthorsson G, Simpson RJ, Watts J, Jacob M, Tavares IA, et al. Uncoupling of intestinal mitochondrial oxidative phosphorylation and inhibition of cyclooxygenase are required for the development of NSAID-enteropathy in the rat. Aliment Pharmacol Ther. 2000;14:639–50. doi: 10.1046/j.1365-2036.2000.00723.x. [DOI] [PubMed] [Google Scholar]
- 160.Berkes J, Viswanathan VK, Savkovic SD, Hecht G. Intestinal epithelial responses to enteric pathogens: effects on the tight junction barrier, ion transport, and inflammation. Gut. 2003;52:439–51. doi: 10.1136/gut.52.3.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Wu Z, Milton D, Nybom P, Sjo A, Magnusson KE. Vibrio cholerae hemagglutinin/protease (HA/protease) causes morphological changes in cultured epithelial cells and perturbs their paracellular barrier function. Microb Pathog. 1996;21:111–23. doi: 10.1006/mpat.1996.0047. [DOI] [PubMed] [Google Scholar]
- 162.Mel SF, Fullner KJ, Wimer-Mackin S, Lencer WI, Mekalanos JJ. Association of protease activity in Vibrio cholerae vaccine strains with decreases in transcellular epithelial resistance of polarized T84 intestinal epithelial cells. Infect Immun. 2000;68:6487–92. doi: 10.1128/iai.68.11.6487-6492.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Wu Z, Nybom P, Magnusson KE. Distinct effects of Vibrio cholerae haemagglutinin/protease on the structure and localization of the tight junction-associated proteins occludin and ZO-1. Cell Microbiol. 2000;2:11–7. doi: 10.1046/j.1462-5822.2000.00025.x. [DOI] [PubMed] [Google Scholar]
- 164.Fasano A, Baudry B, Pumplin DW, Wasserman SS, Tall BD, Ketley JM, et al. Vibrio cholerae produces a second enterotoxin, which affects intestinal tight junctions. Proc Natl Acad Sci U S A. 1991;88:5242–6. doi: 10.1073/pnas.88.12.5242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Fasano A, Fiorentini C, Donelli G, Uzzau S, Kaper JB, Margaretten K, et al. Zonula occludens toxin modulates tight junctions through protein kinase C-dependent actin reorganization, in vitro. J Clin Invest. 1995;96:710–20. doi: 10.1172/JCI118114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Schmidt E, Kelly SM, van der Walle CF. Tight junction modulation and biochemical characterisation of the zonula occludens toxin C-and N-termini. FEBS Lett. 2007;581:2974–80. doi: 10.1016/j.febslet.2007.05.051. [DOI] [PubMed] [Google Scholar]
- 167.Fasano A, Uzzau S, Fiore C, Margaretten K. The enterotoxic effect of zonula occludens toxin on rabbit small intestine involves the paracellular pathway. Gastroenterology. 1997;112:839–46. doi: 10.1053/gast.1997.v112.pm9041245. [DOI] [PubMed] [Google Scholar]
- 168.Wang W, Uzzau S, Goldblum SE, Fasano A. Human zonulin, a potential modulator of intestinal tight junctions. J Cell Sci. 2000;113(Pt 24):4435–40. doi: 10.1242/jcs.113.24.4435. [DOI] [PubMed] [Google Scholar]
- 169.Knutton S, Lloyd DR, McNeish AS. Adhesion of enteropathogenic Escherichia coli to human intestinal enterocytes and cultured human intestinal mucosa. Infect Immun. 1987;55:69–77. doi: 10.1128/iai.55.1.69-77.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Jarvis KG, Giron JA, Jerse AE, McDaniel TK, Donnenberg MS, Kaper JB. Enteropathogenic Escherichia coli contains a putative type III secretion system necessary for the export of proteins involved in attaching and effacing lesion formation. Proc Natl Acad Sci U S A. 1995;92:7996–8000. doi: 10.1073/pnas.92.17.7996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Spitz J, Yuhan R, Koutsouris A, Blatt C, Alverdy J, Hecht G. Enteropathogenic Escherichia coli adherence to intestinal epithelial monolayers diminishes barrier function. Am J Physiol. 1995;268:G374–9. doi: 10.1152/ajpgi.1995.268.2.G374. [DOI] [PubMed] [Google Scholar]
- 172.Muza-Moons MM, Schneeberger EE, Hecht GA. Enteropathogenic Escherichia coli infection leads to appearance of aberrant tight junctions strands in the lateral membrane of intestinal epithelial cells. Cell Microbiol. 2004;6:783–93. doi: 10.1111/j.1462-5822.2004.00404.x. [DOI] [PubMed] [Google Scholar]
- 173.Philpott DJ, McKay DM, Sherman PM, Perdue MH. Infection of T84 cells with enteropathogenic Escherichia coli alters barrier and transport functions. Am J Physiol. 1996;270:G634–45. doi: 10.1152/ajpgi.1996.270.4.G634. [DOI] [PubMed] [Google Scholar]
- 174.Yuhan R, Koutsouris A, Savkovic SD, Hecht G. Enteropathogenic Escherichia coli-induced myosin light chain phosphorylation alters intestinal epithelial permeability. Gastroenterology. 1997;113:1873–82. doi: 10.1016/s0016-5085(97)70006-4. [DOI] [PubMed] [Google Scholar]
- 175.Fujita K, Katahira J, Horiguchi Y, Sonoda N, Furuse M, Tsukita S. Clostridium perfringens enterotoxin binds to the second extracellular loop of claudin-3, a tight junction integral membrane protein. FEBS Lett. 2000;476:258–61. doi: 10.1016/s0014-5793(00)01744-0. [DOI] [PubMed] [Google Scholar]
- 176.Katahira J, Sugiyama H, Inoue N, Horiguchi Y, Matsuda M, Sugimoto N. Clostridium perfringens enterotoxin utilizes two structurally related membrane proteins as functional receptors in vivo. J Biol Chem. 1997;272:26652–8. doi: 10.1074/jbc.272.42.26652. [DOI] [PubMed] [Google Scholar]
- 177.Singh U, Mitic LL, Wieckowski EU, Anderson JM, McClane BA. Comparative biochemical and immunocytochemical studies reveal differences in the effects of Clostridium perfringens enterotoxin on polarized CaCo-2 cells versus Vero cells. J Biol Chem. 2001;276:33402–12. doi: 10.1074/jbc.M104200200. [DOI] [PubMed] [Google Scholar]
- 178.Singh U, Van Itallie CM, Mitic LL, Anderson JM, McClane BA. CaCo-2 cells treated with Clostridium perfringens enterotoxin form multiple large complex species, one of which contains the tight junction protein occludin. J Biol Chem. 2000;275:18407–17. doi: 10.1074/jbc.M001530200. [DOI] [PubMed] [Google Scholar]
- 179.Sonoda N, Furuse M, Sasaki H, Yonemura S, Katahira J, Horiguchi Y, et al. Clostridium perfringens enterotoxin fragment removes specific claudins from tight junction strands: Evidence for direct involvement of claudins in tight junction barrier. J Cell Biol. 1999;147:195–204. doi: 10.1083/jcb.147.1.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Chakrabarti G, McClane BA. The importance of calcium influx, calpain and calmodulin for the activation of CaCo-2 cell death pathways by Clostridium perfringens enterotoxin. Cell Microbiol. 2005;7:129–46. doi: 10.1111/j.1462-5822.2004.00442.x. [DOI] [PubMed] [Google Scholar]
- 181.Chakrabarti G, Zhou X, McClane BA. Death pathways activated in CaCo-2 cells by Clostridium perfringens enterotoxin. Infect Immun. 2003;71:4260–70. doi: 10.1128/IAI.71.8.4260-4270.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Meddings J. The significance of the gut barrier in disease. Gut. 2008;57:438–40. doi: 10.1136/gut.2007.143172. [DOI] [PubMed] [Google Scholar]
- 183.Buhner S, Buning C, Genschel J, Kling K, Herrmann D, Dignass A, et al. Genetic basis for increased intestinal permeability in families with Crohn’s disease: role of CARD15 3020insC mutation? Gut. 2006;55:342–7. doi: 10.1136/gut.2005.065557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Takeuchi K, Maiden L, Bjarnason I. Genetic aspects of intestinal permeability in inflammatory bowel disease. In: Chadwick D, Goode J, editors. Inflammatory bowel disease-crossroads of microbes, epithelium and immune systems. Chichester: Wiley; 2004. pp. 151–63. [PubMed] [Google Scholar]
- 185.Mankertz J, Schulzke J. Altered permeability in inflammatory bowel disease: pathophysiology and clinical implications. Curr Opin Gastroenterol. 2007;23:379–83. doi: 10.1097/MOG.0b013e32816aa392. [DOI] [PubMed] [Google Scholar]
- 186.Heyman M, Desjeux JF. Cytokine-induced alteration of the epithelial barrier to food antigens in disease. Ann N Y Acad Sci. 2003;202:304–11. doi: 10.1111/j.1749-6632.2000.tb05258.x. [DOI] [PubMed] [Google Scholar]
- 187.Heymann M. Gut barrier dysfunction in food allergy. Eur J Gastroenterol Hepatol. 2005;17:1279–85. doi: 10.1097/00042737-200512000-00003. [DOI] [PubMed] [Google Scholar]
- 188.Bjarnason I, Peters TJ. In vitro determination of small intestinal permeability: demonstration of a persistent defect in patients with coeliac disease. Gut. 1984;25:145–50. doi: 10.1136/gut.25.2.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Schulzke JD, Bentzel CJ, Schulzke I, Riecken EO, Fromm M. Epithelial tight junction structure in the jejunum of children with acute and treated celiac sprue. Pediatr Res. 1998;43:435–41. doi: 10.1203/00006450-199804000-00001. [DOI] [PubMed] [Google Scholar]
- 190.Wyatt J, Vogelsang H, Hubl W, Waldhoer T, Lochs H. Intestinal permeability and the prediction of relapse in Crohn’s disease. Lancet. 1993;341:1437–9. doi: 10.1016/0140-6736(93)90882-h. [DOI] [PubMed] [Google Scholar]
- 191.D’Inca R, Di Leo V, Corrao G, Martines D, D’Odorico A, Mestriner C, et al. Intestinal permeability test as a predictor of clinical course in Crohn’s disease. Am J Gastroenterol. 1999;94:2956–60. doi: 10.1111/j.1572-0241.1999.01444.x. [DOI] [PubMed] [Google Scholar]
- 192.Ventura MT, Polimeno L, Amoruso AC, Gatti F, Annoscia E, Marinaro M, et al. Intestinal permeability in patients with adverse reactions to food. Dig Liver Dis. 2006;38:732–6. doi: 10.1016/j.dld.2006.06.012. [DOI] [PubMed] [Google Scholar]
- 193.Laudat A, Arnaud P, Napoly A, Brion F. The intestinal permeability test applied to the diagnosis of food allergy in pediatrics. West Indian Medical Journal. 1994;43:87–8. [PubMed] [Google Scholar]
- 194.Lacaille F, Laurent J, Bousquet J. Life-threatening food allergy in a child treated with FK506. J Pediatr Gastroenterol Nutr. 1997;25:228–9. doi: 10.1097/00005176-199708000-00019. [DOI] [PubMed] [Google Scholar]
- 195.Asante-Korang A, Boyle GJ, Webber SA, Miller SA, Fricker FJ. Experience of FK506 immune suppression in pediatric heart transplantation: a study of long-term adverse effects. J Heart Lung Transplant. 1996;15:415–22. [PubMed] [Google Scholar]
- 196.Granot E, Yakobovich E, Bardenstein R. Tacrolimus immunosuppression - an association with asymptomatic eosinophilia and elevated total and specific IgE levels. Pediatr Transplant. 2006;10:690–3. doi: 10.1111/j.1399-3046.2006.00542.x. [DOI] [PubMed] [Google Scholar]
- 197.Fisher A, Schwartz M, Mor E, Sheiner P, Emre S, Guy S, et al. Gastrointestinal toxicity associated with FK 506 in liver transplant recipients. Transplant Proc. 1994;26:3106–7. [PubMed] [Google Scholar]
- 198.Gabe SM, Bjarnason I, Tolou-Ghamari Z, Tredger JM, Johnson PG, Barclay GR, et al. The effect of tacrolimus (FK506) on intestinal barrier function and cellular energy production in humans. Gastroenterology. 1998;115:67–74. doi: 10.1016/s0016-5085(98)70366-x. [DOI] [PubMed] [Google Scholar]
- 199.Boyle RJ, Hardikar W, Tang ML. The development of food allergy after liver transplantation. Liver Transpl. 2005;11:326–30. doi: 10.1002/lt.20368. [DOI] [PubMed] [Google Scholar]
- 200.Legendre C, Caillat-Zucman S, Samuel D, Morelon S, Bismuth H, Bach JF, et al. Transfer of symptomatic peanut allergy to the recipient of a combined liver-and-kidney transplant. N Engl J Med. 1997;337:822–4. doi: 10.1056/NEJM199709183371204. [DOI] [PubMed] [Google Scholar]
- 201.Chehade M, Nowak-Wegrzyn A, Kaufman SS, Fishbein TM, Tschernia A, LeLeiko NS. De novo food allergy after intestinal transplantation: a report of three cases. J Pediatr Gastroenterol Nutr. 2004;38:545–7. doi: 10.1097/00005176-200405000-00017. [DOI] [PubMed] [Google Scholar]
- 202.Ozdemir O, Arrey-Mensah A, Sorensen RU. Development of multiple food allergies in children taking tacrolimus after heart and liver transplantation. Pediatr Transplant. 2006;10:380–3. doi: 10.1111/j.1399-3046.2005.00474.x. [DOI] [PubMed] [Google Scholar]
- 203.Madsen KL, Yanchar NL, Sigalet DL, Reigel T, Fedorak RN. FK506 increases permeability in rat intestine by inhibiting mitochondrial function. Gastroenterology. 1995;109:107–14. doi: 10.1016/0016-5085(95)90274-0. [DOI] [PubMed] [Google Scholar]
- 204.Madara JL, Parkos C, Colgan S, Nusrat A, Atisook K, Kaoutzani P. The movement of solutes and cells across tight junctions. Ann N Y Acad Sci. 1992;664:47–60. doi: 10.1111/j.1749-6632.1992.tb39748.x. [DOI] [PubMed] [Google Scholar]
- 205.Liu J, Farmer JD, Jr, Lane WS, Friedman J, Weissman I, Schreiber SL. Calcineurin is a common target of cyclophilin-cyclosporin A and FKBP-FK506 complexes. Cell. 1991;66:807–15. doi: 10.1016/0092-8674(91)90124-h. [DOI] [PubMed] [Google Scholar]
- 206.Forbes EE, Groschwitz K, Abonia JP, Brandt EB, Cohen E, Blanchard C, et al. IL-9- and mast cell-mediated intestinal permeability predisposes to oral antigen hypersensitivity. J Exp Med. 2008;205:897–913. doi: 10.1084/jem.20071046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Andre C, Andre F, Colin L, Cavagna S. Measurement of intestinal permeability to mannitol and lactulose as a means of diagnosing food allergy and evaluating therapeutic effectiveness of disodium cromoglycate. Ann Allergy. 1987;59:127–30. [PubMed] [Google Scholar]
- 208.Li XM, Schofield BH, Huang CK, Kleiner GI, Sampson HA. A murine model of IgE-mediated cow’s milk hypersensitivity. J Allergy Clin Immunol. 1999;103:206–14. doi: 10.1016/s0091-6749(99)70492-6. [DOI] [PubMed] [Google Scholar]
- 209.Brandt EB, Strait RT, Hershko D, Wang Q, Muntel EE, Scribner TA, et al. Mast cells are required for experimental oral allergen-induced diarrhea. J Clin Invest. 2003;112:1666–77. doi: 10.1172/JCI19785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Berin MC, Kiliaan AJ, Yang PC, Groot JA, Kitamura Y, Perdue MH. The influence of mast cells on pathways of transepithelial antigen transport in rat intestine. J Immunol. 1998;161:2561–6. [PubMed] [Google Scholar]
- 211.Clayburgh DR, Shen L, Turner JR. A porous defense: the leaky epithelial barrier in intestinal disease. Lab Invest. 2004;84:282–91. doi: 10.1038/labinvest.3700050. [DOI] [PubMed] [Google Scholar]
- 212.Secondulfo M, de Magistris L, Fiandra R, Caserta L, Belletta M, Tartaglione MT, et al. Intestinal permeability in Crohn’s disease patients and their first degree relatives. Dig Liver Dis. 2001;33:680–5. doi: 10.1016/s1590-8658(01)80045-1. [DOI] [PubMed] [Google Scholar]
- 213.Soderholm JD, Olaison G, Lindberg E, Hannestad U, Vindels A, Tysk C, et al. Different intestinal permeability patterns in relatives and spouses of patients with Crohn’s disease: an inherited defect in mucosal defence? Gut. 1999;44:96–100. doi: 10.1136/gut.44.1.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Peeters M, Geypens B, Claus D, Nevens H, Ghoos Y, Verbeke G, et al. Clustering of increased small intestinal permeability in families with Crohn’s disease. Gastroenterology. 1997;113:802–7. doi: 10.1016/s0016-5085(97)70174-4. [DOI] [PubMed] [Google Scholar]
- 215.Hilsden RJ, Meddings JB, Sutherland LR. Intestinal permeability changes in response to acetylsalicylic acid in relatives of patients with Crohn’s disease. Gastroenterology. 1996;110:1395–403. doi: 10.1053/gast.1996.v110.pm8613043. [DOI] [PubMed] [Google Scholar]
- 216.Irvine EJ, Marshall JK. Increased intestinal permeability precedes the onset of Crohn’s disease in a subject with familial risk. Gastroenterology. 2000;119:1740–4. doi: 10.1053/gast.2000.20231. [DOI] [PubMed] [Google Scholar]
- 217.Olson TS, Reuter BK, Scott KG, Morris MA, Wang XM, Hancock LN, et al. The primary defect in experimental ileitis originates from a nonhematopoietic source. J Exp Med. 2006;203:541–52. doi: 10.1084/jem.20050407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Resta-Lenert S, Smitham J, Barrett KE. Epithelial dysfunction associated with the development of colitis in conventionally housed mdr1a−/− mice. Am J Physiol Gastrointest Liver Physiol. 2005;289:G153–62. doi: 10.1152/ajpgi.00395.2004. [DOI] [PubMed] [Google Scholar]
- 219.Su L, Shen L, Clayburgh DR, Nalle SC, Sullivan EA, Meddings JB, et al. Targeted Epithelial Tight Junction Dysfunction Causes Immune Activation and Contributes to Development of Experimental Colitis. Gastroenterology. 2008 doi: 10.1053/j.gastro.2008.10.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Hugot JP, Chamaillard M, Zouali H, Lesage S, Cezard JP, Belaiche J, et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature. 2001;411:599–603. doi: 10.1038/35079107. [DOI] [PubMed] [Google Scholar]
- 221.Ogura Y, Bonen DK, Inohara N, Nicolae DL, Chen FF, Ramos R, et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature. 2001;411:603–6. doi: 10.1038/35079114. [DOI] [PubMed] [Google Scholar]
- 222.Gitter AH, Bendfeldt K, Schulzke JD, Fromm M. Leaks in the epithelial barrier caused by spontaneous and TNF-alpha-induced single-cell apoptosis. Faseb J. 2000;14:1749–53. doi: 10.1096/fj.99-0898com. [DOI] [PubMed] [Google Scholar]
- 223.Kucharzik T, Walsh SV, Chen J, Parkos CA, Nusrat A. Neutrophil transmigration in inflammatory bowel disease is associated with differential expression of epithelial intercellular junction proteins. Am J Pathol. 2001;159:2001–9. doi: 10.1016/S0002-9440(10)63051-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Zeissig S, Burgel N, Gunzel D, Richter J, Mankertz J, Wahnschaffe U, et al. Changes in expression and distribution of claudin 2, 5 and 8 lead to discontinuous tight junctions and barrier dysfunction in active Crohn’s disease. Gut. 2007;56:61–72. doi: 10.1136/gut.2006.094375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Madara JL, Stafford J. Interferon-gamma directly affects barrier function of cultured intestinal epithelial monolayers. J Clin Invest. 1989;83:724–7. doi: 10.1172/JCI113938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Mullin JM, Laughlin KV, Marano CW, Russo LM, Soler AP. Modulation of tumor necrosis factor-induced increase in renal (LLC-PK1) transepithelial permeability. Am J Physiol. 1992;263:F915–24. doi: 10.1152/ajprenal.1992.263.5.F915. [DOI] [PubMed] [Google Scholar]
- 227.Baert FJ, D’Haens GR, Peeters M, Hiele MI, Schaible TF, Shealy D, et al. Tumor necrosis factor alpha antibody (infliximab) therapy profoundly down-regulates the inflammation in Crohn’s ileocolitis. Gastroenterology. 1999;116:22–8. doi: 10.1016/s0016-5085(99)70224-6. [DOI] [PubMed] [Google Scholar]
- 228.Suenaert P, Bulteel V, Lemmens L, Noman M, Geypens B, Van Assche G, et al. Anti-tumor necrosis factor treatment restores the gut barrier in Crohn’s disease. Am J Gastroenterol. 2002;97:2000–4. doi: 10.1111/j.1572-0241.2002.05914.x. [DOI] [PubMed] [Google Scholar]
- 229.Madara JL, Trier JS. Structural abnormalities of jejunal epithelial cell membranes in celiac sprue. Lab Invest. 1980;43:254–61. [PubMed] [Google Scholar]
- 230.Smecuol E, Bai JC, Vazquez H, Kogan Z, Cabanne A, Niveloni S, et al. Gastrointestinal permeability in celiac disease. Gastroenterology. 1997;112:1129–36. doi: 10.1016/s0016-5085(97)70123-9. [DOI] [PubMed] [Google Scholar]
- 231.van Elburg RM, Uil JJ, Mulder CJ, Heymans HS. Intestinal permeability in patients with coeliac disease and relatives of patients with coeliac disease. Gut. 1993;34:354–7. doi: 10.1136/gut.34.3.354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Batt RM, Carter MW, McLean L. Morphological and biochemical studies of a naturally occurring enteropathy in the Irish setter dog: a comparison with coeliac disease in man. Res Vet Sci. 1984;37:339–46. [PubMed] [Google Scholar]
- 233.Hall EJ, Batt RM. Abnormal permeability precedes the development of a gluten sensitive enteropathy in Irish setter dogs. Gut. 1991;32:749–53. doi: 10.1136/gut.32.7.749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Lammers KM, Lu R, Brownley J, Lu B, Gerard C, Thomas K, et al. Gliadin induces an increase in intestinal permeability and zonulin release by binding to the chemokine receptor CXCR3. Gastroenterology. 2008;135:194–204. e3. doi: 10.1053/j.gastro.2008.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Drago S, El Asmar R, Di Pierro M, Grazia Clemente M, Tripathi A, Sapone A, et al. Gliadin, zonulin and gut permeability: Effects on celiac and non-celiac intestinal mucosa and intestinal cell lines. Scand J Gastroenterol. 2006;41:408–19. doi: 10.1080/00365520500235334. [DOI] [PubMed] [Google Scholar]
- 236.Fasano A, Not T, Wang W, Uzzau S, Berti I, Tommasini A, et al. Zonulin, a newly discovered modulator of intestinal permeability, and its expression in coeliac disease. Lancet. 2000;355:1518–9. doi: 10.1016/S0140-6736(00)02169-3. [DOI] [PubMed] [Google Scholar]
- 237.Clemente MG, De Virgiliis S, Kang JS, Macatagney R, Musu MP, Di Pierro MR, et al. Early effects of gliadin on enterocyte intracellular signalling involved in intestinal barrier function. Gut. 2003;52:218–23. doi: 10.1136/gut.52.2.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Montalto M, Cuoco L, Ricci R, Maggiano N, Vecchio FM, Gasbarrini G. Immunohistochemical analysis of ZO-1 in the duodenal mucosa of patients with untreated and treated celiac disease. Digestion. 2002;65:227–33. doi: 10.1159/000063817. [DOI] [PubMed] [Google Scholar]
- 239.Pizzuti D, Bortolami M, Mazzon E, Buda A, Guariso G, D’Odorico A, et al. Transcriptional downregulation of tight junction protein ZO-1 in active coeliac disease is reversed after a gluten-free diet. Dig Liver Dis. 2004;36:337–41. doi: 10.1016/j.dld.2004.01.013. [DOI] [PubMed] [Google Scholar]
- 240.Wolters VM, Wijmenga C. Genetic background of celiac disease and its clinical implications. Am J Gastroenterol. 2008;103:190–5. doi: 10.1111/j.1572-0241.2007.01471.x. [DOI] [PubMed] [Google Scholar]
- 241.Mooradian AD, Morley JE, Levine AS, Prigge WF, Gebhard RL. Abnormal intestinal permeability to sugars in diabetes mellitus. Diabetologia. 1986;29:221–4. doi: 10.1007/BF00454879. [DOI] [PubMed] [Google Scholar]
- 242.Carratu R, Secondulfo M, de Magistris L, Iafusco D, Urio A, Carbone MG, et al. Altered intestinal permeability to mannitol in diabetes mellitus type I. J Pediatr Gastroenterol Nutr. 1999;28:264–9. doi: 10.1097/00005176-199903000-00010. [DOI] [PubMed] [Google Scholar]
- 243.Secondulfo M, Riegler G, De Magistris L, Belletta M, Fiandra R, Caserta L, et al. Intestinal permeability assessment before and after ileal pouch-anal anastomosis. Minerva Gastroenterol Dietol. 2004;50:155–63. [PubMed] [Google Scholar]
- 244.Secondulfo M, Iafusco D, Carratu R, deMagistris L, Sapone A, Generoso M, et al. Ultrastructural mucosal alterations and increased intestinal permeability in non-celiac, type I diabetic patients. Dig Liver Dis. 2004;36:35–45. doi: 10.1016/j.dld.2003.09.016. [DOI] [PubMed] [Google Scholar]
- 245.Neu J, Reverte CM, Mackey AD, Liboni K, Tuhacek-Tenace LM, Hatch M, et al. Changes in intestinal morphology and permeability in the biobreeding rat before the onset of type 1 diabetes. J Pediatr Gastroenterol Nutr. 2005;40:589–95. doi: 10.1097/01.mpg.0000159636.19346.c1. [DOI] [PubMed] [Google Scholar]
- 246.Watts T, Berti I, Sapone A, Gerarduzzi T, Not T, Zielke R, et al. Role of the intestinal tight junction modulator zonulin in the pathogenesis of type I diabetes in BB diabetic-prone rats. Proc Natl Acad Sci U S A. 2005;102:2916–21. doi: 10.1073/pnas.0500178102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Sapone A, de Magistris L, Pietzak M, Clemente MG, Tripathi A, Cucca F, et al. Zonulin upregulation is associated with increased gut permeability in subjects with type 1 diabetes and their relatives. Diabetes. 2006;55:1443–9. doi: 10.2337/db05-1593. [DOI] [PubMed] [Google Scholar]
- 248.Saunders PR, Kosecka U, McKay DM, Perdue MH. Acute stressors stimulate ion secretion and increase epithelial permeability in rat intestine. Am J Physiol. 1994;267:G794–9. doi: 10.1152/ajpgi.1994.267.5.G794. [DOI] [PubMed] [Google Scholar]
- 249.Kiliaan AJ, Saunders PR, Bijlsma PB, Berin MC, Taminiau JA, Groot JA, et al. Stress stimulates transepithelial macromolecular uptake in rat jejunum. Am J Physiol. 1998;275:G1037–44. doi: 10.1152/ajpgi.1998.275.5.G1037. [DOI] [PubMed] [Google Scholar]
- 250.Mazzon E, Sturniolo GC, Puzzolo D, Frisina N, Fries W. Effect of stress on the paracellular barrier in the rat ileum. Gut. 2002;51:507–13. doi: 10.1136/gut.51.4.507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Santos J, Saunders PR, Hanssen NP, Yang PC, Yates D, Groot JA, et al. Corticotropin-releasing hormone mimics stress-induced colonic epithelial pathophysiology in the rat. Am J Physiol. 1999;277:G391–9. doi: 10.1152/ajpgi.1999.277.2.G391. [DOI] [PubMed] [Google Scholar]
- 252.Santos J, Bayarri C, Saperas E, Nogueiras C, Antolin M, Mourelle M, et al. Characterisation of immune mediator release during the immediate response to segmental mucosal challenge in the jejunum of patients with food allergy. Gut. 1999;45:553–8. doi: 10.1136/gut.45.4.553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Santos J, Benjamin M, Yang PC, Prior T, Perdue MH. Chronic stress impairs rat growth and jejunal epithelial barrier function: role of mast cells. Am J Physiol Gastrointest Liver Physiol. 2000;278:G847–54. doi: 10.1152/ajpgi.2000.278.6.G847. [DOI] [PubMed] [Google Scholar]
- 254.Castagliuolo I, Wershil BK, Karalis K, Pasha A, Nikulasson ST, Pothoulakis C. Colonic mucin release in response to immobilization stress is mast cell dependent. Am J Physiol. 1998;274:G1094–100. doi: 10.1152/ajpgi.1998.274.6.G1094. [DOI] [PubMed] [Google Scholar]
- 255.Levenstein S, Prantera C, Varvo V, Scribano ML, Andreoli A, Luzi C, et al. Stress and exacerbation in ulcerative colitis: a prospective study of patients enrolled in remission. Am J Gastroenterol. 2000;95:1213–20. doi: 10.1111/j.1572-0241.2000.02012.x. [DOI] [PubMed] [Google Scholar]
- 256.Collins SM. Stress and the Gastrointestinal Tract IV. Modulation of intestinal inflammation by stress: basic mechanisms and clinical relevance. Am J Physiol Gastrointest Liver Physiol. 2001;280:G315–8. doi: 10.1152/ajpgi.2001.280.3.G315. [DOI] [PubMed] [Google Scholar]
- 257.Ringel Y, Drossman DA. Psychosocial aspects of Crohn’s disease. Surg Clin North Am. 2001;81:231–52. x. doi: 10.1016/s0039-6109(05)70283-8. [DOI] [PubMed] [Google Scholar]
- 258.Million M, Tache Y, Anton P. Susceptibility of Lewis and Fischer rats to stress-induced worsening of TNB-colitis: protective role of brain CRF. Am J Physiol. 1999;276:G1027–36. doi: 10.1152/ajpgi.1999.276.4.G1027. [DOI] [PubMed] [Google Scholar]
- 259.Qiu BS, Vallance BA, Blennerhassett PA, Collins SM. The role of CD4+ lymphocytes in the susceptibility of mice to stress-induced reactivation of experimental colitis. Nat Med. 1999;5:1178–82. doi: 10.1038/13503. [DOI] [PubMed] [Google Scholar]
- 260.Bennett EJ, Tennant CC, Piesse C, Badcock CA, Kellow JE. Level of chronic life stress predicts clinical outcome in irritable bowel syndrome. Gut. 1998;43:256–61. doi: 10.1136/gut.43.2.256. [DOI] [PMC free article] [PubMed] [Google Scholar]