

Significance
Despite the widespread use and success of tamoxifen for treating ER-positive breast cancers, overcoming resistance to this drug remains an unmet need in clinical breast oncology. The results presented in this study demonstrate that overexpression of a novel gene, MACROD2, can mediate tamoxifen resistance and estrogen independent growth in human breast cancers, and that amplification of MACROD2 in primary breast tumors is associated with worse overall survival.
Keywords: breast cancer, tamoxifen, resistance, MACROD2, ER positive
Abstract
Tamoxifen is effective for treating estrogen receptor-alpha (ER) positive breast cancers. However, few molecular mediators of tamoxifen resistance have been elucidated. Here we describe a previously unidentified gene, MACROD2 that confers tamoxifen resistance and estrogen independent growth. We found MACROD2 is amplified and overexpressed in metastatic tamoxifen-resistant tumors. Transgene overexpression of MACROD2 in breast cancer cell lines results in tamoxifen resistance, whereas RNAi-mediated gene knock down reverses this phenotype. MACROD2 overexpression also leads to estrogen independent growth in xenograft assays. Mechanistically, MACROD2 increases p300 binding to estrogen response elements in a subset of ER regulated genes. Primary breast cancers and matched metastases demonstrate MACROD2 expression can change with disease evolution, and increased expression and amplification of MACROD2 in primary tumors is associated with worse overall survival. These studies establish MACROD2 as a key mediator of estrogen independent growth and tamoxifen resistance, as well as a potential novel target for diagnostics and therapy.
The selective estrogen receptor modulator (SERM) tamoxifen is a highly effective drug for the prevention and treatment of estrogen receptor-alpha (ER) positive breast cancers (1). However, resistance to this drug remains a clinically important problem. The molecular mediators of tamoxifen resistance have not been fully elucidated. In part, this is due to the heterogeneous nature of breast cancers, resulting in multiple mechanisms of resistance. For example, past studies have demonstrated that tamoxifen resistance is mediated by differential expression of nuclear hormone receptor coregulators (2, 3), growth factor signaling crosstalk (4–7), regulation of microRNAs (8), cyclin dependent kinases (CKDs) (9), CDK inhibitors (10, 11), and more recently, acquired somatic mutations and alterations in ER (12–17). Further insight into the molecular mediators of tamoxifen and hormone therapy resistance would have great impact on the ability to target genes and pathways that could overcome drug resistance and lead to improved clinical outcomes.
In this study we describe a previously unidentified gene, MACROD2, which is amplified and overexpressed in a subset of breast cancers. MACROD2 belongs to a family of genes containing a macro domain, an evolutionarily conserved protein motif (18), whose functional role until recently has been unclear. Studies have demonstrated that MACROD2 deacetylates O-acetyl-ADP ribose, a signaling molecule generated by the deacetylation of acetylated lysine residues in histones and other proteins (19). More recent work demonstrates that MACRO domain containing proteins are involved with mono-ADP ribosylation, and can regulate cell signaling pathways and modify proteins involved with gene transcription (20). Interestingly, MACROD1 (LRP16) has been implicated in modulating ER and androgen receptor (AR) signaling in prior studies (21, 22). Additionally, recent reports suggest that the locus encompassing the MACROD2 gene at chromosome 20p12.1 may be a cancer-specific fragile site leading to frequent somatic deletions (23). Notably, breast cancers were not prone to fragile site deletions in these studies. Here we show that MACROD2 is amplified and overexpressed in human breast cancers, leading to tamoxifen resistance and estrogen independent growth, and that patients with primary breast cancers with overexpression/amplification of MACROD2 have worse survival. Thus, our study identifies MACROD2 as a new mediator of ER signaling and tamoxifen resistance with potential clinical implications.
Results
MACROD2 Is Amplified in a Subset of Tamoxifen-Resistant Breast Cancers.
We previously generated tamoxifen-resistant (TamR) clones derived from the ER-positive breast cancer cell line MCF-7 after long term culture and demonstrated that loss of the CDK inhibitor p21 could mediate resistance to this SERM (10). We reasoned that additional tamoxifen resistant clones, which retained p21 expression, acquired resistance through additional mechanisms and that common copy number (CN) alterations within these clones could help identify molecular mediators of this phenotype. Using single nucleotide polymorphism (SNP) arrays, we identified regions of genomic gains and losses in three independently derived TamR clones compared with parental MCF-7 cells. As shown in SI Appendix, Fig. S1A, all three clones had varying regions of copy number alterations, some of which were unique for a given clone. A total of 16 regions of shared CN gains or losses were identified (SI Appendix, Table S1).
A region on chromosome 20p12.1 (SI Appendix, Fig. S1B) demonstrated the highest increase in CN gain across the three TamR clones. This locus, containing the genes SEL1L2, MACROD2, and FLRT3, was further investigated. Using quantitative PCR (qPCR) with primers within the 20p12.1 locus along with primers within an invariant chromosome 20 locus as a reference control, we observed a 6- to 12-fold increase (P < 0.05) in DNA copy number compared with parental MCF-7 cells, consistent with amplification of this region (Fig. 1A). We next evaluated the expression of the three genes within the 20p12.1 locus. Using quantitative real-time reverse transcriptase PCR (qRT-PCR), we found that expression of MACROD2 was increased in all three TamR clones, whereas SEL1L2 and FLRT3 appeared to have unchanged levels of expression (Fig. 1B). Western blot confirmed overexpression of MACROD2 protein in the TamR clones, whereas FLRT3 and SEL1L2 protein expression remained equivalent to parental MCF-7 cells (Fig. 1C). These results strongly suggest that amplification of MACROD2 leads to its increased gene expression in TamR clones.
Fig. 1.

Increased copy number of MACROD2 in tamoxifen-resistant MCF-7 cell lines. (A) Increase in copy number of the MACROD2 locus was verified by qPCR performed using multiple primer sets. Shown are 2 different primer sets contained within the locus relative to control primers located within an invariant locus on chromosome 20. qPCR was performed in triplicate within each PCR assay and each assay was performed at least 4 times. *P < 0.05. (B) RNA was harvested from the TamR lines and parental MCF-7 cells and used for qRT-PCR analysis. Transcripts located within the region of amplification were tested for levels using primers within the three genes: MACROD2, FLRT3, and SEL1L2. The samples were normalized to β-actin and TamR clones’ relative expression of these three genes was compared and normalized to MCF-7 gene expression. *P < 0.01. (C) Western blot analysis was performed on harvested whole cell lysates demonstrating an increase in MACROD2 protein levels in the TamR cell lines with no change in protein levels of FLRT3 or SEL1L2. GAPDH antibody was used as a loading control.
To verify that amplification of this region was also present in actual human breast cancers, we evaluated liver metastases from five ER-positive breast cancer patients. These patients (patients 3, 5, 6, 8, and 10) had documented tamoxifen resistance, and were part of a rapid autopsy series (24). Single metastatic breast cancer lesions and adjacent normal tissues were analyzed for the presence of chromosome 20p12.1 CN gain using qPCR. Interestingly, we found that in three of the five patient samples, there was a marked reduction in CN, indicating a possible loss of this genomic locus. Conversely, patients 5 and 10 showed a significant CN gain of this locus, indicating an increase CN gain of the 20p12.1 region (Fig. 2A). We then performed immunohistochemistry (IHC) staining of these five patients’ primary breast cancers and matching metastatic lesions from multiple sites. Patient 10’s liver lesion used for qPCR was unavailable for this analysis; however other sites of disease were available and analyzed. As seen in Fig. 2B and SI Appendix, Fig. S2 and Table S2, MACROD2 labeling was positive in three out of five primary breast cancer samples and was present in multiple metastatic sites from all five patients, although there was variability in expression between metastatic sites of disease within each patient. This finding is consistent with the known concept of tumor heterogeneity within a primary tumor and metastatic sites (25). These findings are also in accord with recent data demonstrating that acquired ER mutations can be found in liver metastatic lesions, but not pulmonary metastases within the same patient (15), and additional studies revealing that ER mutations are relatively rare in primary breast cancers but are more common after acquired resistance to endocrine therapies (12–14, 16). Gene expression qRT-PCR data from the prior analysis in Fig. 1B correlated with IHC results for the metastatic sites queried in these five patients. These results demonstrate that MACROD2 overexpression is present in primary breast cancers and can increase in metastatic sites of disease after development of resistance to tamoxifen therapy.
Fig. 2.

Increased copy number of MACROD2 in metastatic lesions from patients with tamoxifen-resistant disease. (A) Quantitative PCR was performed on genomic DNA isolated from frozen tissue specimens obtained from breast cancer patients with documented tamoxifen resistance. Liver metastases (Tu) and adjacent normal tissue (Nl) were analyzed for increased MACROD2 copy number using primers within the amplified region and normalizing to an invariant locus on chromosome 20p. *P < 0.05. (B) FFPE samples were used for MACROD2 immunohistochemical labeling as described in the text. Shown is patient 3’s primary breast cancer (Upper) scored as negative for nuclear MACROD2 labeling and a soft tissue metastatic rib lesion (Lower) from patient 3, scored as positive for nuclear MACROD2 labeling. Magnification: 400×.
MACROD2 Overexpression Leads to Tamoxifen Resistance, and Gene Knock Down Reverses This Resistance.
To determine whether increased gene expression of MACROD2 in TamR clones mediates tamoxifen resistance, we overexpressed the full length MACROD2 cDNA in two ER-positive breast cancer cell lines, MCF-7 and T47D. Western blot analysis verified overexpression of MACROD2 relative to parental and empty vector controls (Fig. 3A). We then exposed the MACROD2 overexpressing cell lines to tamoxifen for 7 d and verified relative resistance to tamoxifen in both MCF-7– and T47D-derived clones (Fig. 3B). Interestingly, MACROD2 overexpressing cell lines were growth-stimulated by tamoxifen, similar to prior reports that tamoxifen can often act as an ER agonist when resistance occurs (10). To definitively demonstrate that MACROD2 was mediating tamoxifen resistance along with tamoxifen-stimulated growth, TamR and MACROD2 overexpressing cell lines were exposed to estrogen, tamoxifen, or both. As shown in Fig. 3C, both TamR and MACROD2 overexpressing clones demonstrated a tamoxifen resistant phenotype, and when grown in tamoxifen alone, both MACROD2 overexpressing clones and TamR cell lines appeared to grow preferentially with tamoxifen. As expected, the parental and empty vector control cells did not show growth with tamoxifen compared with the vehicle control but did demonstrate the anticipated growth response to estrogen that was blocked with the addition of tamoxifen.
Fig. 3.

MACROD2 overexpression in human breast cancer cell lines mediates tamoxifen resistance and growth. (A) Whole cell lysates were harvested from cell lines as indicated and used for Western blots. EV, empty vector control. GAPDH antibody staining was used for a loading control. (B) MCF-7, T47D and their derivate cell lines were seeded in assay media and exposed to vehicle (ethanol) or 1 μM 4-OH tamoxifen for 7 d as described in the text. EV, empty vector control. *P < 0.01. (C) MCF-7 and the derivative cell lines were seeded in triplicate and exposed to estrogen (1.25 nM 17-β-estradiol), tamoxifen (1 μM 4-OH tamoxifen), or a combination of both drugs for 7 d as described in the text. EV, empty vector control. *P < 0.01.
We next sought to determine whether knock down of MACROD2 gene expression by RNAi could reverse tamoxifen resistance. This reversal was accomplished through the use of short hairpin RNAi (shRNA) constructs against the MACROD2 transcript in TamR cell lines. We generated two constructs, shRNA3 and shRNA5, which were effective in reducing expression of MACROD2 when stably expressed in TamR clones (Fig. 4A). We then assessed tamoxifen sensitivity by growing the MACROD2 knock down and control cell lines in the presence or absence of tamoxifen. As shown in Fig. 4 B and C, shRNA knock down resulted in reversal of tamoxifen resistance compared with control shRNA cell lines or parental TamR clones. There was, however, partial rather than full restoration of tamoxifen sensitivity, which may be due to the inability of RNAi to completely abrogate gene expression. Taken together with our cDNA overexpression experiments, these results strongly suggest that overexpression of MACROD2 can mediate a tamoxifen resistance phenotype.
Fig. 4.

Gene knock down of MACROD2 reverses tamoxifen resistance in TamR clones. (A) The shRNA constructs (shRNA3 and shRNA5) were used to stably knock down MACROD2 gene expression in TamR clones along with a control shRNA. Verification of knock down was performed by Western blot for MACROD2 in whole cell lysates. GAPDH was used as a loading control. (B and C) MCF-7 and the derivative cell lines were seeded in triplicate and exposed to vehicle control (ethanol) or tamoxifen (1uM 4-OH tamoxifen) for 7 d as described in the text. EV, empty vector control. *P < 0.05. Representative results are shown in B with quantification relative to controls and depicted graphically in C.
MACROD2 Overexpression Increases Gene Expression of ER Regulated Genes and p300 Binding to Estrogen Response Elements.
MCF-7 cells are a frequently used model for ER-positive estrogen-dependent breast cancer cell growth. However, TamR clones demonstrated a relatively reduced response to exogenous estrogens and indeed were capable of cell proliferation in the absence of estrogen and tamoxifen. Prior reports demonstrated that the MACROD2 homolog MACROD1 (LRP16), functions as an ER coactivator in MCF-7 cells (21). We reasoned that MACROD2 overexpression may function similarly as an ER coactivator and that its overexpression may lead to increased expression of ER regulated genes, even in the absence of estrogen. We initially surveyed selected candidate ER-regulated genes using RT-PCR and demonstrated that in the absence of estrogen and tamoxifen, gene expression was increased in TamR clones (Fig. 5A). We hypothesized that MACROD2 may directly or indirectly bind regulatory elements in these estrogen regulated genes, and/or increase binding of other known ER coactivators. We therefore performed chromatin immunoprecipitation (ChIP) using anti-MACROD2, ER and p300 (an ER coactivator) antibodies. For these experiments, we used parental MCF-7 cells as well as two TamR clones grown under serum-starved conditions in the presence of ethanol (vehicle control), estrogen or tamoxifen. Quantitative real time PCR was used to assess relative fold changes in DNA binding using primers for promoter and enhancer regions with known estrogen response elements (EREs) (26). As seen in Fig. 5B and SI Appendix, Fig. S3, there was variability in DNA binding using our anti-MACROD2 antibody for the regions analyzed in estrogen and tamoxifen growth conditions relative to vehicle controls. This variability may reflect differences in clonal populations and/or the fact that there are currently no validated MACROD2 antibodies for ChIP. Indeed, in our hands commercial antibodies did not have specificity for MACROD2 by Western blot, necessitating the use of our own antibody for these studies. On the other hand, ER binding was for the most part consistent with its known role in binding EREs in several ER regulated genes. Strikingly, in TamR cells stimulated with tamoxifen, a consistent pattern of increased p300 binding was seen in all four promoter regions compared with parental MCF-7 cells (Fig. 5B). This same consistent pattern of increased p300 binding upon tamoxifen stimulation was seen in other regulatory elements of the known ER response genes PGR (enhancer region), SMAD, SBNO2, P2RY2, ABCA3, and GREB1, whereas estrogen induced variable results in p300 binding (SI Appendix, Fig. S3). The single exception to this trend was the analysis of NRIP1. Because p300 is a well known coactivator and mediator of ER regulated gene expression, taken together, these results suggest that overexpression of MACROD2 may lead to increased expression of ER regulated genes by facilitating augmented binding of p300 to EREs.
Fig. 5.

MACROD2 overexpression can induce estrogen regulated genes and increases p300 binding to estrogen response elements in response to tamoxifen. (A) The parental MCF-7 and TamR clones were seeded in assay media without estrogen and tamoxifen and then RNA was harvested from these cells and RT-PCR was performed on a subset of estrogen responsive genes (shown). GAPDH was used as a loading control. (B) Parental MCF-7 cells and TamR cells were seeded in assay conditions and then vehicle (ethanol) or tamoxifen was added to the cells. The cells were then harvested and subjected to chromatin immunoprecipitation (ChIP) as described in the text using antibodies against MACROD2, ER, p300, and a control [IgG(−)]. Quantitative PCR was then performed using primers that encompass estrogen response elements in promoter or enhancer regions of known ER target genes (primers in SI Appendix, Table S5). Results are representative of duplicate samples comparing quantitative real time PCR results after controlling for input DNA and then analyzing relative fold differences between tamoxifen and vehicle control. Parental MCF-7 and two representative TamR clones are shown.
MACROD2 Overexpression Alters Protein Expression of ER and Non-ER Regulated Genes and Leads to Estrogen Independent Growth in Vivo.
Given MACROD2’s recently described function in ADP ribosylation (20), and the fact that ER coactivators can have both estrogen dependent and independent mechanisms of mediating tumor growth (27) we sought to examine a broader array of signaling pathways that regulate cell growth. We therefore subjected TamR and parental MCF-7 cells to reverse phase protein array (RPPA) analysis. As shown in SI Appendix, Fig. S4A, the overall expression pattern was similar between TamR clones, yet was strikingly distinct from parental MCF-7 cells. Interestingly, several growth promoting genes and enzymes were differentially expressed in TamR clones relative to parental MCF-7 cells (SI Appendix, Fig. S4B), suggesting MACROD2 overexpression may mediate its effects via ER and non-ER regulated genes.
To further characterize MACROD2 estrogen independent growth, we assessed TamR proliferation as xenografts in athymic nude mice. It is well established that MCF-7 cells require estrogen supplementation to grow as xenografts (28, 29). Therefore, we inoculated female athymic nude mice with the TamR clones as well as their MACROD2 knock down counterparts without exogenous estrogen supplementation. Xenograft tumors did not form in control mice inoculated with parental MCF-7 cells (SI Appendix, Table S3). However, as seen in Fig. 6A, we observed extensive in vivo tumor growth for parental TamR clones compared with TamR clones expressing MACROD2 shRNA knock down. Histologic analysis of TamR xenografts demonstrated poorly differentiated tumors with numerous mitoses (SI Appendix, Fig. S5). These results strongly suggest that overexpression of MACROD2 can lead to an estrogen independent growth phenotype.
Fig. 6.
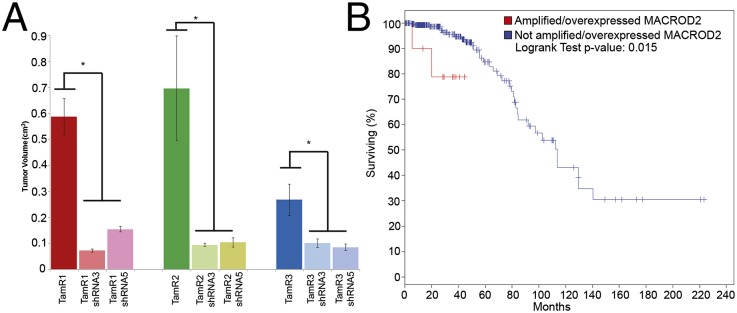
MACROD2 overexpression results in estrogen-independent tumor formation in vivo, and overexpression in primary breast cancers is associated with worse survival in luminal A/B tumors. (A) TamR clones and their shRNA-expressing counterparts (shRNA3 or shRNA5) were inoculated into female athymic nude mice (2 × 106 cells per mouse) in reduced growth factor Matrigel. After 42 d, mice were killed and tumor volume was measured. Results are representative of three independent experiments with 10 mice per group. *P < 0.05. (B) MACROD2 amplification and overexpression (greater than twofold) were used as parameters to query The Cancer Genome Atlas (TCGA) breast cancer database, limited to luminal A/B (i.e., ER positive) tumors as described in the text. Shown is a Kaplan–Meier estimate of overall survival between patients with luminal A/B primary breast cancers with amplification or overexpression of MACROD2 (red) and those without (blue).
Cell Proliferation Mediated by MACROD2 Overexpression Is Dependent on ER and Is Not Sufficient for Growth Factor Independent Growth in ER Negative Cells.
Although MACROD2 overexpression led to tamoxifen resistance and estrogen independent growth, its ability to enhance gene expression of ER regulated genes suggested these effects may require ER and other cofactors to mediate cell proliferation. To address this, we first used pharmacologic means to reduce ER expression with the selective estrogen receptor down modulator, fulvestrant. As seen in SI Appendix, Fig. S6A, fulvestrant successfully reduced levels of ER in MCF-7 and TamR cell lines as shown by Western blot, although interestingly, this effect was reversed with the addition of tamoxifen. Cell lines were then subjected to proliferation assays to determine if reduction of ER could affect growth of TamR cell lines. As shown in SI Appendix, Fig. S6B, fulvestrant did reduce cell proliferation relative to tamoxifen stimulated growth, although this effect was partial. This partial effect could be due to other growth factors regulated by MACROD2 and/or that pharmacologic reduction of ER was not 100%. To further address this question, we then overexpressed the MACROD2 cDNA in the ER negative, nontumorigenic cell line, MCF-10A (30). This cell line requires exogenous growth factors for continuous proliferation, including epidermal growth factor (EGF), and we have previously characterized that gene targeted oncogenic mutations can lead to EGF independent growth (31). As demonstrated in SI Appendix, Fig. S6C, transient and stable transfection of MCF-10A led to detectable MACROD2 protein by Western blot. However, when comparing EGF-independent growth relative to EGF dependent growth, (SI Appendix, Fig. S6D), there was no discernible differences between parental MCF-10A, empty vector control or cells transfected with MACROD2. Moreover, gene expression of ER regulated genes was not increased in MACROD2 overexpressing MCF-10A cells (SI Appendix, Fig. S6E), despite the fact that transfection of ER in MCF-10A cells can lead to increased expression of these genes (32). Collectively, these results suggest that MACROD2 is dependent on ER and possibly other cofactors to mediate its proliferative effects.
Primary Luminal Breast Cancers with Overexpression or Amplification of MACROD2 Have Worse Prognosis.
Finally, to determine the clinical significance of MACROD2 overexpression in human breast cancers, we queried The Cancer Genome Atlas (TCGA) database for luminal human breast cancers that demonstrated overexpression and/or increased gene copy number of MACROD2. Using the cBioPortal for Cancer Genomics platform (33), ∼4% of luminal A or B (i.e., ER positive) primary breast cancers demonstrated increased expression or amplification of MACROD2 in this analysis. An overall survival Kaplan–Meier estimate demonstrated a statistically significant difference between patients whose tumors had overexpression/amplification of MACROD2 compared with those that did not (P = 0.015) (Fig. 6B). Because this analysis represented fewer than 200 patients, we wished to extend these findings to additional larger datasets. The METABRIC database incorporates almost 2,000 women and clinical follow up with a history of primary breast cancers (34). Importantly, ER/PR status, copy number variations and gene expression analysis is available on these patients’ tumors as well as survival outcomes. We queried these data to determine whether our initial results could be confirmed. As shown in SI Appendix, Fig. S7, there were statistically significant differences in survival in patients whose tumors demonstrated amplification/overexpression of MACROD2, but almost exclusively in women with ER and/or PR-positive disease. These results provide additional evidence that MACROD2 overexpression is biologically relevant, has prognostic significance for ER-positive breast cancers, and may serve as a predictive marker and future target of therapy.
Discussion
Although great strides have been made in treating ER-positive breast cancers using endocrine therapies, drug resistance remains a formidable clinical problem. Indeed, there has been renewed interest in understanding and uncovering genetic effectors of endocrine therapy resistance with the recent discovery of ER mutations and translocations that are found at relatively high frequency in metastases but are rare in primary breast tumors. Notably, these studies suggest the emergence of ER mutations/alterations after treatment and progression on endocrine therapies. However, ER mutations/alterations do not account for all mechanisms of hormone resistance, and the ability to identify additional mediators of resistance remains of high clinical importance. In this study we have identified MACROD2 as a mediator of tamoxifen resistance and estrogen independent growth. We conclude this on the following bases: First, independently derived tamoxifen-resistant MCF-7 clones (TamR) have overexpression and amplification of MACROD2. Second, MACROD2 overexpression is observed in human breast cancer samples from patients with tamoxifen-resistant disease. Third, overexpression of MACROD2 in two separate ER-positive breast cancer cell lines leads to tamoxifen resistance, and gene knock down by stable shRNA reverses this phenotype. Fourth, MACROD2 overexpression leads to estrogen independent growth in vivo, and up-regulates growth promoting genes that are both ER-regulated and non-ER regulated. Finally, overexpression or amplification of MACROD2 in ER-positive (luminal A/B) primary breast tumors has a significantly worse outcome compared with breast cancer patients whose primary tumors do not have overexpressed/amplified MACROD2. This result suggests that these patients may have de novo resistance to tamoxifen and perhaps other endocrine therapies. Of note, ESR1 (ER) mutations were not found in TamR clones further underscoring the various mechanisms leading to tamoxifen resistance (SI Appendix, Fig. S8).
As mentioned, MACROD2 is a relatively newly characterized gene and paradoxically, some cancer sequencing/genomic studies have shown that it is frequently deleted, leading to the hypothesis that it may function as a tumor suppressor (23). Interestingly, the Sanger Institute demonstrated that MACROD2 is not a general common fragile site (35), but suggested that it may be a cancer-specific fragile site similar to a recent report (23), although in the latter study breast cancers were not prone to a high frequency of MACROD2 deletion. In contrast, our study demonstrates that MACROD2 overexpression and amplification occur in breast cancers, leading to tamoxifen resistance and estrogen-independent growth, properties more consistent with an oncogene. It may be that as a cancer specific fragile site, MACROD2 is lost without any selective pressure for its retention. However, in the case of ER-positive breast cancers treated with tamoxifen, the fragility of this locus allows for amplification, so that drug resistant clones can emerge. Along those lines, data from TCGA would suggest that overexpression/amplification of MACROD2 occurs at relatively low frequency in primary breast cancers, but the results from our study suggest that metastatic sites of disease display a higher frequency of MACROD2 overexpression, which may mediate tamoxifen resistance, and also estrogen independent growth. A limitation of our study is the relatively small number of patients examined, as it is difficult to obtain metastatic biopsies from patients with clinical follow up. However, because MACROD2 is amplified, it may be possible in the future to quickly identify and “track” MACROD2 amplified metastatic disease using a liquid biopsy approach recently described by Bardelli and colleagues (36). Regardless, the significantly worse outcomes in patients whose primary breast cancers display overexpression/amplification of MACROD2, including the large dataset from the METABRIC study, is consistent with the idea that MACROD2 may herald intrinsic resistance to tamoxifen therapy with a more aggressive cancer phenotype.
Our gene expression and ChIP data suggest that MACROD2 can mediate cell growth and proliferation through ER-dependent and -independent mechanisms. Given MACROD2’s recently described role in mono-ADP ribosylation and other enzymatic functions, as well as the fact that MACROD2 overexpression increases p300 coactivator binding to EREs, it is likely that MACROD2 affects gene expression via transcriptional regulation and epigenetic modifications. With the recent interest and success of epigenetic therapies for cancer treatment, it is tempting to speculate that MACROD2 may be a “druggable” protein, and that its overexpression may help identify patients whose tumors have intrinsic resistance to tamoxifen and a high risk phenotype. Thus, the discovery of a previously unidentified gene, MACROD2, and its functional role in drug resistance, may lead to improved systemic therapies and predictive markers for the treatment of ER-positive breast cancers.
Materials and Methods
Cell Culture and Transfections.
Cell lines used have been described (31). Overexpression and shRNA constructs used for transfection and assays were performed as described (37, 38).
Cell proliferation Assays.
Cell proliferation assays were performed as described (37, 39).
Xenograft Assays.
Xenograft assays were performed as described (37).
In Silico Data Analysis.
Expression data and prognostic survival curves were generated using cBioPortal (33) using the TCGA database for breast cancers and selecting for luminal A/B tumors which demonstrated either 2× overexpression or amplification of MACROD2. The METABRIC database has been previously described (34). Upon IRB approval, these data were accessed through Synapse (synapse.sagebase.org), and used for correlating MACROD2 amplification/overexpression with survival.
Additional methods are provided in SI Appendix, SI Materials and Methods.
Supplementary Material
Acknowledgments
This work was supported by Department of Defense Breast Cancer Research Program BC083057 (to M.M.), BC100972 (to B.G.B.), and BC102278 (to J.L.); Flight Attendant Medical Research Institute (FAMRI) (J.L. and B.H.P.); The Avon Foundation (B.H.P.); The Stetler Fund (J.A.B.); Susan G. Komen for the Cure (J.L.); and NIH CA088843 (to J.L. and B.H.P.), GM007309 (to G.M.W. and D.J.Z.), CA168180 (to R.L.C.), CA167939 (to S.C.), and CA009071-S1 (to K.C.). We acknowledge the support of the NIH Cancer Center Support Grant, P30 CA006973. We also thank and acknowledge the support of the Sandy Garcia Charitable Foundation, the Commonwealth Foundation, the Santa Fe Foundation, the Breast Cancer Research Foundation, the Health Network Foundation, the Marcie and Ellen (ME) Foundation, and The Robin Page/Lebor Foundation.
Footnotes
Conflict of interest statement: B.H.P. is a paid consultant for GlaxoSmithKline and Novartis. B.H.P. is a paid member of the scientific advisory boards of Horizon Discovery, LTD and Loxo Oncology. Under separate licensing agreements between Horizon Discovery, LTD and The Johns Hopkins University, B.H.P. is entitled to a share of royalties received by the University on sales of products. The terms of this arrangement are being managed by the Johns Hopkins University in accordance with its conflict of interest policies. All other authors declare no potential conflicts.
*This Direct Submission article had a prearranged editor.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1408650111/-/DCSupplemental.
References
- 1.Jordan VC. Selective estrogen receptor modulation: Concept and consequences in cancer. Cancer Cell. 2004;5(3):207–213. doi: 10.1016/s1535-6108(04)00059-5. [DOI] [PubMed] [Google Scholar]
- 2.Shang Y, Brown M. Molecular determinants for the tissue specificity of SERMs. Science. 2002;295(5564):2465–2468. doi: 10.1126/science.1068537. [DOI] [PubMed] [Google Scholar]
- 3.Reiter R, Oh AS, Wellstein A, Riegel AT. Impact of the nuclear receptor coactivator AIB1 isoform AIB1-Delta3 on estrogenic ligands with different intrinsic activity. Oncogene. 2004;23(2):403–409. doi: 10.1038/sj.onc.1207202. [DOI] [PubMed] [Google Scholar]
- 4.Osborne CK, et al. Role of the estrogen receptor coactivator AIB1 (SRC-3) and HER-2/neu in tamoxifen resistance in breast cancer. J Natl Cancer Inst. 2003;95(5):353–361. doi: 10.1093/jnci/95.5.353. [DOI] [PubMed] [Google Scholar]
- 5.Kurokawa H, et al. Inhibition of HER2/neu (erbB-2) and mitogen-activated protein kinases enhances tamoxifen action against HER2-overexpressing, tamoxifen-resistant breast cancer cells. Cancer Res. 2000;60(20):5887–5894. [PubMed] [Google Scholar]
- 6.Jin K, et al. The HOXB7 protein renders breast cancer cells resistant to tamoxifen through activation of the EGFR pathway. Proc Natl Acad Sci USA. 2012;109(8):2736–2741. doi: 10.1073/pnas.1018859108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fagan DH, Uselman RR, Sachdev D, Yee D. Acquired resistance to tamoxifen is associated with loss of the type I insulin-like growth factor receptor: Implications for breast cancer treatment. Cancer Res. 2012;72(13):3372–3380. doi: 10.1158/0008-5472.CAN-12-0684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bergamaschi A, Katzenellenbogen BS. Tamoxifen downregulation of miR-451 increases 14-3-3ζ and promotes breast cancer cell survival and endocrine resistance. Oncogene. 2012;31(1):39–47. doi: 10.1038/onc.2011.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Iorns E, et al. Identification of CDK10 as an important determinant of resistance to endocrine therapy for breast cancer. Cancer Cell. 2008;13(2):91–104. doi: 10.1016/j.ccr.2008.01.001. [DOI] [PubMed] [Google Scholar]
- 10.Abukhdeir AM, et al. Tamoxifen-stimulated growth of breast cancer due to p21 loss. Proc Natl Acad Sci USA. 2008;105(1):288–293. doi: 10.1073/pnas.0710887105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Planas-Silva MD, Weinberg RA. Estrogen-dependent cyclin E-cdk2 activation through p21 redistribution. Mol Cell Biol. 1997;17(7):4059–4069. doi: 10.1128/mcb.17.7.4059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jeselsohn R, et al. Emergence of constitutively active estrogen receptor-α mutations in pretreated advanced estrogen receptor-positive breast cancer. Clin Cancer Res. 2014;20(7):1757–1767. doi: 10.1158/1078-0432.CCR-13-2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Toy W, et al. ESR1 ligand-binding domain mutations in hormone-resistant breast cancer. Nat Genet. 2013;45(12):1439–1445. doi: 10.1038/ng.2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Robinson DR, et al. Activating ESR1 mutations in hormone-resistant metastatic breast cancer. Nat Genet. 2013;45(12):1446–1451. doi: 10.1038/ng.2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Merenbakh-Lamin K, et al. D538G mutation in estrogen receptor-α: A novel mechanism for acquired endocrine resistance in breast cancer. Cancer Res. 2013;73(23):6856–6864. doi: 10.1158/0008-5472.CAN-13-1197. [DOI] [PubMed] [Google Scholar]
- 16.Li S, et al. Endocrine-therapy-resistant ESR1 variants revealed by genomic characterization of breast-cancer-derived xenografts. Cell Reports. 2013;4(6):1116–1130. doi: 10.1016/j.celrep.2013.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang QX, Borg A, Wolf DM, Oesterreich S, Fuqua SA. An estrogen receptor mutant with strong hormone-independent activity from a metastatic breast cancer. Cancer Res. 1997;57(7):1244–1249. [PubMed] [Google Scholar]
- 18.Feijs KL, Forst AH, Verheugd P, Lüscher B. Macrodomain-containing proteins: Regulating new intracellular functions of mono(ADP-ribosyl)ation. Nat Rev Mol Cell Biol. 2013;14(7):443–451. doi: 10.1038/nrm3601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen D, et al. Identification of macrodomain proteins as novel O-acetyl-ADP-ribose deacetylases. J Biol Chem. 2011;286(15):13261–13271. doi: 10.1074/jbc.M110.206771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rosenthal F, et al. Macrodomain-containing proteins are new mono-ADP-ribosylhydrolases. Nat Struct Mol Biol. 2013;20(4):502–507. doi: 10.1038/nsmb.2521. [DOI] [PubMed] [Google Scholar]
- 21.Han WD, et al. Estrogenically regulated LRP16 interacts with estrogen receptor alpha and enhances the receptor’s transcriptional activity. Endocr Relat Cancer. 2007;14(3):741–753. doi: 10.1677/ERC-06-0082. [DOI] [PubMed] [Google Scholar]
- 22.Yang J, et al. The single-macro domain protein LRP16 is an essential cofactor of androgen receptor. Endocr Relat Cancer. 2009;16(1):139–153. doi: 10.1677/ERC-08-0150. [DOI] [PubMed] [Google Scholar]
- 23.Rajaram M, et al. Two distinct categories of focal deletions in cancer genomes. PLoS ONE. 2013;8(6):e66264. doi: 10.1371/journal.pone.0066264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cimino-Mathews A, et al. Androgen receptor expression is usually maintained in initial surgically resected breast cancer metastases but is often lost in end-stage metastases found at autopsy. Hum Pathol. 2012;43(7):1003–1011. doi: 10.1016/j.humpath.2011.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gerlinger M, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366(10):883–892. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lin CY, et al. Whole-genome cartography of estrogen receptor alpha binding sites. PLoS Genet. 2007;3(6):e87. doi: 10.1371/journal.pgen.0030087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Torres-Arzayus MI, Zhao J, Bronson R, Brown M. Estrogen-dependent and estrogen-independent mechanisms contribute to AIB1-mediated tumor formation. Cancer Res. 2010;70(10):4102–4111. doi: 10.1158/0008-5472.CAN-09-4080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Levenson AS, Jordan VC. MCF-7: The first hormone-responsive breast cancer cell line. Cancer Res. 1997;57(15):3071–3078. [PubMed] [Google Scholar]
- 29.Kyprianou N, English HF, Davidson NE, Isaacs JT. Programmed cell death during regression of the MCF-7 human breast cancer following estrogen ablation. Cancer Res. 1991;51(1):162–166. [PubMed] [Google Scholar]
- 30.Soule HD, et al. Isolation and characterization of a spontaneously immortalized human breast epithelial cell line, MCF-10. Cancer Res. 1990;50(18):6075–6086. [PubMed] [Google Scholar]
- 31.Gustin JP, et al. Knockin of mutant PIK3CA activates multiple oncogenic pathways. Proc Natl Acad Sci USA. 2009;106(8):2835–2840. doi: 10.1073/pnas.0813351106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Abukhdeir AM, et al. Physiologic estrogen receptor alpha signaling in non-tumorigenic human mammary epithelial cells. Breast Cancer Res Treat. 2006;99(1):23–33. doi: 10.1007/s10549-006-9177-0. [DOI] [PubMed] [Google Scholar]
- 33.Gao J, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6(269):pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Curtis C, et al. METABRIC Group The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012;486(7403):346–352. doi: 10.1038/nature10983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bignell GR, et al. Signatures of mutation and selection in the cancer genome. Nature. 2010;463(7283):893–898. doi: 10.1038/nature08768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bardelli A, et al. Amplification of the MET receptor drives resistance to anti-EGFR therapies in colorectal cancer. Cancer Disc. 2013;3(6):658–673. doi: 10.1158/2159-8290.CD-12-0558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Konishi H, et al. Knock-in of mutant K-ras in nontumorigenic human epithelial cells as a new model for studying K-ras mediated transformation. Cancer Res. 2007;67(18):8460–8467. doi: 10.1158/0008-5472.CAN-07-0108. [DOI] [PubMed] [Google Scholar]
- 38.Konishi H, et al. A PCR-based high-throughput screen with multiround sample pooling: Application to somatic cell gene targeting. Nat Protoc. 2007;2(11):2865–2874. doi: 10.1038/nprot.2007.409. [DOI] [PubMed] [Google Scholar]
- 39.Bachman KE, et al. p21(WAF1/CIP1) mediates the growth response to TGF-beta in human epithelial cells. Cancer Biol Ther. 2004;3(2):221–225. doi: 10.4161/cbt.3.2.666. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.