

Significance
Double-strand break (DSB) repair sits at the core of genome stability maintenance. Abnormalities in the repair pathway often lead to the development of cancers and resistance to anticancer therapies. Tumor suppressor p53-binding protein 1 (53BP1), a protein critical in regulating DSB repair, is reduced in advanced breast tumors. Furthermore, low levels of 53BP1 correlated with poor prognosis and resistance to chemotherapy. Thus, the protein level of 53BP1 is important for therapeutic response. However, mechanisms regulating 53BP1 protein levels are poorly understood. Here we report a previously unidentified mechanism regulating the protein level of 53BP1, specifically, ubiquitin-conjugating enzyme H7 (UbcH7)-dependent ubiquitination and proteasome-dependent degradation. We further propose an innovative hypothesis that increasing the protein level of 53BP1 enhances the effect of radiotherapy or chemotherapy through suppressing faithful DSB repair.
Keywords: DNA damage response, UbcH7, 53BP1, protein degradation, DSB repair
Abstract
DNA double-strand break (DSB) repair is not only key to genome stability but is also an important anticancer target. Through an shRNA library-based screening, we identified ubiquitin-conjugating enzyme H7 (UbcH7, also known as Ube2L3), a ubiquitin E2 enzyme, as a critical player in DSB repair. UbcH7 regulates both the steady-state and replicative stress-induced ubiquitination and proteasome-dependent degradation of the tumor suppressor p53-binding protein 1 (53BP1). Phosphorylation of 53BP1 at the N terminus is involved in the replicative stress-induced 53BP1 degradation. Depletion of UbcH7 stabilizes 53BP1, leading to inhibition of DSB end resection. Therefore, UbcH7-depleted cells display increased nonhomologous end-joining and reduced homologous recombination for DSB repair. Accordingly, UbcH7-depleted cells are sensitive to DNA damage likely because they mainly used the error-prone nonhomologous end-joining pathway to repair DSBs. Our studies reveal a novel layer of regulation of the DSB repair choice and propose an innovative approach to enhance the effect of radiotherapy or chemotherapy through stabilizing 53BP1.
Prompt response to double-strand breaks (DSBs) caused by, for example, ionization radiation (IR), requires sequential and coordinated assembly of DNA damage response (DDR) proteins at damage sites (1). Recent research findings reveal key roles of the tumor suppressor p53-binding protein 1 (53BP1) and BRCA1 in the decision making of DSB repair. 53BP1, together with Rif1, suppress BRCA1-dependent homologous recombination (HR), thereby promoting nonhomologous end-joining (NHEJ) in G1 phase (2–6). Conversely, BRCA1 antagonizes 53BP1/Rif1, favoring HR in S and G2 phases (7, 8). In the absence of BRCA1 or with enhanced retention of 53BP1 at DSB sites, cells primarily use the error-prone NHEJ to repair DSBs throughout the cell cycle, which leads to gene rearrangement, cell death, and increased sensitivity to anticancer therapies (9–11). Consistently, BRCA1-null mice are early embryonic lethal (12, 13) and codepletion of TP53BP1 rescued the lethality phenotype of BRCA1-null mice (12–14).
Low expression level of 53BP1 was found to be associated with poor clinical outcome in triple negative breast cancer patients with BRCA1 mutation (12, 15), as well as resistance to genotoxins and poly(ADP-ribose) polymerase inhibitors (12, 16, 17). This finding is probably because loss of 53BP1 restored HR and promoted cell survival (12–14). Reduced expression of 53BP1 was also observed in tumors from the brain (18), lymph node (19), and pancreas (20). These data indicate that loss of 53BP1 might be a common mechanism for advanced tumors to evade from radiotherapy or chemotherapy. However, molecular mechanisms controlling the protein level of 53BP1 remain less well understood.
Here we show that UbcH7, an E2 enzyme involved in the ubiquitin (Ub) pathway, controls the protein stability of 53BP1, thereby determining the DSB repair choice. Loss of UbcH7 stabilizes 53BP1, forcing cells to choose NHEJ, but not HR, to repair DSBs, which poses a significant threat to cells treated with DNA damage, especially S-phase genotoxins, such as camptothecin (CPT), a topoisomerase 1 (Top1) inhibitor. The ternary CPT-Top1-DNA complex places a roadblock in the path of advancing DNA replication forks, leading to replication fork collapse and generation of one-ended DSBs. Such one-ended DSBs require HR, but not NHEJ, to repair (8). In contrast, repair of one-ended DSBs by NHEJ leads to radial chromosomes and cell death (12–14). Therefore, stabilization of 53BP1 by UbcH7 depletion increased the sensitivity of cancers cells to CPT and other DNA damaging agents. Our data suggest a novel strategy in enhancing the anticancer effect of radiotherapy or chemotherapy through stabilizing or increasing 53BP1.
Results
Screening of Novel Ub Genes in DDR.
Given the importance of protein ubiquitination in DDR (21, 22), we intended to identify novel Ub genes in DDR and DSB repair through a lentiviral shRNA-based screening with two validated shRNA vectors for each gene. We focused on E2s because of the feasibility of targeting ∼30 genes and included E3 ligases with known roles in DDR (e.g., RNF8 and RNF168, and so forth). We reasoned that if a Ub gene is involved in DDR, then depletion of that gene should impair Chk1 phosphorylation at Ser-345, a gold standard of DDR activation (23). After two rounds of screening (Fig. S1), we observed that cells depleted of UbcH7 exhibited significantly less Chk1 phosphorylation than control cells by DNA damage (Fig. 1A, lanes 2 and 4). The level of phosphorylated Chk1 correlated with the knock down effect of UbcH7 (Fig. 1A, lanes 2, 4, and 6), indicating that UbcH7 might be a rate-limiting factor for Chk1 phosphorylation. UbcH7 depletion stabilized Chk1 (Fig. 1A), consistent with previous reports (24, 25).
Fig. 1.

Depletion of UbcH7 impairs Chk1 phosphorylation induced by DNA damage. (A) A549 control or UbcH7-depleted cells (from two individual shRNA vectors) were treated with 2 mM hydroxyurea (HU) for 4 h, and immunoblotted with anti–pS345-Chk1 and anti-UbcH7 antibodies. The same membrane for pS345-Chk1 was stripped and reblotted with anti-Chk1 antibodies. (B) HEK293T cells were infected with shRNA lentiviral vector for control or UbcH7 for 24 h, transfected with RNAi-resistant Flag-UbcH7 WT or C86S mutant for an additional 48 h, treated with 500 nM CPT for 4 h, and immunoblotted as in A. (C) A549 control or UbcH7-depleted cells grown on glass coverslips were treated with 5 Gy IR and released for 1, 4, 15, and 24 h. The cells were fixed and immunostained with anti-RPA antibodies. Representative images are shown. (Scale bar: 10 μm.) (D) Quantitation of RPA foci number per cell from C. Data represent mean and SD from at least 50 cells. *P < 0.05. (E) Parallel cell samples from C were stained with anti-53BP1 antibodies and representative images were shown. (Scale bar: 10 μm.) (F) Quantitation of 53BP1 foci per cell from E. Data represent mean and SD from at least 50 cells. *P < 0.05.
UbcH7 was initially identified as the E2 enzyme that interacts with the E3 ligase, E6AP (26–28). UbcH7 can form a complex with BRCA1 in vitro, but failed to support the BRCA1-mediated Ub ligase activity (29). A recent study suggested that depletion of UbcH7 increased 53BP1 retention at DSBs sites (30). However, how exactly UbcH7 regulates DDR and DSB repair is unknown.
Depletion of UbcH7 Impaired Chk1 Phosphorylation.
We also observed reduced Chk1 phosphorylation in a number of cell lines with transient UbcH7 depletion by different DNA damaging agents (Fig. 1B and Fig. S2 A–C). When we assessed the time dependence of Chk1 phosphorylation, we noticed a slight delay in the onset of Chk1 phosphorylation by CPT (Fig. S2D, lanes 3 and 8); however, it appears more significant that UbcH7-depleted cells failed to maintain Chk1 phosphorylation (Fig. S2D, lanes 4, 5, and 9–10). These results indicate that UbcH7-depleted cells have defects in maintaining Chk1 phosphorylation.
To understand if the Chk1 phosphorylation defects are dependent on the enzymatic activity of UbcH7, we transfected UbcH7 knockdown HEK293T cells with shRNA-resistant Flag-UbcH7 WT or a catalytically dead (C86S) mutant, and examined CPT-induced Chk1 phosphorylation. The results showed that the UbcH7 WT, but not the C86S mutant, rescued Chk1 phosphorylation by CPT (Fig. 1B, lanes 2, 4, and 6). The level of phosphorylated Chk1 was even lower in cells expressing the C86S mutant (Fig. 1B, lanes 4 and 8), indicating that C86S may function as a dominant-negative mutant of endogenous UbcH7. These data suggest that DNA damage-induced Chk1 phosphorylation depends on UbcH7’s enzymatic activity.
Depletion of UbcH7 Inhibited DSB End Resection.
Long stretches of single-strand DNA (ssDNA) are key for the maintenance of Chk1 phosphorylation and for the initiation of HR (23, 31). In DSB, ssDNA is generated through the 5′ to 3′ end resection of DSBs. Therefore, we asked if UbcH7-depleted cells are defective in DSB end resection by evaluating foci formation of RPA, the ssDNA-binding protein (31).
UbcH7 is primarily expressed in the cytoplasm with diffuse nuclear staining, and the signal is lost with UbcH7 stable silencing (Fig. S2E). UbcH7 did not form foci after IR or other DNA damaging agents, suggesting that UbcH7 does not directly participate in DDR initiation. However, UbcH7-depleted cells formed significantly fewer RPA foci than control cells (Fig. 1 C and D, maximum difference measured at 15 h). 53BP1 inhibits HR through inhibiting DSB end resection (7). Therefore, we assessed 53BP1 foci by IR. The results showed that UbcH7-depleted cells displayed more 53BP1 foci than control cells, especially at earlier time points (Fig. 1 E and F). UbcH7-depleted cells also formed more large 53BP1 foci (Fig. S3A), consistent with a recent report (30). Importantly, overexpression of UbcH7 WT, but not the C86S mutant, reversed the increased 53BP1 foci in UbcH7 depleted cells (Fig. S3B). These results suggest that UbcH7 depletion suppressed DSB end resection, likely because of the increased and enlarged 53BP1 foci formation at DSB sites.
Loss of UbcH7 Impaired the Long-Term Cell Viability After DNA Damage.
We then asked whether depletion of UbcH7 would affect cell viability. We found no difference in growth between control and UbcH7-depleted cells under nonstressful conditions (Fig. 2A). These findings are similar to previous studies of UbcM4, the mouse ortholog of UbcH7 (32). These data suggest that UbcH7 is not required for normal cell proliferation. However, UbcH7 depletion significantly reduced the long-term cell viability by DNA damage, including UV light and IR (Fig. 2 B and C; see also CPT in Fig. 6C). Again, expressing the UbcH7 WT but not the C86S mutant reversed the increased sensitivity of UbcH7-depleted cells to DNA damage (Fig. S3C). These results suggest that loss of UbcH7 is deleterious to cell viability in the presence of DNA damage.
Fig. 2.
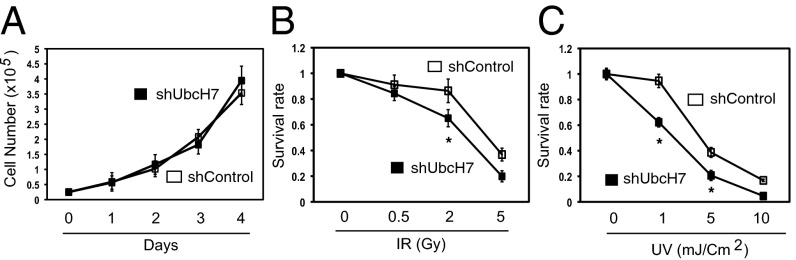
Depletion of UbcH7 increased cellular sensitivity to DNA damaging agents. (A) Equal number of viable cells from A549 control or UbcH7 depletion was plated in 12-well plates at day 0. The cell number was counted for the following 4 d. Data represent mean and SD from three wells. (B and C) A549 control or UbcH7-depleted cells were treated with increasing doses of IR or UV, reseeded at a density of 10,000 viable cells per well of six-well plates in triplicate. Viable cells after 10 d of culture were measured by crystal violate assay. Data represent mean and SD. *P < 0.05.
Fig. 6.

UbcH7-depleted cells were extremely sensitive to CPT. (A) A549 control or UbcH7-depleted cells infected with sh53BP1 lentivirus were treated with 100 nM CPT for 12 h before NHEJ assay. Data represent mean and SD from three replicates. (B) HR assay in U2-OS DR cells after treatment with 100 nM CPT for 12 h. Data represent mean and SD from three replicates. (C) Control, UbcH7, or both UbcH7- and 53BP1-depleted cells were treated with indicated concentrations of CPT for 4 h, resuspended in drug-free media, and 10,000 viable cells were replated into six-well plates in triplicate. Viable cells after 7 d of culture were measured as described in SI Materials and Methods. Data represent mean and SD from three replicates. *P < 0.05. (D) HEK293T cells were transfected with GFP, GFP-53BP1 WT, or the S28A mutant for 48 h, treated with 500 nM CPT for 6 h, and released into drug-free media for additional 24 h. Cells were then treated with 4 μg/mL propidium iodide (PI) for 5 min and cell death was assessed by visualizing both PI+ and GFP+ cells under live cell fluorescent microscope. Data represent mean and SD from three parallel experiments. *P < 0.05. (E) Working model for the role of UbcH7 in DSB repair and cell survival.
UbcH7 Regulates Ubiquitination and Proteasome-Dependent Degradation of 53BP1.
Because UbcH7 is an E2 enzyme, we hypothesized that it regulates DSB repair through controlling ubiquitination and degradation of key DDR proteins. To this end, we monitored protein levels of a number of DDR proteins in the presence or absence of UbcH7. We repeatedly observed that 53BP1 is significantly elevated in UbcH7-depleted cells (Fig. 3A). On the other hand, the cofactor for 53BP1, Rif1, showed no change (Fig. 3A). We further showed that the increased expression of 53BP1 is mostly specific to UbcH7, but not other E2s (Fig. S4). We then measured the mRNA level of TP53BP1 by quantitative PCR (qPCR). The results showed comparable levels of TP53BP1 between control and UbcH7-depleted cells (Fig. 3B), suggesting that UbcH7 controls the level of 53BP1 via posttranscriptional regulation.
Fig. 3.

UbcH7 regulates ubiquitination and proteasome-dependent degradation of 53BP1. (A) DDR protein expression in A549 control or UbcH7-depleted cells. Numbers represent the relative intensity of 53BP1 analyzed by the ImageJ software. (B) The mRNA levels of TP53BP1 in A549 control or UbcH7-depleted cells were analyzed by qPCR. Data represent mean and SD from three independent experiments using β-actin as the internal control. (C) A549 control or UbcH7-depleted cells were treated with 300 μM CHX for 0, 2, 4, and 8 h and expression of indicated proteins was analyzed. Numbers represent the relative intensity of 53BP1. (D) The protein band intensity of 53BP1 in C was analyzed and the intensity of 53BP1 at 0 h treatment was set as 1. Data represent mean and SD from at least three blots. *P < 0.05. (E) A549 control cells were treated or not with 500 nM CPT for 6 h, lysed, and immunoprecipitated (IPed) with anti-53BP1 antibodies, and probed with indicated antibodies. Protein expression in WCE was also examined. The relative band intensities of 53BP1 and UbcH7 were analyzed. (F) A549 control or UbcH7-depleted cells were treated with 300 μM CHX in the presence or absence of 25 μM MG132 for 6 h, and ubiquitinated 53BP1 was analyzed as described in SI Materials and Methods. The same membrane after anti-Ub blotting was stripped and reprobed with anti-53BP1 antibodies. The arrowhead in the anti-Ub blot indicates the position of 53BP1 proteins.
We subsequently assessed the protein stability of 53BP1 in the presence of cycloheximide (CHX) to block de novo protein synthesis. The results showed that depletion of UbcH7 significantly increased the protein stability of 53BP1 (Fig. 3 C and D). The stability of the known UbcH7 target, Chk1, was also increased (Fig. 3C). Again, overexpression of the UbcH7 WT, but not the C86S mutant, reversed the increased protein stability of 53BP1 (Fig. S5A). To understand how UbcH7 regulates the protein stability of 53BP1, we asked if UbcH7 forms a complex with 53BP1. Indeed, we detected UbcH7 from protein complexes immunoprecipitated by the anti-53BP1 antibody (Fig. 3E, lane 2 in IP). Interestingly, we observed a twofold increase in the interaction between 53BP1 and UbcH7 after CPT treatment (Fig. 3E, lanes 2 and 3 in IP). These data suggest that UbcH7 interacts with 53BP1 and DNA damage, or at least CPT treatment, increases such an interaction.
To understand if 53BP1 undergoes proteasome-dependent degradation, we cotreated cells with CHX and the proteasome inhibitor, MG132. Cotreatment with MG132 almost completely blocked degradation of 53BP1 by CHX in control cells [Fig. 3F, lanes 2 and 3 in whole-cell extracts (WCE)]. Importantly, 53BP1 was stable in UbcH7 depleted cells (Fig. 3F, lanes 5 and 6 in WCE). We further confirmed that 53BP1 is ubiquitinated in A549 control cells (Fig. 3F, lanes 1–3 in IP). However, the levels of ubiquitinated 53BP1 proteins were significantly lower in UbcH7-depleted cells than in control cells, despite the finding that more 53BP1 proteins were pulled down (Fig. 3F). Taken together, these results suggest that UbcH7 regulates ubiquitination and proteasome-dependent degradation of 53BP1.
UbcH7 Depletion Enhances NHEJ While Suppressing HR for DSB Repair.
53BP1 endorses NHEJ while suppressing HR for DSB repair (7). Because UbcH7-depleted cells expressed more 53BP1 proteins and formed more 53BP1 foci by IR, we measured DSB repair in these cells. We first assessed IR-induced foci formation of Ligase IV, the key NHEJ repair protein. UbcH7-depleted cells exhibited more Ligase IV foci than control cells (Fig. 4 A and B) without altering the levels of Ligase IV proteins (Fig. 5D). Second, we measured NHEJ using a cell-based assay and the results showed that NHEJ was significantly increased in UbcH7-depleted cells (Fig. 4C), consistent with the results of 53BP1 and Ligase IV foci formation.
Fig. 4.

Depletion of UbcH7 enhanced NHEJ while suppressing HR for DSB repair. (A) A549 control or UbcH7-depleted cells were treated with 5 Gy IR, fixed and stained with anti-Ligase IV antibodies. Representative images are shown. (Scale bar: 10 μm.) (B) Quantitation results of Ligase IV foci per cell from A. Data represent mean and SD from at least 50 cells. *P < 0.05. (C) A549 control or UbcH7-depleted cells were infected with sh53BP1 and NHEJ assay was performed as described in SI Materials and Methods. Data represent mean and SD from three replicates. *P < 0.05. (D) Protein expression from samples in C. (E) U2-OS DR cells were infected with shRNA virus for control or UbcH7 with or without sh53BP1 and HR assay was performed as described in SI Materials and Methods. Data represent mean and SD from three replicates. (F) Protein expression from samples in E.
Fig. 5.

Replicative stresses induce UbcH7-dependent degradation of 53BP1. (A) A549 control or UbcH7-depleted cells were infected with lentiviral sh53BP1 for 3 d, treated with 500 nM CPT for 6 h, and protein expression was examined. (B) A549 control or UbcH7-depleted cells were treated with 500 nM CPT for 0, 2, 4, and 8 h and immunoblotted as in A. (C) The protein band intensity of 53BP1 in B was quantitated and the relative intensity at 0 h treatment was set as 1.0. Data represent mean and SD from three experiments. *P < 0.05. (D) A549 control or UbcH7-depleted cells were treated with 5 Gy IR and released for the indicated times and immunoblotted as in A. (E) A549 control or UbcH7-depleted cells were treated with 500 nM CPT in the presence or absence of 25 μM MG132 for 6 h, and immunoblotted as in A. (F) HEK293T cells were transfected with GFP-53BP1 WT or the S28A mutant for 48 h, treated with 500 nM CPT for 0, 4, or 8 h, and immunoblotted with indicated antibodies.
UbcH7-depleted cells also expressed high levels of Chk1. To preclude the possibility that increased Chk1 contributed to the enhanced NHEJ, we assessed the effect of overexpression of a Chk1 K436R mutant on NHEJ. We previously showed that this Chk1 mutant is resistant to ubiquitination and degradation (33), mimicking the situation of increased Chk1 protein levels in UbcH7-depleted cells. Our analysis suggests that increasing the protein level of Chk1 alone does not promote NHEJ (Fig. S5 B and C).
Subsequently, we measured HR using the well-established in vivo HR reporter assay (34). Transient depletion of UbcH7 in U2-OS cells also stabilized 53BP1 (Fig. 4F) and these cells displayed reduced HR compared with control cells (Fig. 4E), although the inhibition in HR by UbcH7 depletion was less severe than depletion of BRCA1 (Fig. S5D). Finally, to verify that 53BP1 is critical for the DSB repair shift in UbcH7-depleted cells, we used lentiviral sh53BP1 to reduce the increased expression of 53BP1 by UbcH7 depletion (Fig. 4 D and F). We observed that inhibiting 53BP1 expression reversed both the enhanced NHEJ and the suppressed HR by UbcH7 depletion (Fig. 4 C–E and Fig. S5D). Although depletion of several other E2s affected NHEJ or HR, UbcH7 seems to be the only one that induces DSB repair shift, correlating with its impact on the level of 53BP1 (Figs. S4 and S6). Taken together, these data suggest that 53BP1 is the key downstream target of UbcH7 in DSB repair.
Increased 53BP1 Is Responsible for Chk1 Phosphorylation Defects by UbcH7 Depletion.
To understand if the increased expression of 53BP1 is also responsible for Chk1 phosphorylation defects, we infected A549 control or UbcH7-depleted cells with sh53BP1 lentivirus, treated cells with CPT, and monitored Chk1 phosphorylation. The results showed that codepletion of 53BP1 rescued the Chk1 phosphorylation defects in UbcH7-depleted cells (Fig. 5A, lanes 2, 6, and 8). These changes are unlikely because of cell cycle arrest, as only marginal changes in the cell cycle profile were observed (Fig. S7).
Replicative Stresses Induce UbcH7-Dependent Degradation of 53BP1.
During analysis, we repeatedly observed that CPT treatment reduced the level of 53BP1 (Figs. 3E and 5A). We further showed that CPT treatment induced a time-dependent reduction in the levels of 53BP1 proteins in control, but not in UbcH7-depleted cells (Fig. 5 B and C). This finding is consistent with the increased interaction between 53BP1 and UbcH7 after CPT treatment (Fig. 3E), indicating that CPT treatment induces degradation of 53BP1.
To try to expand this observation, we monitored the level of 53BP1 after IR. However, unlike CPT, IR treatment did not induce obvious decrease in 53BP1 (Fig. 5D). We further observed that agents that specifically induce replicative stress, including CPT, HU, and aphidicolin—but not others, like etoposide, taxol, or IR—reduced the protein level of 53BP1 (Fig. S8 A–D). The CPT-induced degradation of 53BP1 in control, but not in UbcH7-depleted cells, is blocked by cotreatment with MG132 (Fig. 5E). We also observed the same results for overexpressed GFP-53BP1 WT (Fig. S8E, lanes 1 and 2). Furthermore, we confirmed that CPT induces ubiquitination of 53BP1 (Fig. S8F). These lines of findings suggest that replicative stress induces ubiquitination and proteasome-dependent degradation of 53BP1 in a UbcH7-dependent manner.
To understand how replicative stress induces 53BP1 degradation, we asked if phosphorylation of 53BP1 is involved. 53BP1 undergoes ATM/ATR-dependent phosphorylation at 28 S/T-Q sites at its N terminus, and such phosphorylation plays an important role in regulating the DSB repair function of 53BP1 (35). We compared the protein stability of 53BP1 WT and a mutant with all these 28 S/T phosphorylation sites mutated to A (designated as the S28A mutant) in the presence of CPT. The results show that CPT induced time-dependent degradation of the GFP-53BP1 WT, but not the S28A mutant (Fig. 5F). As a positive control (24), Chk1 was degraded after 8 h of CPT treatment in cells expressing both the WT and the S28A mutant (Fig. 5F), indicating a functional degradation system in these cells. These data suggest that phosphorylation is involved in replicative stress induced degradation of 53BP1.
Degradation of 53BP1 Is Required for the Repair of CPT-Induced DSBs.
CPT and its analogs are widely used in the clinic (36, 37); they are specifically toxic to S phase cells and the lethality of CPTs stems from their ability to generate DSBs. Such DSBs are one-ended as a result of collision of replication forks with the ternary CPT-Top1-DNA complex and require HR, but not NEHJ, to repair because of the lack of a partner end (8). Failure to do so leads to aberrant chromosomes and eventually cell death (12–14). Therefore, cells need to suppress NHEJ while promoting HR in response to CPTs for survival (8). However, molecular mechanisms underlying the DSB repair choice (i.e., favoring HR while suppressing NHEJ) are unclear. We hypothesize that cells suppress NHEJ in response to CPT through inducing UbcH7-dependent degradation of 53BP1.
If our hypothesis were correct, we would predict that CPT inhibits NHEJ while enhancing HR in a way dependent on the UbcH7/53BP1 axis. Indeed we observed that CPT reduced NHEJ and enhanced HR by roughly 50% and 30%, respectively, in control cells (Fig. 6 A and B), consistent with the reduction in the protein level of 53BP1 (Fig. 5A). Such changes in NHEJ and HR were not observed in UbcH7-depleted cells (Fig. 6 A and B). However, codepletion of 53BP1 allowed CPT to restore its inhibition and promotion in NHEJ and HR, respectively, in UbcH7-depleted cells (Figs. 6 A and B). If 53BP1 is critical in the DSB repair shift, we would expect that overexpression of 53BP1 WT should enhance NHEJ while suppressing HR, as did the UbcH7-depleted cells. To address this issue, we transfected Flag-53BP1 WT or the S28A mutant in regular U2-OS and the U2-OS DR cells to measure NHEJ and HR, respectively. The results showed that overexpressing the WT, and to a much lesser extent the S28A mutant, induced NHEJ while suppressing HR (Fig. S9 A and B). There results suggest that despite the increased protein stability, the S28A mutant is functionally impaired in shifting DSB repair, consistent with previous report that phosphorylation at the N terminus is important for 53BP1’s DSB repair function (35). Taken together, these data suggest a critical role of the protein level of 53BP1 (WT) in the decision making of repairing CPT-induced DSBs.
UbcH7-Depleted Cells Are Extremely Sensitive to CPT.
Because UbcH7-depleted cells repair CPT-induced DSBs by NHEJ, they should be much more sensitive to CPT than control cells. The results showed that UbcH7-depleted cells indeed exhibited significantly increased sensitivity to CPT compared with control cells (Fig. 6C). Importantly, codepletion of 53BP1 with UbcH7 restored the cellular sensitivity to the level of control cells (Fig. 6C).
We noticed that the increased sensitivity of UbcH7 depletion to CPT is much greater than that to IR or UV (Figs. 2 and 6C), reinforcing the idea that DSBs generated by CPT rely more on HR to repair. These findings also suggest that stabilizing 53BP1 increases the sensitivity of cancer cells to DSB-inducing agents, particularly those that perturb DNA replication. To test this idea, we overexpressed GFP, GFP-53BP1 WT, or the S28A mutant in cells and exposed them to CPT. Our data showed that overexpression of 53BP1 WT, and to a much lesser degree the S28A mutant, significantly increased the cell killing of CPT (Fig. 6D), despite the fact that the S28A mutant expressed at similar levels as the WT (Fig. S9C). On the other hand, GFP control cells survived CPT treatment (Fig. 6D), indicating repair of CPT-induced damage in these cells. Together, these findings suggest that UbcH7 depletion particularly increases the sensitivity of cancer cells to replication stresses that generate one-ended DSBs.
Discussion
In this study, we identify a novel role of the E2 enzyme UbcH7 in DSB repair. We propose a model for the role of UbcH7 in DSB repair through controlling the steady-state level of 53BP1, the key player in the decision making of DSB repair (Fig. 6E). Based on our model, we expect to observe an inverse correlation between UbcH7 and 53BP1 in cells. To test this idea, we analyzed the levels of UbcH7 and 53BP1 proteins in a panel of eight breast cell lines. The results indeed showed an inverse correlation between these two proteins (Fig. S10A), especially in triple-negative breast cancer cells, with a Pearson correlation coefficient R value at −0.7251 (Fig. S10B). These data suggest that the UbcH7/53BP1 axis may play an important role in the pathophysiology of human cancers and lay groundwork for further testing this idea using human tumor tissues in the future. It will be interesting to test this model in normal cells, as well as in stem cells in future trials.
Several studies reported the screening of genes in regulating the recruitment of 53BP1 to DNA damage sites (38–40). Depletion of UbcH7 was reported to increase the size of 53BP1 foci (30), similar to our observation. Our results suggest that this is likely because UbcH7-depleted cells express elevated levels of 53BP1, allowing the formation of more and larger 53BP1 foci at DSB sites. The E2 enzyme RAD6 indirectly regulates 53BP1 foci through modulating RNF168 (41). Here, our results revealed that regulating 53BP1 levels has as strong impact as regulating the foci formation of 53BP1 on the DSB repair choice.
As an E2 enzyme, UbcH7 regulates a wide range of substrates, including Chk1 (24, 25). Our data strongly suggest that it is 53BP1, but not Chk1 or other substrates that contributed to the DSB repair function of UbcH7. First, codepletion of 53BP1 almost completely reversed the DSB repair shift induced by UbcH7 depletion. Second, overexpression of 53BP1 WT, but not Chk1, induced the same phenotype as UbcH7 depletion. Third, Chk1 was reported to promote HR (42); yet, UbcH7-depleted cells displayed significantly reduced HR despite the increased levels of Chk1 proteins.
The de-ubiquitin enzyme, USP28, stabilizes 53BP1 (43), supporting our conclusion that protein ubiquitination regulates the stability of 53BP1. The endosomal/lysosomal protease Cathepsin L cleaves 53BP1, especially in the absence of A-type lamins (44). Here we provided insights that phosphorylation of 53BP1 is involved in its degradation. Interesting future investigations should include the identification of the E3 ligases responsible for 53BP1 ubiquitination and determine how exactly phosphorylation coordinates with replicative stress—or at least CPT—induced 53BP1 degradation. In addition, it is would be worthwhile to test the interplay between Cathepsin L- and UbcH7-dependent 53BP1 degradation. In all, advances in basic mechanisms and cancer therapeutic potentials in the current report will facilitate our understanding of DSB repair choice and cell survival to DNA damage.
Materials and Methods
shRNA Screening.
Lentiviral shRNA was constructed in A549 cells to screen Ub genes whose depletion impaired DNA damage-induced Chk1 phosphorylation. We used immunoblotting to assess Chk1 phosphorylation using the specific anti–pS345-Chk1 antibodies. We repeated the screening process two times, and potential positive hits were further screened for at least two more times.
DSB Repair Assays.
A plasmid (the pEGFP-C1 vector) and the well-established U2-OS DR reporter cell line (34) were used to measure NHEJ and HR, respectively. Both rely on the production of GFP to be measured by FACS analysis.
See SI Materials and Methods for more detailed discussion.
Supplementary Material
Acknowledgments
We thank Daniel Durocher, Jiri Lukas, Maria Jasin, and Xiaochun Yu for reagents; and Rebekah Dumm and Alicia Zhang for technical assistance. This work was supported by the National Institutes of Health/National Cancer Institute R00 Career Development Award (CA126173), and is currently supported by National Institutes of Health/National Cancer Institute Grant R01 CA163214 (to Y.Z.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1408538111/-/DCSupplemental.
References
- 1.Harper JW, Elledge SJ. The DNA damage response: Ten years after. Mol Cell. 2007;28(5):739–745. doi: 10.1016/j.molcel.2007.11.015. [DOI] [PubMed] [Google Scholar]
- 2.Zimmermann M, Lottersberger F, Buonomo SB, Sfeir A, de Lange T. 53BP1 regulates DSB repair using Rif1 to control 5′ end resection. Science. 2013;339(6120):700–704. doi: 10.1126/science.1231573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chapman JR, et al. RIF1 is essential for 53BP1-dependent nonhomologous end joining and suppression of DNA double-strand break resection. Mol Cell. 2013;49(5):858–871. doi: 10.1016/j.molcel.2013.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Di Virgilio M, et al. Rif1 prevents resection of DNA breaks and promotes immunoglobulin class switching. Science. 2013;339(6120):711–715. doi: 10.1126/science.1230624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Feng L, Fong KW, Wang J, Wang W, Chen J. RIF1 counteracts BRCA1-mediated end resection during DNA repair. J Biol Chem. 2013;288(16):11135–11143. doi: 10.1074/jbc.M113.457440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Escribano-Díaz C, et al. A cell cycle-dependent regulatory circuit composed of 53BP1-RIF1 and BRCA1-CtIP controls DNA repair pathway choice. Mol Cell. 2013;49(5):872–883. doi: 10.1016/j.molcel.2013.01.001. [DOI] [PubMed] [Google Scholar]
- 7.Panier S, Boulton SJ. Double-strand break repair: 53BP1 comes into focus. Nat Rev Mol Cell Biol. 2014;15(1):7–18. doi: 10.1038/nrm3719. [DOI] [PubMed] [Google Scholar]
- 8.Goodarzi AA, Jeggo PA. The repair and signaling responses to DNA double-strand breaks. Adv Genet. 2013;82:1–45. doi: 10.1016/B978-0-12-407676-1.00001-9. [DOI] [PubMed] [Google Scholar]
- 9.Saberi A, et al. RAD18 and poly(ADP-ribose) polymerase independently suppress the access of nonhomologous end joining to double-strand breaks and facilitate homologous recombination-mediated repair. Mol Cell Biol. 2007;27(7):2562–2571. doi: 10.1128/MCB.01243-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McGlynn P, Lloyd RG. Recombinational repair and restart of damaged replication forks. Nat Rev Mol Cell Biol. 2002;3(11):859–870. doi: 10.1038/nrm951. [DOI] [PubMed] [Google Scholar]
- 11.Tang J, et al. Acetylation limits 53BP1 association with damaged chromatin to promote homologous recombination. Nat Struct Mol Biol. 2013;20(3):317–325. doi: 10.1038/nsmb.2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bouwman P, et al. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat Struct Mol Biol. 2010;17(6):688–695. doi: 10.1038/nsmb.1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bunting SF, et al. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell. 2010;141(2):243–254. doi: 10.1016/j.cell.2010.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cao L, et al. A selective requirement for 53BP1 in the biological response to genomic instability induced by Brca1 deficiency. Mol Cell. 2009;35(4):534–541. doi: 10.1016/j.molcel.2009.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Neboori HJ, et al. Low p53 binding protein 1 (53BP1) expression is associated with increased local recurrence in breast cancer patients treated with breast-conserving surgery and radiotherapy. Int J Radiat Oncol Biol Phys. 2012;83(5):e677–e683. doi: 10.1016/j.ijrobp.2012.01.089. [DOI] [PubMed] [Google Scholar]
- 16.Jaspers JE, et al. Loss of 53BP1 causes PARP inhibitor resistance in Brca1-mutated mouse mammary tumors. Cancer Discov. 2013;3(1):68–81. doi: 10.1158/2159-8290.CD-12-0049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Oplustilova L, et al. Evaluation of candidate biomarkers to predict cancer cell sensitivity or resistance to PARP-1 inhibitor treatment. Cell Cycle. 2012;11(20):3837–3850. doi: 10.4161/cc.22026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Squatrito M, Vanoli F, Schultz N, Jasin M, Holland EC. 53BP1 is a haploinsufficient tumor suppressor and protects cells from radiation response in glioma. Cancer Res. 2012;72(20):5250–5260. doi: 10.1158/0008-5472.CAN-12-0045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Takeyama K, et al. Integrative analysis reveals 53BP1 copy loss and decreased expression in a subset of human diffuse large B-cell lymphomas. Oncogene. 2008;27(3):318–322. doi: 10.1038/sj.onc.1210650. [DOI] [PubMed] [Google Scholar]
- 20.Ausborn NL, et al. 53BP1 expression is a modifier of the prognostic value of lymph node ratio and CA 19-9 in pancreatic adenocarcinoma. BMC Cancer. 2013;13:155. doi: 10.1186/1471-2407-13-155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Messick TE, Greenberg RA. The ubiquitin landscape at DNA double-strand breaks. J Cell Biol. 2009;187(3):319–326. doi: 10.1083/jcb.200908074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jackson SP, Durocher D. Regulation of DNA damage responses by ubiquitin and SUMO. Mol Cell. 2013;49(5):795–807. doi: 10.1016/j.molcel.2013.01.017. [DOI] [PubMed] [Google Scholar]
- 23.Zhang Y, Hunter T. Roles of Chk1 in cell biology and cancer therapy. Int J Cancer. 2014;134(5):1013–1023. doi: 10.1002/ijc.28226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang YW, et al. Genotoxic stress targets human Chk1 for degradation by the ubiquitin-proteasome pathway. Mol Cell. 2005;19(5):607–618. doi: 10.1016/j.molcel.2005.07.019. [DOI] [PubMed] [Google Scholar]
- 25.Whitcomb EA, Dudek EJ, Liu Q, Taylor A. Novel control of S phase of the cell cycle by ubiquitin-conjugating enzyme H7. Mol Biol Cell. 2009;20(1):1–9. doi: 10.1091/mbc.E08-01-0036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Robinson PA, et al. A human ubiquitin conjugating enzyme, L-UBC, maps in the Alzheimer’s disease locus on chromosome 14q24.3. Mamm Genome. 1995;6(10):725–731. doi: 10.1007/BF00354295. [DOI] [PubMed] [Google Scholar]
- 27.Moynihan TP, et al. Characterization of a human ubiquitin-conjugating enzyme gene UBE2L3. Mamm Genome. 1996;7(7):520–525. doi: 10.1007/s003359900155. [DOI] [PubMed] [Google Scholar]
- 28.Nuber U, Schwarz S, Kaiser P, Schneider R, Scheffner M. Cloning of human ubiquitin-conjugating enzymes UbcH6 and UbcH7 (E2-F1) and characterization of their interaction with E6-AP and RSP5. J Biol Chem. 1996;271(5):2795–2800. doi: 10.1074/jbc.271.5.2795. [DOI] [PubMed] [Google Scholar]
- 29.Brzovic PS, et al. Binding and recognition in the assembly of an active BRCA1/BARD1 ubiquitin-ligase complex. Proc Natl Acad Sci USA. 2003;100(10):5646–5651. doi: 10.1073/pnas.0836054100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gudjonsson T, et al. TRIP12 and UBR5 suppress spreading of chromatin ubiquitylation at damaged chromosomes. Cell. 2012;150(4):697–709. doi: 10.1016/j.cell.2012.06.039. [DOI] [PubMed] [Google Scholar]
- 31.Cimprich KA, Cortez D. ATR: An essential regulator of genome integrity. Nat Rev Mol Cell Biol. 2008;9(8):616–627. doi: 10.1038/nrm2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pringa E, Meier I, Müller U, Martinez-Noel G, Harbers K. Disruption of the gene encoding the ubiquitin-conjugating enzyme UbcM4 has no effect on proliferation and in vitro differentiation of mouse embryonic stem cells. Biochim Biophys Acta. 2000;1494(1-2):75–82. doi: 10.1016/s0167-4781(00)00221-9. [DOI] [PubMed] [Google Scholar]
- 33.Zhang YW, et al. The F box protein Fbx6 regulates Chk1 stability and cellular sensitivity to replication stress. Mol Cell. 2009;35(4):442–453. doi: 10.1016/j.molcel.2009.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pierce AJ, Johnson RD, Thompson LH, Jasin M. XRCC3 promotes homology-directed repair of DNA damage in mammalian cells. Genes Dev. 1999;13(20):2633–2638. doi: 10.1101/gad.13.20.2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Callen E, et al. 53BP1 mediates productive and mutagenic DNA repair through distinct phosphoprotein interactions. Cell. 2013;153(6):1266–1280. doi: 10.1016/j.cell.2013.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stewart CF. Topoisomerase I interactive agents. Cancer Chemother Biol Response Modif. 2001;19:85–128. [PubMed] [Google Scholar]
- 37.Bailly C. Topoisomerase I poisons and suppressors as anticancer drugs. Curr Med Chem. 2000;7(1):39–58. doi: 10.2174/0929867003375489. [DOI] [PubMed] [Google Scholar]
- 38.Moudry P, et al. Nucleoporin NUP153 guards genome integrity by promoting nuclear import of 53BP1. Cell Death Differ. 2012;19(5):798–807. doi: 10.1038/cdd.2011.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Moudry P, et al. Ubiquitin-activating enzyme UBA1 is required for cellular response to DNA damage. Cell Cycle. 2012;11(8):1573–1582. doi: 10.4161/cc.19978. [DOI] [PubMed] [Google Scholar]
- 40.Butler LR, et al. The proteasomal de-ubiquitinating enzyme POH1 promotes the double-strand DNA break response. EMBO J. 2012;31(19):3918–3934. doi: 10.1038/emboj.2012.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu C, et al. RNF168 forms a functional complex with RAD6 during the DNA damage response. J Cell Sci. 2013;126(Pt 9):2042–2051. doi: 10.1242/jcs.122945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sørensen CS, et al. The cell-cycle checkpoint kinase Chk1 is required for mammalian homologous recombination repair. Nat Cell Biol. 2005;7(2):195–201. doi: 10.1038/ncb1212. [DOI] [PubMed] [Google Scholar]
- 43.Zhang D, Zaugg K, Mak TW, Elledge SJ. A role for the deubiquitinating enzyme USP28 in control of the DNA-damage response. Cell. 2006;126(3):529–542. doi: 10.1016/j.cell.2006.06.039. [DOI] [PubMed] [Google Scholar]
- 44.Grotsky DA, et al. BRCA1 loss activates cathepsin L-mediated degradation of 53BP1 in breast cancer cells. J Cell Biol. 2013;200(2):187–202. doi: 10.1083/jcb.201204053. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.