

Abstract
The function of a protein is determined by its intrinsic activity in the context of its subcellular distribution. Membranes localize proteins within cellular compartments and govern their specific activities. Discovering such membrane-protein interactions is important for understanding biological mechanisms, and could uncover novel sites for therapeutic intervention. Here we present a method for detecting membrane interactive proteins and their exposed residues that insert into lipid bilayers. Although the development process involved analysis of how C1b, C2, ENTH, FYVE, Gla, pleckstrin homology (PH) and PX domains bind membranes, the resulting Membrane Optimal Docking Area (MODA) method yields predictions for a given protein of known three dimensional structures without referring to canonical membrane-targeting modules. This approach was tested on the Arf1 GTPase, ATF2 acetyltransferase, von Willebrand factor A3 domain and Neisseria gonorrhoeae MsrB protein, and further refined with membrane interactive and non-interactive FAPP1 and PKD1 pleckstrin homology domains, respectively. Furthermore we demonstrate how this tool can be used to discover unprecedented membrane binding functions as illustrated by the Bro1 domain of Alix, which was revealed to recognize lysobisphosphatidic acid (LBPA). Validation of novel membrane-protein interactions relies on other techniques such as nuclear magnetic resonance spectroscopy (NMR) which was used here to map the sites of micelle interaction. Together this indicates that genome-wide identification of known and novel membrane interactive proteins and sites is now feasible, and provides a new tool for functional annotation of the proteome.
Keywords: peripheral membrane protein, membrane interaction interface, phospholipid interaction, bilayer insertion, lipid site identification, protein structure annotation
Introduction
Recruitment of soluble proteins to membranes constitutes a critical element in a variety of cellular processes including signal transduction, protein sorting, trafficking and cytoskeletal rearrangement. Recruitment to a membrane concentrates proteins at their sites of action, and can activate and position their binding sites to orchestrate complex events. Hence the underlying principles are of prime importance for understanding biological mechanisms.
Proteins are targeted to membranes by a variety of forces, including covalent attachment of a lipid anchor and binding to transmembrane proteins. However, many proteins mediate direct and reversible interaction with membrane surfaces, allowing them to circulate between the bilayer and aqueous environments depending on whether their lipid and protein ligands are exposed. The paradigms of such peripheral membrane proteins include FYVE, PH and PX domains, with many other peripheral membrane proteins being investigated and also remaining to be discovered.
The information about whether and how a protein interacts with a membrane is embedded in its sequence and structure, but robust tools to detect this at an accurate predictive level have not been available. The lack of such information inevitably increases the time and effort required to show whether and how a protein binds membranes. This prevents genome wide analysis of the repertoire of membrane interactions, and hinders the investigation and exploitation of many biologically relevant protein mechanisms. Clearly, convenient and accurate prediction of membrane binding propensity is increasingly necessary and important for bringing structural, molecular and cellular biology together in order to reduce the costs and experimental time required for finding membrane interaction sites.
Predicting membrane binding propensity is confounded by several factors. Although several structural motifs that bind membranes have been identified, their functions are often poorly conserved. For example only about 10% of PH domains have been estimated to bind membranes, with many others binding instead to proteins (Lemmon 2007). Moreover, even those families whose abilities to bind membranes are well-conserved, like PX domains, exhibit diverse lipid specificities and membrane affinities (Lemmon 2003). Third, some proteins employ surfaces of large domains such as enzymes rather than small lipid binding domains to target membranes. Thus a robust computational solution should: 1) identify residues that mediate both weak and strong membrane interactions, 2) detect membrane insertion elements irrespective of lipid specificity, 3) not be unduly biased towards known membrane-interacting domains but also identify novel peripheral membrane proteins. While this may challenge the long standing dogma that peripheral membrane interactions are largely electrostatic (Murray et al. 2001), we argue that while electrostatics may guide initial docking, stable and specific membrane interaction necessarily requires hydrophobic insertion into the bilayer.
Direct interaction with the bilayer may involve several modes, including (i) non-specific superficial electrostatic interaction, (ii) insertion of hydrophobic groups into the bilayer, (iii) specific binding to a particular species of lipid and (iv) recognition and induction of membrane curvature and (dis)order. Several structural modules that target membranes by specifically binding to selected membrane lipids have been described (Cho and Stahelin 2005). These include the so-called C1 domains whose targeting to the membrane is governed by diacylglycerol (DAG) binding; C2 domains which bind phosphatidylserine (PS), phosphatidylcholine (PC), or various phosphoinositide (PI) types; PX domains which bind a spectrum of PI ligands; FYVE domains, most of which specifically interact with PI3P; and PH, ENTH/ANTH, BAR, FERM, and Tubby domains which are recruited to membranes through reversible bilayer binding.
While anionic lipid binders form the largest and the best-studied class of peripheral membrane proteins, they do not cover all possible mechanisms of membrane targeting. Less studied mechanisms include non-specific lipid interactions or specific binding to other types of lipids, that maybe neutral, positively charged, or zwitterionic: phosphatidylcholine (PC) or phosphatidyletanolamine (PE), sphingolipids/glycosphingolipids, steroids, etc., which may be distinctive of protein localization to a particular kind of cellular membrane. Such structural and compositional diversity of lipid binding sites on the surface of the peripheral proteins complicates their identification.
Electrostatics, the only long-range interatomic interaction, is widely recognized as an important contributor to protein-membrane binding. Intracellular membranes contain a varying degree of anionic lipids, and many peripheral proteins contain cationic patches, facilitating their membrane translocation by means of electrostatic attraction. However, it is also known that electrostatic interactions are not sufficient to anchor peripheral proteins at membrane surfaces (Murray et al. 2001). The variations in lipid composition and net charge of different membranes suggest that electrostatics-based membrane recruitment is only important for a particular subset of peripheral proteins, and that other forces might be crucial for other classes. Indeed, the role of electrostatics was shown to be a minor contributor to membrane targeting of bee venom phospholipase A2 (Bollinger et al. 2004; Ghomashchi et al. 1998; Lin et al. 1998). This and other secreted enzymes function on the exoplasmic side of the cellular membrane where positively charged lipids, like phosphatidylcholine and sphingomyelin, are abundant. In other words, a cationic patch alone cannot provide a reliable discrimination of peripheral proteins.
Peripheral membrane proteins can be categorized by the reach of their membrane insertion motifs (Cho and Stahelin 2005). While so “H-” and “I-types” possess hydrophobic motifs that can interact the hydrocarbons of membranes bilayers, “S-type” proteins display predominantly superficial interactions. There are few S-type proteins including some PH, FERM, and C2 domain where electrostatics and polar lipid headgroup binding are the major determinants for membrane association.
In other words, none of the modes of membrane interaction is universally applicable or can be used alone to discriminate a particular peripheral membrane protein. It is rather a careful combination of the various features that may help identify reversible membrane binders within the universe of soluble proteins.
Analysis of compositional determinants of reversible membrane binding led us to the conclusion that they are roughly similar to protein-protein interaction determinants. Consistent with this observation, two previously published methods for predicting protein interfaces, ODA (Fernandez-Recio et al. 2005), and PIER (Kufareva et al. 2007), were able to predict potential membrane-inserting residues within 20 reversible membrane binding protein sites.
To improve membrane-interface prediction accuracy, we adapted the PIER algorithm (Figure 1). This yielded a fast computational method for detecting potential peripheral membrane proteins and individual membrane inserting residues on their surface. The resulting method is solely based on a 3D structure and does not refer to homology with known peripheral membrane modules or electrostatics analysis. It is based on the statistical preferences of the sub-residue atomic groups and on protein curvature analysis. When tested on a set of known peripheral proteins, the method achieved the precision of 64% at 50% recall at the residue level, with 7% precision being expected from random prediction. Using the method we were able to identify several novel potential peripheral proteins, which were consequently validated using NMR spectroscopy and micelle titrations, and evaluated by liposome binding and subcellular localization in some cases. Together this provides a general approach for the discovery of novel membrane interacting proteins.
Figure 1.
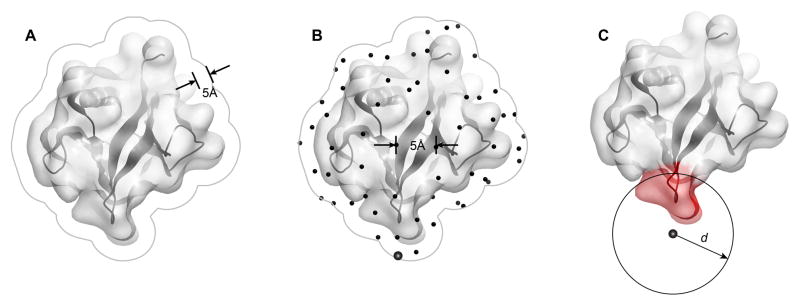
Protein surface patch generation procedure used by MODA, as illustrated using the PH domain of human B lymphocyte adapter protein Bam32 (PDB 1 fao). The generation of surface patches involves (A) expanding the solvent accessible surface by 5 Å to represent the overall protein shape rather than fine atomic details and to avoid leakage through thin sections of protein, (B) generating an even distribution of points across the surface from which to predict the membrane binding propensity, and (C) assigning each point to a patch consisting of all the proximal solvent accessible atoms within distance d Å from the point, where d and the learning algorithm are as described for the PIER program (Kufareva et al., 2007) using patch predictors which were defined from the known membrane interaction sites.
Methods and Materials
Protocols for Identifying Membrane Docking Sites
The MODA algorithm
The steps of the MODA algorithm represent an improved version of the previously published PIER algorithm for prediction of protein interaction sites (Kufareva et al. 2007). Briefly, the surface of the protein of interest is covered by a series of overlapping two-level patches by first building a set of evenly distributed points at the average distance of 5 Å from one another and from the protein surface, and then by defining each patch as the set of all protein surface atoms located within 14 Å (for level 1, i.e. larger patches) or 10 Å (for level 2, i.e. smaller patches) from its master point (Figure 1). Next, surface atoms are assigned their per-atom membrane propensity values calculated as the atom solvent accessible surface area (SASA) multiplied by an atom type specific weight (derivation of atom types and weights is described below). Next, these values are averaged over the level 1 (larger) patches. Finally, patch membrane propensity scores are transferred to the individual surface residues by assigning each residue the weighted average of all level 2 (smaller) patches encompassing it; the weights are proportional to the relative SASA that the residue contributes to each patch.
Definition and optimization of MODA parameters
For the purpose of MODA prediction, each atom is treated as belonging to one of 12 previously defined types (Kufareva et al. 2007). These 12 types constitute a statistically significant aggregation of 32 types of heavy atoms that occur in natural amino acids and differ by chemical element, formal charge, sp-, sp2-, or sp3-hybridization, and the number and the type of covalently bound heavy atoms.
The type-specific weights for MODA were obtained by Nelder-Mead simplex optimization of the area under recall-precision curve on a set of well-characterized peripheral membrane-binding proteins exhibiting a total of 191 membrane interactive residues (see below for training set definition). The protocol for optimizing the MODA algorithm was initiated from 24 random starting sets, of which 22 of them converged at the global maximum, while the other 2 ended up at local maximums that were incomparably lower. Figure 2 contains the parameter plot, where each curve represents a set of parameters derived from a single start. It shows that convergence in the simulations was observed for all atomic groups except groups 5 and 6 [S(Cys) and S(Met)]; these weights were assigned to 0 in the final version of the algorithm.
Figure 2.

Sub-residue atomic group weights for membrane-propensity score calculations. Each curve represents a set of parameters obtained from a single start of simplex optimization. The inconsistent points (red boxes) indicate insufficient data for particular groups.
Dataset for MODA training and parameterization
The MODA method was trained using a set of validated peripheral membrane proteins for which membrane inserting residues were known. Monotopic membrane proteins or close homologs of the proteins already in the set were not included. For proteins with more than one 3D structure in PDB, only the highest resolution structure was retained. This yielded a set of 20 proteins that are listed in Table I and represented in Figure 3. The assignment of the membrane interacting residues was based on available biophysical or biochemical evidence from published NMR, electron paramagnetic resonance (EPR), fluorescence, surface plasmon resonance (SPR) or mutagenesis studies as follows:
Table 1.
The structures of peripheral membrane proteins used as a training set for MODA.
| Protein Name | Domain | PDB | Membrane Inserting Residues | Reference |
|---|---|---|---|---|
| PKCα | C2 | 1dsyA | 188:190;247;249:253 | (Kohout et al. 2003; Verdaguer et al. 1999) |
| Cytosolic phospholipase A2 | C2 | 1cjyA | 33:36; 38:39; 62:65; 95:98 | (Dessen et al. 1999; Frazier et al. 2002; Xu et al. 1998) |
| Synaptotagmin-1 | C2 | 1rsyA | 173:177; 232:238 | (Chae et al. 1998; Frazier et al. 2003; Sutton et al. 1995) |
| Synaptotagmin-1 | C2 | 1tjxA | 305:307; 367:369 | (Cheng et al. 2004; Rufener et al. 2005) |
| Sec14 | Sec14 | 1auaA | 218:221; 223:224; 227:228; 230:231 | (Sha and Luo 1999; Sha et al. 1998) |
| Epsin-1 | ENTH | 1h0aA | 1:7; 9:16 | (Ford et al. 2002) |
| Coagulation factor V | C | 1czsA | 2090:2093; 2142:2147 | (Macedo-Ribeiro et al. 1999) |
| Coagulation factor VIII | C | 1d7pM | 2217:2220; 2268:2272 | (Pratt et al. 1999) |
| EEA1 | FYVE | 1jocA, B | 1352:1353; 1365:1371 | (Dumas et al. 2001; Kutateladze et al. 2004; Kutateladze and Overduin 2000) |
| HRS | FYVE | 1dvpA | 171:175 | (Mao et al. 2000; Stahelin et al. 2002) |
| VPS27 | FYVE | 1vfyA | 181:189 | (Misra and Hurley 1999; Stahelin et al. 2002) |
| Thrombin | Gla | 1nl2A | 44:53; 59 | (Falls et al. 2001; Huang et al. 2003) |
| p47-phox | PX | 1o7kA | 64:69; 77:85 | (Karathanassis et al. 2002; Stahelin et al. 2003) |
| p40-phox | PX | 1h6hA | 35:36; 58; 60; 92:98 | (Bravo et al. 2001; Stahelin et al. 2003) |
| Sgk3 | PX | 1xteA | 76:82 | (Xing et al. 2004) |
| SNX3 | PX | 1ocuA | 112:118 | (Zhou et al. 2003) |
| PKC-δ | Cys2 | 1ptrA | 239:243; 250; 252:254 | (Wang et al. 2001; Xu et al. 1997; Zhang et al. 1994) |
| Cytochrome P450 | P450 | 1dt6A | 30:31; 33:34; 36:41; 43:45; 60:63; 68:69; 376:377; 379 | (Headlam et al. 2003; Williams et al. 2000) |
| PI-PLC | PI-PLC | 1gymA | 73:75; 77:78; 268:274 | (Feng et al. 2002; Heinz et al. 1996) |
| Ganglioside GM2 activator protein | GM2-AP | 1tjjA | 90:95; 160:163 | (Wright et al. 2003) |
Figure 3.

Prediction of the membrane-inserting residues for the 20 known peripheral membrane proteins. Each panel presents two side-by-side views of the same protein, with residues on the left colored based on experimental data (blue – negative, red - positive), and on the right based on MODA’s predicted membrane-propensity scores (white – low, green - high). Heteroatoms are shown in magenta sticks, and metal ions in CPK.
The membrane-docking surface of a protein kinase C (PKCα) C2 domain was mapped in a combined fluorescence and EPR study (Kohout et al. 2003).
Membrane interaction of C2 domain of cytosolic phospholipase A2 was determined using NMR (Xu et al. 1998) and EPR spectroscopy (Frazier et al. 2002).
Membrane insertion of C2A domain of synaptotagmin I has been probed using NMR (Chae et al. 1998) and EPR spectroscopy (Frazier et al. 2003).
Membrane docking and insertion of the second C2 domain from synaptotagmin I (C2B) has been studied by site-directed spin labeling and EPR spectroscopy (Rufener et al. 2005).
The membrane interacting site of PI transfer protein Sec14 was mapped based on X-ray crystallographic studies (Sha and Luo 1999).
Membrane penetration by the epsin ENTH domain upon PI binding has been studied by X-ray crystallographic, monolayer penetration and electron microscopy studies (Ford et al. 2002).
Membrane interacting sites of C2 domains of coagulation factors V and VIII were mapped based on X-ray crystallographic studies (Macedo-Ribeiro et al. 1999; Pratt et al. 1999).
Binding of PtdIns(3)P, PS and PC molecules and micelles by the EEA1 FYVE domain has been characterized by NMR studies (Kutateladze et al. 2004).
Membrane interaction sites of FYVE domains of Vps27p and Hrs have been characterized by SPR and monolayer penetration analyses (Stahelin et al. 2002).
Membrane interaction of Gla domain of Prothrombin has been determined by fluorescence studies (Falls et al. 2001).
Membrane interactions of PX domains of NCF-1 and NCF-4 (p47phox and p40phox) have been characterized by SPR and monolayer penetration studies (Stahelin et al. 2003).
Membrane interacting sites of PX domains of sorting nexin 3 and Sgk3 were mapped based on X-ray crystallographic studies (Xing et al. 2004; Zhou et al. 2003).
Membrane interactions of the PKCd C1b domains were mapped by mutagenesis and vesicle binding (Wang et al. 2001) and NMR (Xu et al. 1997).
Membrane binding of cytochrome P450 has been characterized by site-directed labelling and fluorescence studies (Headlam et al. 2003).
Membrane binding of PI-specific phospholipase C has been characterized by kinetic, fluorescence and vesicle binding studies (Feng et al. 2002).
Membrane interacting site of ganglioside GM2-activator protein was mapped based on X-ray crystallographic studies (Wright et al. 2003).
Together this was deemed to form a representative albeit limited set of established peripheral membrane proteins with established bilayer binding properties. Their interaction surfaces were taken to embody the core elements that generally facilitate membrane docking, and hence were used to train the MODA algorithm to predict novel functional sites of lipid bilayer interaction.
Interpretation of MODA predictions
Prediction of membrane interaction sites from protein structures using the MODA tool, which is available at moda.ucsd.edu, is based on a set of numerical membrane propensity scores which are assigned to surface residues of the protein in question. There scores typically range between 0 and 50. Because in the training process, the area under recall-precision curve was optimized rather than the fraction of true positives at any fixed MODA value cut-off, there is no uniform threshold above which the residue is considered positive; instead, higher values represent higher likelihood of that residue being involved in membrane interaction. Empirical observations with MODA indicated that scores above 40 are highly correlated with membrane interaction sites while scores between 20 and 40 represent marginal signal and should be treated with caution.
Protocols for Validating Membrane Docking Sites
Protein purification
The test proteins were expressed and purified from E. coli grown in M9 media supplemented with 15N ammonium chloride. They were either purified using published protocols or standard purification methods, with the final sample always in conditions used for assigning the protein’s resonances (Amor et al. 2002; Nishida et al. 2002; Thureau et al. 2004; Tyler et al. 2006).
NMR spectroscopy
To map the binding sites we recorded 1H,15N resolved NMR spectra of the proteins (Kay et al. 1992) on a Varian INOVA 600 spectrometer. Backbone amide resonance assignments were gathered from Biological Magnetic Resonance Data Bank (BMRB). The backbone assignments of the Neisseria gonorrhoeae MsrB protein were validated using a 1H,15N NOESY-HSQC experiment (Zhang et al. 1994). Micelle binding was characterized by monitoring chemical shift changes in the 1H,15N HSQC spectra as detergent micelles composed of diheptanoyl phosphocholine (DHPC, purchased from Avanti Lipids), dodecylphosphocholine (DPC, Avanti Lipids), diheptanoyl phosphate (DHPA, Avanti lipids), 3-((3-cholamidopropyl)dimethylammonio)-1-propanesulfonic acid (CHAPS) (Sigma) or dimethyl heptylphosphocholine (FCI09, Anatrace) were added stepwise to values above their critical micelle concentrations (cmc), correcting for any effects of monomeric detergent molecule interactions. The normalized change was calculated based on the following equation: , where Δ is the normalized chemical shift variation, δH is the observed 1H chemical shift change and δN is the observed 15N chemical shift change, respectively.
Determination of membrane orientation
The membrane-solvent boundary was modeled by a 80×80×80 Å3 orthogonal grid map M with grid step of 1 Å. Grid values were set to 1 for all grid points with z ≥ 0 (aqueous environment) and to −1 for all grid points with z < 0 (membrane environment). Rigid body position/orientation of each protein in the map was found using the ICM program’s Monte-Carlo optimization (Abagyan et al. 1994; Abagyan and Totrov 1994) of the following function:
where Δi represents normalized chemical shift of the backbone amide nitrogen of i-th residue, (xi, yi, zi) are its Cartesian coordinates, M(xi, yi, zi) is the grid value in the corresponding point, and c is a solvent-correction coefficient introduced in such a way that fully solvated state of the protein is slightly more preferable than fully buried state.
Results
The comparison between the sites predicted by MODA and those which have been experimentally verified shows how membrane interaction sites can be accurately predicted from protein structure alone. Although this does not necessarily mean that experimental validation is not required, this offers a tremendous opportunity to discover many novel membrane interacting sites in diverse proteins, either singly or at a structural proteome-wide level.
Identification of Novel Peripheral Membrane Proteins
To demonstrate the predictive power of the MODA algorithm, the method was applied to a set of 521 unique protein domain structures selected by the following criteria:
X-ray crystal structure of resolution no more than 3.5 Å available in PDB,
NMR chemical shift assignments of residues available from the BMRB, and
experimentally tractable structural domains of up to 250 residues.
From this set, the following candidates were selected due to pronounced MODA-positive signals within their structures and their suitability for NMR-based validation of micelle binding residues:
ADP-ribosylation factor 1 (Arf1, PDB 1HUR)
Acetyltransferase At1g77540 (At1g, PDB 1XMT)
von Willebrand factor (VWF) A3-Domain (PDB 2ADF)
Methionine sulfoxide reductase (MsrB) C-terminal domain (PDB 1L1D)
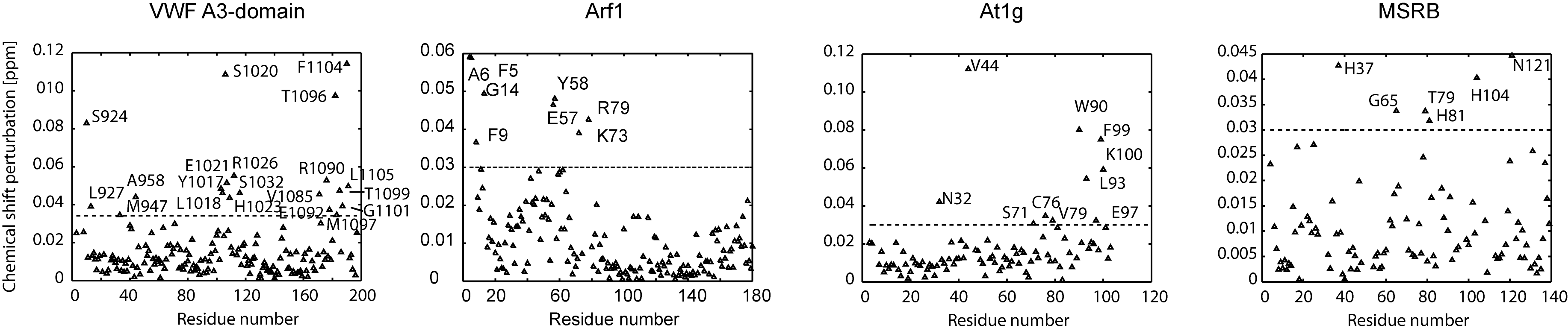
Protein binding to a bilayer was assessed by monitoring chemical shift changes in the 1H,15N resolved NMR spectra of 15N-labelled proteins (200 μM) upon addition of membrane-mimicking detergent micelles. As the behaviour of membrane-associated proteins can be idiosyncratic (Poget and Girvin 2007), various detergents were tested by thermal shift and NMR assays in order to identify the most suitable micelle system for maintaining a given protein’s stability and spectral quality at concentrations suitable for mapping interaction sites. The binding regions were mapped by monitoring progressive perturbations of amide NMR signals of assigned residues. The membrane docking position of each protein was determined by rigid-body Monte Carlo optimization in a bi-layer map representing membrane/solvent boundary. The validity of MODA prediction was assessed by comparing the set of MODA-positive residues inserted into the membrane phase within the model and the experimentally mapped set of micelle-interacting residues.
Prediction of Novel Membrane Binding Sites
Arf1 regulates membrane trafficking within eukaryotic cells. Members of this family of GTPases bind reversibly to membranes by nucleotide-dependent exposure of a myristoylated N-terminal amphipathic α-helix. The membrane interaction by this element of the human Arf1 structure (Amor et al. 2002) was confirmed by the NMR-monitored micelle titration (Figure 4B) as well as by MODA prediction. In addition, another interacting region was identified on the protein surface consisting of residues E57, Y58, K73 and R79, which all displayed significant chemical shift variation upon addition of a mixture of DPC and CHAPS to the solution (Supplementary Figure 1). In the case of Arf1 the initial MODA prediction was valid with 100% precision and 43% recall at the residue level. The N-terminal helix is also known to bind bicelles (Liu et al. 2010), while the second region overlaps a binding site for the AP-1 clathrin adaptor with which has been shown to docks to liposome (Ren et al. 2013), inferring a particularly complex binding mechanism.
Figure 4.

(A)–(D) Superimposed 1H,15N-resolved NMR spectra obtained in micelle titration of the predicted membrane binding proteins: (A) VWF A3-domain, (B) Arf1 (C) At1g and (D) MsrB. (E)–(F) Membrane docking positions for each of the same proteins, respectively. The membrane-solvent boundary is represented by the grey plane. Backbone amides with significant chemical shifts are shown in CPK and are colored by their normalized chemical shift perturbations. The residues with high predicted membrane propensity scores based on MODA analysis are shown in green sticks.
The ATF2 protein encoded by the Arabidopsis thaliana At1g77540 gene is present in the nucleus and peroxisomes (Reumann 2011), and acts as an acetyltransferase against histones, although its specific biological substrate remains unknown (Tyler et al. 2006). The purified protein exhibited perturbations in the amide resonances of eight residues when dimethyl heptylphosphocholine (FCI09) was added step-wise to the NMR sample (Supplementary Figure 1). Mapping these residues onto the structure of the protein with subsequent membrane docking yielded the orientation shown in Figure 4C. The MODA prediction was confirmed to be valid with 88% precision and 27% recall at the residue level. Biological evaluation of this putative membrane interaction region, which includes part of the active site of this understudied protein, will require further analysis.
The bilayer interaction site of the VWF A3 domain was tested by mapping NMR signal perturbations obtained upon titration with DHPC micelles (Supplementary Figure 1) onto the crystal structure (Staelens et al. 2006). This demonstrates a complicating role of conformational flexibility, as it suggests that the C-terminal α-helix of this domain is displaced upon detergent interaction. Indeed, this amphipathic helix makes extensive contacts with helix I in crystals, thus masking five other residues which exhibit large chemical shift perturbations. Consequently, we inferred that the position this helix adopts in crystallized form is not likely to be representative of the membrane bound state. Hence, this helix was not included in membrane docking optimization, which yielded an optimal rigid body position (Figure 4A). The MODA prediction was confirmed with 94% precision and 55% recall. Further biological studies are warranted given that this putative membrane interaction region overlaps sites involved in collagen and an antithrombotic antibody binding (Romijn et al. 2001; Staelens et al. 2006).
The Neisseria gonorrhoeae MsrB protein domain was prepared for micelle interaction experiments, and a set of residues showed significant NMR chemical shift changes upon addition of mixed DHPC/DHPA micelles (Supplementary Figure 1). The membrane docked position of the crystal structure (Lowther et al. 2002) is shown in Figure 4D. The MODA prediction is valid with 100% precision and a recall of 54% at the residue level, and is consistent with the known localization of the protein to bacterial outer membrane (Skaar et al. 2002).
Together these four examples show that novel and diverse membrane binding sites predicted by MODA can be validated by NMR-based analysis of interactions using phospholipid-like detergents that do not compromise protein stability or spectral quality and have suitably low critical micelle concentrations. The benefits of the NMR method for experimental validation include the ability to detect potential conformational changes as well as lipid and micelle specificities. Nonetheless, the ability of membrane and protein partners to compete for binding in vivo will usually require detailed further investigation, and demonstrates the complexity of biological interactions which often employ coincidence detection of multiple ligands. Hence, other approaches such as vesicle binding and cell-based fluorescence assays combined with site-directed mutagenesis and competitor studies could also provide useful and convenient validation, particularly for larger proteins.
Positive and Negative Examples: PH domains
The ability of MODA to discriminate between proteins that can and cannot bind membranes was tested using a well-characterized superfamily. The PH domain is one of the most common structural domains in mammalian proteomes (Lemmon 2007), being present in more than 320 human proteins. The domain is known to mediate a variety of functions, including targeting cellular membranes by specific binding to PIs, and many others binding to proteins (Lemmon 2007). These two functional types of PH proteins can be represented by the phosphoinositol 4-phosphate adaptor protein 1 (FAPP1) and the serine/threonine-protein kinase D1 (PKD1) proteins, respectively. The solution structure of the FAPP-1 PH domain was solved using NMR spectroscopy (Lenoir et al. 2010), and the interaction site for Arf1 was mapped (He et al. 2011). The PH domain of PKD1 was modeled by homology from NMR structure of PKD2 (PDB 2COA) based on its 65% sequence identity. Running MODA on the two models revealed a membrane interface on FAPP-1 PH domain (residues K7-P17, Y72 and K74) next to the Arf1 binding site (Liu et al. 2014), but no site for PKD1-PH domain. The predictions from MODA were confirmed experimentally with titrations of DPC:CHAPS micelles (8mM) (Figure 5). The perturbed FAPP1 PH residues were situated within a hydrophobic loop encompassing W8-W15 and the adjacent β7 strand L63, H70 and Y72, whereas no significant chemical shift changes were measured for PKD1-PH after a similar titration. The FAPP1-bilayer interaction is supported by a PtdIns(4)P-containing vesicle affinity of 230 nM, as well as lipid monolayer binding and membrane tubulation activity that is abrogated by mutation of the inserted hydrophobic residues (Lenoir et al. 2010). Together this illustrates how the MODA predictions based on similar folds can accurately discriminate between membrane binding and non-binding variants by their structural motifs alone, even using homology models.
Figure 5.

Micelle titrations of membrane binding vs non-binding PH domains. Superimposed 1H,15N-resolved NMR spectra of the FAPP1-PH domain (A) and PKD1-PH domain (B) are shown at the start (black) and the end (red) of the titration, where a final concentration of 8mM DPC:CHAPS (3:1) had been added. The perturbed peaks seen in the FAPP1-PH indicate protein residues interacting with micelles, while the lack of significant chemical shift perturbations in the PKD1-PH spectra indicate a lack of binding.
Discovering Novel Lipid Recognition Functions
The Alix protein is involved in the penetration of pathogenic agents in host cells, and is resident in the late endosome (Bissig and Gruenberg 2014). However the site of membrane interaction was unknown. Furthermore there were no obvious structural homologues of Alix domains which were known to interact with membranes. Hence MODA was used for de novo prediction of possible sites on its structural domains including Bro1 (Bissig et al. 2013). Using the crystal structures, a major and minor potential site of interaction in the Alix Bro1 domain were identified between residues K101-K110 and Q232-D235, respectively. The major site contained part of a hydrophobic loop encircled by cationic residues (Figure 6), and hence resembled motifs commonly involved in intracellular membrane interaction. Further investigation confirmed the major site as being where Alix binds membrane in a calcium-dependent fashion, as mutations of residues L104 and F105 to glutamic acid residues in the Alix Bro1 domain abrogate calcium-dependent liposome binding, endosomal membrane localisation and cellular functions of Alix including viral nucleocapsid delivery into the cytoplasm during infection (Bissig et al. 2013).
Figure 6.

Membrane binding site of Bro1 Domain of Alix. The structure of the Bro1 domain (Fisher et al. 2007) is shown as a ribbon model with the residues predicted by MODA to interact with membrane in red. Sidechains of residues that were mutated for validation of the LBPA binding site are depicted (Bissig et al. 2013).
Conclusions
With perhaps half of all proteins interacting with membranes, predicting their membrane binding sites is of widespread importance to biological research. Here we have demonstrated not only recapitulation of known sites but also accurate prediction of novel membrane binding sites. Moreover, identifying such docking sites allows three dimensional positioning of protein structures in bilayer models, allowing mechanistic inferences to be made. Given the MODA tool’s speed, the potential to query big data such as large sets of protein structures for membrane interactive surfaces is now possible, and precise bilayer docking areas can be mapped on individual structures for subsequent bio-evaluation. This sidesteps the need for laborious manual curation and experimental assays of membrane proteins, although validation is still recommended for maximum certainty. This method could uncover the broader diversity of protein folds and forces used to engage and manipulate a range of lipid bilayers, and could allow entirely new pockets to be exploited for therapeutic intervention into diseases and conditions.
Supplementary Material
Figure S1: Chemical shift perturbations for backbone amide resonances of each residue of the test proteins corresponding to those shown on the structures in Figure 4. Residues exhibiting significantly perturbed amide peaks upon micelle addition are labelled in the panels of the VWF-A3 domain, Arf1, At1g and MsrB proteins, respectively.
{kind=link}
Acknowledgments
We thank the Welcome Trust, HWB-NMR and the EU-funded bio-NMR project for NMR facility support. The authors thank Chris Edwards for MODA server support. Support to M.O. and J.G. was provided by the PRISM EU Sixth Framework Program, to M.O. from BBSRC and Cancer Research UK, and to J.G. from the Swiss National Science Foundation, the NCCR in Chemical Biology and LipidX from the Swiss SystemsX.ch initiative, evaluated by the Swiss National Science Foundation. I.K. and R.A. received support from NIH grants R01 GM071872, U01 GM094612, and U54 GM094618.
References
- Abagyan R, Frishman D, Argos P. Recognition of distantly related proteins through energy calculations. Proteins. 1994;19(2):132–140. doi: 10.1002/prot.340190206. [DOI] [PubMed] [Google Scholar]
- Abagyan R, Totrov M. Biased probability Monte Carlo conformational searches and electrostatic calculations for peptides and proteins. J Mol Biol. 1994;235(3):983–1002. doi: 10.1006/jmbi.1994.1052. [DOI] [PubMed] [Google Scholar]
- Amor JC, Seidel RD, 3rd, Tian F, Kahn RA, Prestegard JH. 1H, 15N and 13C assignments of full length human ADP ribosylation factor 1 (ARF1) using triple resonance connectivities and dipolar couplings. J Biomol NMR. 2002;23(3):253–254. doi: 10.1023/a:1019839607202. [DOI] [PubMed] [Google Scholar]
- Bissig C, Gruenberg J. ALIX and the multivesicular endosome: ALIX in Wonderland. Trends Cell Biol. 2014;24(1):19–25. doi: 10.1016/j.tcb.2013.10.009. [DOI] [PubMed] [Google Scholar]
- Bissig C, Lenoir M, Velluz MC, Kufareva I, Abagyan R, Overduin M, Gruenberg J. Viral infection controlled by a calcium-dependent lipid-binding module in ALIX. Dev Cell. 2013;25(4):364–373. doi: 10.1016/j.devcel.2013.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bollinger JG, Diraviyam K, Ghomashchi F, Murray D, Gelb MH. Interfacial binding of bee venom secreted phospholipase A2 to membranes occurs predominantly by a nonelectrostatic mechanism. Biochemistry. 2004;43(42):13293–13304. doi: 10.1021/bi049390i. [DOI] [PubMed] [Google Scholar]
- Bravo J, Karathanassis D, Pacold CM, Pacold ME, Ellson CD, Anderson KE, Butler PJ, Lavenir I, Perisic O, Hawkins PT, Stephens L, Williams RL. The crystal structure of the PX domain from p40(phox) bound to phosphatidylinositol 3-phosphate. Mol Cell. 2001;8(4):829–839. doi: 10.1016/s1097-2765(01)00372-0. S1097-2765(01)00372-0. [DOI] [PubMed] [Google Scholar]
- Chae YK, Abildgaard F, Chapman ER, Markley JL. Lipid binding ridge on loops 2 and 3 of the C2A domain of synaptotagmin I as revealed by NMR spectroscopy. J Biol Chem. 1998;273(40):25659–25663. doi: 10.1074/jbc.273.40.25659. [DOI] [PubMed] [Google Scholar]
- Cheng Y, Sequeira SM, Malinina L, Tereshko V, Sollner TH, Patel DJ. Crystallographic identification of Ca2+ and Sr2+ coordination sites in synaptotagmin I C2B domain. Protein Sci. 2004;13(10):2665–2672. doi: 10.1110/ps.04832604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho W, Stahelin RV. Membrane-protein interactions in cell signaling and membrane trafficking. Annu Rev Biophys Biomol Struct. 2005;34:119–151. doi: 10.1146/annurev.biophys.33.110502.133337. [DOI] [PubMed] [Google Scholar]
- Dessen A, Tang J, Schmidt H, Stahl M, Clark JD, Seehra J, Somers WS. Crystal structure of human cytosolic phospholipase A2 reveals a novel topology and catalytic mechanism. Cell. 1999;97(3):349–360. doi: 10.1016/s0092-8674(00)80744-8. S0092-8674(00)80744-8. [DOI] [PubMed] [Google Scholar]
- Dumas JJ, Merithew E, Sudharshan E, Rajamani D, Hayes S, Lawe D, Corvera S, Lambright DG. Multivalent endosome targeting by homodimeric EEA1. Mol Cell. 2001;8(5):947–958. doi: 10.1016/s1097-2765(01)00385-9. S1097-2765(01)00385-9. [DOI] [PubMed] [Google Scholar]
- Falls LA, Furie BC, Jacobs M, Furie B, Rigby AC. The omega-loop region of the human prothrombin gamma-carboxyglutamic acid domain penetrates anionic phospholipid membranes. J Biol Chem. 2001;276(26):23895–23902. doi: 10.1074/jbc.M008332200. [DOI] [PubMed] [Google Scholar]
- Feng J, Wehbi H, Roberts MF. Role of tryptophan residues in interfacial binding of phosphatidylinositol-specific phospholipase C. J Biol Chem. 2002;277(22):19867–19875. doi: 10.1074/jbc.M200938200. [DOI] [PubMed] [Google Scholar]
- Fernandez-Recio J, Totrov M, Skorodumov C, Abagyan R. Optimal docking area: a new method for predicting protein-protein interaction sites. Proteins. 2005;58(1):134–143. doi: 10.1002/prot.20285. [DOI] [PubMed] [Google Scholar]
- Fisher RD, Chung HY, Zhai Q, Robinson H, Sundquist WI, Hill CP. Structural and biochemical studies of ALIX/AIP1 and its role in retrovirus budding. Cell. 2007;128(5):841–852. doi: 10.1016/j.cell.2007.01.035. [DOI] [PubMed] [Google Scholar]
- Ford MG, Mills IG, Peter BJ, Vallis Y, Praefcke GJ, Evans PR, McMahon HT. Curvature of clathrin-coated pits driven by epsin. Nature. 2002;419(6905):361–366. doi: 10.1038/nature01020. [DOI] [PubMed] [Google Scholar]
- Frazier AA, Roller CR, Havelka JJ, Hinderliter A, Cafiso DS. Membrane-bound orientation and position of the synaptotagmin I C2A domain by site-directed spin labeling. Biochemistry. 2003;42(1):96–105. doi: 10.1021/bi0268145. [DOI] [PubMed] [Google Scholar]
- Frazier AA, Wisner MA, Malmberg NJ, Victor KG, Fanucci GE, Nalefski EA, Falke JJ, Cafiso DS. Membrane orientation and position of the C2 domain from cPLA2 by site-directed spin labeling. Biochemistry. 2002;41(20):6282–6292. doi: 10.1021/bi0160821. bi0160821. [DOI] [PubMed] [Google Scholar]
- Ghomashchi F, Lin Y, Hixon MS, Yu BZ, Annand R, Jain MK, Gelb MH. Interfacial recognition by bee venom phospholipase A2: insights into nonelectrostatic molecular determinants by charge reversal mutagenesis. Biochemistry. 1998;37(19):6697–6710. doi: 10.1021/bi972525i. [DOI] [PubMed] [Google Scholar]
- He J, Scott JL, Heroux A, Roy S, Lenoir M, Overduin M, Stahelin RV, Kutateladze TG. Molecular basis of phosphatidylinositol 4-phosphate and ARF1 GTPase recognition by the FAPP1 pleckstrin homology (PH) domain. J Biol Chem. 2011;286(21):18650–18657. doi: 10.1074/jbc.M111.233015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Headlam MJ, Wilce MC, Tuckey RC. The F-G loop region of cytochrome P450scc (CYP11A1) interacts with the phospholipid membrane. Biochim Biophys Acta. 2003;1617(1–2):96–108. doi: 10.1016/j.bbamem.2003.09.007. S000527360300292X. [DOI] [PubMed] [Google Scholar]
- Heinz DW, Ryan M, Smith MP, Weaver LH, Keana JF, Griffith OH. Crystal structure of phosphatidylinositol-specific phospholipase C from Bacillus cereus in complex with glucosaminyl(alpha 1-->6)-D-myo-inositol, an essential fragment of GPI anchors. Biochemistry. 1996;35(29):9496–9504. doi: 10.1021/bi9606105. [DOI] [PubMed] [Google Scholar]
- Huang M, Rigby AC, Morelli X, Grant MA, Huang G, Furie B, Seaton B, Furie BC. Structural basis of membrane binding by Gla domains of vitamin K-dependent proteins. Nat Struct Biol. 2003;10(9):751–756. doi: 10.1038/nsb971. [DOI] [PubMed] [Google Scholar]
- Karathanassis D, Stahelin RV, Bravo J, Perisic O, Pacold CM, Cho W, Williams RL. Binding of the PX domain of p47(phox) to phosphatidylinositol 3,4-bisphosphate and phosphatidic acid is masked by an intramolecular interaction. EMBO J. 2002;21(19):5057–5068. doi: 10.1093/emboj/cdf519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kay HH, Hawkins SR, Gordon JD, Wang Y, Ribeiro AA, Spicer LD. Comparative analysis of normal and growth-retarded placentas with phosphorus nuclear magnetic resonance spectroscopy. Am J Obstet Gynecol. 1992;167(2):548–553. doi: 10.1016/s0002-9378(11)91451-3. [DOI] [PubMed] [Google Scholar]
- Kohout SC, Corbalan-Garcia S, Gomez-Fernandez JC, Falke JJ. C2 domain of protein kinase C alpha: elucidation of the membrane docking surface by site-directed fluorescence and spin labeling. Biochemistry. 2003;42(5):1254–1265. doi: 10.1021/bi026596f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kufareva I, Budagyan L, Raush E, Totrov M, Abagyan R. PIER: protein interface recognition for structural proteomics. Proteins. 2007;67(2):400–417. doi: 10.1002/prot.21233. [DOI] [PubMed] [Google Scholar]
- Kutateladze TG, Capelluto DG, Ferguson CG, Cheever ML, Kutateladze AG, Prestwich GD, Overduin M. Multivalent mechanism of membrane insertion by the FYVE domain. J Biol Chem. 2004;279(4):3050–3057. doi: 10.1074/jbc.M309007200. [DOI] [PubMed] [Google Scholar]
- Kutateladze TG, Overduin M. Sequence-specific 1H, 15N and 13C resonance assignments of the EEA1 FYVE domain. J Biomol NMR. 2000;17(1):89–90. doi: 10.1023/a:1008394731942. [DOI] [PubMed] [Google Scholar]
- Lemmon MA. Phosphoinositide recognition domains. Traffic. 2003;4(4):201–213. doi: 10.1034/j.1600-0854.2004.00071.x. 071. [DOI] [PubMed] [Google Scholar]
- Lemmon MA. Pleckstrin homology (PH) domains and phosphoinositides. Biochem Soc Symp. 2007;(74):81–93. doi: 10.1042/BSS0740081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenoir M, Coskun U, Grzybek M, Cao X, Buschhorn SB, James J, Simons K, Overduin M. Structural basis of wedging the Golgi membrane by FAPP pleckstrin homology domains. EMBO Rep. 2010;11(4):279–284. doi: 10.1038/embor.2010.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y, Nielsen R, Murray D, Hubbell WL, Mailer C, Robinson BH, Gelb MH. Docking phospholipase A2 on membranes using electrostatic potential-modulated spin relaxation magnetic resonance. Science. 1998;279(5358):1925–1929. doi: 10.1126/science.279.5358.1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Kahn RA, Prestegard JH. Dynamic structure of membrane-anchored Arf*GTP. Nature structural & molecular biology. 2010;17(7):876–881. doi: 10.1038/nsmb.1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Kahn RA, Prestegard JH. Interaction of Fapp1 with Arf1 and PI4P at a membrane surface: an example of coincidence detection. Structure. 2014;22(3):421–430. doi: 10.1016/j.str.2013.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowther WT, Weissbach H, Etienne F, Brot N, Matthews BW. The mirrored methionine sulfoxide reductases of Neisseria gonorrhoeae pilB. Nat Struct Biol. 2002;9(5):348–352. doi: 10.1038/nsb783. [DOI] [PubMed] [Google Scholar]
- Macedo-Ribeiro S, Bode W, Huber R, Quinn-Allen MA, Kim SW, Ortel TL, Bourenkov GP, Bartunik HD, Stubbs MT, Kane WH, Fuentes-Prior P. Crystal structures of the membrane-binding C2 domain of human coagulation factor V. Nature. 1999;402(6760):434–439. doi: 10.1038/46594. [DOI] [PubMed] [Google Scholar]
- Mao Y, Nickitenko A, Duan X, Lloyd TE, Wu MN, Bellen H, Quiocho FA. Crystal structure of the VHS and FYVE tandem domains of Hrs, a protein involved in membrane trafficking and signal transduction. Cell. 2000;100(4):447–456. doi: 10.1016/s0092-8674(00)80680-7. S0092-8674(00)80680-7. [DOI] [PubMed] [Google Scholar]
- Misra S, Hurley JH. Crystal structure of a phosphatidylinositol 3-phosphate-specific membrane-targeting motif, the FYVE domain of Vps27p. Cell. 1999;97(5):657–666. doi: 10.1016/s0092-8674(00)80776-x. S0092-8674(00)80776-X. [DOI] [PubMed] [Google Scholar]
- Murray D, McLaughlin S, Honig B. The role of electrostatic interactions in the regulation of the membrane association of G protein beta gamma heterodimers. J Biol Chem. 2001;276(48):45153–45159. doi: 10.1074/jbc.M101784200. [DOI] [PubMed] [Google Scholar]
- Nishida N, Miyazawa M, Sumikawa H, Sakakura M, Shimba N, Takahashi H, Terasawa H, Suzuki E, Shimada I. Backbone 1H, 13C, and 15N resonance assignments of the von Willebrand factor A3 domain. J Biomol NMR. 2002;24(4):357–358. doi: 10.1023/a:1021669129988. 5110699. [DOI] [PubMed] [Google Scholar]
- Poget SF, Girvin ME. Solution NMR of membrane proteins in bilayer mimics: small is beautiful, but sometimes bigger is better. Biochim Biophys Acta. 2007;1768(12):3098–3106. doi: 10.1016/j.bbamem.2007.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratt KP, Shen BW, Takeshima K, Davie EW, Fujikawa K, Stoddard BL. Structure of the C2 domain of human factor VIII at 1.5 A resolution. Nature. 1999;402(6760):439–442. doi: 10.1038/46601. [DOI] [PubMed] [Google Scholar]
- Ren X, Farias GG, Canagarajah BJ, Bonifacino JS, Hurley JH. Structural basis for recruitment and activation of the AP-1 clathrin adaptor complex by Arf1. Cell. 2013;152(4):755–767. doi: 10.1016/j.cell.2012.12.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reumann S. Toward a definition of the complete proteome of plant peroxisomes: Where experimental proteomics must be complemented by bioinformatics. Proteomics. 2011;11(9):1764–1779. doi: 10.1002/pmic.201000681. [DOI] [PubMed] [Google Scholar]
- Romijn RA, Bouma B, Wuyster W, Gros P, Kroon J, Sixma JJ, Huizinga EG. Identification of the collagen-binding site of the von Willebrand factor A3-domain. J Biol Chem. 2001;276(13):9985–9991. doi: 10.1074/jbc.M006548200. [DOI] [PubMed] [Google Scholar]
- Rufener E, Frazier AA, Wieser CM, Hinderliter A, Cafiso DS. Membrane-bound orientation and position of the synaptotagmin C2B domain determined by site-directed spin labeling. Biochemistry. 2005;44(1):18–28. doi: 10.1021/bi048370d. [DOI] [PubMed] [Google Scholar]
- Sha B, Luo M. PI transfer protein: the specific recognition of phospholipids and its functions. Biochim Biophys Acta. 1999;1441(2–3):268–277. doi: 10.1016/s1388-1981(99)00162-6. S1388-1981(99)00162-6. [DOI] [PubMed] [Google Scholar]
- Sha B, Phillips SE, Bankaitis VA, Luo M. Crystal structure of the Saccharomyces cerevisiae phosphatidylinositol-transfer protein. Nature. 1998;391(6666):506–510. doi: 10.1038/35179. [DOI] [PubMed] [Google Scholar]
- Skaar EP, Tobiason DM, Quick J, Judd RC, Weissbach H, Etienne F, Brot N, Seifert HS. The outer membrane localization of the Neisseria gonorrhoeae MsrA/B is involved in survival against reactive oxygen species. Proc Natl Acad Sci U S A. 2002;99(15):10108–10113. doi: 10.1073/pnas.152334799.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staelens S, Hadders MA, Vauterin S, Platteau C, De Maeyer M, Vanhoorelbeke K, Huizinga EG, Deckmyn H. Paratope determination of the antithrombotic antibody 82D6A3 based on the crystal structure of its complex with the von Willebrand factor A3-domain. J Biol Chem. 2006;281(4):2225–2231. doi: 10.1074/jbc.M508191200. [DOI] [PubMed] [Google Scholar]
- Stahelin RV, Burian A, Bruzik KS, Murray D, Cho W. Membrane binding mechanisms of the PX domains of NADPH oxidase p40phox and p47phox. J Biol Chem. 2003;278(16):14469–14479. doi: 10.1074/jbc.M212579200. [DOI] [PubMed] [Google Scholar]
- Stahelin RV, Long F, Diraviyam K, Bruzik KS, Murray D, Cho W. Phosphatidylinositol 3-phosphate induces the membrane penetration of the FYVE domains of Vps27p and Hrs. J Biol Chem. 2002;277(29):26379–26388. doi: 10.1074/jbc.M201106200. [DOI] [PubMed] [Google Scholar]
- Sutton RB, Davletov BA, Berghuis AM, Sudhof TC, Sprang SR. Structure of the first C2 domain of synaptotagmin I: a novel Ca2+/phospholipid-binding fold. Cell. 1995;80(6):929–938. doi: 10.1016/0092-8674(95)90296-1. 0092-8674(95)90296-1. [DOI] [PubMed] [Google Scholar]
- Thureau A, Olry A, Coudevylle N, Azza S, Boschi-Muller S, Branlant G, Cung MT. 1H, 13C and 15N resonance assignment of the methionine sulfoxide reductase B from Neisseria meningitidis. J Biomol NMR. 2004;30(2):223–224. doi: 10.1023/B:JNMR.0000048851.95591.80. [DOI] [PubMed] [Google Scholar]
- Tyler RC, Bitto E, Berndsen CE, Bingman CA, Singh S, Lee MS, Wesenberg GE, Denu JM, Phillips GN, Jr, Markley JL. Structure of Arabidopsis thaliana At1g77540 protein, a minimal acetyltransferase from the COG2388 family. Biochemistry. 2006;45(48):14325–14336. doi: 10.1021/bi0612059.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdaguer N, Corbalan-Garcia S, Ochoa WF, Fita I, Gomez-Fernandez JC. Ca(2+) bridges the C2 membrane-binding domain of protein kinase Calpha directly to phosphatidylserine. EMBO J. 1999;18(22):6329–6338. doi: 10.1093/emboj/18.22.6329.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang QJ, Fang TW, Nacro K, Marquez VE, Wang S, Blumberg PM. Role of hydrophobic residues in the C1b domain of protein kinase C delta on ligand and phospholipid interactions. J Biol Chem. 2001;276(22):19580–19587. doi: 10.1074/jbc.M010089200. [DOI] [PubMed] [Google Scholar]
- Williams PA, Cosme J, Sridhar V, Johnson EF, McRee DE. Microsomal cytochrome P450 2C5: comparison to microbial P450s and unique features. J Inorg Biochem. 2000;81(3):183–190. doi: 10.1016/s0162-0134(00)00102-1. S0162-0134(00)00102-1. [DOI] [PubMed] [Google Scholar]
- Wright CS, Zhao Q, Rastinejad F. Structural analysis of lipid complexes of GM2-activator protein. J Mol Biol. 2003;331(4):951–964. doi: 10.1016/s0022-2836(03)00794-0. S0022283603007940. [DOI] [PubMed] [Google Scholar]
- Xing Y, Liu D, Zhang R, Joachimiak A, Songyang Z, Xu W. Structural basis of membrane targeting by the Phox homology domain of cytokine-independent survival kinase (CISK-PX) J Biol Chem. 2004;279(29):30662–30669. doi: 10.1074/jbc.M404107200. [DOI] [PubMed] [Google Scholar]
- Xu GY, McDonagh T, Yu HA, Nalefski EA, Clark JD, Cumming DA. Solution structure and membrane interactions of the C2 domain of cytosolic phospholipase A2. J Mol Biol. 1998;280(3):485–500. doi: 10.1006/jmbi.1998.1874. [DOI] [PubMed] [Google Scholar]
- Xu RX, Pawelczyk T, Xia TH, Brown SC. NMR structure of a protein kinase C-gamma phorbol-binding domain and study of protein-lipid micelle interactions. Biochemistry. 1997;36(35):10709–10717. doi: 10.1021/bi970833a. [DOI] [PubMed] [Google Scholar]
- Zhang O, Kay LE, Olivier JP, Forman-Kay JD. Backbone 1H and 15N resonance assignments of the N-terminal SH3 domain of drk in folded and unfolded states using enhanced-sensitivity pulsed field gradient NMR techniques. J Biomol NMR. 1994;4(6):845–858. doi: 10.1007/BF00398413. [DOI] [PubMed] [Google Scholar]
- Zhou CZ, Li de La Sierra-Gallay I, Quevillon-Cheruel S, Collinet B, Minard P, Blondeau K, Henckes G, Aufrere R, Leulliot N, Graille M, Sorel I, Savarin P, de la Torre F, Poupon A, Janin J, van Tilbeurgh H. Crystal structure of the yeast Phox homology (PX) domain protein Grd19p complexed to phosphatidylinositol-3-phosphate. J Biol Chem. 2003;278(50):50371–50376. doi: 10.1074/jbc.M304392200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: Chemical shift perturbations for backbone amide resonances of each residue of the test proteins corresponding to those shown on the structures in Figure 4. Residues exhibiting significantly perturbed amide peaks upon micelle addition are labelled in the panels of the VWF-A3 domain, Arf1, At1g and MsrB proteins, respectively.