

Significance
Nucleoside analogues include important drugs that target DNA polymerases and cause chain termination. However, how ganciclovir, the first line of treatment for human cytomegalovirus infections, does this is unknown. Ganciclovir resistance is a serious clinical problem. Many ganciclovir-resistant isolates contain substitutions in the 3′–5′ exonuclease domain of the catalytic subunit of viral DNA polymerase. How these mutations confer resistance is a long-standing question. This study shows that both wild-type and exonuclease mutant polymerases incorporate ganciclovir into DNA and continue synthesis, but whereas the wild-type enzyme excises nucleotides two positions downstream of incorporated ganciclovir, the mutant enzymes do not, permitting chain extension. These results show how a therapeutically important drug causes chain termination and explain an unusual mechanism of drug resistance.
Keywords: human cytomegalovirus, DNA polymerase, 3′–5′ exonuclease, ganciclovir, drug resistance
Abstract
Many antiviral and anticancer drugs are nucleoside analogs that target polymerases and cause DNA chain termination. Interestingly, ganciclovir (GCV), the first line of therapy for human cytomegalovirus (HCMV) infections, induces chain termination despite containing the equivalent of a 3′-hydroxyl group. Certain HCMV GCV resistance (GCVr) mutations, including ones associated with treatment failures, result in substitutions in the 3′–5′ exonuclease (Exo) domain of the catalytic subunit of the viral DNA polymerase (Pol). To investigate how these mutations confer resistance, we overexpressed and purified wild-type (WT) HCMV Pol and three GCVr Exo mutants. Kinetic studies provided little support for resistance being due to effects on Pol binding or incorporation of GCV-triphosphate. The mutants were defective for Exo activity on all primer templates tested, including those with primers terminating with GCV, arguing against the mutations increasing excision of the incorporated drug. However, although the WT enzyme terminated DNA synthesis after incorporation of GCV-triphosphate and an additional nucleotide (N+1), the Exo mutants could efficiently synthesize DNA to the end of such primer templates. Notably, the Exo activity of WT Pol rapidly and efficiently degraded N+2 primer templates to N+1 products that were not further degraded. On N+1 primer templates, WT Pol, much more than the Exo mutants, converted the incoming deoxynucleoside triphosphate to its monophosphate, indicative of rapid addition and removal of incorporated nucleotides (“idling”). These results explain how GCV induces chain termination and elucidate a previously unidentified mechanism of antiviral drug resistance.
Nucleoside analog drugs have been a mainstay of antiviral and anticancer therapy ever since their development in the late 20th century. In general, these compounds are phosphorylated intracellularly to drug triphosphates (TPs) that mimic natural nucleotides and inhibit polymerases, thereby abrogating genome replication. In many cases, drug TPs also are incorporated into DNA, obligatorily causing chain termination, because the analogs lack a structural equivalent of a 3′-hydroxyl group.
Ganciclovir (GCV) and its prodrug valganciclovir are nucleoside analogs that are first-line therapies against human cytomegalovirus (HCMV), a common opportunistic pathogen responsible for a variety of diseases, particularly in immunocompromised patients and newborns (1). GCV is also under clinical investigation in combination with gene delivery of an enzyme to phosphorylate it as an anticancer therapy (2). The mechanism of GCV action against HCMV (reviewed in ref. 3) entails preferential phosphorylation in HCMV-infected cells by the virus-encoded UL97 kinase, followed by conversion of GCV-monophosphate to GCV-TP by cellular kinases. As is true with most other antiviral nucleoside analogs, GCV-TP is both a competitive inhibitor and a substrate for the viral DNA polymerase (4, 5). Interestingly, GCV consists of guanine linked to an acyclic sugar moiety lacking the equivalent of a 2′-position, but it does contain the equivalent of a 3′-hydroxyl group, so it is not an obligate chain terminator. Nevertheless, HCMV DNA polymerase, like other family B DNA polymerases, terminates DNA synthesis after incorporating GCV and, somewhat surprisingly, an additional nucleotide (4, 6–8), and this is associated with the synthesis of short chains of DNA in infected cells (9). How GCV incorporation results in this pattern of chain termination is poorly understood.
HCMV can become resistant to GCV by means of mutations affecting the viral UL97 kinase or the catalytic subunit (Pol) of the viral DNA polymerase, and both kinds of mutations have been associated with failures in HCMV therapy, with resistance being particularly problematic in patients with viruses containing both UL97 and pol mutations (10). In the case of HCMV pol mutations that confer GCV resistance, although some mutations affect conserved motifs known to be involved in polymerase substrate recognition and catalysis in related enzymes, roughly half affect residues in or near motifs conserved among 3′–5′ exonuclease (Exo) domains of DNA polymerases (11). In the closely related HSV Pol, these motifs lie within a separate structural Exo domain (12). How these mutations confer GCV resistance is not yet known.
We considered three hypotheses for how the Exo mutations might confer GCV resistance: (i) GCV resistance might be caused by effects on DNA polymerase activity that alter binding or incorporation of GCV-TP. This mechanism holds for herpes simplex virus (HSV) pol mutations conferring resistance to acyclovir (13), and the HIV M184V mutation affecting reverse transcriptase that decreases incorporation of 3-thiacytidine TP (14–16). Although the HCMV mutations in question alter the Exo domain, there are precedents in the HSV system for Exo mutations affecting HSV DNA polymerase activity and susceptibilities to certain polymerase inhibitors (17–19). If this hypothesis were correct, we would expect GCV-resistant (GCVr) Exo mutants to show increased apparent Km or decreased apparent kcat values for GCV-TP incorporation relative to wild-type (WT) Pol. (ii) Resistance might be caused by increased excision of incorporated GCV moieties by Exo, as suggested by Chou et al. (20), akin to the mechanism by which mutant HIV reverse transcriptases (although they lack Exo activity) excise nucleoside analogs such as 3′ azidothymidine from the ends of primers by an ATP-dependent mechanism (16, 21–23). In support of this possibility, Kariya et al. reported a GCV-resistant clinical HCMV isolate whose DNA polymerase displayed higher Exo activity than the parental sensitive strain (24). If the increased excision hypothesis were correct, we would expect to detect higher Exo activity of the Exo mutants on GCV-terminated primer templates. (iii) Resistance might be due to decreased Exo activity that overcomes chain termination after incorporation of GCV and one additional nucleotide such that polymerization continues. Although, to our knowledge, there is no precedent for drug resistance conferred by this mechanism, Exo-deficient bacteriophage and cellular polymerases are known to synthesize longer DNA chains on certain primer templates (25–29). Consistent with this possible mechanism, Cihlar et al. have reported that a GCVr Exo mutant exhibits much reduced Exo activity (30). This hypothesis predicts that Exo mutants, but not WT Pol, would continue DNA synthesis even after incorporating GCV and an additional nucleotide.
To test these ideas, we overexpressed and purified WT HCMV Pol and three HCMV GCVr Exo mutant Pols, each with a substitution in a different conserved Exo motif, and compared the WT and mutant enzymes with respect to GCV-TP binding and incorporation, Exo activity on GCV-containing primer templates, and DNA synthesis after incorporation of GCV and an additional nucleotide.
Results
Do the Exo Mutations Decrease the Ability of the Enzyme to Bind or Incorporate GCV-TP into DNA?
To investigate how Exo mutations confer GCV resistance, we used recombinant baculoviruses to overexpress WT HCMV Pol and three different Exo mutants—D301N, F412V, and L545S—as glutathione-S-tranferase fusion proteins, and purified the enzymes. Each mutant protein contains a substitution in or near one of three conserved motifs that are important for Exo activity of diverse DNA polymerases (31–33). Two of these substitutions—D301N and L545S—have been found in clinical GCVr isolates, and one—F412V—was selected in cell culture (20, 34). The mutations have been found to confer 3–5-fold resistance to GCV (11, 34, 35).
We first investigated whether the mutant enzymes were affected in their ability to bind GCV-TP or to incorporate it into DNA. To test this hypothesis, we measured apparent Km and kcat values for incorporation of dGTP or GCV-TP into DNA by WT or mutant Pols using a 40-mer hairpin primer template T1 (Fig. 1), radiolabeled on its 5′ end, which was designed to accept dGTP or a dGTP analog such as GCV-TP as the first incorporated nucleotide opposite a C on the template strand. We also measured apparent Ki values for GCV-TP inhibition of dGTP incorporation using the same primer template. We took a steady-state kinetics approach used previously to examine acyclovir-TP incorporation into similar primer templates by HSV Pol (8, 13, 36). To reduce any contribution of dissociation of polymerase from primer template to the rate of incorporation, we omitted the presumptive HCMV polymerase processivity subunit, UL44, in these assays. Rates of incorporation were saturable by both TPs and fit well to the Michaelis–Menten equation (examples in Fig. S1), permitting derivation of apparent Km and kcat values (Table 1). For WT Pol, the apparent Km value for GCV-TP was higher than that for dGTP, and the apparent kcat value for GCV-TP was lower than that for dGTP, as expected given GCV’s lack of a 2'-position equivalent and its greater flexibility. If the mutations affect the binding or incorporation of GCV-TP to confer resistance, we would expect to observe increased apparent Km or decreased apparent kcat for GCV-TP incorporation relative to WT Pol, without corresponding effects on these values for dGTP incorporation. WT Pol and the three mutant Pols exhibited similar apparent Km values for dGTP (Table 1). Similarly, WT Pol and the three mutants exhibited only slight differences in apparent Km values for GCV-TP and apparent Ki values for GCV-TP inhibition of dGTP incorporation, with the apparent Km and Ki values being similar to each other, as expected (Table 1). Two of the mutants—D301N and F412V—exhibited 2–4.4-fold lower apparent kcat values for dGTP than WT Pol (Table 1), which may relate to the slower replication of the D301N mutant virus (20). The three mutant Pols exhibited 1.7–2-fold decreases in apparent kcat for GCV-TP, relative to WT Pol. Thus, for D301N and F412V, the decreases in this parameter were less than those for dGTP and do not explain GCV resistance. Among the three mutants, then, only L545S exhibited a change in kinetic parameters that might help explain GCV resistance, but even then the twofold decrease in kcat was less than the 3.5–5-fold resistance exhibited by L545S virus (34, 35). Taken together, these results provide little support for the hypothesis that the Exo mutations confer GCV resistance by affecting binding or incorporation of GCV-TP.
Fig. 1.

Primer templates used in this study. g, GCV.
Table 1.
Apparent kinetic constants for incorporation of dGTP and GCV-TP by WT and mutant Pols
| dGTP | GCV-TP | |||||
| Pol | Motif | Km, μM | kcat, min−1 | Km, μM | kcat, min−1 | Ki, μM |
| WT | 0.44 ± 0.05 | 22 ± 0.70 | 5.5 ± 1.2 | 1.7 ± 0.17 | 4.5 ± 0.50 | |
| D301N | Exo I | 0.45 ± 0.09 | 11 ± 0.60 | 7.8 ± 1.5 | 1.0 ± 0.07 | 4.4 ± 0.60 |
| F412V | Exo II | 0.45 ± 0.06 | 5.0 ± 0.20 | 5.7 ± 1.8 | 1.0 ± 0.07 | 5.1 ± 0.90 |
| L545S | Exo III | 0.46 ± 0.07 | 20 ± 0.80 | 5.8 ± 1.3 | 0.87 ± 0.06 | 4.8 ± 0.70 |
Apparent Km values were calculated from assays using radiolabeled primer template by fitting data points to the Michaelis–Menten equation using GraphPad Prism (Version 6). Apparent kcat values were determined by dividing apparent Vmax values by the enzyme concentrations. Apparent Ki values were derived from Lineweaver–Burk plots. Data are mean ± SEs on the basis of two independent replicates.
Do the Exo Mutations Increase the Ability of the 3′–5′ Exo to Excise GCV?
We next investigated whether Exo mutations confer GCV resistance by increasing excision of incorporated GCV. If this hypothesis were true, we would expect to observe increased degradation of GCV-terminated primer template by the mutants. To this end, we incubated each mutant enzyme with radiolabeled synthetic primer template T2, which terminates with GCV (Fig. 1), in the absence of TPs and in the presence of the presumptive processivity subunit UL44. Incubation with WT Pol and UL44 resulted in the production of degradation products within 1 min and degradation of full-length primer template that continued for at least 30 min (Fig. 2 and Fig. S2). (Without UL44, degradation was less extensive; Fig. S2.) In contrast, incubation with any of the mutant enzymes resulted in little or no degradation of the primer template and few if any degradation products, even after 30 min (Fig. 2). Similar results were obtained with radiolabeled primer template T1 (Fig. 1), which terminates with dC, although low levels of degradation by L545S could be seen (Fig. S3). We conclude that the Exo mutations do not confer GCV resistance by increasing excision of incorporated GCV; in fact, they substantially reduce Exo activity.
Fig. 2.

Exo mutants fail to degrade a GCV-terminated primer template. Radiolabeled primer template T2 (Fig. 1) was incubated with each WT and mutant Pol (indicated at the top of each panel) in the absence of TPs and the presence of UL44 at 37 °C for the times indicated above each lane, and the products were analyzed by gel electrophoresis and autoradiography. Note that the amount of full-length primer template continued to diminish over the course of incubation, whereas amounts of degradation products increase with time (see also Fig. S2, which shows a plot of the decrease in full-length primer template and an autoradiogram of the entire gel from which this figure is taken).
Do the Exo Mutations Permit Incorporation of GCV-TP and Then Extension of Primer Templates?
We then asked if the Exo mutations somehow allow the polymerase to incorporate GCV-TP into DNA and then, rather than chain-terminating after incorporation of the next nucleotide (N+1 position), extend the primer to produce full-length product. To investigate this hypothesis, we incubated radiolabeled primer template T1 with WT and each Exo mutant in the presence of UL44, GCV-TP, and dATP/dTTP/dCTP (no dGTP). Consistent with a previous report using enzyme isolated from HCMV-infected cells (4), WT Pol efficiently terminated DNA synthesis after incorporating GCV-TP plus one additional nucleotide (in this case, dCTP), with very little production of larger products (Fig. 3). Similarly, WT Pol incorporated just one additional nucleotide (dCTP) into primer template T2 (Fig. 1), which terminates with GCV (Fig. S4), and failed to extend primer template T3 (Fig. 1), which terminates at the N+1 position (Fig. S5). In contrast, after the three Exo mutants incorporated GCV-TP into primer template T1, although there was some termination following incorporation of dCTP into the N+1 position, most DNA synthesis continued to the end of the template (Fig. 3). These full-length products were not the result of misincorporation of dATP, dTTP, or dCTP instead of GCV-TP, as no synthesis was observed in the absence of GCV-TP (Fig. S6B). Additionally, full-length products from reactions containing GCV-TP exhibited increased electrophoretic mobility relative to dG-containing full-length products (Fig. S6 B and C). Moreover, the Exo mutants synthesized full-length products from GCV-terminated primer template T2, and these products comigrated with full-length products synthesized from primer template T1 in the presence of GCV-TP and dATP/dTTP/dCTP (Fig. S6C), and migrated faster than products from primer template T5 (Fig. S6A), which is identical to T2 except for containing dG instead of GCV. Thus, we conclude that these faster migrating full-length products contain GCV. (In reactions using Exo mutants, particularly with D301N and F412V, we also detected species larger than full-length; Fig. 3 and Fig. S6 B and C.) We speculate that these species may arise due to nontemplated addition of a nucleotide, which occurs with other Exo-deficient polymerases (37), or possibly to slippage of the enzymes on the run of six dT residues at the end of the template (Fig. 1).
Fig. 3.
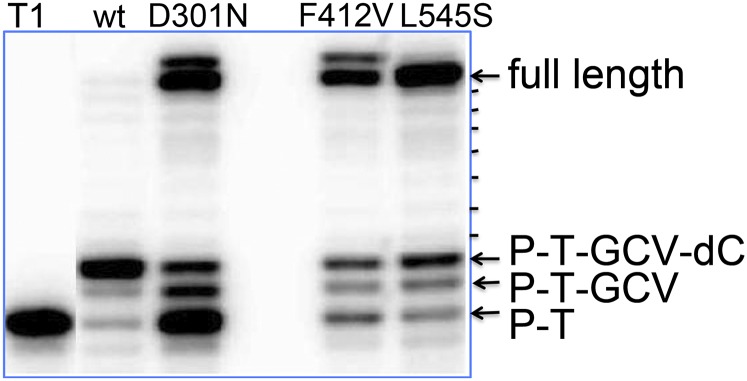
DNA extension by WT and Exo mutants following GCV incorporation. Radiolabeled primer template T1 (Fig. 1) was incubated with GCV-TP, dATP, dCTP, and dTTP, and each of the indicated Pols in the presence of UL44 at 37 °C for 10 min, and the products were analyzed alongside untreated T1 by polyacrylamide gel electrophoresis and autoradiography. Leftmost lane, untreated radiolabeled T1. For the remaining lanes, the Pol used is indicated at the top of each lane. The arrows to the right of the panel indicate the major species observed. P-T, unmodified primer template T1; P-T-GCV, T1 with GCV added; P-T-GCV-dC, T1 with GCV and dC added. The dashes to the right of the panel indicate minor products.
Taken together, these results recapitulate GCV-resistant DNA synthesis by the mutant Pols in vitro and support the hypothesis that GCVr Exo mutations confer resistance by making the polymerase less likely to terminate following incorporation of GCV plus the next nucleotide (N+1 position).
Why Does WT Pol Terminate at the N+1 Position Whereas Exo Mutants Continue DNA Synthesis?
In the course of addressing this question, we investigated whether WT Pol or the Exo mutants can degrade a synthetic primer template (T3; Fig. 1) that contains dC at the primer terminus preceded by GCV. As expected from our earlier observations, none of the Exo mutants efficiently degraded this N+1 primer template, even after 30 min of incubation (Fig. 4A). Remarkably, WT Pol was also unable to degrade this primer template detectably over this time period (Fig. 4A). This suggested (i) that once GCV and the N+1 nucleotide are incorporated, WT Pol cannot “go back,” but is committed to this primer template and (ii) that WT Pol might be unable to effectively extend this primer because its Exo activity would remove the next incorporated nucleotide rapidly (“idling”) (25, 38). In contrast, the Exo mutants would not idle and thus be able to continue extension. The ability of other Exo-deficient DNA polymerases to extend certain primer templates has been attributed to idling (25–29). As a first test of this hypothesis, we used L545S Pol to incorporate dATP into radiolabeled T3 primer template to produce an N+2 primer template T4 (Fig. 1). (Conversion of T3 to T4 was not 100% efficient, and a small amount of a larger product was also present, but the major species was N+2; Fig. 4B.) Incubation of this primer template with WT Pol plus UL44 in the absence of TPs resulted in near complete removal of a single nucleotide within 1 min and complete removal within 5 min, but no further degradation, creating an N+1 primer template (Fig. 4B). This excision by WT Pol was much more rapid and efficient than excision of GCV or dC from primer templates T2 or T1, respectively (compare Fig. 4B with Fig. 2 and Fig. S3), and the failure to further degrade the resulting N+1 primer template was consistent with our results using the T3 primer template (Fig. 4A). As expected, Exo mutant Pols were much less able to degrade the N+2 primer template, even after 20 min of incubation, although we could detect generation of product with L545S and possibly F412V (Fig. 4B).
Fig. 4.

Degradation and idling by WT and mutant Pols on primer templates containing internally incorporated GCV. (A and B) Radiolabeled primer template T3, which contains GCV plus dC (A), or T4, whose major species contains GCV plus dC plus dA (B), was incubated with WT or the indicated mutant Pol in the absence of TPs and the presence of UL44 for various times, and the products were analyzed by polyacrylamide gel electrophoresis and autoradiography. The enzymes used are indicated at the top of each panel, and the times incubated in minutes at the top of each lane. Lanes in B marked T3 contain untreated T3 as a control and size marker. (C) Unlabeled primer template T3 was incubated with dATP plus [α-32P]-dATP using each of the Pols indicated at the top of the panel in the presence of UL44 at 37 °C for 10 min, and the products were analyzed by TLC and autoradiography. Co., T3 incubated with dATP plus [α-32P]-dATP without any protein. The positions of dATP and dAMP were determined by visualizing unlabeled standards under UV light and are indicated by arrows to the right of the panel.
Idling implies that the polymerase incorporates a dNTP, which is then converted to dNMP following excision. Thus, we would expect to detect generation of the relevant dNMP by WT Pol, much more than Exo mutant Pols, on an N+1 primer template. To test this hypothesis, we incubated WT and each mutant Exo Pol with synthetic T3 primer template and [α-32P] dATP, and assayed for generation of radiolabeled dAMP using TLC. In this idling assay, WT HCMV Pol efficiently generated dAMP. Almost no detectable dAMP was generated by mutants D301N and F412V, whereas mutant L545S generated considerably (∼sixfold) less dAMP than WT Pol (Fig. 4C). These results strongly suggest that the Exo mutants can continue DNA synthesis after incorporating GCV and an additional nucleotide due to elimination of idling.
Discussion
To date, most if not all mutations in viral polymerases that confer resistance to nucleoside analogs either reduce binding and/or incorporation of drug TPs or increase excision of incorporated drug (e.g., refs. 13–16, 21–23). However, we found little evidence that the HCMV pol mutations studied here, which affect conserved motifs in the Exo domain of the enzyme, confer resistance by either of these mechanisms. Instead, we found that like WT HCMV Pol, Exo mutants incorporate GCV-TP and the next dNTP into DNA, but unlike WT Pol, instead of terminating synthesis at that point (N+1 position), the mutant Pols continue DNA synthesis to the end of the primer template.
Of the three Exo mutants, only L545S exhibited a greater change in a kinetic parameter—apparent kcat—for GCV-TP than for dGTP. Interestingly, this mutant also appeared to exhibit somewhat higher Exo activity and idling than the other two mutants. Thus, it is possible that this mutant’s resistance is due both to decreased incorporation of GCV-TP and to more efficient extension of GCV-containing primer templates.
We further found that (i) the WT enzyme can rapidly and efficiently convert an N+2 primer template to N+1, but cannot degrade an N+1 primer template and (ii) the WT enzyme, much more than the Exo mutants, idles at the N+1 position. Taken together, our results support a model for how GCV induces chain termination at the N+1 position and for how the Exo mutants overcome this termination (Fig. 5). In this model, following incorporation of GCV-TP, WT Pol can incorporate the next dNTP to generate the N+1 product. As shown by Foti et al. (39), who solved the NMR structure of a 10-bp oligonucleotide containing GCV, although the GCV base pairs well to dC, it induces distortions in the DNA backbone at the N+1 and N+2 positions. We postulate that these distortions result in (i) WT Pol being unable to degrade the N+1 primer template and (ii) WT Pol degrading an N+2 primer template to N+1 before it can extend it further (idling). The net result is termination at the N+1 position. In contrast, the Exo mutants do not rapidly degrade the N+2 product, and thus can extend it, leading to continued DNA synthesis and GCV resistance (Fig. 5B).
Fig. 5.

Model for chain termination induced by GCV for WT Pol (A) and for chain extension by Exo mutant Pols after incorporating GCV (B). The template strand is indicated at the top with the primer strand growing from left to right. Open boxes represent natural deoxynucleosides. Hatched boxes represent GCV. Black dots represent phosphates, with lines representing nucleoside–phosphate and phosphate–phosphate bonds. The asymmetric lines connecting GCV to the next incorporated nucleotide (N+1) and the N+1 nucleotide to the N+2 nucleotide indicate the distortion in the sugar–phosphate backbone induced by GCV (39). Black vertical arrows pointing downward represent incorporation, and those pointing upward represent excision. Open arrows represent repeated incorporation and removal of nucleotides at the end of the primer (idling). X’s represent steps that are blocked. See text for further description.
Our results showing that Exo mutations eliminate idling and permit chain extension from GCV-containing primers are similar to results in other systems in which Exo mutations permit synthesis of longer chains of DNA (26–29). However, in those systems, the barrier to chain extension is usually a hairpin or an adduct in the template strand rather than an altered nucleoside in the primer strand.
The Exo mutations that we studied here and other GCVr Exo mutations also confer resistance to cidofovir (CDV), a nucleoside analog that is approved as a treatment for HCMV infections (40). A more orally available prodrug of CDV, brincidofovir (CMX-001), is currently in clinical trials for HCMV infections (41). Like GCV, CDV is not an obligate chain terminator. Interestingly, although incorporation of a single CDV residue by HCMV DNA polymerase does not cause chain termination, incorporation of two successive residues or incorporation of CDV residues on both sides of a natural nucleotide results in chain termination (42). We speculate that idling by WT Pol after incorporation of two closely apposed CDV residues is responsible for chain termination by CDV, much as it is for chain termination after incorporation of GCV and one nucleotide, and therefore that Exo mutants would be able to continue DNA synthesis after incorporation of CDV, conferring CDV resistance. We speculate that a defect in excision could be a mechanism of resistance to other antiviral and anticancer nucleoside analogs that are nonobligate chain terminators.
This mechanism of GCV (and likely CDV) resistance for HCMV Exo mutants has an interesting implication: Replicated viral DNA would contain internally incorporated drug. A question remaining then is the fate of the incorporated drug in Exo mutant-infected cells. Does the resistant virus replication machinery copy drug-containing DNA? If so, what bases are inserted? It is possible that incorporated GCV could increase mutation frequencies, which would already be expected to be high with Exo mutants (43). Alternatively, would DNA repair remove the drug? If so, what repair mechanisms would operate? These questions are under investigation. Regardless, as incorporation of nucleoside analogs is generally thought to be deleterious, the mechanism of drug resistance that we have defined, where internal incorporation allows DNA synthesis, is highly unusual.
Materials and Methods
Construction of Recombinant Baculoviruses.
HCMV pol (UL54) coding sequences were amplified by PCR from pBAC/AD169 bacmid DNA (a generous gift of Dong Yu and Thomas Shenk, Princeton University, Princeton, NJ) using primers that contain terminal CpoI sites and KOD hot start DNA polymerase (EMD Biosciences). Sequences of oligonucleotides used in this study are provided in Table S1. The PCR products were digested with CpoI (Fermentas) and inserted into pFASTBAC1_Cpo_GST_UL97 (kindly provided by Jeremy Kamil, Harvard Medical School, Boston) following removal of UL97 sequences, resulting in the plasmid pGST-WT Pol. A similar plasmid for each GST-pol mutant was constructed via site-directed mutagenesis of pGST-WT Pol using the QuikChange method according to the manufacturer’s instructions (Stratagene). By using a Bac-to-Bac baculovirus expression system kit (Invitrogen), each plasmid was transformed into DH10Bac-competent Escherichia coli to generate a corresponding recombinant bacmid, and then Spodoptera frugiperda (Sf9) cells (Invitrogen), cultured in sf-900II serum-free medium (Invitrogen), supplemented with 10 μg/mL gentamicin, were transfected with the recombinant bacmids to produce recombinant baculoviruses expressing each Pol tagged at its amino terminus with GST. Viruses were titrated using a BacPAK baculovirus rapid titer kit (Clontech). The pol genes of the recombinant baculoviruses were sequenced to ensure that the desired mutation and no other was introduced.
Purification of HCMV Pol.
Each WT and mutant HCMV Pol was overexpressed in 2 × 109 Sf9 cells infected with the corresponding recombinant baculovirus at a multiplicity of infection of 5. Cells were harvested at 70 h postinfection and were pelleted by centrifugation at 15,000 × g. Cell pellets were washed with Dulbecco’s PBS (Cellgro) and resuspended in 50 mM Tris, 10% (vol/vol) glycerol, 0.1% Triton X-100, 50 mM EDTA, 2 mM DTT, 150 mM NaCl, and one Roche complete protease inhibitor mixture tablet per 100 mL. NaCl was added to the lysate to make the final concentration 0.5 M. Following centrifugation at 18,000 × g for 1 h at 4 °C, the supernatant (filtered through a 0.45-μm cellulose acetate membrane if cloudy) was loaded onto a glutathione Sepharose 4 fast flow resin column (GE Healthcare) that had been equilibrated with buffer A (50 mM Tris, 10% glycerol, 50 mM EDTA, 2 mM DTT, 250 mM NaCl). The column was washed with 10 column volumes of buffer B (buffer A plus 0.05% Triton X-100), and then proteins were eluted with five column volumes of buffer B containing 10 mM glutathione. Fractions that contained GST-tagged proteins (detected using SDS polyacrylamide gel electrophoresis) were pooled and passed through a 1-mL HiTrap heparin HP column (GE Healthcare) that had been equilibrated with five column volumes of buffer C (buffer B, but containing 50 mM NaCl) on an ÄKTApurifier system (GE Healthcare). The column was washed with buffer C, and the protein was eluted by a linear gradient of 50–2,000 mM NaCl in buffer C. Fractions containing proteins were collected and concentrations were estimated by Bradford assay (BIO-RAD).
Enzyme Assays.
Primer templates (Fig. 1) were purchased from Integrated DNA Technologies (T1) or ChemGenes (T5), synthesized by ChemGenes using GCV phosphoramidite prepared as described by Marshalko et al. (44) (T2, T3), or synthesized from T3 by incubation with L545S Pol and dATP using conditions described below, then purified by phenol-chloroform extraction (T4). Polymerase, Exo, and idling assays were performed using conditions and methods described previously (4). All reactions were performed in 10-μL volumes and contained 2.5–4 pmol of the indicated primer template, either unlabeled or radiolabeled using [γ-32P]-ATP and T4 polynucleotide kinase as indicated; WT or Exo mutant Pol, with or without a twofold molar excess of UL44ΔC290 (45) (kindly provided by Gloria Komazin-Meredith, Harvard Medical School, Boston), as indicated; and 50 mM Tris (pH 8.0), 1 mM DTT, 100 mM KCl, and 40 μg/mL BSA. Polymerase assays for kinetic studies used radiolabeled primer templates, 6–30 fmol of each Pol without UL44ΔC290, and also contained various concentrations of GCV-TP (Trilink Biotechnologies) or dGTP. Polymerase assays for testing chain extension also used radiolabeled primer templates and contained 700 fmol of each Pol with UL44ΔC290, 25 μΜ GCV-TP or dGTP, and 25 μΜ each of dCTP, dATP, and dTTP. Exo assays used radiolabeled primer templates and 360 fmol of each Pol with UL44ΔC290, and no TPs. Idling assays used 720 fmol of each Pol with UL44ΔC290, unlabeled primer template, and 250 pmol dATP including 10 μCi [α-32P]-dATP, based on a previously reported method (29). For all assays, reactions were initiated by adding 10 mM MgCl2 and quenched after incubation at 37 °C for 10 min (polymerase assays for testing chain extension and idling assays) or for the times indicated [polymerase assays for kinetic studies and Exo assays] using 10 μL of stopping buffer (0.05% bromophenol blue, 0.05% xylene cyanol, and 10 mM EDTA in formamide for polymerase and Exo assays; 25 mM EDTA, 1% SDS, 5 mM dATP, and 5 mM dAMP for idling assays). The stopped polymerase and Exo reactions were heated to 100 °C for 3 min, quickly cooled on ice, and the products fractionated on a 20% (mass/vol) denaturing polyacrylamide gel. The gel images were quantified using a phosphorimager (BIO-RAD). Polymerase reactions for kinetic studies, with any of the enzymes studied, were linear for 10 min using dGTP as a substrate (Fig. S7) and were stopped at 5 min, and those using GCV-TP as a substrate were linear for at least 15 min (Fig. S7) and were stopped at 12 or 15 min. Polymerase kinetic constants were measured based on a method previously described (13). For apparent Km and Vmax, at each substrate concentration, the rate was calculated by measuring the fraction of primer template converted to product. Apparent Km and Vmax values were determined by fitting the data to the Michaelis–Menten equation using GraphPad Prism (Version 6), and apparent kcat values were calculated by dividing Vmax values by the enzyme concentration. Apparent Ki values were determined by using various concentrations of dGTP and 3 μM GCV-TP (at this concentration, no detectable incorporation of GCV-TP into primer template could be observed after 5 min of incubation) and Lineweaver–Burk analysis. Stopped idling assay reactions were analyzed by TLC on polyethyyleneimine/cellulose (Sigma) in 0.1 M phosphate buffer (pH 7.0), followed by phosphorimager analysis.
Supplementary Material
Acknowledgments
We thank Gloria Komazin-Meredith, Jeremy Kamil, and Dong Yu for kind provision of reagents; Suresh Srivastava of ChemGenes Corporation for expertise in the synthesis of GCV-containing oligonucleotides; and Charles Richardson and Blair Strang for very helpful suggestions. The contributions of Alexander Nussbaum to our understanding of the chemical methodology are gratefully remembered. This work was supported by National Institutes of Health Grant R01 AI019838.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1405981111/-/DCSupplemental.
References
- 1.Mocarski ES, Shenk T, Griffiths PD, Pass RF. Cytomegaloviruses. In: Knipe DM, et al., editors. Fields Virology. Lippincott Williams & Wilkins; Philadelphia: 2013. pp. 1960–2014. [Google Scholar]
- 2.Castro MG, Lowenstein PR. Neuro-oncology: The long and winding road—Gene therapy for glioma. Nat Rev Neurol. 2013;9(11):609–610. doi: 10.1038/nrneurol.2013.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Coen DM, Richman DD. Antiviral agents. In: Knipe DM, et al., editors. Fields Virology. Lippincott Williams & Wilkins; Philadelphia: 2013. pp. 338–373. [Google Scholar]
- 4.Reid R, Mar E-C, Huang E-S, Topal MD. Insertion and extension of acyclic, dideoxy, and ara nucleotides by herpesviridae, human α and human β polymerases. A unique inhibition mechanism for 9-(1,3-dihydroxy-2-propoxymethyl)guanine triphosphate. J Biol Chem. 1988;263(8):3898–3904. [PubMed] [Google Scholar]
- 5.Mar E-C, Chiou J-F, Cheng Y-C, Huang E-S. Inhibition of cellular DNA polymerase α and human cytomegalovirus-induced DNA polymerase by the triphosphates of 9-(2-hydroxyethoxymethyl)guanine and 9-(1,3-dihydroxy-2-propoxymethyl)guanine. J Virol. 1985;53(3):776–780. doi: 10.1128/jvi.53.3.776-780.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ilsley DD, Lee S-H, Miller WH, Kuchta RD. Acyclic guanosine analogs inhibit DNA polymerases α, δ, and ε with very different potencies and have unique mechanisms of action. Biochemistry. 1995;34(8):2504–2510. doi: 10.1021/bi00008a014. [DOI] [PubMed] [Google Scholar]
- 7.Frank KB, Chiou J-F, Cheng Y-C. Interaction of herpes simplex virus-induced DNA polymerase with 9-(1,3-dihydroxy-2-propoxymethyl)guanine triphosphate. J Biol Chem. 1984;259(3):1566–1569. [PubMed] [Google Scholar]
- 8.Reardon JE. Herpes simplex virus type 1 and human DNA polymerase interactions with 2′-deoxyguanosine 5′-triphosphate analogues. Kinetics of incorporation into DNA and induction of inhibition. J Biol Chem. 1989;264(32):19039–19044. [PubMed] [Google Scholar]
- 9.Hamzeh FM, Lietman PS. Intranuclear accumulation of subgenomic noninfectious human cytomegalovirus DNA in infected cells in the presence of ganciclovir. Antimicrob Agents Chemother. 1991;35(9):1818–1823. doi: 10.1128/aac.35.9.1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gilbert C, Bestman-Smith J, Boivin G. Resistance of herpesviruses to antiviral drugs: Clinical impacts and molecular mechanisms. Drug Resist Updat. 2002;5(2):88–114. doi: 10.1016/s1368-7646(02)00021-3. [DOI] [PubMed] [Google Scholar]
- 11.Lurain NS, Chou S. Antiviral drug resistance of human cytomegalovirus. Clin Microbiol Rev. 2010;23(4):689–712. doi: 10.1128/CMR.00009-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu S, et al. Crystal structure of the herpes simplex virus 1 DNA polymerase. J Biol Chem. 2006;281(26):18193–18200. doi: 10.1074/jbc.M602414200. [DOI] [PubMed] [Google Scholar]
- 13.Huang L, et al. The enzymological basis for resistance of herpesvirus DNA polymerase mutants to acyclovir: Relationship to the structure of α-like DNA polymerases. Proc Natl Acad Sci USA. 1999;96(2):447–452. doi: 10.1073/pnas.96.2.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Feng JY, Anderson KS. Mechanistic studies examining the efficiency and fidelity of DNA synthesis by the 3TC-resistant mutant (184V) of HIV-1 reverse transcriptase. Biochemistry. 1999;38(29):9440–9448. doi: 10.1021/bi990709m. [DOI] [PubMed] [Google Scholar]
- 15.Sarafianos SG, et al. Lamivudine (3TC) resistance in HIV-1 reverse transcriptase involves steric hindrance with β-branched amino acids. Proc Natl Acad Sci USA. 1999;96(18):10027–10032. doi: 10.1073/pnas.96.18.10027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sarafianos SG, et al. Structure and function of HIV-1 reverse transcriptase: Molecular mechanisms of polymerization and inhibition. J Mol Biol. 2009;385(3):693–713. doi: 10.1016/j.jmb.2008.10.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gibbs JS, et al. Polymerization activity of an α-like DNA polymerase requires a conserved 3′-5′ exonuclease active site. Mol Cell Biol. 1991;11(9):4786–4795. doi: 10.1128/mcb.11.9.4786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kühn FJ, Knopf CW. Herpes simplex virus type 1 DNA polymerase. Mutational analysis of the 3′-5′-exonuclease domain. J Biol Chem. 1996;271(46):29245–29254. doi: 10.1074/jbc.271.46.29245. [DOI] [PubMed] [Google Scholar]
- 19.Hwang YT, Smith JF, Gao L, Hwang CB. Mutations in the Exo III motif of the herpes simplex virus DNA polymerase gene can confer altered drug sensitivities. Virology. 1998;246(2):298–305. doi: 10.1006/viro.1998.9201. [DOI] [PubMed] [Google Scholar]
- 20.Chou S, Lurain NS, Thompson KD, Miner RC, Drew WL. Viral DNA polymerase mutations associated with drug resistance in human cytomegalovirus. J Infect Dis. 2003;188(1):32–39. doi: 10.1086/375743. [DOI] [PubMed] [Google Scholar]
- 21.Meyer PR, Matsuura SE, So AG, Scott WA. Unblocking of chain-terminated primer by HIV-1 reverse transcriptase through a nucleotide-dependent mechanism. Proc Natl Acad Sci USA. 1998;95(23):13471–13476. doi: 10.1073/pnas.95.23.13471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Arion D, Kaushik N, McCormick S, Borkow G, Parniak MA. Phenotypic mechanism of HIV-1 resistance to 3′-azido-3′-deoxythymidine (AZT): Increased polymerization processivity and enhanced sensitivity to pyrophosphate of the mutant viral reverse transcriptase. Biochemistry. 1998;37(45):15908–15917. doi: 10.1021/bi981200e. [DOI] [PubMed] [Google Scholar]
- 23.Tu X, et al. Structural basis of HIV-1 resistance to AZT by excision. Nat Struct Mol Biol. 2010;17(10):1202–1209. doi: 10.1038/nsmb.1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kariya M, Mori S, Eizuru Y. Comparison of human cytomegalovirus DNA polymerase activity for ganciclovir-resistant and -sensitive clinical strains. Antiviral Res. 2000;45(2):115–122. doi: 10.1016/s0166-3542(00)00063-2. [DOI] [PubMed] [Google Scholar]
- 25.Roth AC, Nossal NG, Englund PT. Rapid hydrolysis of deoxynucleoside triphosphates accompanies DNA synthesis by T4 DNA polymerase and T4 accessory proteins 44/62 and 45. J Biol Chem. 1982;257(3):1267–1273. [PubMed] [Google Scholar]
- 26.Tabor S, Richardson CC. Selective inactivation of the exonuclease activity of bacteriophage T7 DNA polymerase by in vitro mutagenesis. J Biol Chem. 1989;264(11):6447–6458. [PubMed] [Google Scholar]
- 27.Belguise-Valladier P, Maki H, Sekiguchi M, Fuchs RP. Effect of single DNA lesions on in vitro replication with DNA polymerase III holoenzyme. Comparison with other polymerases. J Mol Biol. 1994;236(1):151–164. doi: 10.1006/jmbi.1994.1125. [DOI] [PubMed] [Google Scholar]
- 28.Khare V, Eckert KA. The 3′ → 5′ exonuclease of T4 DNA polymerase removes premutagenic alkyl mispairs and contributes to futile cycling at O6-methylguanine lesions. J Biol Chem. 2001;276(26):24286–24292. doi: 10.1074/jbc.M011025200. [DOI] [PubMed] [Google Scholar]
- 29.Garg P, Stith CM, Sabouri N, Johansson E, Burgers PM. Idling by DNA polymerase δ maintains a ligatable nick during lagging-strand DNA replication. Genes Dev. 2004;18(22):2764–2773. doi: 10.1101/gad.1252304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cihlar T, Fuller MD, Mulato AS, Cherrington JM. A point mutation in the human cytomegalovirus DNA polymerase gene selected in vitro by cidofovir confers a slow replication phenotype in cell culture. Virology. 1998;248(2):382–393. doi: 10.1006/viro.1998.9299. [DOI] [PubMed] [Google Scholar]
- 31.Bernad A, Blanco L, Lázaro JM, Martín G, Salas M. A conserved 3′–5′ exonuclease active site in prokaryotic and eukaryotic DNA polymerases. Cell. 1989;59(1):219–228. doi: 10.1016/0092-8674(89)90883-0. [DOI] [PubMed] [Google Scholar]
- 32.Simon M, Giot L, Faye G. The 3′ to 5′ exonuclease activity located in the DNA polymerase δ subunit of Saccharomyces cerevisiae is required for accurate replication. EMBO J. 1991;10(8):2165–2170. doi: 10.1002/j.1460-2075.1991.tb07751.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Morrison A, Bell JB, Kunkel TA, Sugino A. Eukaryotic DNA polymerase amino acid sequence required for 3′–5′ exonuclease activity. Proc Natl Acad Sci USA. 1991;88(21):9473–9477. doi: 10.1073/pnas.88.21.9473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cihlar T, Fuller MD, Cherrington JM. Characterization of drug resistance-associated mutations in the human cytomegalovirus DNA polymerase gene by using recombinant mutant viruses generated from overlapping DNA fragments. J Virol. 1998;72(7):5927–5936. doi: 10.1128/jvi.72.7.5927-5936.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gilbert C, Azzi A, Goyette N, Lin S-X, Boivin G. Recombinant phenotyping of cytomegalovirus UL54 mutations that emerged during cell passages in the presence of either ganciclovir or foscarnet. Antimicrob Agents Chemother. 2011;55(9):4019–4027. doi: 10.1128/AAC.00334-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Reardon JE, Spector T. Herpes simplex virus type 1 DNA polymerase. Mechanism of inhibition by acyclovir triphosphate. J Biol Chem. 1989;264(13):7405–7411. [PubMed] [Google Scholar]
- 37.Clark JM, Joyce CM, Beardsley GP. Novel blunt-end addition reactions catalyzed by DNA polymerase I of Escherichia coli. J Mol Biol. 1987;198(1):123–127. doi: 10.1016/0022-2836(87)90462-1. [DOI] [PubMed] [Google Scholar]
- 38.Topal MD, DiGuiseppi SR, Sinha NK. Molecular basis for substitution mutations. Effect of primer terminal and template residues on nucleotide selection by phage T4 DNA polymerase in vitro. J Biol Chem. 1980;255(24):11717–11724. [PubMed] [Google Scholar]
- 39.Foti M, et al. Solution structure of a DNA decamer containing the antiviral drug ganciclovir: Combined use of NMR, restrained molecular dynamics, and full relaxation matrix refinement. Biochemistry. 1997;36(18):5336–5345. doi: 10.1021/bi962604e. [DOI] [PubMed] [Google Scholar]
- 40.Safrin S, Cherrington J, Jaffe HS. Clinical uses of cidofovir. Rev Med Virol. 1997;7(3):145–156. doi: 10.1002/(sici)1099-1654(199709)7:3<145::aid-rmv196>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 41.Marty FM, et al. CMX001-201 Clinical Study Group CMX001 to prevent cytomegalovirus disease in hematopoietic-cell transplantation. N Engl J Med. 2013;369(13):1227–1236. doi: 10.1056/NEJMoa1303688. [DOI] [PubMed] [Google Scholar]
- 42.Xiong X, Smith JL, Chen MS. Effect of incorporation of cidofovir into DNA by human cytomegalovirus DNA polymerase on DNA elongation. Antimicrob Agents Chemother. 1997;41(3):594–599. doi: 10.1128/aac.41.3.594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hwang YT, Liu B-Y, Coen DM, Hwang CB. Effects of mutations in the Exo III motif of the herpes simplex virus DNA polymerase gene on enzyme activities, viral replication, and replication fidelity. J Virol. 1997;71(10):7791–7798. doi: 10.1128/jvi.71.10.7791-7798.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Marshalko SJ, Schweitzer BI, Beardsley GP. Chiral chemical synthesis of DNA containing (S)-9-(1,3-dihydroxy-2-propoxymethyl)guanine (DHPG) and effects on thermal stability, duplex structure, and thermodynamics of duplex formation. Biochemistry. 1995;34(28):9235–9248. doi: 10.1021/bi00028a037. [DOI] [PubMed] [Google Scholar]
- 45.Appleton BA, Loregian A, Filman DJ, Coen DM, Hogle JM. The cytomegalovirus DNA polymerase subunit UL44 forms a C clamp-shaped dimer. Mol Cell. 2004;15(2):233–244. doi: 10.1016/j.molcel.2004.06.018. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.