

Abstract
A comparison of 10 most popular Multiple Sequence Alignment (MSA) tools, namely, MUSCLE, MAFFT(L-INS-i), MAFFT (FFT-NS-2), T-Coffee, ProbCons, SATe, Clustal Omega, Kalign, Multalin, and Dialign-TX is presented. We also focused on the significance of some implementations embedded in algorithm of each tool. Based on 10 simulated trees of different number of taxa generated by R, 400 known alignments and sequence files were constructed using indel-Seq-Gen. A total of 4000 test alignments were generated to study the effect of sequence length, indel size, deletion rate, and insertion rate. Results showed that alignment quality was highly dependent on the number of deletions and insertions in the sequences and that the sequence length and indel size had a weaker effect. Overall, ProbCons was consistently on the top of list of the evaluated MSA tools. SATe, being little less accurate, was 529.10% faster than ProbCons and 236.72% faster than MAFFT(L-INS-i). Among other tools, Kalign and MUSCLE achieved the highest sum of pairs. We also considered BALiBASE benchmark datasets and the results relative to BAliBASE- and indel-Seq-Gen-generated alignments were consistent in the most cases.
Keywords: Multiple Sequence Alignment Tools, comparative study of MSA tools, sum of pairs score, column score, evolutionary parameters
Introduction
Multiple Sequence Alignments (MSAs) have become highly scrutinized and a fundamental approach in several research domains in molecular biology and bioinformatics such as studies of epidemiology and virulence,1 drug design,2 recon-struction of phylogenetic tree, prediction of 3D structure, identifying conserved regions,3–5 and finding molecular function.6–8 Dozens of algorithms have been developed as a part of an attempt to improve the accuracy of alignments, but still there is not a single MSA method that may generate accurate alignments for all types of test cases.9
Manually refined repositories of MSAs such as BAliBASE,10 PREFAB,11 and SABmark12 are good sources of accurate alignments to gauge performance of various MSA programs, but they have a number of disadvantages such as due to the small size they do not cover the full range of scenarios of protein evolution and due to the uncertain positional homology assessing accuracy of the alignments becomes difficult.13 In addition, because of the lack of evolutionary history among the sequences, benchmark alignments cannot be used to test phylogenetic software applications. The developers may also be misguided to develop algorithms to resolve the problems that are highlighted only in the manually curated alignment sets.14 Finally, high-level expertise is required to generate benchmark alignments.
Simulated/true alignments are an alternative to benchmark alignments for comparing MSA tools. The major motivation of simulated sequences is that their true evolutionary history is known, which is very useful to generate accurate alignments and phylogenetic trees. Second, the user can generate simulated alignments comprising varying insertion rate, deletion rate, sequence length, indel size, and number of sequences. Third, as compared to benchmark alignments, it is very easy for the end user to generate simulated alignments. Simulated sequences also have some drawbacks. First, because of dependency of all observations drawn from true alignments on simplifications and assumptions of the model used to reconstruct these alignments, the simulated sequences cannot give an explanation for all evolutionary aspects. The other potential threat is the use of simulation settings more close to the strategy of some MSA methods than others. For example, the selected model of sequence evolution might be similar to the fundamental model of a particular MSA tool and thus provide it with an excessive advantage.15 Keeping in view disadvantages of simulated sequences, there is a need to compare results of true alignments with the results of benchmark sequences. However, the easy construction of simulated alignments is one of the major motivations to apply them for comparative study of MSA methods.
Several sequence simulators, with their own strengths and weaknesses, are available. ROSE16 generates MSA for DNA, RNA, and protein sequences, and true evolutionary history is also logged. ROSE incorporates indels linearly in accordance with the evolutionary distance and length distribution. Indel models with nonlinear indel probabilities cannot be shown in ROSE. SIMPROT17 is another simulation tool that incorporates indels; however, it does not support the feature of root sequence as an input, the conservation of motifs or modifying amino acid frequencies among subsequences. It also does not generate alignments for nucleotide sequences. MySSP18 simulates DNA sequences using different models of DNA evolution such as Jukes-Cantor,19 Kimura two-parameter,20 equal input, and Hasegawa–Kishino–Yano.21 It incorporates the features of indels, nonstationary patterns and output of ancestral sequences. Indel-Seq-Gen−2.1.03 (iSGv2.0)22 generates highly divergent DNA sequences and protein families by incorporating a number of indel models. It can also model coding and noncoding DNA evolutions. iSGv2.0 is a new tool for generating simulated datasets. iSGv2.0 improves upon iSGv1.0 through the addition of motif conservation, lineage-specific evolution, using indel tracking, PROSITE-like regular expressions, and subsequence length constraints, as well as coding and noncoding DNA evolution. The authors in their original article claimed that iSGv2.0 has unique features for generating highly divergent protein sequences with the incorporation of indels.
In the current era, a large number of MSA methods are available. Comparative studies9,14,23 of MSA programs showed that none of them were capable to generate accurate alignments for all test cases. The choice of an MSA method is based on the sequences to be aligned. In this article, we present a comparative study of 10 of the most often used MSA methods based on a different approach. The selected MSA programs are T-Coffee,24 MAFFT(FF-TNS-2), MAFFT(L-INS-i),25,26 MUSCLE,11,27 Kalign,28,29 Dialign-TX,30 Multalin,31 Clustal Omega,32 ProbCons,33 and SATe.34,35 Our study also focused on the significance of some implementations embedded in the each program’s algorithm. Using various evolutionary parameters, simulated alignments were constructed through iSG. As a part of input to iSG, simulated trees were generated under birth–death model using TreeSim36 package integrated in R, which is a suite of software tools for data interpretation and graphical view.37 Birth–death model allows species to speciate with constant rate b, and go extinct with a constant rate d. Consequently, simulated tree grows at the rate of b−d. In order to ensure the net growth of the phylogenetic tree, the birth rate should be higher than the death rate. iSG produced both the aligned and unaligned sequences. Unaligned sequences were used as an input to the selected MSA programs to generate MSAs, which were then compared with the alignments produced by iSG. We also present a comparison of the results obtained on the simulated alignments and the results obtained on BALi-BASE benchmark alignments. This comparison, with a few exceptions, confirmed that the simulated alignments may be used as an alternative for the comparative study of MSA tools.
Results
Simulated sequences and alignments
Simulated sequences and alignments generated by iSG were used to study the effect of sequence length, indel size, deletion rate, and insertion rate on alignment accuracy. Four hundred known alignments and corresponding sequence files with no indels were constructed based on the trees generated by R. The two most popular scores, ie, sum of pairs (SPS) and column score (CS) were applied to measure quality of the alignments.
MSA tool evaluation: overall alignment accuracy
For each of the 400 reference alignments in the simulated dataset, the 10 MSA methods were applied, resulting in a total of 4000 test alignments. These 4000 test alignments consisted of 1000 alignments with varying indel size, 1000 alignments with varying sequence length, 1000 alignments with varying deletion rate, and 1000 alignments with varying insertion rate. The varying evolutionary parameters are shown in Table 1. The overall accuracy of these alignments was measured using average sum-of-pairs scores. The experiment confirmed previous findings9,14 in the sense that ProbCons outperformed all other MSA tools (Fig. 1). SATe, which was not tested in the previous studies, was in the second position and MAFFT(L-INS-i) was in the third position. Among other tools, Kalign achieved the highest score. T-Coffee and MAFFT(FF-TNS-2) generated the least quality alignments. One-way analysis of variance (ANOVA) showed 0.002 significant level, which means that there is a significant difference among SPS of the alignments generated by MSA tools. Multiple Comparisons Table (MCT) developed using Tukey post hoc test confirmed our results that ProbCons, SATe, and MAFFT(L-INS-i) were the most accurate tools. ANOVA and MCT are attached as Supplementary file (ANOVA-MCT).
Table 1.
Four sets of evolutionary parameters.
| VARYING DELETION RATES | VARYING INSERTION RATES | ||||||
|---|---|---|---|---|---|---|---|
| SEQUENCE LENGTH | INDEL SIZE | INSERTION RATE | DELETION RATE | SEQUENCE LENGTH | INDEL SIZE | INSERTION RATE | DELETION RATE |
| 500 | 20 | 0.000002 | 0.002 | 500 | 20 | 0.001 | 0.000002 |
| 500 | 20 | 0.000002 | 0.002222222 | 500 | 20 | 0.001333333 | 0.000002 |
| 500 | 20 | 0.000002 | 0.002857143 | 500 | 20 | 0.001666667 | 0.000002 |
| 500 | 20 | 0.000002 | 0.005 | 500 | 20 | 0.002222222 | 0.000002 |
| 500 | 20 | 0.000002 | 0.006666667 | 500 | 20 | 0.002857143 | 0.000002 |
| 500 | 20 | 0.000002 | 0.01 | 500 | 20 | 0.005 | 0.000002 |
| 500 | 20 | 0.000002 | 0.013 | 500 | 20 | 0.006666667 | 0.000002 |
| 500 | 20 | 0.000002 | 0.05 | 500 | 20 | 0.013333 | 0.000002 |
| 500 | 20 | 0.000002 | 0.1 | 500 | 20 | 0.02 | 0.000002 |
| 500 | 20 | 0.000002 | 0.2 | 500 | 20 | 0.04 | 0.000002 |
| VARYING INDEL SIZES | VARYING SEQUENCE LENGTHS | ||||||
| SEQUENCE LENGTH | INDEL SIZE | INSERTION RATE | DELETION RATE | SEQUENCE LENGTH | INDEL SIZE | INSERTION RATE | DELETION RATE |
| 500 | 5 | 0.000002 | 0.000002 | 10 | 20 | 0.000002 | 0.000002 |
| 500 | 10 | 0.000002 | 0.000002 | 50 | 20 | 0.000002 | 0.000002 |
| 500 | 20 | 0.000002 | 0.000002 | 150 | 20 | 0.000002 | 0.000002 |
| 500 | 40 | 0.000002 | 0.000002 | 300 | 20 | 0.000002 | 0.000002 |
| 500 | 80 | 0.000002 | 0.000002 | 500 | 20 | 0.000002 | 0.000002 |
| 500 | 160 | 0.000002 | 0.000002 | 750 | 20 | 0.000002 | 0.000002 |
| 500 | 320 | 0.000002 | 0.000002 | 1200 | 20 | 0.000002 | 0.000002 |
| 500 | 740 | 0.000002 | 0.000002 | 1500 | 20 | 0.000002 | 0.000002 |
| 500 | 850 | 0.000002 | 0.000002 | 2000 | 20 | 0.000002 | 0.000002 |
| 500 | 950 | 0.000002 | 0.000002 | 2500 | 20 | 0.000002 | 0.000002 |
Note: In each of the four sets, three parameters are kept constant and one is varying (highlighted).
Figure 1.
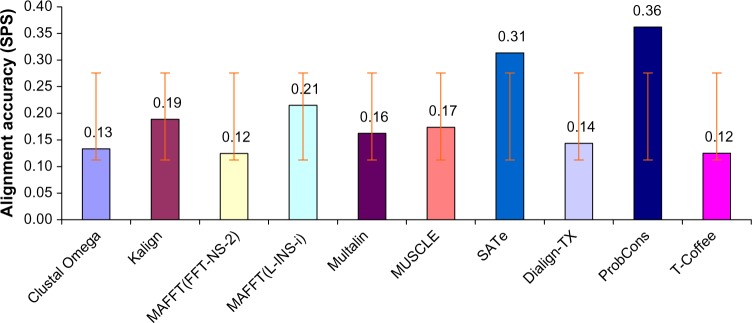
Overall alignment quality measured using SPS. Error bars correspond to one standard deviation. ProbCons maintained its first position but MAFFT (L-INS-i) loosed the second position, which was occupied by the SATe.
MSA tool evaluation: effect of indel size
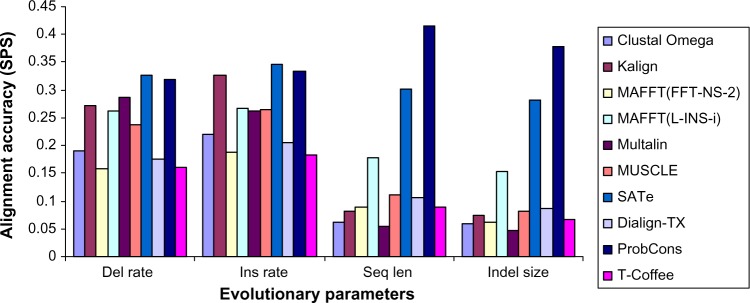
To evaluate the effect of indel size, we generated 1000 alignments (100 alignments by each MSA method) with varying indel size (5–950). This study showed that alignment quality was much less dependent on indel size (line charts of Figs. 2A and 2B), but nevertheless evaluation of effect of indel size on alignment quality measured using SPS (Fig. 2A) showed that ProbCons was the top performer. SATe and MAFFT(L-INS-i) were in the second and third positions, respectively. Among other MSA tools, Dialign-TX achieved the highest SPS. Multalin generated alignments with the lowest SPS. Study of the effect of indel size on alignment quality measured using CS (Fig. 2B) showed that ProbCons, SATe, and MAFFT(L-INS-i) were in the first, second, and third positions, respectively. Among other MSA programs, T-Coffee was the top performer. Majority of the MSA tools such as Clustal Omega, Multalin, Kalign, MAFFT(FFT-NS-2), MUSCLE, and Dialign-TX achieved very low CS.
Figure 2.

Effect of varying indel size on alignment quality. Indel size did not affect alignment quality (line charts); however, ProbCons was the top performer. MAFFT(L-INS-i) and SATe achieved second and third positions, respectively.
MSA tool evaluation: effect of sequence length
To study the effect of increasing sequence length on alignment quality, another dataset having 1000 alignments (100 alignments by each MSA method) of varying sequence lengths (30–2500 residues) were generated. This experiment showed that sequence length had a weaker effect on alignment (line charts of Figs. 3A and 3B), but nevertheless study of the effect of sequence length measured using SPS (Fig. 3A) showed that ProbCons achieved the highest average scores. SATe and MAFFT(L-INS-i) were consistently in the second and third positions, respectively. Among other MSA tools, MUSCLE and Multalin achieved the highest and lowest SPS, respectively. Evaluation of effect of sequence length measured using CS (Fig. 3B) showed that ProbCons was in the top, followed by SATe and MAFFT (L-INS-i), whereas other MSA programs achieved a low CS.
Figure 3.

Effect of increasing sequence length on alignment accuracy. Line charts A and b show that sequence length had a weaker effect on performance of all MSA methods; however, ProbCons outperformed all other MSA tools. SATe and MAFFT(L-INS-i) were in the second and third positions, respectively. Among other MSA tools, MUSCLE and MAFFT(FFT-NS-2) gave good SPS and CS, respectively.
MSA tool evaluation: effect of deletion rate
Effect of deletion rate on the alignment quality was studied by generating alignments with varying deletion rates (Table 1). Similar to the studies of effect of indel size and sequence length on alignment quality, effect of deletion rate was investigated by developing a dataset consisting of 1000 alignments (100 alignments by each MSA method). This study showed different results. First, a tradeoff between increasing deletion rate and alignment quality was observed. High deletion rate affected accuracy of almost all MSA tools (line charts of Figs. 4A and 4B). Second, SATe achieved the highest average sum of pairs scores (Fig. 4A) and column scores (Fig. 4B). In case of alignment quality measured using SPS, ProbCons and Multalin were in the second and third positions, respectively. Among other MSA tools, Kalign and MUSCLE were the best performers. The smallest accuracy was shown by MAFFT(FFT-NS-2). In the case of alignment quality measured using CS, MAFFT(L-INS-i) and Multalin were in the second and third positions, respectively.
Figure 4.

Effect of increasing deletion rate on alignment quality. The study showed different results. Firstly, SATe beated ProbCons. Second, a higher deletion rate had significant effect on alignment quality (line charts A and b). In case of alignment quality measured using CS, SATe, MAFFT(L-INS-i), and Multalin were in the first, second, and third positions, respectively.
MSA tool evaluation: effect of insertion rate
To investigate the effect of increasing insertion rate, the fourth dataset consisting of 1000 alignments (100 alignments by each MSA method) with varying insertion rate (Table 1) was generated. Results of this study were similar to the study of “effect of deletion rate on alignment quality”, which showed that performance of all MSA tools was highly dependent on the insertion rate (line charts of Figs. 5A and 5B). Evaluation of effect of insertion rate measured using SPS (Fig. 5A) showed that SATe generated the most accurate alignments. ProbCons, Kalign, and MAFFT(L-INS-i) were in the second, third, and fourth positions, respectively. T-Coffee achieved the lowest SPS. Study of insertion rate on alignment quality measured using CS (Fig. 5B) showed that SATe outperformed all other MSA programs. MUSCLE, MAFFT(L-INS-i), and ProbCons were in the second, third, and fourth positions respectively. T-Coffee achieved the lowest CS.
Figure 5.

Effect of increasing insertion rate on the alignment accuracy. Insertion rate had significant effect on alignment quality. Performance of almost all MSA tools was poor on high insertion rate (line charts). SATe achieved the highest average SPS. ProbCons and Kalign were in the second and third positions, respectively. Accuracy of T-Coffee was the lowest. Study of insertion rate measured using CS showed that SATe, MUSCLE, and MAFFT(L-INS-i) were in first, second, and third positions, respectively. ProbCons was in the fourth position.
MSA tool evaluation: time consumed by each MSA method
Results showed that, overall, accuracy of ProbCons was the highest; however, it was also the slowest tool. SATe, being the second more accurate tool, was 529% faster than ProbCons. MAFFT(L-INS-i) consumed more time than SATe and less time than ProbCons. However, MUSCLE was the fastest tool. Figure 6 shows time spent in seconds by each MSA method.
Figure 6.

Efficiency of the MSA tools. ProbCons spent maximum time. SATe was little less accurate but 529.10% faster than ProbCons and 236.72% faster than MAFFT(L-INS-i). MUSCLE was the fastest tool. It took only 375 seconds.
User guidance for choosing MSA tools
It was important to rank MSA methods based on the experiments conducted in this study. Results are summarized in Figure 7. ProbCons, SATe, and MAFFT(L-INS-i) were the best tools for sequences with varying indel size and sequence length. For sequences with varying insertion rate, SATe, ProbCons, and Kalign achieved the highest SPS. In case of sequences with varying deletion rate, SATe, ProbCons, and Multalin outperformed other MSA tools. On the whole, SATe, based on its overall alignment quality and processing speed, was the best tool.
Figure 7.
User guidance for choosing MSA tools. ProbCons, SATe, and MAFFT(L-INS-i) are the best tools for sequences with varying indel size and sequence length. For sequences with varying insertion rate, SATe, ProbCons, and Kalign achieved the highest SPS. In case of sequences with varying deletion rate, SATe, ProbCons, and Multalin outperformed other MSA tools.
Comparison of results obtained on simulated data with the results obtained on benchmark sequences
In order to determine that the results obtained on iSG simulated alignments were also applicable to the benchmark alignments, we measured accuracy of the 10 MSA tools using six reference test cases (RV11, RV12, RV20, RV30, RV40, and RV50) available in the version 3 of the BALiBASE (ftp://ftp-igbmc.u-strasbg.fr/pub/BAliBASE3). RV11 contains sequences having equal distances. RV12 comprised sequences with orphans. RV20 consists of sequences from deviating subfamilies. RV30 comprised sequences from families with highly diverged sequences. RV40 contains sequences with N/C terminal extensions. RV50 comprised sequences with large insertions.
Overall, the results obtained on BALiBASE benchmark alignments were similar to those obtained on the true alignments (Fig. 8). PronCons and MAFFT(L-INS-i) gave similar performances on BALiBASE reference sets RV11, RV20, RV30, and RV50. In case of BALiBASE reference set RV12, ProbCons outperformed MAFFT(L-INS-i); however, on RV40, MAFFT(L-INS-i) performed better than all other MSA methods. SATe, except on RV11 (where it gave performance equal to ProbCons and MAFFT(L-INS-i)), outperformed all MSA tools on all the reference sets. Almost all MSA tools performed much better, but overall the trends were similar. ProbCons, SATe, and MAFFT(L-INS-i) were consistently the best tools. However, in contrast to the results obtained on simulated sequences, SATe, Clustal Omega, and T-Coffee showed different performances. SATe outperformed all other MSA tools, and T-Coffee showed a better performance than MUSCLE. Clustal Omega outperformed Kalign, Multalin, and Dialign-TX. Other MSA tools were almost consistent in their performances. Time spent on BALiBASE benchmark alignments by all MSA tools was also very similar to the time spent on simulated alignments (Fig. 9). In case of both datasets, SATe was faster than ProbCons and T-Coffee. However, some differences were also observed. Kalign, with a minute difference, consumed less time than MUSCLE. MAFFT(L-INS-i) and Dialign-TX were faster than SATe and Multalin, respectively. Overall, the findings found from comparison of results obtained on simulated data with the results obtained on benchmark sequences were consistent and confirmed previous findings14 in the sense that ProbCons and MAFFT(L-INS-i) were the best tools.
Figure 8.

Overall alignment quality comparison between the results obtained on simulated data and results obtained on benchmark sequences. With the exception of SATe, T-Coffee, and Clustal Omega, which performed better in case of benchmark alignments, the results were similar to the results obtained on simulated data and confirmed the previous findings.
Figure 9.

Efficiency comparison between the results obtained on simulated data and results obtained on benchmark sequences. Major findings were almost similar. SATe was faster than ProbCons and T-Coffee. However, Kalign, MAFFT(L-INS-i), and Dialign-TX showed better efficiency than MUSCLE, SATe, and Multalin, respectively, on benchmark alignments.
Discussion
We used 4000 alignments to test whether the MSA methods have potential to generate high-quality alignments. Accuracy and efficiency of the latest versions of MSA methods (which were based on various algorithms and techniques) were evaluated with their default parameters/configurations. Different parameter settings may improve their performance. We intentionally generated alignments comprising very high insertion rate, deletion rate, indel size, and sequence length. Overall alignment quality investigation showed that in case of simulated sequences as well as BAliBASE’s v3.0 reference sets,10 ProbCons,33 SATe,34,35 and MAFFT(L-INS-i)25,26 were the best performers. The same results have been reported by the previous studies.9,14,23 Among other MSA methods, in case of simulated alignments, Kalign28,29 and MUSCLE11,27 achieved the highest SPS,38 with MUSCLE being the most efficient method, and in the case of BALiBASE benchmark datasets, T-Coffee generated the most accurate alignments, but it was consistently slower than MUSCLE.
Studies of the effect of indel size and sequence length measured using SPS and CS showed that they have the least effect on the performance of MSA tools; however, ProbCons, SATe, and MAFFT(L-INS-i) were in the first, second, and third positions, respectively. These results confirmed findings of the previous studies.14,39 Investigation of the effect of deletion rate on alignment quality showed that performance of MSA methods was significantly low on higher deletion rates. Nuin et al reported the same findings.14 SATe achieved the highest SPS and CS. In case of alignment measured using SPS, ProbCons and Multalin were the second and third top performers. In case of alignment quality measured using CS, Multalin, MUSCLE, and MAFFT(L-INS-i) were in second, third, and fourth positions, respectively. Study of the effect of insertion rate on alignment quality also confirmed the previous findings14 in the sense that alignment quality is significantly dependent on the insertion rate. In case of alignment quality measured using SPS and CS, SATe outperformed all other MSA methods. Alignment quality measured using SPS showed that Kalign and ProbCons were in the second and third positions, respectively. Alignment accuracy measured using CS showed that MUSCLE, MAFFT(L-INS-i), and ProbCons were in the second, third, and fourth positions, respectively. For the both evolutionary parameters, ie, deletion rate and insertion rate, T-Coffee achieved the lowest SPS and CS.
All MSA methods performed much better with BALi-BASE benchmark alignments than simulated datasets but the overall trends were similar. Our findings generally confirmed results of the previous studies.9,14,23 One distinguished finding was the fact that when simulated datasets were used, SATe outperformed MAFFT(L-INS-i). Among the best MSA tools, SATe was also the fastest tool. Original articles of MAFFT(L-INS-i)25,26 and ProbCons33 placed them on the top with the best accuracy on benchmark alignments. Our results also proved the claim of the authors of Kalign28,29 that its efficiency is very close to the fast mode of MUSCLE11,27 and MAFFT(FFT-NS-2)25,26 but accuracy is comparable to other MSA tools.
Our study highlighted strengths and weakness of all the MSA tools. ProbCons, which is based on consistency approach,23 outperformed other MSA tools when alignments with varying sequence length and indel size were tested. SATe, which is based on an iterative divide and conquer approach,34 outperformed all MSA methods when alignments comprising high insertion or deletion rate were investigated. However, overall, there is a minor difference between ProbCons and SATe. MAFFT(L-INS-i), which also adopted consistency approach in its algorithm,23 was in the third position in all test cases. Among other MAS tools, overall, Kalign and Multalin whose alignment generating process is based on progressive alignment approach were the better alternatives.28,29,31 However, in case of high sequence lengths and indel sizes, they did not generate high-quality alignments. Their performance was good when alignments with high insertion rates and deletion rates were used. Algorithm used by Clustal Omega is based on Hidden Markov Model approach32 and Dialign-TX is based on consistency-based algorithm.30 Overall, both tools performed very poorly, and especially in the case of big indel sizes and large sequence lengths, they generated low-quality alignments. MUSCLE, which is based on iterative approach,27 performed better when alignments comprising high deletion rates and insertion rates were provided. In case of benchmark datasets, T-Coffee, which is based on consistency approach, performed better than MUSCLE. Performance of MUSCLE and MAFFT(FFT-NS-2) was consistent for all test cases.
Materials and Methods
Figure 10 describes all steps of the methodology adopted in this research work.
Figure 10.
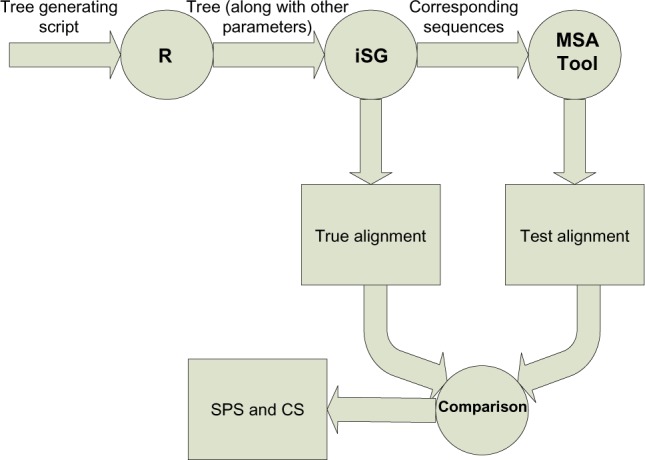
Methodology of the comparative study.
Construction of simulated trees
TreeSim package of R was used to generate 10 simulated trees comprising different number of taxa (10, 25, 75, 200, 350, 500, 600, 700, 850, and 1000) under the birth–death model.
Construction of simulated alignments
iSGv2.0 was used to construct four datasets. Each of the four datasets consisted of 100 alignments; 100 with varying deletion rate, 100 with varying insertion rate, 100 with varying indel size, and 100 with varying sequence length. Thus, a total of 400 known alignments were generated.
Construction of test alignments
Each of the MSA methods was applied to generate 100 alignments with varying deletion rate, 100 alignments with varying insertion rate, 100 alignments with varying indel size, and 100 alignments with varying sequence length. Thus, a total of 400 alignments were generated by each MSA method, resulting in a grand total of 4000 alignments. Table 1 shows the four parameters and their varying values (base pairs) used to construct the true alignments. Each of the four sets had one varying (highlighted) and three constant parameters.
Alignment accuracy assessment procedure
The most common practice of measuring accuracy of MSA programs is to compute SPS and CS by comparing an alignment generated by an MSA tool with a reference alignment.38 SPS is calculated by counting the correctly aligned residue pairs. It measures the ability of MSA tools to align some, if not all, of the sequences in an alignment.40 Let an alignment of N sequences comprise M columns. The cth column can be designated as Ac1, Ac2,…., AcN. For each pair of residues Acj and Ack, we define Scjk such that Scjk = 1, if Acj and Ack are in the same column of reference alignment. The score for cth column (Sc) can be defined as follows.
The sum of pair score for the full alignment can be computed as
Cr denotes number of columns and Src represents the score of the cth column in reference alignment.
Column score examines the ability of MSA tools of aligning all the columns correctly.39 It is computed by dividing the “matched” columns between test and reference alignments with the total number of “considered” columns in the test alignment. Cc = 1 if a column of a (test) alignment matches with the column of reference alignment otherwise it is zero.
Accuracy of the MSA methods were measured using SPS and CS. MQAT, which is an interactive tool for computing quality scores of several alignments simultaneously,41 was used to calculate SPS and CS.
Statistical analysis
For each of the four datasets (based on four evolutionary parameters), average SPS and average CS were computed from the results produced by the 10 MSA methods. One-way ANOVA was performed to determine the level of significance for accuracy of all MSA tools. In order to find the significant difference between specific MSA tools, we developed MCT using Tukey post hoc test. The positive or negative mean differences indicate the significant difference between the MSA tools. The P < 0.05 was used as the level of significance.
MSA methods evaluated
The 10 MSA tools were selected based on two parameters: (1) the underlying algorithms and (2) their popularity. Table 2 describes the MSA tools with their versions, main algorithms, and URL for download. All these MSA methods were run using default parameters.
Table 2.
MSA methods used in this article.
| MSA METHOD | VERSION | MAIN ALGORITHM | AVAILABILITY |
|---|---|---|---|
| T-Coffee [22] | 10.00.r1613 | Consistency (multi-core usage capable) | http://www.tcoffee.org/ |
| ProbCons [32] | 1.12 | Consistency | http://probcons.stanford.edu/download.html |
| Dialign-TX [29] | 1.0.2 | Consistency | http://dialign-tx.gobics.de/download |
| Kalign [27, 28] | 2.0 | Progressive (Wu-Manber) | http://msa.sbc.su.se/cgi-bin/msa.cgi |
| Multalin [30] | 5.4.1 | Progressive | http://multalin.toulouse.inra.fr/multalin/ |
| MAFFT(L-INS-i) [23,24] | 7.0 | Consistency | http://mafft.cbrc.jp/alignment/software/ |
| Clustal Omega [31] | 1.2.0 | Hidden Markov Model | http://www.clustal.org/omega/ |
| MUSCLE3.8.31 [25,26] | 3.8.31 | Iterative | http://www.drive5.com/MUSCLE/downloads.htm |
| MAFFT(FFT-NS-2) [23,24] | 7.0 | Iterative | http://mafft.cbrc.jp/alignment/software/ |
| SATe [33,24] | 2.2.7 | Iterative divide and conquer | http://phylo.bio.ku.edu/software/sate/sate.html |
Computing machine
A computing machine with Core i7 3.34 GHz processor, 8 GB RAM, and Fedora OS was used for comparative study of MSA methods.
Conclusions
Our study discloses that SATe was the best tool, based on its overall alignment accuracy and efficiency. Overall, the results showed that ProbCons was consistently on the top of the list of the evaluated MSA tools, but it was a very slow tool. SATe, being little less accurate, was 529.10% faster than ProbCons and 236.72% faster than MAFFT(L-INS-i). Among other tools, Kalign and MUSCLE gave the highest SPS and CS, respectively. A comparison on the results obtained on simulated alignments and the results obtained on BALiBASE benchmark alignments showed the similar trends. Our analysis allows the user to establish with more detail the strengths and weaknesses of each MSA tool and its algorithmic approach. iSG was also confirmed to be an appropriate choice to test alignment quality. It allows a user to construct large simulated datasets in seconds, with full control of its characteristics.
Availability
Simulated trees, unaligned sequences for all the true alignments, true alignments generated by iSG and test alignments constructed by all the MSA tools are available at www.ivistmsa.com.
Supplementary File
ANOVA-MCT (DOC)
Acknowledgments
We are highly thankful to the members of Institute of Biochemistry and Biotechnology, University of Veterinary and Animal Sciences Lahore, Pakistan for discussions and technical support. We are also very thankful to our colleagues at Virtual University of Pakistan especially Syed Shah Mohammad (Lecturer CS), Abdul Basit (Lecturer CS), and Gulzar Ahmad Siddiqui (Lecturer Statistics) for their support in the statistical analysis of the comparative study and write-up of the article.
Footnotes
Author Contributions
Conceived the concept: MTP. Designed the experiments: MEB, NN, AN. Contributed to the design of the study: MS. Performed the experiments: ARA, TH, UW, NA. Analyzed the data: MTP, SQ, MEB. Wrote the first draft of the manuscript: MTP, MA. Contributed to the writing of the manuscript: MS. Made critical revisions and approved final version: MEB. All authors reviewed and approved of the final manuscript.
ACADEMIC EDITOR: Jike Cui, Associate Editor
FUNDING: Authors disclose no funding sources.
COMPETING INTERESTS: Authors disclose no potential conflicts of interest.
Paper subject to independent expert blind peer review by minimum of two reviewers. All editorial decisions made by independent academic editor. Upon submission manuscript was subject to anti-plagiarism scanning. Prior to publication all authors have given signed confirmation of agreement to article publication and compliance with all applicable ethical and legal requirements, including the accuracy of author and contributor information, disclosure of competing interests and funding sources, compliance with ethical requirements relating to human and animal study participants, and compliance with any copyright requirements of third parties. This journal is a member of the Committee on Publication Ethics (COPE).
REFERENCES
- 1.Bao Y, Bolotov P, Dernovoy D, et al. The influenza virus resource at the National Center for Biotechnology Information. J Virol. 2008;82:596–601. doi: 10.1128/JVI.02005-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kuipers RK, Joosten HJ, van Berkel WJ, et al. 3DM: systematic analysis of heterogeneous superfamily data to discover protein functionalities. Proteins. 2010;78:2101–13. doi: 10.1002/prot.22725. [DOI] [PubMed] [Google Scholar]
- 3.Kim J, Ma J. PSAR: measuring multiple sequence alignment reliability by probabilistic sampling. Nucl Acids Res. 2011;39(15):6359–68. doi: 10.1093/nar/gkr334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Siepel A, Bejerano G, Pedersen JS, et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005;15(8):1034–50. doi: 10.1101/gr.3715005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Roskin KM, Diekhans M, Haussler D. Scoring two-species local alignments to try to statistically separate neutrally evolving from selected DNA segments; Proceedings of the seventh annual international conference on computational molecular biology; ACM Press; 2003. pp. 257–66. [Google Scholar]
- 6.Levasseur A, Pontarotti P, Poch O, Thompson JD. Strategies for reliable exploitation of evolutionary concepts in high throughput biology. Evol Bioinform Online. 2008;4:121–37. doi: 10.4137/ebo.s597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wong KM, Suchard MA, Huelsenbeck JP. Alignment uncertainty and genomic analysis. Science. 2008;319:473–6. doi: 10.1126/science.1151532. [DOI] [PubMed] [Google Scholar]
- 8.Loytynoja A, Goldman N. Phylogeny-aware gap placement prevents errors in sequence alignment and evolutionary analysis. Science. 2008;320:1632–5. doi: 10.1126/science.1158395. [DOI] [PubMed] [Google Scholar]
- 9.Thompson JD, Linard B, Lecompte O, Poch O. A comprehensive benchmark study of multiple sequence alignment methods: current challenges and future perspectives. PLoS One. 2011;6(3):e18093. doi: 10.1371/journal.pone.0018093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thompson JD, Koehl P, Ripp R, Poch O. BAliBASE 3.0: latest developments of the multiple sequence alignment benchmark. Proteins. 2005;61:127–36. doi: 10.1002/prot.20527. [DOI] [PubMed] [Google Scholar]
- 11.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32(5):1792–7. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Walle IV, Lasters I, Wyns L. SABmark-a benchmark for sequence alignment that covers the entire known fold space. Bioinformatics. 2005;21(7):1267–8. doi: 10.1093/bioinformatics/bth493. [DOI] [PubMed] [Google Scholar]
- 13.Karplus K, Hu B. Evaluation of protein multiple alignments by SAM-T99 using the BAliBASE multiple alignment test set. Bioinformatics. 2001;17(8):713–20. doi: 10.1093/bioinformatics/17.8.713. [DOI] [PubMed] [Google Scholar]
- 14.Nuin PA, Wang Z, Tillier ER. The accuracy of several multiple sequence alignment programs for proteins. BMC Bioinformatics. 2006;7:471. doi: 10.1186/1471-2105-7-471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Iantorno S, Gori K, Goldman N, Gil M, Dessimoz C. Who watches the watchmen? An appraisal of benchmarks for multiple sequence alignment. In: Russell DJ, editor. Multiple Sequence Alignment Methods. Vol. 1069. Clifton, NY: Humana Press; 2014. pp. 59–73. [DOI] [PubMed] [Google Scholar]
- 16.Stoye J, Evers D, Meyer F. Rose: generating sequence families. Bioinformatics. 1998;14:157–63. doi: 10.1093/bioinformatics/14.2.157. [DOI] [PubMed] [Google Scholar]
- 17.Pang A, Smith AD, Nuin PAS, Tillier ERM. SIMPROT: using an empirically determined indel distribution in simulations of protein evolution. BMC Bioinformatics. 2005;6:236. doi: 10.1186/1471-2105-6-236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rosenberg MS. MySSP: Non-stationary evolutionary sequence simulation, including indels. Evol Bioinform Online. 2005;1:81–3. [PMC free article] [PubMed] [Google Scholar]
- 19.Jukes TH, Cantor CR. Evolution of protein molecules. In: Munro HN, editor. Mammalian Protein Metabolism. New York: Academic Press; 1969. pp. 21–132. [Google Scholar]
- 20.Kimura M. A simple method for estimating evolutionary rates of base subsitutions through comparative studies of nucleotide sequences. J Mol Evol. 1980;16:111–20. doi: 10.1007/BF01731581. [DOI] [PubMed] [Google Scholar]
- 21.Hasegawa M, Kishino H, Yano T. Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. J Mol Evol. 1985;22:160–74. doi: 10.1007/BF02101694. [DOI] [PubMed] [Google Scholar]
- 22.Strope CL, Abel K, Scott SD, Moriyama EN. Biological sequence simulation for testing complex evolutionary hypotheses: indel-Seq-Gen version 2.0. Mol Biol Evol. 2009;26(11):2581–93. doi: 10.1093/molbev/msp174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pais FSM, Ruy PC, Oliveira G, Coimbra RS. Assessing the efficiency of multiple sequence alignment programs. Algorithms Mol Biol. 2014;9:4. doi: 10.1186/1748-7188-9-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Notredame C, Higgins DG, Heringa J. T-Coffee: a novel method for fast and accurate multiple sequence alignment. J Mol Biol. 2000;302(1):205–17. doi: 10.1006/jmbi.2000.4042. [DOI] [PubMed] [Google Scholar]
- 25.Katoh K, Misawa K, Kuma K, Miyata T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002;30(14):3059–66. doi: 10.1093/nar/gkf436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30:772–80. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Edgar RC. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics. 2004;5:113. doi: 10.1186/1471-2105-5-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lassmann T, Frings OS, Sonnhammer EL. Kalign2: high-performance multiple alignment of protein and nucleotide sequences allowing external features. Nucleic Acids Res. 2009;37:858–65. doi: 10.1093/nar/gkn1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lassmann T, Sonnhammer E. Kalign–an accurate and fast multiple sequence alignment algorithm. BMC Bioinformatics. 2005;6:298. doi: 10.1186/1471-2105-6-298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Subramanian AR, Kaufmann M, Morgenstern B. DIALIGN-TX: greedy and progressive approaches for segment-based multiple sequence alignment. Algorithms Mol Biol. 2008;3:6. doi: 10.1186/1748-7188-3-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Corpet F. Multiple sequence alignment with hierarchical clustering. Nucl Acids Res. 1988;16(22):10881–90. doi: 10.1093/nar/16.22.10881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sievers F, Wilm A, Dineen DG, et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol. 2011;7:539. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Do CB, Mahabhashyam MS, Brudno M, Batzoglou S. ProbCons: probabilistic consistency-based multiple sequence alignment. Genome Res. 2005;15:330–40. doi: 10.1101/gr.2821705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu K, Raghavan S, Nelesen S, Linder CR, Warnow T. Rapid and accurate large scale coestimation of sequence alignments and phylogenetic trees. Science. 2009;324(5934):1561–4. doi: 10.1126/science.1171243. [DOI] [PubMed] [Google Scholar]
- 35.Liu K, Warnow T, Holder MT, et al. SATé-II: very fast and accurate simultaneous estimation of multiple sequence alignments and phylogenetic trees. Syst Biol. 2012;61(1):90–106. doi: 10.1093/sysbio/syr095. [DOI] [PubMed] [Google Scholar]
- 36.Cusimano N, Stadler T, Renner SS. A new method for handling missing species in diversification analysis applicable to randomly or non-randomly sampled phylogenies. Syst Biol. 2012;61:785–92. doi: 10.1093/sysbio/sys031. [DOI] [PubMed] [Google Scholar]
- 37.R Development Core Team . R: a language and environment for statistical computing. R Foundation for Statistical Computing; Vienna, Austria: 2008. Available from: http://www.R-project.org. [Google Scholar]
- 38.Catherine LA, Cory LS, Etsuko NM. SuiteMSA: visual tools for multiple sequence alignment comparison and molecular sequence simulation. BMC Bioinformatics. 2011;12:184. doi: 10.1186/1471-2105-12-184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lassmann T, Sonnhammer E. Quality assessment of multiple alignment programs. FEBS Lett. 2002;529:126–30. doi: 10.1016/s0014-5793(02)03189-7. [DOI] [PubMed] [Google Scholar]
- 40.Thompson JD, Plewniak F, Poch O. A comprehensive comparison of multiple sequence alignment programs. Nucleic Acids Res. 1999;27(13):2682–90. doi: 10.1093/nar/27.13.2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Perez MT, Babar ME, Nadeem A, et al. MQAT: an efficient quality assessment tool for large multiple sequence alignments. Life Sci J. 2013;10(9s):9–16. [Google Scholar]