

Abstract
Objectives
We investigated the efficacy of the novel antimicrobial agent SMT19969 in treating simulated Clostridium difficile infection using an in vitro human gut model.
Methods
Concentrations of the predominant cultivable members of the indigenous gut microfloras and C. difficile (total and spore counts) were determined by viable counting. Cytotoxin titres were determined using cell cytotoxicity and expressed as log10 relative units (RU). Clindamycin was used to induce simulated C. difficile PCR ribotype 027 infection. Once high-level cytotoxin titres (≥4 RU) were observed, SMT19969 was instilled for 7 days. Two SMT19969 dosing regimens (31.25 and 62.5 mg/L four times daily) were evaluated simultaneously in separate experiments. MICs of SMT19969 were determined against 30 genotypically distinct C. difficile ribotypes.
Results
SMT19969 was 7- and 17-fold more active against C. difficile than metronidazole and vancomycin, respectively, against a panel of genotypically distinct isolates (P < 0.05). Both SMT19969 dosing regimens demonstrated little antimicrobial activity against indigenous gut microflora groups except clostridia. SMT19969 inhibited C. difficile growth and repressed C. difficile cytotoxin titres in the gut model.
Conclusions
These data suggest that SMT19969 is a narrow-spectrum and potent antimicrobial agent against C. difficile. Additional studies evaluating SMT19969 in other models of C. difficile infection are warranted, with human studies to place these gut model observations in context.
Keywords: Clostridium difficile, antibiotics, cytotoxin, microflora, gut model, NAP1/027
Introduction
Clostridium difficile infection (CDI) is the leading cause of infectious nosocomial diarrhoea. It is the aetiological agent of pseudomembranous colitis and is implicated in ∼30% of cases of antibiotic-associated diarrhoea.1,2 Most antimicrobials have been implicated, but clindamycin, third-generation cephalosporins and aminopenicillins are particularly noted for their propensity to induce CDI.3 Despite improved clinical management strategies for CDI, healthcare costs for treating CDI remain high and have been estimated in the USA at US$1.1–3.2 billion.4,5 Following the emergence of C. difficile PCR ribotype 027, the incidence of CDI in the UK increased markedly, but has subsequently declined. Metronidazole and vancomycin were considered for many years to be similarly effective, but later studies indicated that the former antimicrobial agent is inferior, particularly in severe CDI.6–10 Fidaxomicin is active in vitro and in vivo against C. difficile,11,12 and although a fidaxomicin-resistant C. difficile was isolated from a recurrent CDI patient (MIC of 16 mg/L) during Phase III clinical trials,11 the significance of this result remains to be determined. Recurrent CDI remains a therapeutic challenge, despite the recent availability of fidaxomicin, which is less efficacious against CDI due to PCR ribotype 027.6
We have previously described an in vitro human gut model of CDI that yields results consistent with in vivo data. The aim of this study was to evaluate the effects of the investigational drug SMT19969 on clindamycin-induced growth and toxin production in this gut model, using an epidemic C. difficile NAP1/027 strain.
Materials and methods
C. difficile strains
The C. difficile PCR ribotype 027 strain (CD 210) evaluated in the in vitro human gut model was isolated during an outbreak of CDI at the Maine Medical Centre (Portland, ME, USA) in 2005 and was supplied courtesy of Dr Rob Owens. A panel of clinical C. difficile strains were evaluated for their antimicrobial susceptibilities to metronidazole, vancomycin and SMT19969, comprising 30 genotypically distinct PCR ribotypes (PCR ribotypes: 002, 003, 005, 009, 010, 011, 014, 015, 017, 018, 019, 014/20, 023, 026, 027, 035, 044, 045, 046, 050, 056, 060, 063, 064, 067, 078, 085, 103, 106 and 153) and also an internal control strain (E4, ribotype 010) previously demonstrated to be intermediately susceptible to metronidazole.13
Antimicrobial susceptibility testing
Antimicrobial susceptibilities were determined using a previously described (Wilkins Chalgren) agar incorporation method; this technique can detect reduced susceptibility of C. difficile to metronidazole.14
In vitro human gut model
We have described previously the use of a triple-stage chemostat human gut model to study the interplay between antimicrobial agents, the indigenous gut microflora and C. difficile.15 The gut model was validated against physicochemical and microbiological measurements from the intestinal contents of sudden death victims.16 The gut model is, however, limited by its inability to simulate immunological and secretory events that occur in the human colon. The model comprises three pH-maintained (pH 5.5 ± 0.2, vessel 1; pH 6.2 ± 0.2, vessel 2; pH 6.8 ± 0.2, vessel 3) fermentation vessels, top-fed by growth medium at a controlled rate (dilution rate = 0.015 h−1). The gut model is inoculated with a faecal emulsion (∼10% w/v in pre-reduced PBS) prepared from C. difficile-negative faeces of five healthy elderly (>65 years) volunteers. Faecal donors were in good health and had received no antimicrobial therapy for at least 3 months prior to commencement of this study.
Experimental design
Time periods for this experiment are displayed in Figure 1. SMT19969 was evaluated in separate experiments: (i) 31.25 mg/L four times daily dosing regimen for 7 days; and (ii) 62.5 mg/L four times daily for 7 days. These dosing regimens were selected to mimic 1× and 2× the clinical regimen for vancomycin in treating CDI. Following inoculation of the gut model with faecal emulsion (day 0), the media pump was started and no further interventions were made for 13 days (period A). Gut microflora were enumerated every 2 days. C. difficile spores (107 cfu) were prepared as described previously12 and inoculated into vessel 1 on day 14 (period B). Viable counts of C. difficile and the indigenous gut microflora and C. difficile cytotoxin titres (Vero cell cytotoxicity assay) were monitored daily. After 7 days a further inoculum of C. difficile spores was instilled into vessel 1, followed by 33.9 mg/L of clindamycin (four times daily for 7 days, period C), which was instilled to reflect the concentration observed in faeces of patients and volunteers.17 Instillation of SMT19969 commenced once high-level cytotoxin titres ≥4 log10-relative units (RU) were observed on at least two consecutive days (period E). Following cessation of antimicrobial agent instillation, gut microflora populations and C. difficile cytotoxin titres were monitored for a further 14 days (period F).
Figure 1.
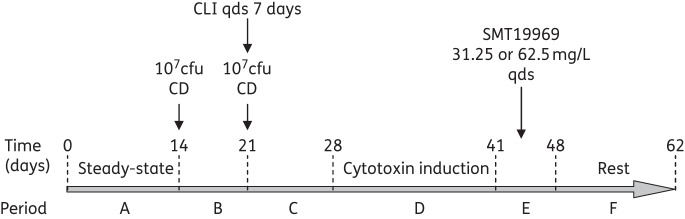
Time scheme in gut model experiments with SMT19969. CLI, clindamycin; CD, C. difficile spores; qds, four times daily.
Enumeration of gut microflora and C. difficile cytotoxin titres
Gut bacterial populations and C. difficile concentrations were determined as described previously.18 Gut microflora populations cultured were total facultative anaerobes, total anaerobes (facultative + obligate), lactose-fermenting Enterobacteriaceae (LFE), enterococci, lactobacilli, bifidobacteria, total Clostridium spp., Bacteroides fragilis group (BFG), C. difficile total viable counts (vegetative C. difficile + spores) and C. difficile spore viable counts. C. difficile cytotoxin production was monitored using a Vero cell cytotoxicity assay as described previously.18 Indigenous gut microflora populations from vessel 1 of the gut models were not determined; only C. difficile total viable counts, spore counts and cytotoxin titres were quantified. Possible emergence of C. difficile with reduced susceptibility to SMT19969 was monitored by inoculating aliquots from the model onto Brazier's CCEY agar incorporating 0.5 mg/L (4× MIC) of SMT19969 throughout the experiment.
Determination of SMT19969 and clindamycin concentrations
Concentrations of clindamycin achieved in each of the vessels of the gut model were determined using a microbiological bioassay with the indicator organism Kochuria rhizophila ATCC 9341 as described previously.18 Concentrations of SMT19969 achieved in each of the vessels of the gut model were determined using a microbiological bioassay with the indicator organism Enterococcus faecalis ATCC 29212. Samples (1 mL) for assay of antimicrobial concentrations in gut model contents were removed daily during the gut model experiments and stored at −20°C (bioassay and HPLC) and assayed retrospectively. Briefly, for bioassays performed with E. faecalis, the indicator organism was inoculated onto a fresh Columbia blood agar plate and incubated aerobically for 24 h at 37°C. One millilitre of a standard suspension (0.5 McFarland, 1 × 107 cfu) in sterile saline was inoculated into 100 mL molten Mueller–Hinton agar and mixed gently by inversion. Bioassay dishes were incubated in air at 37°C overnight, and zone inhibition diameters measured with callipers.
Statistical analysis of susceptibility data
MIC data were log2-transformed and assessed for normality and homogeneity of variance using Minitab version 16 and SPSS version 20. Non-parametric testing (Kruskal–Wallis and post hoc tests) for significance was performed using these statistical analysis packages. P < 0.05 was considered to be statistically significant.
Results
Antimicrobial susceptibility testing
SMT19969 was 7- and 17-fold more active than metronidazole and vancomycin, respectively (P < 0.05), against the panel of 30 genotypically distinct C. difficile PCR ribotypes; metronidazole was twice as active as vancomycin (P < 0.05) (Table 1).
Table 1.
Antibiotic susceptibilities (mg/L) of C. difficile to metronidazole (MTZ), vancomycin (VAN) and SMT19969
| MTZ | VAN | SMT19969 | |
|---|---|---|---|
| Geometric mean MIC | 0.70 | 1.53 | 0.090 |
| MIC50 | 0.50 | 2.00 | 0.125 |
| MIC90 | 2.00 | 2.00 | 0.125 |
In vitro gut model experiments
Observations during equilibration period
The results observed in vessel 3 of the gut model are shown given their close similarity to data for vessel 2. Additionally, only the gut microflora viable counts from the highest dosage (250 mg/L/day SMT19969) gut model are depicted in this study due to the close similarity between the two experiments. Viable counts of all bacterial groups increased during the steady-state period compared with those observed on the first day of the experiment (Figures 2 and 3). Gut microfloras in vessels 2 and 3 of the gut models were dominated by obligate anaerobes, particularly Bifidobacterium spp. and BFG, with lower observed viable counts of facultatively anaerobic bacterial groups. Viable counts of indigenous gut microfloras were largely stable by the end of this period.
Figure 2.

Mean viable counts (±SE) of obligate anaerobe bacterial groups (log10 cfu/mL) in vessel 3 of the SMT19969 250 mg/L/day gut model (period E). TA, total anaerobes (obligate and facultative).
Figure 3.

Mean viable counts (±SE) of facultative anaerobe bacterial groups (log10 cfu/mL) in vessel 3 of the SMT19969 250 mg/L/day gut model (period E). TFA, total facultative anaerobes.
Observations following instillation of C. difficile spores
Single inocula of C. difficile spores (2 × 107 cfu) were instilled into the gut models on day 14 of each experiment. Although some minor fluctuations in viable counts of the indigenous gut microflora were observed following instillation of C. difficile spores, no consistent trend was seen across both gut models. C. difficile viable counts in vessel 1 of both gut models declined at a similar rate after the initial inoculation of C. difficile spores and there was no evidence of spore germination, proliferation or cytotoxin production (Figures 4 and 5). C. difficile viable counts in vessels 2 and 3 of the gut models increased during the initial stages of the period, as a consequence of washout from vessel 1, then declined for the remainder of the period. There was no evidence of C. difficile germination, proliferation or cytotoxin production (Figures 4 and 5).
Figure 4.

C. difficile total viable counts, spore counts (log10 cfu/mL) and cytotoxin titres (RU) in vessel 3 of the gut model dosed with 250 mg/L/day SMT19969 (period E).
Figure 5.

C. difficile total viable counts, spore counts (log10 cfu/mL) and cytotoxin titres (RU) in vessel 3 of the gut model dosed with 125 mg/L/day SMT19969 (period E).
Observations following instillation of clindamycin
Instillation of clindamycin facilitated similar alterations in viable counts of gut microfloras in both gut models. A sustained reduction in viable counts of lactobacilli was observed following clindamycin instillation (1.5–2.0 log10 cfu/mL), while transient minor declines in viable counts of some other groups of obligate anaerobes was observed (Figures 3 and 4). Marked declines in viable counts of bifidobacteria were observed during clindamycin instillation (6.0–7.0 log10 cfu/mL) in both gut models. Additionally, BFG viable counts declined by 1.0 log10 cfu/mL (Figure 2). Conversely, viable counts of Enterococcus spp. increased markedly following cessation of clindamycin instillation (Figure 3). C. difficile remained quiescent during clindamycin instillation in both gut models; populations comprised principally spores, and cytotoxin was undetectable (Figures 4 and 5). Bacterial groups deleteriously affected by clindamycin instillation largely recovered by the end of the cytotoxin induction period. LFE viable counts increased markedly during the cytotoxin induction period.
C. difficile germination and proliferation were observed 8 days (vessel 2, day 35) and 9 days (vessel 3, day 36) after cessation of clindamycin instillation (Figures 4 and 5). Cytotoxin was demonstrated in vessel 1 of both gut models 11 days after cessation of clindamycin instillation. Cytotoxin titres increased to 4 RU in vessels 2 and 3 of both gut models, but remained at 2 RU in vessel 1. Instillation of SMT19969 commenced 15 days after cessation of clindamycin instillation (day 42) in both gut models.
Clindamycin concentrations detected in the gut models
Mean clindamycin concentrations (mg/L) observed during the instillation period in the 125 and 250 mg/L/day gut models were 48.4 and 47.7 (vessel 1), 35.2 and 39.9 (vessel 2), and 36.2 and 38.8 (vessel 3), respectively. Clindamycin was quickly washed out of the gut model vessels following cessation of instillation, and was undetectable 6 days after cessation in all vessels of both experiments.
Effect of SMT19969 on the indigenous gut microflora and C. difficile
Both SMT19969 dosing regimens demonstrated little antimicrobial activity against the indigenous gut microflora in the two gut models (Table 2). Total Clostridium spp. was the only indigenous gut microflora group for which viable counts declined following SMT19969 instillation (Figure 2). Modest increases in viable counts of LFE were observed following instillation of both dosing regimens. Vegetative forms of C. difficile (SMT19969 MIC = 0.125 mg/L) declined during the SMT19969 instillation period (∼1.5 log10 cfu/mL over 7 days) in all vessels of both gut models (Figures 4 and 5). Following cessation of SMT19969 administration, continued declines in C. difficile counts by up to a further 2 log10 cfu/mL were observed in the 250 mg/L/day model (Figure 4). C. difficile cytotoxin titres declined rapidly following commencement of SMT19969 instillation, with overall declines of 2, 4 and 3 RU observed in vessels 1, 2 and 3, respectively, from days 42 to 48. Following cessation of SMT19969 instillation, C. difficile vegetative populations remained detectable and increased slightly by the end of the experiment. Cytotoxin was undetectable for the final 6 and 5 days of the 125 and 250 mg/L/day gut models, respectively.
Table 2.
Overall change in viable counts (log10 cfu/mL) of gut microflora groups and C. difficile, and cytotoxin titres (CYT, RU) during SMT19969 dosing (days 42–48)
| Bacterial group | 125 mg/L/day |
250 mg/L/day |
||
|---|---|---|---|---|
| vessel 2 | vessel 3 | vessel 2 | vessel 3 | |
| TFA | NC | NC | NC | NC |
| LFE | +1.0 | +0.6 | +0.6 | +0.6 |
| BFG | NC | NC | NC | NC |
| Bifidobacteria | NC | NC | NC | NC |
| Lactobacilli | NC | NC | NC | NC |
| Clostridia | −2.0 | −1.7 | −2.0 | −1.4 |
| Enterococci | NC | NC | NC | NC |
| Total anaerobes | NC | NC | NC | NC |
| CD TVC | −1.5 | −1.5 | −1.7 | −1.4 |
| CD SP | −1.3 | NC | −1.1 | −0.9 |
| CD CYT | −4.0 | −3.0 | −4.0 | −3.0 |
TFA, total facultative anaerobes; TVC, total viable count; SP, spore count; CD, C. difficile; NC, no substantial change (±0.5 log10 cfu/mL).
Isolation of C. difficile on breakpoint agars
No C. difficile were isolated from SMT19969 breakpoint agar (4× MIC) plates during the course of the experiments.
Discussion
The efficacy of existing CDI therapies can be affected by: (i) poor drug pharmacokinetics, e.g. very low concentrations of metronidazole within the colonic lumen; (ii) fear of bacterial resistance emergence (e.g. VRE following vancomycin exposure), although a recent study suggested that these fears may be unfounded;19 and (iii) reduced efficacy against hypervirulent C. difficile ribotypes.6 Emergence of resistance or reduced susceptibility is a concern for metronidazole,14 especially given its aforementioned poor bioavailability, although currently no link between reduced susceptibility and treatment failure has been established.20 Emergence of resistance to fidaxomicin has been documented in a single instance in the clinical setting; however, rigorous serial passage studies and human gut model studies have failed to document similar findings.11 Similarly, serial passage studies with SMT19969 demonstrated a low propensity of resistance development with mutation frequencies <3.09 × 10−9.21
The results of the present study show that SMT19969, a novel, narrow-spectrum, non-absorbable antimicrobial agent, has high potency against C. difficile in standard agar incorporation MIC experiments. SMT19969 MICs were 7- and 17-fold lower than corresponding metronidazole and vancomycin MICs and are amongst the most potent anti-C. difficile antimicrobial agents reported. The narrow spectrum of antibacterial activity demonstrated from SMT19969 susceptibility testing studies22 was reinforced in the present gut model experiments, in that the only indigenous microflora group adversely affected following SMT19969 instillation was Clostridium spp. (2 log10 cfu/mL decline). This observed anticlostridial activity reflects prior in vitro susceptibility studies for this genus,23 particularly Clostridium innocuum, which is often found within the gut models in high concentrations.
The gut model has proven a valuable tool in the drug development setting, both to assess the likelihood of CDI induction by antibiotics, and to determine the efficacies of novel antimicrobial agents for treating CDI. Indeed, clinical observations have reflected in vitro observations in predicting suboptimal efficacy of the toxin-binding polymer tolevamer,9,24 the superiority of fidaxomicin compared with vancomycin in treating recurrent CDI and the low propensity of certain clinical antimicrobial agents to induce CDI.25–27 Prior gut model studies showed that metronidazole and vancomycin adversely affect the anaerobic gut microflora (BFG and/or bifidobacteria) and shift the balance from anaerobe- to facultative-anaerobe-dominated microflora (LFE and/or lactobacilli and/or enterococci), which is a similar microflora shift to that observed for antimicrobial agents with a recognized propensity to induce CDI (clindamycin, ceftriaxone and cefotaxime). In the present study, neither SMT19969 regimen adversely affected the normal, anaerobe-dominated microflora in the gut model vessels, which are analogous to the distal end of the colon, which may confer this antimicrobial an advantage over existing therapies. Fidaxomicin was recently demonstrated to inhibit Bifidobacterium spp. in three of four gut model experiments and facilitated a microflora dominated by LFE, with reduced BFG concentrations.12 Despite these microflora changes, fidaxomicin instillation was not associated with recurrent simulated CDI in the gut model; it was suggested that the persistence of supra-MIC of fidaxomicin prevents C. difficile spore germination and subsequent simulated recurrence. Metronidazole was demonstrated as an ineffective treatment for simulated CDI in the gut model in prior studies, where poor bioactive concentrations of drug within the gut model and signs of recurrent simulated CDI were observed.12
Instillation of SMT19969 into both gut models elicited an immediate decline in cytotoxin production. Cytotoxin titres declined substantially by the end of period E, and were at the limit of detection 2 days later in both high and low SMT19969 dosage models. The decline in C. difficile total viable counts observed following SMT19969 instillation was incremental over several days and indicated a bacteriostatic mechanism of action for this antibiotic under these experimental conditions. However, in standard in vitro testing SMT19969 demonstrates bactericidal activity with >5 log reduction in cfu/mL at 24 h.28 These declines in C. difficile concentrations following SMT19969 instillation in the gut model are similar to, or better than, declines observed following instillation of vancomycin into the gut model,29,30 although in the present study total viable counts remained above spore counts in the SMT19969 dosing period. In a recent in vivo study using the hamster model of CDI, SMT19969 elicited complete clearance of viable C. difficile following commencement of dosing (R. Vickers, unpublished results), and in the Phase I clinical trial of SMT19969, clostridial viable counts were below the lower limits of detection for viable counting (whereas other gut microflora groups were unaffected).31 The differences between the present in vitro study and recent in vivo studies with SMT19969 probably reflect a suboptimal formulation used in the present experiments that has been shown to result in significantly reduced efficacy in the hamster model of CDI (R. Vickers, unpublished results). Visible precipitates of SMT19969 were observed following instillation of the drug and persisted for the duration of the experiment in both gut models. Poor solubility also hampered efforts to determine the dissolved (bioavailable) fraction of SMT19969, and therefore some caution must be exercised when interpreting the antimicrobial activity of SMT19969 in these gut model studies. Cytotoxin titres increased slightly during period F (rest period), but to a relatively low titre (2 RU), and titres were below the limits of detection for the last 5–6 days of both experiments. The sporadic spikes in total viable count and low-level cytotoxin observed during period F may be due to the release of vegetative C. difficile from biofilms within the gut models; both the organism and its cytotoxins have been demonstrated to be present in the complex, multispecies biofilms within the gut model vessels (G. S. Crowther, M. H. Wilcox and J. Freeman, unpublished results).32 Instillation of SMT19969 curtailed the production of cytotoxin by C. difficile ribotype 027, noting that prior gut model studies have consistently demonstrated a substantially longer (>23 days), but not higher, peak cytotoxin titre by this virulent ribotype.18
The results of the present study, combined with a good Phase I safety and tolerability profile, and superior efficacy to vancomycin in treating CDI in the hamster model,21 indicate that further studies with SMT19969 are warranted to determine its efficacy as a potential CDI therapeutic option. Improvements to the solubility of SMT19969, or reduced dosing to aid dissolution, may facilitate the demonstration of enhanced efficacy in vitro and in vivo in future studies. In particular, an assessment of the efficacy of SMT19969 in preventing recurrent CDI both in vitro and in vivo would help to determine whether this is an area in which this novel antimicrobial agent may outperform other CDI therapies. Indeed, the extremely narrow spectrum of antibacterial activity of SMT19969 is attractive, and theoretically suggests a reduced likelihood of CDI recurrence due to reduced perturbation of colonic colonization resistance.
Funding
This study was initiated and financially supported by Summit plc through a Seeding Drug Discovery Award from the Wellcome Trust (grant number 091055).
Transparency declarations
In the past two years, G. S. C. has received support to attend meetings from Novacta. M. H. W. has received research funding from Actelion, Astellas, BioMerieux, Cubist, Pfizer, Summit Plc. and The Medicines Company and consultancies and/or lecture honoraria from Actelion, Astellas, Astra-Zeneca, Bayer, Cubist, Durata, J&J, Merck, Nabriva, Novacta, Novartis, Optimer, Pfizer, Sanofi-Pasteur, The Medicines Company, VH Squared and Viropharma. All other authors: none to declare.
References
- 1.Bartlett J, Moon N, Chang TW, et al. Role of Clostridium difficile in antibiotic-associated pseudomembranous colitis. Garstroenterology. 1978;75:778–82. [PubMed] [Google Scholar]
- 2.George WL, Rolfe RD, Harding GK, et al. Clostridium difficile and cytotoxin in feces of patients with antimicrobial agent-associated pseudomembranous colitis. Infection. 1982;10:205–8. doi: 10.1007/BF01666910. [DOI] [PubMed] [Google Scholar]
- 3.Owens RC, Donskey CJ, Gaynes RP, et al. Antimicrobial-associated risk factors for Clostridium difficile infection. Clin Infect Dis. 2008;46:S19–31. doi: 10.1086/521859. [DOI] [PubMed] [Google Scholar]
- 4.Kyne L, Hamel MB, Polavaram R, et al. Health care costs and mortality associated with nosocomial diarrhea due to Clostridium difficile. Clin Infect Dis. 2002;34:346–53. doi: 10.1086/338260. [DOI] [PubMed] [Google Scholar]
- 5.O'Brien JA, Lahue BJ, Caro JJ, et al. The emerging infectious challenge of Clostridium difficile-associated disease in Massachusetts hospitals: clinical and economic consequences. Infect Control Hosp Epidemiol. 2007;28:1219–27. doi: 10.1086/522676. [DOI] [PubMed] [Google Scholar]
- 6.Debast SB, Bauer MP, Kuijper EJ, et al. European Society of Clinical Microbiology and Infectious Diseases: update of the treatment guidance document for Clostridium difficile infection. Clin Microbiol Infect. 2014;20(Suppl 2):S1–S26. doi: 10.1111/1469-0691.12418. [DOI] [PubMed] [Google Scholar]
- 7.Nelson RL, Kelsey P, Leeman H, et al. Antibiotic treatment for Clostridium difficile-associated diarrhea in adults. Cochrane Database Syst Rev. 2011;7:CD004610. doi: 10.1002/14651858.CD004610.pub4. [DOI] [PubMed] [Google Scholar]
- 8.Al-Nassir WN, Sethi AK, Nerandzic MM, et al. Comparison of clinical and microbiological response to treatment of Clostridium difficile-associated disease with metronidazole and vancomycin. Clin Infect Dis. 2008;47:56–62. doi: 10.1086/588293. [DOI] [PubMed] [Google Scholar]
- 9.Bouza E, Dryden M, Mohammed R, et al. Results of a phase III trial comparing tolevamer, vancomycin and metronidazole in patients with Clostridium difficile-associated diarrhoea; Hoboken, NJ, USA: Blackwell; Abstracts of the Eighteenth European Congress of Clinical Microbiology and Infectious Diseases, Barcelona, Spain, 2008. Abstract O464, p. S103. [Google Scholar]
- 10.Pepin J, Alary ME, Valiquette L, et al. Increasing risk of relapse after treatment of Clostridium difficile colitis in Quebec, Canada. Clin Infect Dis. 2005;40:1591–7. doi: 10.1086/430315. [DOI] [PubMed] [Google Scholar]
- 11.Goldstein EJ, Babakhani F, Citron D. Antimicrobial activities of fidaxomicin. Clin Infect Dis. 2012;55(Suppl 2):S143–8. doi: 10.1093/cid/cis339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chilton CH, Crowther GS, Freeman J, et al. Successful treatment of simulated Clostridium difficile infection in a human gut model by fidaxomicin first line after vancomycin or metronidazole failure. J Antimicrob Chemother. 2014;69:451–62. doi: 10.1093/jac/dkt347. [DOI] [PubMed] [Google Scholar]
- 13.Brazier JS, Fawley W, Freeman J, et al. Reduced susceptibility of Clostridium difficile to metronidazole. J Antimicrob Chemother. 2001;48:741–2. doi: 10.1093/jac/48.5.741. [DOI] [PubMed] [Google Scholar]
- 14.Baines SD, O'Connor R, Freeman J, et al. Emergence of reduced susceptibility to metronidazole in Clostridium difficile. J Antimicrob Chemother. 2008;62:1046–52. doi: 10.1093/jac/dkn313. [DOI] [PubMed] [Google Scholar]
- 15.Best EL, Freeman J, Wilcox MH. Models for the study of Clostridium difficile infection. Gut Microbes. 2012;3:145–67. doi: 10.4161/gmic.19526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Macfarlane GT, Macfarlane S, Gibson GR. Validation of a three-stage compound continuous culture system for investigating the effect of retention time on the ecology and metabolism of bacteria in the human colon. Microb Ecol. 1998;35:180–7. doi: 10.1007/s002489900072. [DOI] [PubMed] [Google Scholar]
- 17.Brown RB, Martyak SN, Barza M, et al. Penetration of clindamycin phosphate into the abnormal human biliary tract. Ann Intern Med. 1976;84:168–70. doi: 10.7326/0003-4819-84-2-168. [DOI] [PubMed] [Google Scholar]
- 18.Freeman J, Baines SD, Saxton K, et al. Effect of metronidazole on growth and cytotoxin production by epidemic Clostridium difficile PCR ribotypes 001 and 027 in a human gut model. J Antimicrob Chemother. 2007;60:83–91. doi: 10.1093/jac/dkm113. [DOI] [PubMed] [Google Scholar]
- 19.Miller M, Bernard L, Thompson M, et al. Lack of increased colonization with vancomycin-resistant enterococci during preferential use of vancomycin for treatment during an outbreak of healthcare-associated Clostridium difficile infection. Infect Control Hosp Epidemiol. 2010;31:710–5. doi: 10.1086/653613. [DOI] [PubMed] [Google Scholar]
- 20.Johnson S, Sanchez JL, Gerding DN. Metronidazole resistance in Clostridium difficile. Clin Infect Dis. 2000;31:625–6. doi: 10.1086/313955. [DOI] [PubMed] [Google Scholar]
- 21.Vickers R, Tinsley J, Wilson F, et al. SMT19969: In vitro and in vivo evaluation of a novel antibiotic for Clostridium difficile infection; Hoboken, NJ, USA: Blackwell; Abstracts of the Twenty-third European Congress of Clinical Microbiology and Infectious Diseases, Berlin, Germany, 2013. Abstract P1655. [Google Scholar]
- 22.Goldstein EJ, Citron DM, Tyrrell KL, et al. Comparative in vitro activities SMT19969, a new antimicrobial agent, against Clostridium difficile and 350 Gram-positive and Gram-negative aerobic and anaerobic intestinal flora isolates. Antimicrob Agents Chemother. 2013;57:4872–6. doi: 10.1128/AAC.01136-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goldstein EJ, Citron DM, Tyrrell KL, et al. Comparative in vitro activities of SMT19969, a new antimicrobial agent, against 162 strains from 35 less frequently recovered intestinal Clostridium species: implications for Clostridium difficile recurrence. Antimicrob Agents Chemother. 2014;58:1187–91. doi: 10.1128/AAC.02184-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baines SD, Freeman J, Wilcox MH. Tolevamer is not efficacious in the neutralization of cytotoxin in a human gut model of Clostridium difficile infection. Antimicrob Agents Chemother. 2009;53:2202–4. doi: 10.1128/AAC.01085-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baines SD, Freeman J, Wilcox MH. Effects of piperacillin/tazobactam on Clostridium difficile growth and cytotoxin production in a human gut model. J Antimicrob Chemother. 2005;55:974–82. doi: 10.1093/jac/dki120. [DOI] [PubMed] [Google Scholar]
- 26.Baines SD, Saxton K, Freeman J, et al. Tigecycline does not induce proliferation or cytotoxin production by epidemic Clostridium difficile strains in a human gut model. J Antimicrob Chemother. 2006;58:1062–5. doi: 10.1093/jac/dkl364. [DOI] [PubMed] [Google Scholar]
- 27.Baines SD, O'Connor R, Huscroft G, et al. Mecillinam: a low risk antimicrobial agent for induction of Clostridium difficile infection in an in vitro human gut model. J Antimicrob Chemother. 2009;63:838–9. doi: 10.1093/jac/dkp017. [DOI] [PubMed] [Google Scholar]
- 28.Corbett D, Wise A, Payne LJ, et al. SMT19969: In vitro pharmacodynamics against Clostridium difficile; Hoboken, NJ, USA: Blackwell; Abstracts of the Twenty-third European Congress of Clinical Microbiology and Infectious Diseases, Berlin, Germany, 2013. Abstract P1654. [Google Scholar]
- 29.Baines SD, O'Connor R, Saxton K, et al. Activity of vancomycin against epidemic Clostridium difficile strains in a human gut model. J Antimicrob Chemother. 2009;63:520–5. doi: 10.1093/jac/dkn502. [DOI] [PubMed] [Google Scholar]
- 30.Baines SD, O'Connor R, Saxton K, et al. Comparison of oritavancin versus vancomycin as treatments for clindamycin-induced Clostridium difficile PCR ribotype 027 infection in a human gut model. J Antimicrob Chemother. 2008;62:1078–85. doi: 10.1093/jac/dkn358. [DOI] [PubMed] [Google Scholar]
- 31.Best E, Vickers R, Wilcox MH. Effects of a novel antimicrobial (SMT19969) for treatment of Clostridium difficile infection (CDI) on gut flora; Abstracts of the Fifty-third Interscience Conference on Antimicrobial Agents and Chemotherapy, Denver, Colorado, 2013. Abstract K-167, ASM Press, Washington DC, USA. [Google Scholar]
- 32.Crowther GS, Chilton CH, Todhunter SL, et al. Development and validation of a chemostat gut model to study both planktonic and biofilm modes of growth of Clostridium difficile and human microbiota. PLoS One. 2014;9:e88396. doi: 10.1371/journal.pone.0088396. [DOI] [PMC free article] [PubMed] [Google Scholar]