

Abstract
Recent clinical studies indicate that traumatic brain injury (TBI) produces chronic and progressive neurodegenerative changes leading to late neurological dysfunction but little is known about the mechanisms underlying such changes. Microglial-mediated neuroinflammation is an important secondary injury mechanism after TBI. In human studies, microglial activation has been found to persist for many years after the initial brain trauma, particularly after moderate-to-severe TBI. In the present study, adult C57Bl/6 mice were subjected to single moderate-level controlled cortical impact and were followed up by longitudinal T2-weighted magnetic resonance imaging in combination with stereological histological assessment of lesion volume expansion, neuronal loss and microglial activation for up to 1 year after TBI. Persistent microglial activation was observed in the injured cortex through 1 year post-injury, and was associated with progressive lesion expansion, hippocampal neurodegeneration, and loss of myelin. Notably, highly activated microglia that expressed major histocompatibility complex class II (CR3/43), CD68 and NADPH oxidase (NOX2) were detected at the margins of the expanding lesion at 1 year post-injury; biochemical markers of neuroinflammation and oxidative stress were significantly elevated at this time point. These data support emerging clinical TBI findings and provide a mechanistic link between TBI-induced chronic microglial activation and progressive neurodegeneration.
Keywords: Chronic microglial activation, NADPH oxidase, Progressive neurodegeneration, Traumatic brain injury
INTRODUCTION
Traumatic brain injury (TBI) causes cell death and neurological dysfunction through secondary biochemical changes; the latter reflect delayed and potentially reversible molecular and cellular pathophysiological mechanisms (1). These processes are characterized by neuronal cell death, astrocyte activation, infiltration of peripheral monocytes and activation of resident microglia (1).
TBI initiates a complex array of inflammatory responses following TBI (2). There is rapid proliferation and migration of resident microglia to the site of injury in response to extracellular ATP released by the injured tissue (3, 4). Upon activation, microglia undergo marked changes in cell morphology and behavior, i.e. they contract their processes and transform from a resting state with a ramified cellular morphology to an activated state with an amoeboid-like cellular morphology. Activated microglia can secrete many factors including pro- and anti-inflammatory cytokines, chemokines and neurotrophic factors that play an important role in determining the molecular phenotype and functional response of microglia after brain injury (5). Pro-inflammatory molecules, such as interferon-γ and lipopolysaccharide, promote a ‘classical activation’ phenotype (also known as M1 state), which produces high levels of pro-inflammatory cytokines and oxidative metabolites that are essential for host defense and phagocytic activity (6). However, Excessive M1-polarization can lead to exacerbation of injury and progressive tissue destruction. Conversely, anti-inflammatory cytokines, such as interleukin (IL)-4 or IL-10, promote ‘alternative activation’ (M2a state) or ‘acquired deactivated’ (M2c state) microglial phenotypes, respectively (5), which may suppress destructive M1 immune responses and promote repair processes such as angiogenesis and extracellular matrix remodeling after TBI. While much research has focused on the mechanisms underlying the inflammatory response in the acute phase after TBI, the effects of chronic microglial activation after TBI have received more limited attention.
Inflammation with microglial activation is increasingly recognized as a component of many chronic neurodegenerative diseases (7, 8). It has been suggested that damaged-associated molecular pattern molecules released by injured neurons can interact with pattern recognition receptors on activated microglia (e.g. toll-like receptors), thereby triggering a self-perpetuating cycle of injury with prolonged, microglial activation that contributes to neurodegeneration (9). Human and animal studies indicate that microglia are chronically activated for weeks to years after brain trauma (10–14). Persistent microglial activation has been demonstrated in animal models of TBI and is associated with increased expression of IL-1β and tumor necrosis factor (15). A recent clinical study utilizing the PET ligand [11C](R)PK11195 to assess chronic microglial activation in patients who sustained moderate to severe TBI months before demonstrated significantly increased binding bilaterally at sites distant from areas of focal injury, which was correlated with cognitive dysfunction (14). Furthermore, postmortem studies have also demonstrated chronic upregulation of reactive microglia in white matter of the corpus callosum and the frontal lobe of TBI patients from months to many years after the trauma (10, 11, 16). Thus, experimental and clinical evidence now suggest that TBI should not be viewed as a static, acute disorder. Rather, TBI initiates chronic biochemical processes leading to prolonged neuroinflammation and microglial activation that may contribute to late neurological dysfunction. However, the relationship of chronic microglial activation to delayed lesion expansion and selected neurodegeneration in a clinically relevant injury model has not been characterized in detail.
In the present study, we examined effects of controlled cortical impact (CCI) in adult male mice on neuroinflammation, lesion development and hippocampal degeneration through 1 year post-injury. Longitudinal T2-weighted magnetic resonance imaging (MRI) was combined with stereological histological assessment of lesion volume expansion, neuronal loss and markers of classical and alternative microglial activation over time. We show that microglia are classically activated up to 1 year after moderate-level focal TBI and are associated with progressive neurodegeneration; data consistent with emerging clinical TBI findings that implicate chronic microglial activation in the post-traumatic neurodegeneration.
MATERIALS AND METHODS
Controlled Cortical Impact Injury
All surgical procedures were carried out in accordance with protocols approved by University of Maryland School of Medicine Institutional Animal Care and Use Committee. Our custom-designed Controlled Cortical Impact (CCI) injury device consists of a microprocessor-controlled pneumatic impactor with a 3.5-mm-diameter tip (17). Adult male C57Bl/6 mice (3-month-old, 22–25 g; Taconic Farms Inc., Rensselaer, NY) were anesthetized with isoflurane evaporated in a gas mixture containing 70% N2O and 30% O2 and administered through a nose mask (induction at 4% and maintenance at 2%). Depth of anesthesia was assessed by monitoring respiration rate and pedal withdrawal reflexes. Mice were placed on a heated pad, and core body temperature was maintained at 37°C. The head was mounted in a stereotaxic frame, and the surgical site was clipped and cleaned with Nolvasan and ethanol scrubs. A 10-mm midline incision was made over the skull, the skin and fascia were reflected, and a 4-mm craniotomy was made on the central aspect of the left parietal bone. The impounder tip of the injury device was then extended to its full stroke distance (44 mm), positioned to the surface of the exposed dura, and reset to impact the cortical surface. Moderate-level injury was induced using an impactor velocity of 6 m/s and deformation depth of 2 mm as previously described (18). After injury, the incision was closed with interrupted 6–0 silk sutures, anesthesia was terminated, and the animal was placed into a heated cage to maintain normal core temperature for 45 minutes post-injury. All animals were monitored carefully for at least 4 hours after surgery and then daily. Sham animals underwent the same procedure as injured mice except for the impact.
In study 1, a cohort of moderate-level CCI TBI mice (n = 5) were used in longitudinal T2-weighted MRI analysis up to 3 months post-injury (MRI study 1). Another cohort of CCI TBI mice (n = 5) underwent T2-weighted MRI at 6 and 12 months post-injury (MRI study 2). Cortical tissue was harvested for biochemical analysis at 12 months post-injury. A group of age-matched sham-injured mice (n = 6) were used as controls for the biochemical analysis.
In study 2, cohorts of moderate-level CCI TBI mice (n = 6/group) were used for histological analysis at 1, 5, 12 or 52 weeks after injury. Cohorts of sham-injured mice were used as non-injured controls at 1 week and age-matched non-injured controls were used at 52 weeks post-injury (n = 6/group).
Longitudinal In Vivo MRI Analysis
In MRI study 1 (n = 5), lesion volumes were quantified within the first week after TBI, as well as at 2 and 3 months post-injury using T2-weighted imaging protocols as previously described (18). In MRI study 2 (n = 5), lesion volumes were quantified at 6 and 12 months post-injury. Briefly, anesthetized TBI mice were placed in a custom built heated plexiglass mouse holder and a respiratory motion detector was positioned over the thorax to facilitate respiratory gating. The plexiglass holder was placed in the center of the 7 Tesla magnet bore (Bruker Medical Inc., Billerica, MA), and a 72-mm proton-tuned birdcage coil was positioned over the mouse brain. Field homogeneity across the brain was optimized and a sagittal scout images were acquired (RARE i.e. rapid acquisition relaxation enhancement pulse sequence; 3 × 3 cm field of vision; 128 × 128 resolution; repetition time to echo time, 1,500/10 milliseconds with a RARE factor of 8, resulting in effective echo time 40 msec). Multi-slice, multi-echo T2-weighted images were acquired using the following parameters: 3 × 3 cm field of vision, 256 × 256 resolution, 1,500/10 msec repetition time to echo time, 8 RARE factor, echo time 40 msec, 10 × slices, 0.75-mm slice thickness. Signal hyperintensity measurements were made from multi-slice, multi-echo T2-weighted images of the TBI mouse brain in the coronal plane. Briefly, using ImageJ software (Version 1.47) the hyperintense regions of interest on both ipsilateral and contralateral hemispheres were manually traced using the freehand selection tool by an investigator blinded to experimental time point. Slices were manually thresholded based on the intensity value of the contralateral thalamic area, which did not show injury dependent signal intensity changes. Lesion volumes were quantified from the summation of areas of hyperintensity on each slice (total = 10 × slices), multiplied by slice thickness (0.75 mm), for both the ipsilateral and contralateral hemispheres. The contralateral volumes were subtracted from ipsilateral volumes to obtain TBI-induced lesion volumes (expressed in mm3) at each time point.
Immunohistochemistry
Immunohistochemistry was performed on 20- or 60-μm coronal sections and standard techniques were employed. Sections were incubated with anti-Iba1 (1:1000, #019-19741; Wako Chemicals, Richmond, VA), anti-major histocompatibility complex class II (MHCII) (Clone CR3/43, 1:800, #M0775, Dako, Carpinteria, CA) or anti-Ym1 (1:600, #01404, Stem Cell Technologies, Vancouver, Canada) overnight, washed in 1 × phosphate-buffered saline (PBS) (3 times) and incubated with biotinylated anti-rabbit IgG antibody (Vector Laboratories, Burlingame, CA) for 2 hours at room temperature. Sections were incubated in avidin–biotin–horseradish peroxidase solution (Vectastain elite ABC kit, Vector Laboratories) for 1 hour and then reacted with 3,3′-diaminobenzidine (Vector Laboratories) for color development. The Iba1-stained coronal sections were counterstained with cresyl violet prior to mounting for immunohistochemical analysis using a Leica DM4000B microscope.
Neuronal Loss and Lesion Volume Assessment
Cresyl violet-stained 60-μm coronal sections were used for neuronal loss and lesion volume analysis. For TBI-induced hippocampal neuronal cell loss, the optical fractionator method of stereology was employed as previously described (19). Briefly, every fourth 60-μm section between −1.22 mm and −2.54 mm from bregma was analyzed beginning from a random start point (i.e. the section where different hippocampal sub-regions were distinctly visible). A total of 5 sections were analyzed. The optical dissector had a size of 50 μm by 50 μm in the x and y-axis, respectively, with a height of 10 μm and guard-zone of 4 μm from the top of the section. The sampled region for each hippocampal sub-region was demarcated in the injured hemisphere and cresyl-violet neuronal cell bodies were counted using Stereoinvestigator software (MBF Biosciences, Williston, VT). For the Cornu Ammonis (CA) 1 (CA1) and CA3 sub-regions, a grid spacing of 75 μm in the x-axis and 100 μm in the y-axis was used, resulting in an area fraction of 1/12th. For the dentate gyrus sub-region, a grid spacing of 175 μm in the x-axis and 100 μm in the y-axis was used, resulting in an area fraction of 1/28th. The volume of each hippocampal sub-region was measured using the Cavalieri estimator method with a grid spacing of 50 μm. The number of surviving neurons in each hippocampal sub-region was divided by the volume of the region of interest to obtain the cellular density expressed in counts/mm3. According to best stereological practice, all stereological probes were optimized to obtain a Gunderson coefficient of error (CE, m = 1) value of less than 0.10 for the TBI tissue. TBI-induced lesion volume was quantified based on the Cavalieri method of stereology as previously described (19). Briefly, out of the total 96 60 μm sections, every eighth section was analyzed beginning from a random start point (i.e. random selection of a 60 μm section from numbers 1 to 6) and a total of 12 sections were analyzed. Using the Cavalieri estimator with a grid spacing of 100 μm, the missing tissue on the injured hemisphere was outlined in the Stereologer 2000 program software (Systems Planning and Analysis, Alexandria, VA) to quantify the TBI-induced lesion volume at each time point.
Stereological Assessment of Microglia Activation Phenotypes
Classification of microglial activation phenotypes in the sham and TBI cortex was performed as previously described (19). Stereoinvestigator software was used to count the number of microglia in each of the 3 morphological phenotypes (ramified, hypertrophic and bushy) using the optical fractionator method of unbiased stereology. The sampled region was the ipsilateral cortex between −1.22 mm and −2.54 mm from bregma. Every fourth 60-μm section was analyzed beginning from the same random start point that was used for neuronal cell counts. A total of 5 sections were analyzed. The optical dissector had a size of 50 μm by 50 μm in the x and y-axis with a height of 10 μm and guard zone of 4 μm from the top of the section. A grid spacing of 150 μm in the x and y-axis was used, resulting in an area fraction of one-ninth. Microglial phenotypic classification was based on the length and thickness of the projections, the number of branches and the size of the cell body as previously described (20, 21). Briefly, ramified microglia had long thin processes (>650 μm in length), a small cell body volume (<10 μm3) and many branches (from 20 to 30). Hypertrophic microglia had medium length processes (300–550 μm in length), larger cell body volumes (50–75 μm3) and from 20 to 30 branches. Bushy microglia had short thick processes (<200 μm in length), a larger cell body volume (80–100 μm3) and very few branches (<10). Ramified microglia had a non-activated (resting) morphology, whereas hypertrophic and bushy microglia had an activated and amoeboid-like morphology. The region of interest volume was measured using a Cavalieri estimator method with a grid spacing of 100 μm. The number of microglia in each phenotypic class was divided by the volume of the region of interest to obtain the cellular density expressed in cells/mm3. According to best stereological practice, all stereological probes were optimized to obtain a Gunderson coefficient of error (CE, m = 1) value of less than 0.10 for the TBI tissue.
Western Immunoblot and OxyBlot Analysis
At the end of MRI study 2 (52 weeks post-injury), TBI mice (n = 6) were anesthetized (100 mg/kg sodium pentobarbital, i.p.) and transcardially perfused with ice-cold 0.9% saline (100 ml). Ipsilateral cortical tissue was rapidly dissected and snap-frozen on liquid nitrogen for biochemical analysis. For Western immunoblot analysis, cortical tissue (n = 3) was homogenized in RIPA buffer and centrifuged at 15,000 rpm for 15 minutes at 4°C to solubilize proteins, and the concentration was determined using a BCA Protein Assay kit (Thermo Scientific, Rockford, IL). Protein samples (25 μg) were run on SDS polyacrylamide gel electrophoresis and transferred onto nitrocellulose membrane using standard techniques. The blots were probed with anti-Iba1 (1:200, Cat No: 016-20001; Wako Chemicals), anti-glial fibrillary acidic protein ([GFAP], 1:15,000, Cat No: MAB360, Millipore Inc., Billerica, MA), and anti-myelin basic protein ([MBP], 1:5000, Cat No: SMI-94R, Covance Inc., Princeton, NJ), and anti-β-actin (1:5000; Sigma-Aldrich, St. Louis, MO) was used as an endogenous control. Immune complexes were detected with the appropriate horseradish peroxidase-conjugated secondary antibodies (KPL, Inc., Gaithersburg, MD) and visualized using SuperSignal West Dura Extended Duration Substrate (Thermo Scientific). Chemiluminescence was captured on a Kodak Image Station 4000R station (Carestream Health, Rochester, NY) and protein bands were quantified by densitometric analysis using Carestream Molecular Imaging Software. The data presented reflects the intensity of target protein band compared to control and normalized based on the intensity of the endogenous control for each sample (expressed in arbitrary units). Immunoblot detection of carbonyl groups introduced into proteins by oxidative reactions was assessed in cortical tissue (n = 3) using an OxyBlot Protein Oxidation Detection assay as described in the manufacturer’s instructions (Cat No: S7150; Millipore Inc.). The immunoreactive signal intensity in each lane of the OxyBlot was quantified by densitometry and grouped for statistical analysis. For Western and OxyBlot immunoblot analyses, age-matched sham injured cortical tissue (n = 3 each) was used as control.
Immunofluorescence Analysis
Triple immunofluorescence staining was performed on 20-μm coronal sections and standard techniques were employed. Sections were washed with 1 × PBS (3 times), blocked for 1 hour in goat serum containing 0.4% Triton X-100, and incubated overnight at 4°C with the appropriate primary antibodies. Primary antibodies were as follows: rat anti-CD68 [ED1] (1:200, Cat No: MCA1957T, AbD Serotec, Raleigh, NC), rabbit anti-Iba1 (1:200; Wako Chemicals), and mouse anti-gp91phox (1:200, Cat No: 611415, BD Transduction, Franklin Lakes, NJ). Sections were washed with 1× PBS (3 times) and incubated with appropriate Alexa Fluor-conjugated secondary antibodies (Life Technologies, Grand Island, NY). Sections were washed with 1× PBS (3 times) and counterstaining was performed with 4′, 6-diamidino-2-phenylindole (1 μg/ml; Sigma), followed by mounting with glass coverslips with Hydromount (National Diagnostics, Atlanta, GA). Fluorescence microscopy was performed using a LEICA (TCS SP5 II) confocal microscope system (Leica Microsystems, Exton, PA).
Luxol Fast Blue Staining
Myelin loss within the corpus callosum was assessed using Luxol fast blue staining. Twenty-μm coronal sections were incubated in Luxol fast blue (0.1%, Sigma) overnight at 56°C, differentiated in alcohol, dipped in 0.05% lithium carbonate solution and counterstained with cresyl violet (FD NeuroTechnologies, Columbia, MD). Myelin stained blue to green color and neurons stained violet thereby enabling differentiation of myelin and the assessment of fiber tract integrity in the corpus callosum after TBI. Phase contrast images were collected using a Leica DM4000B microscope.
Statistical Analysis
Quantitative data were expressed as mean ± SE. Lesion volume and all stereological analyses were performed by an investigator blinded to post-injury group. Longitudinal T2-weighted MRI lesion volume measurements were analyzed by paired Student t-test. Histological lesion volume, neuronal loss and microglial activation stereological quantification were analyzed by one-way ANOVA, followed by post-hoc adjustments using a Student Newman-Keuls correction. Western and OxyBlot immunoblot analysis were analyzed by Student t-test. Statistical analysis was performed using the GraphPad Prism software, Version 3.02 for Windows (GraphPad Software, San Diego, CA); p < 0.05 was considered statistically significant.
RESULTS
Traumatic Brain Injury Induces Progressive Lesion Expansion and Chronic Hippocampal Neurodegeneration up to 1 Year Post-injury
MRI study 1 demonstrated that lesion volumes increased over time with a 210% and 302% increase in lesion size at 2 and 3 months post-injury, respectively, when compared to lesion sizes quantified within the first week after TBI (Fig. 1A). Similarly, in MRI study 2, the TBI-induced lesion expanded significantly from 17.81 ± 1.99 mm3 at 6 months post-injury to 24.09 ± 1.58 mm3 at 12 months post-injury (Fig. 1A; p = 0.0314, paired Student t-test). The lesion expanded latero-ventrally through the hippocampus at 12 months post-injury as shown in T2-weighted MRI images (Fig. 1B).
Figure 1.
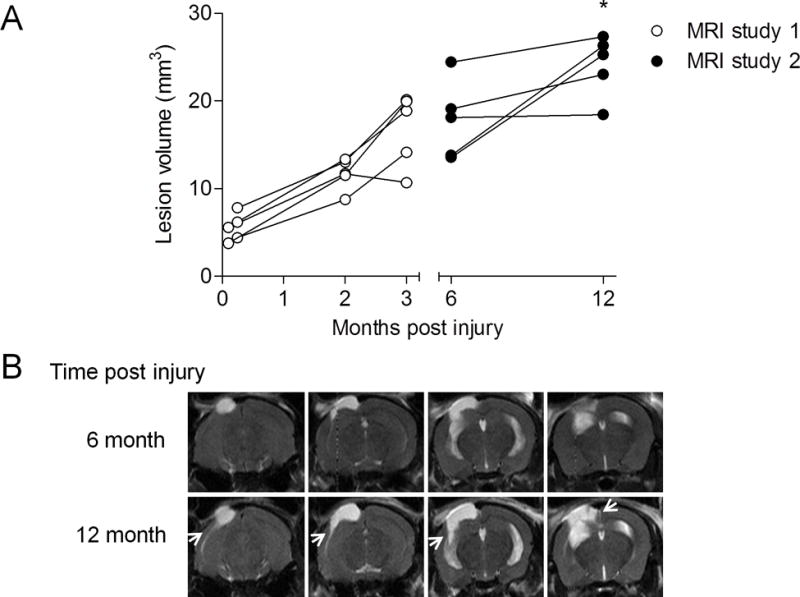
Longitudinal T2-weighted magnetic resonance imaging (MRI) analysis demonstrates progressive lesion expansion up to 1 year after moderate-level controlled cortical impact traumatic brain injury (TBI). Lesion volume was assessed using T2-weighted MRI in moderate-level controlled cortical impact TBI mice. (A) Longitudinal analysis of TBI mice demonstrated expanding lesion volumes over time in MRI studies 1 and 2. There was a significant increase in lesion size in TBI mice at 12 months post-injury when compared with the quantified lesions in the same mice at 6 months post–injury (*p = 0.0314, paired Student t-test). B: Representative T2-weighted MRI images of a TBI mouse imaged at 6 and 12 months post-injury. The TBI lesion expanded latero-ventrally through the cortex and into the hippocampus at 12 months post-injury as shown by hyperintense regions of the expanding lesion (arrows).
In a separate study we assessed TBI lesion volumes by histological analysis of cresyl violet stained coronal sections in cohorts of moderate-level CCI TBI mice that were followed for 1, 5, 12 or 52 weeks post-injury. Stereological analysis revealed significant lesion volume expansion over time resulting in 287%, 309%, and 483% increases in lesion size at 5 and 12, and 52 weeks post-injury, respectively, when compared to the lesion volume at 1 week post-injury (Fig. 2A, B; p < 0.01 for 5 and 12 weeks, p < 0.001 for 52 weeks, one-way ANOVA). Of note, the largest percentage lesion expansion occurred in the 5 TBI week group (188%), and despite a diminished rate of lesion expansion at later time points, there was a significant 56% expansion in lesion size in the 52 week TBI group vs. the 12 week TBI group (p < 0.01). Therefore, the stereological data support the longitudinal T2-weighted MRI analysis that demonstrate that moderate-level CCI TBI produces a significant lesion that expands progressively through the ipsilateral cortex and hippocampus up to 1 year after TBI.
Figure 2.
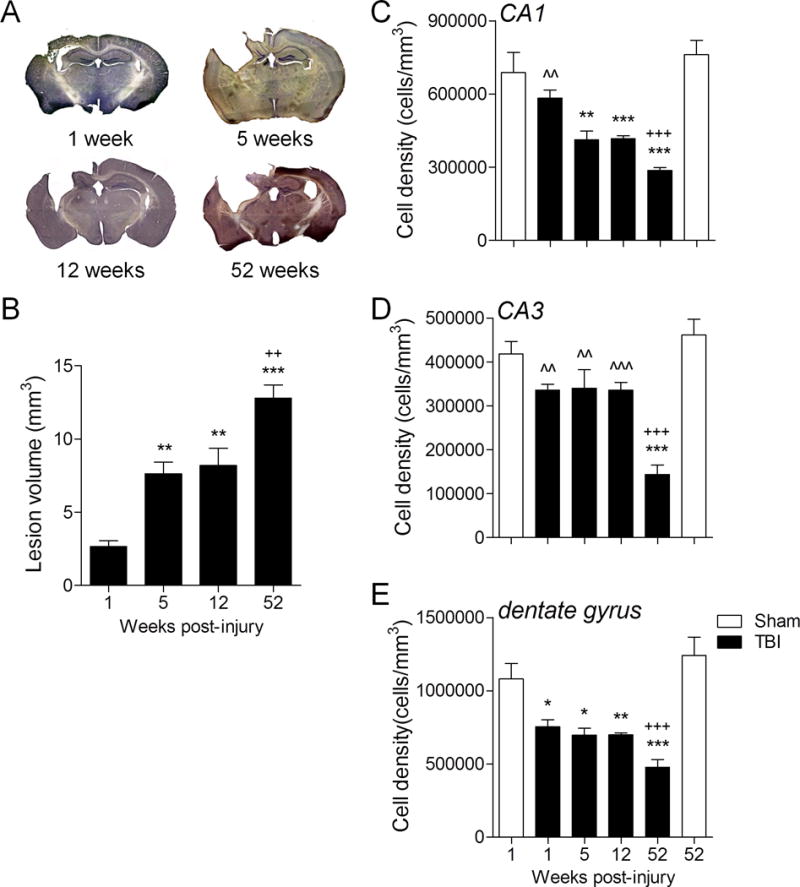
Stereological analysis confirms progressive lesion expansion and demonstrates ongoing hippocampal neurodegeneration up to 1 year post-injury. (A) Representative cresyl violet-stained coronal sections from traumatic brain injury (TBI) mice at 1, 5, 12 and 52 weeks post-injury. (B) Stereological-based lesion volume assessment demonstrated significantly increased lesions at 52 weeks (***p < 0.001), 12 weeks (**p < 0.01) and 5 weeks (**p < 0.01) post-injury vs. 1 week post-injury. In addition, there was a significant increase in lesion volume at 52 weeks post-injury (++p < 0.01) when compared to the 5- and 12-week time points. One-way ANOVA, Student Newman-Keuls post-hoc test; mean ± SE; n = 6/group. (C–E) Stereological quantification of neuronal cell loss in the CA1, CA3 and dentate gyrus sub-regions of the hippocampus at 1, 5, 12 and 52 weeks post-injury, and compared with cell loss in sham (1 week) and sham (52 week)-injured controls. TBI resulted in a significant loss of CA1 neurons in the 5-week (**p < 0.01), 12-week (***p < 0.001) and 52-week (***p < 0.001) TBI groups vs. the sham (1 week) group, and in the 52-week (+++p < 0.001) TBI group vs. the sham (52 week) group. In addition, there was a significant difference in CA1 neuronal densities between the 1-week (ˆˆp < 0.01) TBI and the 52-week TBI groups (C). TBI resulted in a significant loss of CA3 neurons in 52- (***p < 0.001) week TBI group when compared to the sham (1 week) group, and in the 52- (+++p < 0.001) week TBI group when compared to the sham (52 week) group. In addition, there was a significant difference in CA3 neuronal densities between the 1-week (ˆˆp < 0.01), 5-week (ˆˆp < 0.01), and 12-week (ˆˆˆp < 0.001) TBI groups and the 52-week TBI group (D). TBI resulted in a significant loss of neurons in the dentate gyrus in the 1-week (*p < 0.05), 5-week (*p < 0.05), 12-week (**p < 0.01) and 52-week (***p < 0.001) TBI groups vs. the sham (1 week) group, and in the 52-week TBI group when compared to the sham (52 week) group (+++p < 0.001) (E). One-way ANOVA, Student Newman-Keuls post-hoc test; mean ± SE; n = 6/group.
We next assessed post-traumatic neuronal loss in the CA1, CA3 and dentate gyrus sub-regions of the hippocampus in sham-injured and TBI groups using stereological techniques. The analysis revealed that there was a significant loss of CA1 neurons in the 5- and 12-week TBI groups (p < 0.01 for both, one-way ANOVA) when compared to the 1-week sham-injured group (Fig. 2C), and further neuronal loss in the 52-week TBI group when compared to 1-week or 52-week sham-injured groups (p < 0.001 for both). In addition, there was a significant difference in CA1 neuronal density between the 1-week and 52-week TBI groups (p < 0.01). In the CA3 sub-region, there was a significant loss of neurons in the 52-week TBI group vs. 1-week or 52-week sham-injured groups (Fig. 2D, p < 0.001 for both). In addition, CA3 neuronal densities were significantly different at 1 week (p < 0.01), 5 weeks (p < 0.01) and 12 weeks (p < 0.001) vs. the 52-week TBI group. Finally, there was a significant loss of dentate gyrus neurons in the 1-week (p < 0.05), 5-week (p < 0.05), and 12-week (p < 0.01) TBI groups vs. the 1-week sham-injured group (Fig. 2E), with greater neuronal loss in the 52-week TBI group vs. 1-week or 52-week sham-injured groups (p < 0.001 for each). This analysis indicates that moderate-level CCI TBI results in progressive neurodegeneration in the hippocampus with continued neuronal loss in the CA1, CA3 and dentate gyrus sub-fields of the hippocampus through 1 year post-injury.
Microglia Are Chronically Activated in the Cortex, Corpus Callosum and Thalamus up to 1 Year Post-injury
We next assessed post-traumatic microglial activation status in the cortex of sham-injured and TBI mice using stereological techniques. Based on cell morphological features, microglia were classified into 3 categories corresponding to increasing activation status, ramified (resting), hypertrophic (activated), and bushy (highly activated), as previously described (19, 20, 22). We performed stereological assessment of microglial cell number and activation status in the injured cortex of each group. There was a significant decrease in numbers of ramified microglia in the 1-week (p < 0.05), 5-week (p < 0.05) and 12-week (p < 0.01, one-way ANOVA) TBI groups vs. the 1-week sham-injured group (Fig. 3A). There was also a decrease in numbers of ramified microglia in the 52-week TBI group vs. 1-week or 52-week sham-injured groups (p < 0.01 for each). There was a significant increase in the numbers of activated microglia (i.e. cells with hypertrophic and bushy morphologies) in the 1-week, 5-week and 12-week TBI groups (p < 0.001 for each) vs. the 1-week sham-injured group (Fig. 3A, B). Notably, there was a significant increase in the number of activated microglia in the 52-week TBI group vs. 1-week or 52-week sham-injured groups (p < 0.01 for each). The peak in microglial activation occurred at 1 week post-injury, and the numbers of activated microglia in the 5 and 12 week (p < 0.01 for each) and the 52 week (p < 0.001) time points were significantly reduced compared to the 1-week time point. It is important to note that microglia remained chronically activated through 1 year post-injury and did not return to sham-injured levels.
Figure 3.
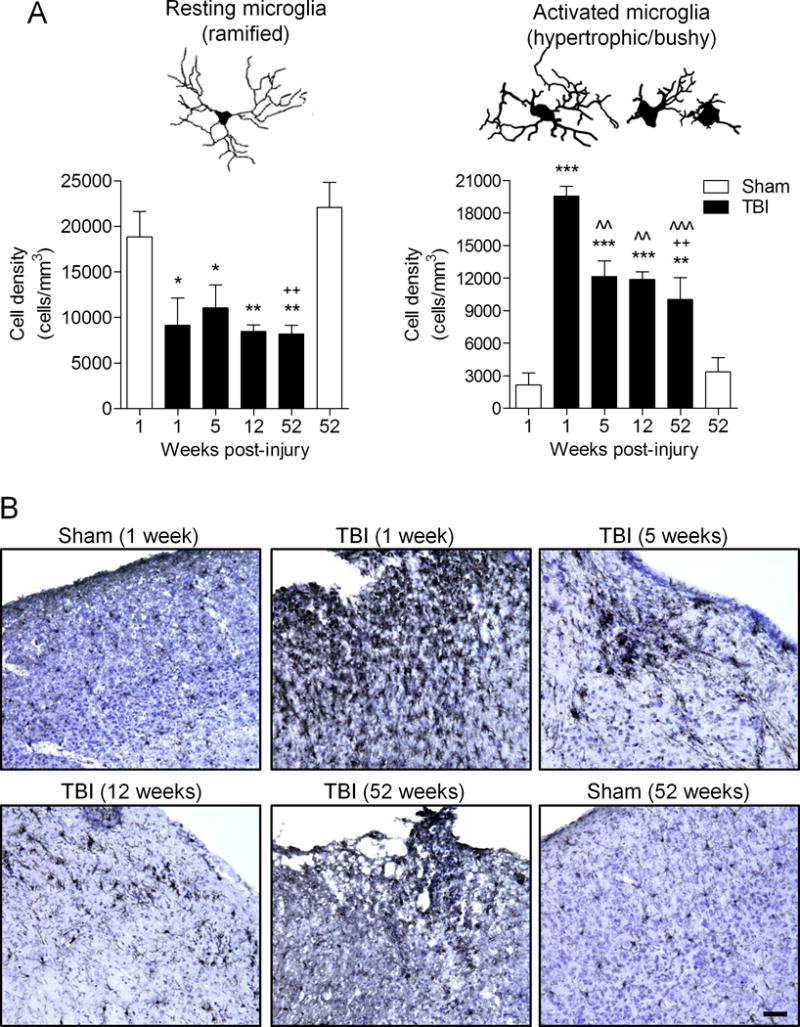
Microglia remain highly activated in the cortex up to 1 year post-injury. Stereological quantification of microglia activation phenotypes (ramified, hypertrophic, and bushy) in the cortex at 1, 5, 12 and 52 weeks post-injury, and compared with microglial activation phenotypes in sham (1 week) and sham (52 week)-injured controls. (A) Neurolucida software reconstructions of ramified, hypertrophic and bushy microglia depict the cell morphological features of each phenotype. Traumatic brain injury (TBI) resulted in a significant reduction in the number of resting microglia (ramified morphology) in the 1-week (*p < 0.05), 5-week (*p < 0.05), 12-week (**p < 0.01) and 52-week (**p < 0.01) TBI groups vs. sham (1 week) group, and in the 52-week (++p < 0.01) TBI group vs. sham (52 week) group. Furthermore, TBI resulted in a significant increase the number of activated microglia (hypertrophic and bushy morphologies) in the 1-week (***p < 0.001), 5-week (***p < 0.001), 12-week (***p < 0.001) and 52-week (**p < 0.01) TBI groups vs. the sham (1 week) group, and in the 52-week (++p < 0.01) TBI group vs. the sham (52 week) group. In addition, there was a significant difference in number of activated microglia in the 5-week (ˆˆp < 0.01), 12-week (ˆˆp < 0.001), and 52-week (ˆˆˆp < 0.001) TBI groups vs. the 1 week TBI group. One-way ANOVA, Student Newman-Keuls post-hoc test; mean ± SE, n = 6/group. (B) Representative Iba1 stained coronal sections of microglia in the injured cortex at each time point are shown. Scale bar = 100 μm.
We then assessed MHCII expression, a key antigen presentation molecule expressed on activated microglia (6), at various time points after TBI. For this analysis, we used the HLA-DP, DQ, DR antigen (CR3/43 clone) antibody for MHCII, which has previously been used to detect chronically activated microglia in postmortem human TBI brain up to 18 years post-trauma (11). We focused on corpus callosum and thalamic regions in the sham-injured and TBI groups. MHCII expression was minimal in the sham-injured control corpus callosum tissue, and staining was absent from fiber tract regions (Fig. 4A). In contrast at 1 week post-injury, MHCII expression was upregulated and dense CR3/43 immunostaining was observed in highly activated microglia within fiber tracts. Although reduced when compared to the 1-week time point, MHCII expression was chronically upregulated in the corpus callosum at 5 weeks, 12 weeks and 52 weeks post-injury and CR3/43 immunostaining was detected along fiber tract regions at these chronic time points. Similar results were obtained in the analysis of the injured thalamus. There was a marked upregulation of MHCII expression in highly activated at 1 week post-injury and MHCII expression was chronically activated in this region through 52 weeks post-injury (Fig. 4B).
Figure 4.
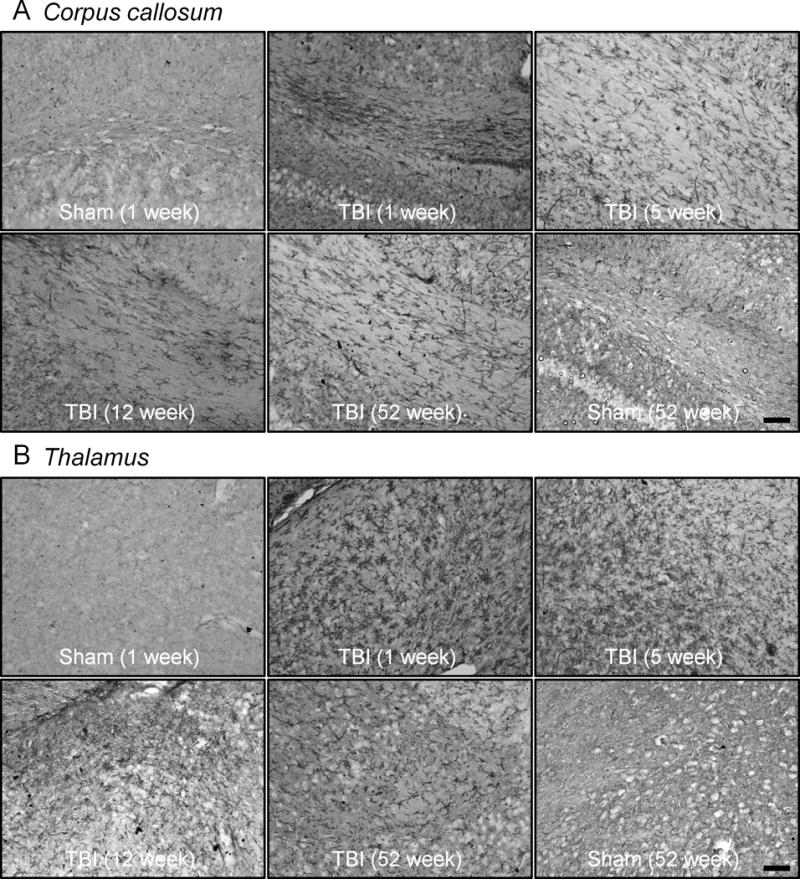
Major histocompatibility complex class II (MHCII) is chronically upregulated in the corpus callosum and thalamus up to 1 year after traumatic brain injury (TBI). Representative immunohistochemical images of MHCII (CR3/43)-stained microglia in the corpus callosum and thalamus at 1, 5, 12 and 52 weeks post-injury. (A) There are highly activated, densely stained MHCII-positive microglia in the fiber tracts of the corpus callosum at 1 week post-injury; MHCII-positive microglia were detected in this region at 5, 12 and 52 weeks post-injury. MHCII staining in the corpus callosum was minimal to absent in sham-injured controls. (B) Similarly, highly activated densely stained MHCII-positive microglia are detected in the thalamus at 1 week post-injury; persistent staining was observed up until 52 weeks post-injury. MHC II staining in the thalamus was minimal to absent in sham-injured controls. Scale bar = 50 μm.
Next, we assessed Ym1 (Chi3-L3) expression, a marker of alternatively activated (M2-polarized) microglia (5), in perilesional cortex and hippocampus at various time points after TBI. When compared with sham-injured controls, Ym1 expression was highly upregulated in the cortex and hippocampus at 1 week post-injury, it was reduced at 5 weeks post-injury, and it was absent at later time-points (12 and 52 weeks post-injury) (Fig. 5A, B), indicating a transient alternative activation phenotype in the acute phase after TBI. Interestingly, during acute time points there were mixed populations of Ym1-positive microglia that were spatially segregated in the injured tissue. For example, within perilesional cortical regions Ym1-positive cells assumed the typical morphology of highly activated amoeboid microglia, whereas at distant sites from the lesion core, such as the hippocampus, Ym1-positive microglia had ramified morphologies with numerous highly branched processes. The transient and heterogenous expression pattern of Ym1-positive microglia after TBI is similar to that of Ym1 expression after stab wound brain injury (23).
Figure 5.
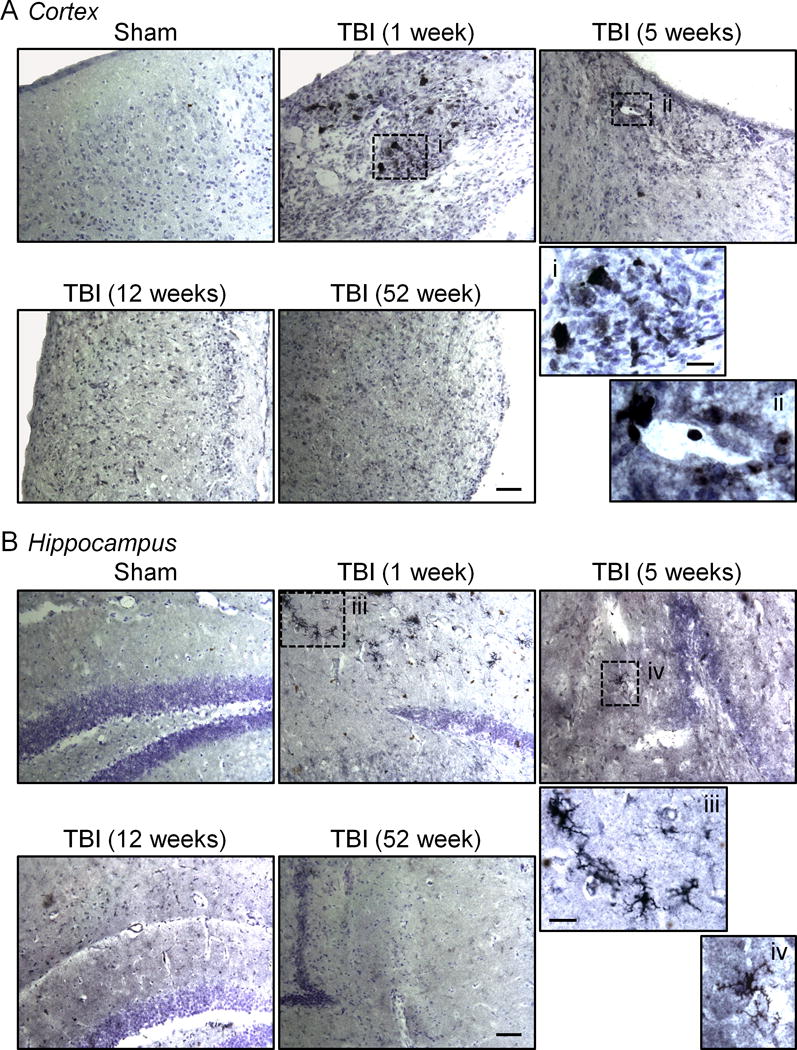
Ym1, a marker of alternatively activated microglia, is transiently expressed at acute time points after traumatic brain injury (TBI). (A, B) Representative immunohistochemical images of Ym1-stained microglia in the cortex (A) and hippocampus (B) at 1, 5, 12 and 52 weeks post-injury. Ym1 expression is highly upregulated in the cortex and hippocampus at 1 week post-injury, reduced at 5 weeks post-injury, and absent at later time-points. At 1 and 5 weeks post-injury, Ym1-positive microglia within the perilesional cortex had a highly activated amoeboid microglial cell morphology (A, inset), whereas in the hippocampus Ym1-positive microglia had ramified morphologies with numerous highly branched processes (B, inset). Ym1 staining was absent in the cortex and hippocampus at 12 and 52 weeks post-injury and in sham-injured controls. Scale bars = 100 μm; inset bar = 25 μm.
Biochemical Markers of Neuroinflammation and Oxidative Stress Were Increased and Myelin was Decreased at 1 Year Post-injury
The cohort of TBI mice from MRI study 2 and age-matched sham-injured controls were killed and the ipsilateral cortex was harvested for biochemical analysis. We performed Western immunoblot analysis for markers of neuroinflammation such as GFAP (astrocyte marker) and Iba1 (microglial marker) in the 52-week TBI cortical tissue. There was a significant increase in GFAP and Iba1 expression in the 52-week TBI group when compared with the age-matched sham-injured group (Fig. 6A; p < 0.05 for both, Student t-test). These data support the prior stereological data on increased numbers of highly activated microglia at chronic time points after TBI and demonstrate the persistent activation of biochemical markers of neuroinflammation up to 1 year post-injury. We performed OxyBlot analysis on the injured cortical tissue to assess oxidative stress-mediated changes in protein carbonyl groups. There was a significant increase in 2,4-dinitrophenylhydrazone (DNP-hydrazone) expression in the 52-week TBI group vs. the age-matched sham-injured group (Fig. 6B; p < 0.05, Student t-test), indicating increased biochemical markers of oxidative stress at 1 year post-injury. Because we previously demonstrated that NADPH oxidase (NOX2), a multi-subunit enzyme complex responsible for the production of both extracellular and intracellular reactive oxygen species by microglia (24), was highly expressed in chronically activated microglia in the TBI cortex (20), we performed triple immunofluorescence staining for highly activated microglia (Iba1/CD68-positive) that expressed NADPH oxidase (gp91phox) in sham-injured 1-week and 52-week TBI samples (Fig. 7). At 1 week post-injury gp91phox was highly upregulated in Iba1/CD68-positive microglia that displayed a hypertrophic/bushy cellular morphology in the cortex adjacent to the expanding lesion, whereas gp91phox and CD68 expression was minimal in the sham-injured control samples (data not shown). Notably, gp91phox expression was also evident in Iba1/CD68-positive microglia at 52 weeks post-injury, and high magnification images clearly demonstrate that reactive microglia within perilesional cortical and thalamic regions of the TBI brain expressed NADPH oxidase (Fig. 7, inset). These data demonstrate the chronic expression of NADPH oxidase (NOX2) in highly activated microglia at 1 year post-injury.
Figure 6.
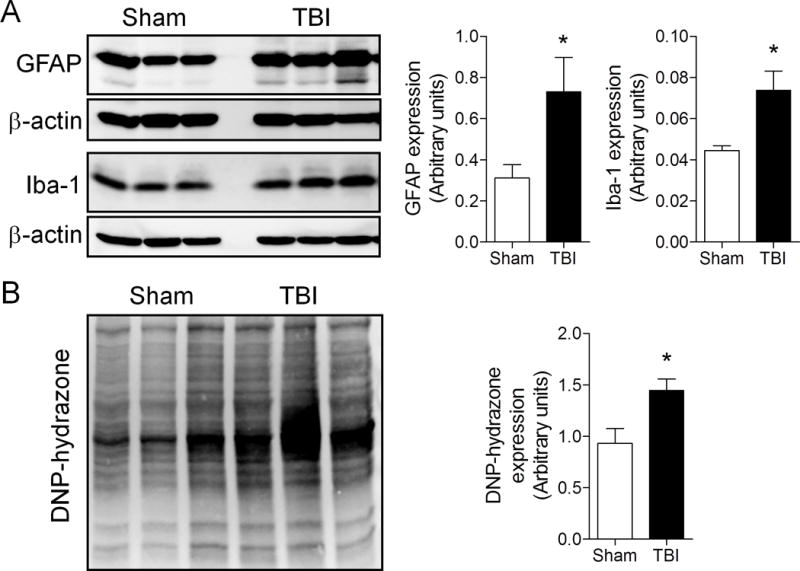
Biochemical markers of neuroinflammation and oxidative stress are increased at 1-year post-traumatic brain injury (TBI). (A) Ipsilateral cortical tissue samples from 52-week TBI mice and age-matched sham-injured controls were harvested for Western immunoblot analysis of markers of astrocytes (glial fibrillary acidic protein, GFAP) and microglial (Iba1) activation. There was a significant increase in both GFAP (*p = 0.040) and Iba1 (*p = 0.019) protein expression in the 52-week TBI group vs. the sham-injured group. Student t-test; mean ± SE, n = 3/group. (B) OxyBlot analysis of ipsilateral cortical tissue from 52-week TBI mice and age-matched sham-injured controls. There was a significant increase in DNP-hydrazone protein expression in the 52-week TBI group vs. the sham-injured group (p = 0.024). Student t-test; mean ± SE, n = 3/group.
Figure 7.
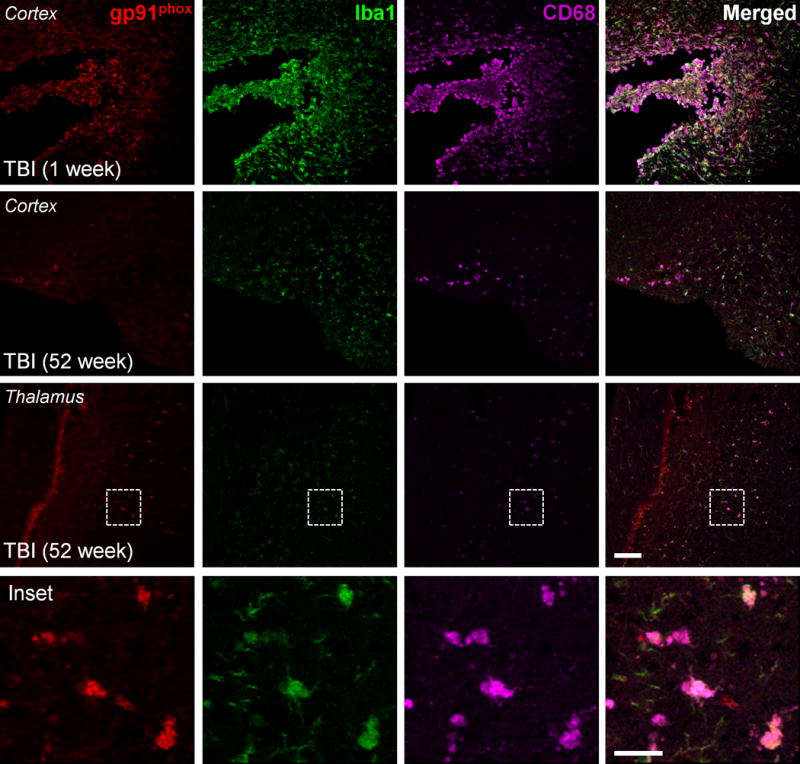
NADPH oxidase is chronically expressed in highly activated microglia up to 1-year post-traumatic brain injury (TBI). Triple immunofluorescence imaging for the NADPH oxidase subunit, gp91phox (red), and microglial activation markers, Iba1 (green) and CD68 (magenta), in 1-week and 52-week post-TBI tissue. gp91phox expression was upregulated in highly activated microglia (Iba1/CD68-positive) in sites of the expanding cortical lesion at 1 week post-injury; persistent gp91phox expression was detected in activated microglia at 52 weeks post-injury. The inset displays high magnification images of gp91phox-positive cells, which colocalized with Iba1 and CD68 at 52 weeks post-injury. Scale bars: main panels = 100 μm, inset panel = 25 μm.
We then performed Western immunoblots for MBP to quantify the loss of myelin at chronic time points. There was a significant reduction in MBP fragments in the 52-week TBI group vs. the age-matched sham-injured control group (Fig. 8A; p < 0.05, Student t-test). In addition, we analyzed myelin integrity in the corpus callosum at 52 weeks post-injury using Luxol fast blue staining. The staining revealed that when compared with age-matched sham injured controls there was a decrease in density and a non-uniform distribution of fibers within the corpus callosum at 52 weeks post-injury (Fig. 8B). Taken together, the biochemical and imaging analysis demonstrate that markers of neuroinflammation and oxidative stress are chronically activated after TBI and these are associated with myelin loss at 1 year post-injury.
Figure 8.
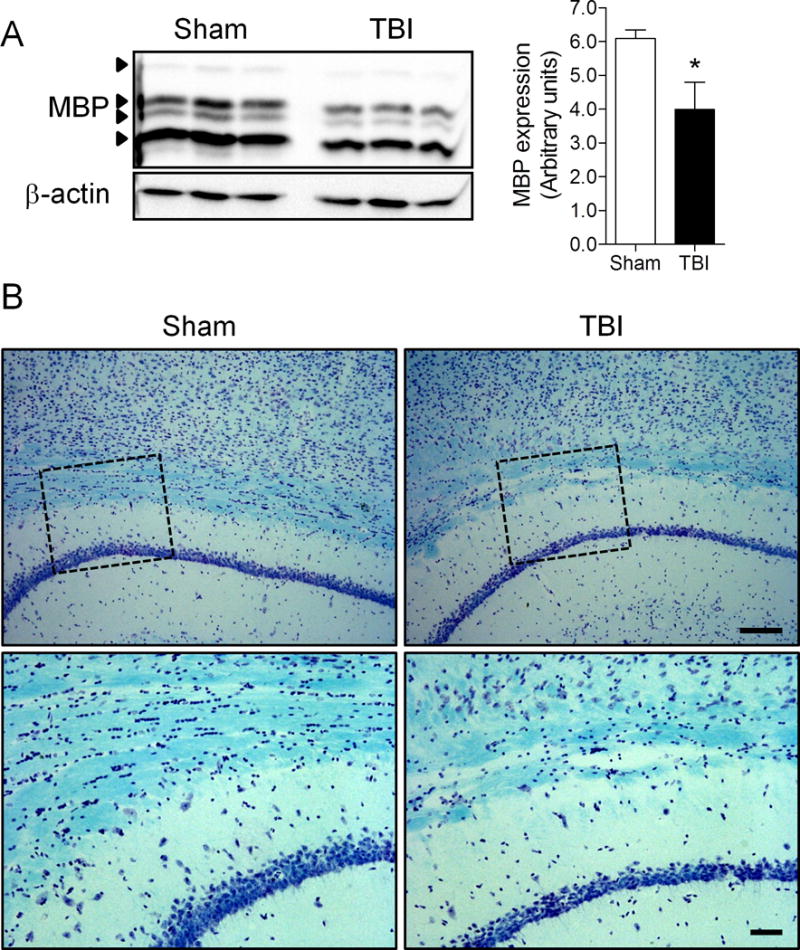
Traumatic brain injury (TBI)-induced myelin loss is increased at 1 year after TBI. (A) Ipsilateral cortical tissue samples from 52-week TBI mice and age-matched sham-injured controls were harvested for Western immunoblot analysis of myelin basic protein (MBP). There was a significant reduction in MBP protein expression (*p = 0.034) in the 52-week TBI group vs. the sham-injured group. Student t-test; mean ± SE, n = 3/group. (B) Luxol fast blue staining was performed to assess myelin integrity in the corpus callosum of 52-week TBI mice and age-matched sham injured control mice. Representative images are shown. There was a decrease in density and a non-uniform distribution of fibers within the corpus callosum in the 52-week TBI group vs. the sham controls.
DISCUSSION
In this study, we demonstrate that ongoing neuroinflammatory processes persist in the ipsilateral cortex, corpus callosum and thalamus up to 1 year after a single moderate-level focal TBI in adult male mice. Specifically, highly activated microglia that express MHCII (CR3/43), CD68 and NADPH oxidase (NOX2) were observed in these sites up to 1 year post-injury; this chronic microglial activation was associated with increased biochemical markers of neuroinflammation and oxidative stress. Notably, progressive cortical lesion expansion, hippocampal neurodegeneration, and loss of myelin were also associated with this chronic neuroinflammatory response.
Until recently TBI was considered to be a static neurodegenerative disorder resulting from an acute traumatic event but more recent clinical studies indicate that trauma may produce chronic and progressive changes. Post-traumatic inflammation may persist for years after the initial brain insult, including areas distant from the trauma site, and can be associated with cognitive changes. In a prospective cohort of more than 200 TBI survivors followed for up to 12 to 14 years, neurobehavioral disability worsened over time in a significant number (25). Neuroimaging studies have demonstrated structural pathology in TBI survivors at sub-acute and chronic time points after TBI (months to years post-trauma), and longitudinal analysis using quantitative MRI demonstrated progressive brain volume loss up to 2.5 years post-injury (26). White matter appears to be particularly vulnerable to ongoing structural damage over time, with longitudinal DTI analysis showing white matter volume loss continuing up to 4 years post-injury (27, 28).
In our study, we performed longitudinal T2 MRI imaging on mice that were subjected to moderate-level CCI TBI and demonstrated progressive lesion expansion in the cortex and subcortical regions through 1 year post-injury. The progressive nature of the degeneration was confirmed by stereological analysis. Post-traumatic lesion volumes at 52 weeks increased nearly 5-fold when compared to 1 week and by more than 50% compared to the 12-week value. These findings are consistent with earlier experimental studies in rats subjected to CCI (29) or lateral fluid percussion (30) brain injury; the latter demonstrated progressive tissue loss and expanding ipsilateral ventricular volumes up to 1 year post-injury. We also performed detailed stereological analysis of post-traumatic neuronal loss in the CA1, CA3 and dentate gyrus sub-regions of the hippocampus in groups of TBI mice at 1, 5, 12 and 52 weeks post-injury. CCI caused progressive neurodegeneration in the CA1, CA3 and dentate gyrus through 1 year post-injury. Furthermore, the loss of hippocampal neurons at chronic time after TBI was associated with long-term deficits in cognitive function in TBI mice as demonstrated by spatial learning and memory impairments persisting up to 11 weeks post-injury in a Morris water maze test (Supplementary Figure). Our data support prior experimental studies that demonstrated that moderate-to-severe TBI can cause long-term deficits in cognitive function that accompanies progressive lesion expansion and continuing neurodegeneration up to 1 year post-injury (31).
It is well established that TBI induces a complex array of acute-phase inflammatory responses as a result of a blood-brain barrier compromise, infiltration of peripheral immune cells, as well as activation of resident microglia within the injured tissue (2). It has long been accepted that this acute phase inflammatory response is essential for recovery and that it resolves over time. However, animal and human studies have increasingly suggested that neuroinflammation may persist in many cases, particularly after moderate-to-severe injury. Recent studies of autopsy-derived material from patients surviving varying intervals following a single moderate-to-severe TBI demonstrates that neuroinflammation persists in many survivors for many months (16), and in some cases for many years after the injury (11). In the latter study, chronic neuroinflammation, demonstrated by the upregulation of MHCII (CR3/43) on reactive amoeboid-like microglia, was detected in white matter regions such as the corpus callosum, and was associated with axonal degeneration and a significant reduction in the corpus callosum thickness with survival more than 1 year post-injury. Consistent with these observations, imaging studies using the PET ligand [11C](R)PK-11195 (PK), which selectively binds translocator protein (18 kDa) on activated microglia and reactive astrocytes (32), reported increased and persistent PK binding in the thalamus, putamen, occipital cortex and posterior limb of the internal capsule in a cohort of severely injured TBI patients with survival times ranging between 11 months and 17 years (14), indicating persistent microglial activation.
Studies of rodent models of moderate-to-severe level CCI by our group and others have replicated the chronic neuroinflammatory response after TBI and demonstrated persistent microglial activation up to 4 months post-injury (20, 33, 34). In the present study, we extended this analysis out through 1 year after TBI. Stereological analysis of microglial cell number and activation state demonstrated that microglia remained highly activated up to 1 year after moderate-level CCI when compared with age-matched sham-injured controls. The peak in microglial activation occurred at 1 week post-injury, but highly activated microglia displaying hypertrophic and bushy cellular morphologies were significantly increased at 5, 12 and 52 weeks post-injury. These data corroborate prior qualitative assessments of microglial activation in the TBI cortex up to 1 year post-injury (12, 13). We also assessed microglial activation at chronic time points and found that MHCII was chronically activated after CCI; peak CR3/43 immunostaining occurred at 1 week post-injury but CR3/43-positive microglia remained highly activated within the fiber tract regions of the corpus callosum and the ipsilateral thalamus up to 1 year post-injury. These data replicate key pathological features of the chronic neuroinflammatory response observed in patients (11).
Biochemical analysis of markers of neuroinflammation and oxidative stress in the injured cortex at 1 year post-injury also showed increased expression of astrocyte and microglial activation markers, i.e. Iba1 and GFAP, respectively. Prolonged expression of reactive astrocytes were reported up to 1 year post-injury following lateral fluid percussion brain injury in rats (30), and have been shown to be associated with increased expression of NFκB at the margins of the expanding lesion within the injured cortex (13). OxyBlot analysis also demonstrated chronic expression of oxidative stress markers in the injured cortex at 1 year post-injury. Furthermore, there was chronic expression of NADPH oxidase (NOX2) in highly activated, CD68-positive microglia at the margins of the expanding cortical lesion at this time point. NADPH oxidase is a common and essential mechanism of microglia-mediated neurotoxicity in neurodegenerative diseases (35). Activation of NADPH oxidase in microglia is neurotoxic, both through the production of extracellular reactive oxygen species that damage neighboring neurons (36), as well as through initiation of redox signaling in microglia that amplifies the pro-inflammatory response (37, 38). NADPH oxidase is upregulated in classically activated/M1-polarized microglia (39), and has been implicated in the pathology of several chronic neurodegenerative diseases, such as Alzheimer disease (40, 41) and Parkinson disease (42). In an experimental Parkinson disease model, NADPH oxidase (NOX2) contributed to persistent microglial activation, reactive oxygen species production, and progressive neurodegeneration of dopaminergic neurons after a single systemic exposure to lipopolysaccharide (5 mg/kg, i.p.); the chronic microglial activation and progressive neurodegeneration was not observed in NOX2−/− mice and was ameliorated in NOX+/+ mice that were given a NOX inhibitor (43). NADPH oxidase also appears to contribute to oxidative stress and neuroinflammation after ischemic brain injury (44, 45) and TBI (46, 47). In this study we show that NADPH oxidase is chronically expressed in highly activated microglia surrounding the lesion site at 1 year post-injury, and that its activity may be a key driver of self-propagating cycles of microglial-mediated neurodegeneration, as in other models of chronic neurodegenerative diseases (35, 43).
We also evaluated the expression of Ym1, a marker of alternatively activated (M2a-polarized) microglia, in the acute and chronic time points after TBI. Ym1 was transiently upregulated in microglia during the acute phase after TBI, with maximal Ym1 expression observed at 1 week post-injury followed by reduced expression at 5 weeks post-injury, and no Ym1 expression detectable at later time points. These preliminary data suggest that TBI results in transient alternative activation (M2a-polarization) of microglia in the acute phase after TBI followed by a shift towards classical activation (M1) during the chronic phase; these data are consistent with M1/M2-polarization dynamics observed after spinal cord contusion injury (48), and focal cerebral ischemic injury (49). Further studies on microglial polarization dynamics after TBI are needed to investigate the timing and impact of an M2 to M1 shift after TBI, and to establish the relationships between neurotoxic (M1) and neuroprotective (M2) microglia activation phenotypes and chronic neurodegenerative responses after TBI.
A limitation of the present study, as well as autopsy studies of human TBI brains (11, 16), is that it is unclear whether persistent neuroinflammation is responsible for chronic neurodegeneration or is induced in response to ongoing neuronal damage and axonal degeneration through other mechanisms. However, in 2 recent studies from our group, pharmacologic or physical interventions that attenuate activation of NADPH oxidase and classical activation of microglia, even when initiated 1 month or more after TBI, limit progressive neurodegeneration and improve neurological recovery (20, 34). These observations provide a mechanistic link between TBI-induced chronic neuroinflammation and progressive neurodegeneration. Given that such changes continue for months to years post-trauma, our delayed intervention studies indicate that the therapeutic window for the treatment of TBI may be significantly longer than traditionally accepted.
Supplementary Material
Acknowledgments
We thank Ms. Marie Hanscom for expert technical support.
This work was supported by grants from the NIH/NINDS to A.I.F. (5R01NS037313, 5R01NS052568).
Footnotes
Conflict of interest statement: No conflicts of interest to declare.
References
- 1.Loane DJ, Faden AI. Neuroprotection for traumatic brain injury: translational challenges and emerging therapeutic strategies. Trends Pharmacol Sci. 2010;31:596–604. doi: 10.1016/j.tips.2010.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kumar A, Loane DJ. Neuroinflammation after traumatic brain injury: opportunities for therapeutic intervention. Brain Behav Immun. 2012;26:1191–201. doi: 10.1016/j.bbi.2012.06.008. [DOI] [PubMed] [Google Scholar]
- 3.Davalos D, Grutzendler J, Yang G, et al. ATP mediates rapid microglial response to local brain injury in vivo. Nature neuroscience. 2005;8:752–8. doi: 10.1038/nn1472. [DOI] [PubMed] [Google Scholar]
- 4.Haynes SE, Hollopeter G, Yang G, et al. The P2Y12 receptor regulates microglial activation by extracellular nucleotides. Nature neuroscience. 2006;9:1512–9. doi: 10.1038/nn1805. [DOI] [PubMed] [Google Scholar]
- 5.Colton CA. Heterogeneity of microglial activation in the innate immune response in the brain. J Neuroimmune Pharmacol. 2009;4:399–418. doi: 10.1007/s11481-009-9164-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lynch MA. The multifaceted profile of activated microglia. Mol Neurobiol. 2009;40:139–56. doi: 10.1007/s12035-009-8077-9. [DOI] [PubMed] [Google Scholar]
- 7.Eikelenboom P, van Exel E, Hoozemans JJ, et al. Neuroinflammation – an early event in both the history and pathogenesis of Alzheimer’s disease. Neurodegener Dis. 2010;7:38–41. doi: 10.1159/000283480. [DOI] [PubMed] [Google Scholar]
- 8.Perry VH, Nicoll JA, Holmes C. Microglia in neurodegenerative disease. Nat Rev Neurol. 2010;6:193–201. doi: 10.1038/nrneurol.2010.17. [DOI] [PubMed] [Google Scholar]
- 9.Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nature reviews. 2007;8:57–69. doi: 10.1038/nrn2038. [DOI] [PubMed] [Google Scholar]
- 10.Gentleman SM, Leclercq PD, Moyes L, et al. Long-term intracerebral inflammatory response after traumatic brain injury. Forensic Sci Int. 2004;146:97–104. doi: 10.1016/j.forsciint.2004.06.027. [DOI] [PubMed] [Google Scholar]
- 11.Johnson VE, Stewart JE, Begbie FD, et al. Inflammation and white matter degeneration persist for years after a single traumatic brain injury. Brain. 2013;136:28–42. doi: 10.1093/brain/aws322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nagamoto-Combs K, McNeal DW, Morecraft RJ, et al. Prolonged microgliosis in the rhesus monkey central nervous system after traumatic brain injury. Journal of neurotrauma. 2007;24:1719–42. doi: 10.1089/neu.2007.0377. [DOI] [PubMed] [Google Scholar]
- 13.Nonaka M, Chen XH, Pierce JE, et al. Prolonged activation of NF-kappaB following traumatic brain injury in rats. Journal of neurotrauma. 1999;16:1023–34. doi: 10.1089/neu.1999.16.1023. [DOI] [PubMed] [Google Scholar]
- 14.Ramlackhansingh AF, Brooks DJ, Greenwood RJ, et al. Inflammation after trauma: Microglial activation and traumatic brain injury. Ann Neurol. 2011;70:374–83. doi: 10.1002/ana.22455. [DOI] [PubMed] [Google Scholar]
- 15.Holmin S, Mathiesen T. Long-term intracerebral inflammatory response after experimental focal brain injury in rat. Neuroreport. 1999;10:1889–91. doi: 10.1097/00001756-199906230-00017. [DOI] [PubMed] [Google Scholar]
- 16.Smith C, Gentleman SM, Leclercq PD, et al. The neuroinflammatory response in humans after traumatic brain injury. Neuropathol Appl Neurobiol. 2013;39:654–66. doi: 10.1111/nan.12008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fox GB, Fan L, Levasseur RA, et al. Sustained sensory/motor and cognitive deficits with neuronal apoptosis following controlled cortical impact brain injury in the mouse. Journal of neurotrauma. 1998;15:599–614. doi: 10.1089/neu.1998.15.599. [DOI] [PubMed] [Google Scholar]
- 18.Loane DJ, Pocivavsek A, Moussa CE, et al. Amyloid precursor protein secretases as therapeutic targets for traumatic brain injury. Nature medicine. 2009;15:377–9. doi: 10.1038/nm.1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kumar A, Stoica BA, Sabirzhanov B, et al. Traumatic brain injury in aged animals increases lesion size and chronically alters microglial/macrophage classical and alternative activation states. Neurobiol Aging. 2013;34:1397–411. doi: 10.1016/j.neurobiolaging.2012.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Byrnes KR, Loane DJ, Stoica BA, et al. Delayed mGluR5 activation limits neuroinflammation and neurodegeneration after traumatic brain injury. J Neuroinflammation. 2012;9:43. doi: 10.1186/1742-2094-9-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Soltys Z, Ziaja M, Pawlinski R, et al. Morphology of reactive microglia in the injured cerebral cortex. Fractal analysis and complementary quantitative methods. J Neurosci Res. 2001;63:90–7. doi: 10.1002/1097-4547(20010101)63:1<90::AID-JNR11>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 22.Kabadi SV, Stoica BA, Byrnes KR, et al. Selective CDK inhibitor limits neuroinflammation and progressive neurodegeneration after brain trauma. J Cereb Blood Flow Metab. 2012;32:137–49. doi: 10.1038/jcbfm.2011.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hung SI, Chang AC, Kato I, et al. Transient expression of Ym1, a heparin-binding lectin, during developmental hematopoiesis and inflammation. J Leukocyte Biol. 2002;72:72–82. [PubMed] [Google Scholar]
- 24.Surace MJ, Block ML. Targeting microglia-mediated neurotoxicity: the potential of NOX2 inhibitors. Cell Mol Life Sci. 2012;69:2409–27. doi: 10.1007/s00018-012-1015-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McMillan TM, Teasdale GM, Stewart E. Disability in young people and adults after head injury: 12–14 year follow-up of a prospective cohort. J Neurol Neurosurg Psychiatry. 2012;83:1086–91. doi: 10.1136/jnnp-2012-302746. [DOI] [PubMed] [Google Scholar]
- 26.Ng K, Mikulis DJ, Glazer J, et al. Magnetic resonance imaging evidence of progression of subacute brain atrophy in moderate to severe traumatic brain injury. Arch Phys Med Rehabil. 2008;89:S35–44. doi: 10.1016/j.apmr.2008.07.006. [DOI] [PubMed] [Google Scholar]
- 27.Bendlin BB, Ries ML, Lazar M, et al. Longitudinal changes in patients with traumatic brain injury assessed with diffusion-tensor and volumetric imaging. Neuroimage. 2008;42:503–14. doi: 10.1016/j.neuroimage.2008.04.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Farbota KD, Sodhi A, Bendlin BB, et al. Longitudinal volumetric changes following traumatic brain injury: a tensor-based morphometry study. J Int Neuropsychol Soc. 2012;18:1006–18. doi: 10.1017/S1355617712000835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dixon CE, Kochanek PM, Yan HQ, et al. One-year study of spatial memory performance, brain morphology, and cholinergic markers after moderate controlled cortical impact in rats. J Neurotrauma. 1999;16:109–22. doi: 10.1089/neu.1999.16.109. [DOI] [PubMed] [Google Scholar]
- 30.Smith DH, Chen XH, Pierce JE, et al. Progressive atrophy and neuron death for one year following brain trauma in the rat. Journal of neurotrauma. 1997;14:715–27. doi: 10.1089/neu.1997.14.715. [DOI] [PubMed] [Google Scholar]
- 31.Pierce JE, Smith DH, Trojanowski JQ, et al. Enduring cognitive, neurobehavioral and histopathological changes persist for up to one year following severe experimental brain injury in rats. Neuroscience. 1998;87:359–69. doi: 10.1016/s0306-4522(98)00142-0. [DOI] [PubMed] [Google Scholar]
- 32.Chen MK, Guilarte TR. Translocator protein 18 kDa (TSPO): molecular sensor of brain injury and repair. Pharmacol Ther. 2008;118:1–17. doi: 10.1016/j.pharmthera.2007.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Acosta SA, Tajiri N, Shinozuka K, et al. Long-term upregulation of inflammation and suppression of cell proliferation in the brain of adult rats exposed to traumatic brain injury using the controlled cortical impact model. PLoS One. 2013;8:e53376. doi: 10.1371/journal.pone.0053376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Piao CS, Stoica BA, Wu J, et al. Late exercise reduces neuroinflammation and cognitive dysfunction after traumatic brain injury. Neurobiol Dis. 2013;54:252–63. doi: 10.1016/j.nbd.2012.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lull ME, Block ML. Microglial activation and chronic neurodegeneration. Neurotherapeutics. 2010;7:354–65. doi: 10.1016/j.nurt.2010.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Qin L, Liu Y, Wang T, et al. NADPH oxidase mediates lipopolysaccharide-induced neurotoxicity and proinflammatory gene expression in activated microglia. J Biol Chem. 2004;279:1415–21. doi: 10.1074/jbc.M307657200. [DOI] [PubMed] [Google Scholar]
- 37.Mander P, Brown GC. Activation of microglial NADPH oxidase is synergistic with glial iNOS expression in inducing neuronal death: a dual-key mechanism of inflammatory neurodegeneration. J Neuroinflammation. 2005;2:20. doi: 10.1186/1742-2094-2-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pawate S, Shen Q, Fan F, et al. Redox regulation of glial inflammatory response to lipopolysaccharide and interferon gamma. J Neurosci Res. 2004;77:540–51. doi: 10.1002/jnr.20180. [DOI] [PubMed] [Google Scholar]
- 39.Liao B, Zhao W, Beers DR, et al. Transformation from a neuroprotective to a neurotoxic microglial phenotype in a mouse model of ALS. Exp Neurol. 2012;237:147–52. doi: 10.1016/j.expneurol.2012.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shimohama S, Tanino H, Kawakami N, et al. Activation of NADPH oxidase in Alzheimer’s disease brains. Biochem Biophys Res Commun. 2000;273:5–9. doi: 10.1006/bbrc.2000.2897. [DOI] [PubMed] [Google Scholar]
- 41.Bruce-Keller AJ, Gupta S, Parrino TE, et al. NOX activity is increased in mild cognitive impairment. Antioxidants & redox signaling. 2010;12:1371–82. doi: 10.1089/ars.2009.2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wu DC, Teismann P, Tieu K, et al. NADPH oxidase mediates oxidative stress in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of Parkinson’s disease. Proc Natl Acad Sci U S A. 2003;100:6145–50. doi: 10.1073/pnas.0937239100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Qin L, Liu Y, Hong JS, et al. NADPH oxidase and aging drive microglial activation, oxidative stress, and dopaminergic neurodegeneration following systemic LPS administration. Glia. 2013;61:855–68. doi: 10.1002/glia.22479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen H, Kim GS, Okami N, et al. NADPH oxidase is involved in post-ischemic brain inflammation. Neurobiol Dis. 2011;42:341–8. doi: 10.1016/j.nbd.2011.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen H, Song YS, Chan PH. Inhibition of NADPH oxidase is neuroprotective after ischemia-reperfusion. J Cereb Blood Flow Metab. 2009;29:1262–72. doi: 10.1038/jcbfm.2009.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dohi K, Ohtaki H, Nakamachi T, et al. Gp91phox (NOX2) in classically activated microglia exacerbates traumatic brain injury. Journal of neuroinflammation. 2010;7:41. doi: 10.1186/1742-2094-7-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang QG, Laird MD, Han D, et al. Critical role of NADPH oxidase in neuronal oxidative damage and microglia activation following traumatic brain injury. PLoS One. 2012;7:e34504. doi: 10.1371/journal.pone.0034504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kigerl KA, Gensel JC, Ankeny DP, et al. Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. J Neurosci. 2009;29:13435–44. doi: 10.1523/JNEUROSCI.3257-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hu X, Li P, Guo Y, et al. Microglia/macrophage polarization dynamics reveal novel mechanism of injury expansion after focal cerebral ischemia. Stroke. 2012;43:3063–70. doi: 10.1161/STROKEAHA.112.659656. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.