

Abstract
BACKGROUND
Relapsed acute lymphoblastic leukemia (ALL) is difficult to treat despite the availability of aggressive therapies. Chimeric antigen receptor–modified T cells targeting CD19 may overcome many limitations of conventional therapies and induce remission in patients with refractory disease.
METHODS
We infused autologous T cells transduced with a CD19-directed chimeric antigen receptor (CTL019) lentiviral vector in patients with relapsed or refractory ALL at doses of 0.76×106 to 20.6×106 CTL019 cells per kilogram of body weight. Patients were monitored for a response, toxic effects, and the expansion and persistence of circulating CTL019 T cells.
RESULTS
A total of 30 children and adults received CTL019. Complete remission was achieved in 27 patients (90%), including 2 patients with blinatumomab-refractory disease and 15 who had undergone stem-cell transplantation. CTL019 cells proliferated in vivo and were detectable in the blood, bone marrow, and cerebrospinal fluid of patients who had a response. Sustained remission was achieved with a 6-month event-free survival rate of 67% (95% confidence interval [CI], 51 to 88) and an overall survival rate of 78% (95% CI, 65 to 95). At 6 months, the probability that a patient would have persistence of CTL019 was 68% (95% CI, 50 to 92) and the probability that a patient would have relapse-free B-cell aplasia was 73% (95% CI, 57 to 94). All the patients had the cytokine-release syndrome. Severe cytokine-release syndrome, which developed in 27% of the patients, was associated with a higher disease burden before infusion and was effectively treated with the anti–interleukin-6 receptor antibody tocilizumab.
CONCLUSIONS
Chimeric antigen receptor–modified T-cell therapy against CD19 was effective in treating relapsed and refractory ALL. CTL019 was associated with a high remission rate, even among patients for whom stem-cell transplantation had failed, and durable remissions up to 24 months were observed. (Funded by Novartis and others; CART19 ClinicalTrials.gov numbers, NCT01626495 and NCT01029366.)
Engineered T-cell therapy is a new strategy for the treatment of relapsed and refractory acute lymphoblastic leukemia (ALL), which is associated with an extremely poor prognosis in adults and remains a leading cause of death from childhood cancer.1–3 In initial proof-of-principle clinical trials involving patients with chronic lymphocytic leukemia (CLL), chimeric antigen receptor–modified T cells that target CD19 produced a durable complete remission in a small number of patients.4–6 Our group and others then extended these findings to relapsed and refractory B-cell ALL, and we found profound responses in a small number of children and adults.7,8
Chimeric antigen receptors are genetically engineered receptors that couple an anti-CD19 single-chain Fv domain to intracellular T-cell signaling domains of the T-cell receptor, thereby redirecting cytotoxic T lymphocytes to cells expressing this antigen. With the use of lentiviral-vector technology for gene transfer and permanent T-cell modification, CTL019 (formerly known as CART19)-engineered T cells express a chimeric antigen receptor in which the T-cell activation signal is provided by the CD3-zeta domain, and the co-stimulatory signal is provided by the CD137 (4-1BB) domain.4
We previously reported a high degree of in vivo expansion of CTL019 cells that resulted in complete remission in two children with relapsed and highly refractory B-cell ALL.8 However, the rate of complete remission in a larger cohort, long-term persistence of chimeric antigen receptor–modified T cells, and the durability of remission remained unknown. We now report the results of CART19 (A Phase I/IIA Study of Redirected Autologous T Cells Engineered to Contain Anti-CD19 Attached to TCRzeta and 4-1BB Signaling Domains in Patients with Chemotherapy Resistant or Refractory CD19+ Leukemia and Lymphoma) showing the efficacy of CTL019 and provide follow-up of up to 2 years in our expanded cohort of 30 patients with relapsed and refractory ALL. Robust expansion of CTL019 cells rapidly induced complete remission in this cohort of patients who were previously considered to have refractory and incurable disease.
METHODS
TRIAL DESIGN AND OVERSIGHT
We conducted pilot clinical trials at Children’s Hospital of Philadelphia and the Hospital of the University of Pennsylvania that were designed to assess the safety and feasibility of CTL019 T-cell therapy in patients with relapsed and refractory CD19+ cancers; the protocols were approved by the respective institutional review boards. All the authors discussed and interpreted the study results and vouch for the data and analyses. All the patients or their parents provided written informed consent. Enrolled patients received CTL019 infusions between April 2012 and February 2014. Additional details regarding the study design are provided in the Supplementary Appendix, available with the full text of this article at NEJM.org.
Leukapheresis products were stimulated with paramagnetic beads coated with antibodies to CD3 and CD28 and transduced with the CD19-BB-zeta transgene as described previously.4,9 After leukapheresis, patients received interim therapy at the discretion of their treating physician. Chemotherapy aimed at depletion of T lymphocytes (Table S1 in the Supplementary Appendix) was administered 1 week before infusion of CTL019 (Table S2 in the Supplementary Appendix), except in three patients who had persistent cytopenias.
PATIENT CHARACTERISTICS
A total of 25 patients, 5 to 22 years of age, were treated at Children’s Hospital of Philadelphia (pediatric trial), and 5 older patients, 26 to 60 years of age, were treated at the Hospital of the University of Pennsylvania (adult trial) (Table 1). Of these patients, 26 had B-cell ALL in a first to fourth relapse, 3 had primary refractory B-cell ALL, and 1 had relapsed T-cell ALL that expressed CD19. Eighteen patients had relapsed disease after allogeneic stem-cell transplantation. Three patients (Patients 2, 3, and 20) had disease that was previously shown to be refractory to blinatumomab (a bispecific antibody with one domain that binds to CD3 on T cells and the other that binds to CD19).
Table 1.
Baseline Characteristics of the Patients.*
| Characteristic | Pediatric Cohort (N = 25) | Adult Cohort (N = 5) | Total (N = 30) |
|---|---|---|---|
| Sex — no. (%) | |||
| Female | 11 (44) | 1 (20) | 12 (40) |
| Male | 14 (56) | 4 (80) | 18 (60) |
| Age at infusion — yr | |||
| Median | 11 | 47 | 14 |
| Range | 5–22 | 26–60 | 5–60 |
| Allogeneic transplantation — no. (%) | 18 (72) | 0 | 18 (60) |
| Primary refractory disease — no. (%) | 0 | 3 (60) | 3 (10) |
| Relapse — no. (%) | |||
| 1 | 3 (12) | 2 (40) | 5 (17) |
| ≥2 | 22 (88) | 22 (73) | |
| Baseline burden of acute lymphoblastic leukemia — no. (%) | |||
| Presence of detectable disease† | 20 (80) | 4 (80) | 24 (80) |
| Morphologic remission‡ | 1 (20) | 1 (3) | |
| Absence of minimal residual disease | 5 (20) | 5 (17) | |
| High-risk cytogenetic factors — no. | |||
| BCR–ABL1 | 2 | ||
| IKZF1 deletion | 2 | ||
| iAMP21 | 1 | ||
| MLL translocation | 1 | ||
| Hypodiploidy | 2 | ||
| CNS status — no.§ | |||
| CNS-1 | 23 | ||
| CNS-2 | 2 | ||
CNS denotes central nervous system, and iAMP21 intrachromosomal amplification of chromosome 21.
Disease was detected by assessment of bone marrow and cerebrospinal fluid morphologic features and measurement of minimal residual disease.
Minimal residual disease was not measured.
CNS status was determined only in the pediatric trial. CNS status was defined as CNS-1 (no detectable blast cells in a sample of cerebrospinal fluid), CNS-2 (blast cells detected in a sample with <5 leukocytes per cubic millimeter and <10 erythrocytes per cubic millimeter), and CNS-3 (blast cells detected in a sample with ≥5 leukocytes per cubic millimeter and <10 erythrocytes per cubic millimeter). The presence of CNS-3 disease was an exclusion criterion.
RESULTS
OUTCOMES
Twenty-seven of 30 patients (90%) were in a morphologic complete remission at the first assessment 1 month after the infusion of CTL019. A test to detect minimal residual disease by means of multiparametric flow cytometry was negative in 22 patients, positive in 3 patients (the levels of minimal residual disease were 0.1% [and subsequently negative at 3 months], 0.09%, and 0.22%, respectively), and not performed in 2 patients. A complete remission was achieved in 2 of 3 patients who had previously been exposed to blinatumomab. The 2 patients in whom blast cells were detected in the cerebrospinal fluid at the time of infusion subsequently had no detectable central nervous system (CNS) leukemia as of the most recent follow-up (6 months), and no CNS relapses were observed.
Seven patients who had a complete remission subsequently had a relapse between 6 weeks and 8.5 months after infusion of CTL019 cells. Three relapses developed after early loss of CTL019-modified T cells at 2 weeks to 3 months, and in these patients the relapsed ALL remained CD19-positive. After recovery of normal B cells at 2 to 3 months, one relapse occurred rapidly at 3 months and two relapses were delayed (they occurred at 6 and 8.5 months). Patient 9, who had minimal residual disease (0.22%) at 1 month, had a relapse with CD19-positive ALL at 6 weeks. The disease rapidly progressed, and the patient died from ALL. This patient had highly refractory disease that was in the fourth relapse at the time of infusion and was not eligible for stem-cell transplantation because of coexisting conditions. In three patients, the loss of the expression of CD19 in leukemia cells resulted in a relapse; one of these patients (Patient 2) had received prior blinatumomab therapy. In these patients, CTL019 cells were not lost at the time of relapse.
Of the 27 patients who had a complete remission, 19 remained in remission: 15 patients received no further therapy, and 4 patients withdrew from the study to receive other therapy. In Patient 11, the myelodysplastic syndrome developed during ALL remission, and the patient withdrew from the study to receive other therapy. The median follow-up was 7 months (range, 1 to 24). No deaths were related to the study treatment. Seven patients died after disease progression or relapse, including 1 patient who died from the myelodysplastic syndrome. At 6 months, the event-free survival rate was 67% (95% confidence interval [CI], 51 to 88) and the overall survival rate was 78% (95% CI, 65 to 95) (Fig. 1A and 1B).
Figure 1. Probability of Event-free and Overall Survival at 6 Months.
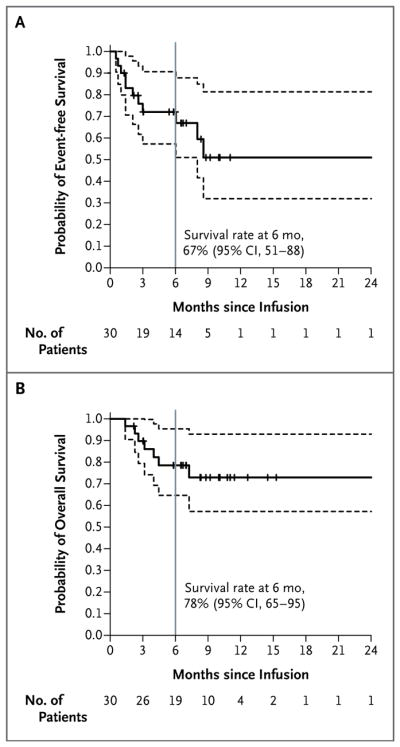
Panel A shows the time to an event after infusion of CTL019. Events were relapse (in seven patients), no response (in three patients), and the myelodysplastic syndrome (in one patient). Tick marks indicate the time of data censoring at the last follow-up or on the date of initiation of alternative therapy (in four patients). The curve in Panel B shows overall survival. Data were censored at the time of the last follow-up. In both panels, dashed lines represent 95% confidence intervals.
IN VIVO EXPANSION AND PERSISTENCE OF CTL019
The CTL019 cells were easily detectable by means of flow cytometry, thus reflecting high in vivo proliferation. In 27 patients who had a response, high peak proportions of CTL019-modified T cells were detected by this method (median, 39.8% of CTL019-positive cells in CD3-positive cells; range, 4.4 to 69.3), whereas in the 3 patients who did not have a response, 0.2%, 0.6%, and 8.2% of CD3-positive cells, respectively, were CTL019-positive at peak levels. CTL019 cells were detectable in the blood by means of flow cytometry for up to 11 months (Fig. 2A and 2B).
Figure 2. Persistence of CTL019.

Panel A shows the results of detection of CTL019-positive T cells detected by means of flow cytometry in peripheral-blood samples. “Confirmed negative” was defined as the first of two consecutive negative measurements (<0.1% CTL019-positive cells in CD3-positive cells). Patients 1 through 25 were participants in the pediatric trial (which included children and young adults 5 to 22 years of age), and Patients 26 through 30 were participants in the adult trial (which included patients 26 to 60 years of age). CTL019-modified T cells were also detected in the cerebrospinal fluid of 17 of 19 patients with specimens that could be evaluated. Panel B shows the Kaplan–Meier curve of the time to the first confirmed negative measurement in peripheral blood and bone marrow. Data were censored at the time of the last follow-up. Dashed lines represent 95% confidence intervals. Panel C shows measurements of CTL019 gene-modified T cells in peripheral blood as assessed by means of quantitative real-time polymerase-chain-reaction (PCR) assay. Genomic DNA was isolated from samples of whole blood obtained at serial time points before and after infusion of CTL019. The horizontal line at 5 copies per microgram of DNA represents the lower limit of quantification of this assay. Data on patients who did not have a response are shown in red. In general, the levels of CTL019 detected by means of quantitative PCR correlated well with the level of CTL019-positive cells detected by means of flow cytometry, with the exception of the levels in 3 patients who did not have a response and whose peak levels measured by means of quantitative PCR (6066, 5982, and 178,481 copies per microgram of genomic DNA, respectively) did not correspond with detection of CTL019 cells by means of flow cytometry or the induction of B-cell aplasia. CTL019 sequences were detected (23 copies of CTL019 cells per microgram of DNA) at month 24 by means of quantitative PCR in the 1 patient who remained in complete remission at the 2-year follow-up. Data at the first time point were obtained before infusion of CTL019 cells. Doses of cells were determined according to the total amount of cells available after manufacturing. The manufacturing goal of 1.5×107 to 5×109 total cells (3×105 to 1×108 cells per kilogram of body weight) was achieved in all treated patients (see Table S2 in the Supplementary Appendix). A split-dose strategy was used to determine safety with 0.1×108 to 1×108 cells per kilogram infused over 1 to 3 days (5×108 to 50×108 cells in patients who weighed 50 kg or more). The transduction efficiency ranged from 5.5 to 45.3%; this yielded a dose of 0.76 to 20.6×106 CTL019 cells per kilogram.
The probability of persistence of CTL019 at 6 months was 68% (95% CI, 50 to 92). CTL019 sequences remained detectable by means of quantitative polymerase-chain-reaction (PCR) assay in patients with sustained remissions until 2 years (Fig. 2C and data not shown). This assay showed very high levels of proliferation of CTL019 cells; all patients had peak levels greater than 5000 copies per microgram of genomic DNA, and 26 patients had peak levels greater than 15,000 copies per microgram of genomic DNA. One patient (Patient 17) received infusions again at 3 months and 6 months because of early loss of CTL019 cells with B-cell recovery, and this patient subsequently had persistence of CTL019. In the patient with the longest remission (2 years), B-cell aplasia (absence of CD19-positive cells) (Fig. 3) continued for a year after the loss of CTL019 cells detectable by flow cytometry, suggesting functional persistence of CTL019 cells below the limits of detection by flow cytometry, whereas CTL019 remained detectable by means of quantitative PCR. The probability of relapse-free B-cell aplasia at 6 months was 73% (95% CI, 57 to 94).
Figure 3. B-Cell Aplasia.
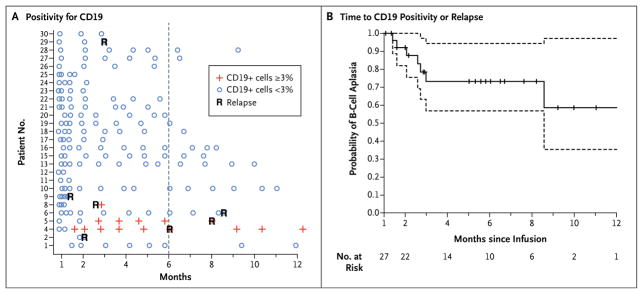
Panel A shows the results of testing to detect the percentage of CD19-positive lymphocytes in peripheral-blood samples by means of flow cytometry. Patients 1 through 25 were participants in the pediatric trial (which included children and young adults 5 to 22 years of age), and Patients 26 through 30 were participants in the adult trial (which included patients 26 to 60 years of age). Negative results were defined as less than 3% of lymphocytes that were positive for CD19. An outlier sample (Patient 16 at month 3) with 4% CD19-positive lymphocytes was discrepant with the measurements on clinical flow cytometry (<1% CD19-positive cells), bone marrow measurements at the same time point, and four subsequent monthly evaluations and was, therefore, considered to be negative. Panel B shows a Kaplan–Meier curve of the time to either CD19 positivity or relapse. Data on patients in remission were censored at the time of the last follow-up (indicated by tick marks). Dashed lines represent 95% confidence intervals. All patients required intravenous immunoglobulin replacement, and no serious infectious complications were observed as a result of B-cell aplasia; however, bronchitis (in one patient), acute otitis media (in two patients), salmonella infection (in one patient), and recurrent urinary tract infections (in one patient) were observed.
CTL019 FOR RELAPSE AFTER ALLOGENEIC STEM-CELL TRANSPLANTATION
In the 18 patients who were treated for relapse of disease after allogeneic stem-cell transplantation, the median donor chimerism at the time of leukapheresis was 100% (range, 68 to 100). No graft-versus-host disease was observed after infusion of CTL019. Event-free survival and overall survival did not differ significantly between the patients who had previously undergone stem-cell transplantation and those who had not undergone stem-cell transplantation (P = 0.21 for event-free survival and P = 0.24 for overall survival).
THERAPY AFTER ADMINISTRATION OF CTL019
Five patients withdrew from the study after the administration of CTL019 to receive other therapy; three of these patients underwent allogeneic stem-cell transplantation while their disease was in remission, and the disease remained in remission 7 to 12 months after the infusion of CTL019. Patient 12, who had undergone a previous stem-cell transplantation, had a post-transplantation relapse of T-cell ALL that aberrantly expressed CD19, was refractory to two intensive reinduction regimens, and entered a morphologic remission after the infusion of CTL019, but the patient had minimal residual disease (0.09%). She subsequently received bortezomib and an infusion of donor lymphocytes, and the disease remained in remission without minimal residual disease at 11 months. In Patient 11, the myelodysplastic syndrome developed and led to overt acute myeloid leukemia with a monosomy 8 clone that also shared cytogenetic features with the original B-cell ALL.
CYTOKINE-RELEASE SYNDROME
A major toxic effect associated with CTL019 is the cytokine-release syndrome, a systemic inflammatory response that is produced by elevated levels of cytokines; these elevations are associated with T-cell activation and proliferation. The cytokine-release syndrome ranges from mild and self-limiting, with high temperatures and myalgias, to severe and life-threatening, with a clinical course that also includes vascular leak, hypotension, respiratory and renal insufficiency, cytopenias, and coagulopathy. Several aspects of the cytokine-release syndrome mirror those of the macrophage activation syndrome.8 All the patients in our studies had the cytokine-release syndrome, which was mild to moderate in 22 of the 30 patients. These patients required hospitalization for febrile neutropenia and received broad-spectrum antibiotics and pain medications. Severe cytokine-release syndrome, which required intensive care with varying degrees of respiratory support (from placement of a nasal cannula to mechanical ventilation), developed in 8 patients (27%), and all these patients required vasopressor support for hypotension. Coagulopathy, with elevated prothrombin and partial-thromboplastin times as well as severe hypofibrinogenemia, was observed in patients who had severe cytokine-release syndrome, although clinical bleeding was rare (observed in 3 patients).
Severe cytokine-release syndrome started a median of 1 day after infusion, whereas cytokine-release syndrome that was not severe started a median of 4 days after infusion (P=0.005). Laboratory markers of systemic inflammation, including C-reactive protein and ferritin levels, were elevated in all the patients. Patients who had severe cytokine-release syndrome had higher peak levels of interleukin-6 than did patients who did not have severe cytokine-release syndrome (P<0.001) (Fig. 4A); they also had higher peak levels of C-reactive protein (P = 0.02), ferritin (P = 0.005), interferon-γ (P<0.001), and soluble interleukin-2 receptor (P<0.001) (Fig. S2 in the Supplementary Appendix). The baseline disease burden (the percentage of blast cells in bone marrow before infusion) correlated with the severity of the cytokine-release syndrome; a higher disease burden was significantly associated with severe cytokine-release syndrome (P = 0.002) (Fig. 4B). Patients with severe cytokine-release syndrome also had higher levels of CTL019-positive CD8 cells (P = 0.012) and CTL019-positive CD3 cells (P=0.026).
Figure 4. Correlates of the Cytokine-Release Syndrome.
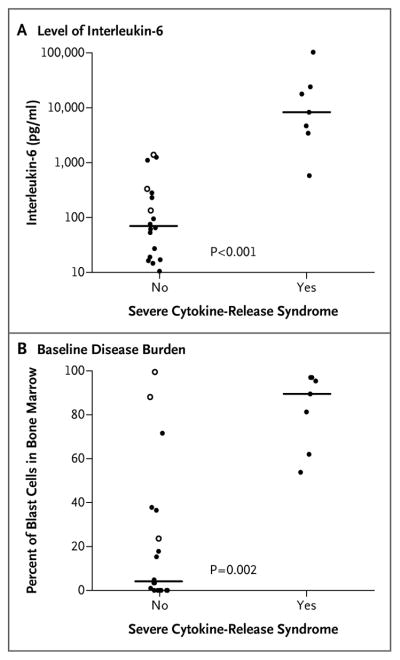
Panel A shows peak levels of interleukin-6 in the first 28 days after infusion of CTL019 cells in patients with severe cytokine-release syndrome as compared with patients with cytokine-release syndrome that was not severe. Severe cytokine-release syndrome was defined as hypotension requiring the use of two or more vasopressors or respiratory failure requiring mechanical ventilation. Panel B shows the severity of cytokine-release syndrome according to the baseline disease burden in bone marrow after chemotherapy and before infusion (in the pediatric trial only). Solid circles indicate complete remission, open circles no response, and horizontal lines medians.
We previously found marked elevation of interleukin-6 levels after CTL019 therapy and rapid reversal of severe cytokine-release syndrome with the interleukin-6–receptor blocking antibody tocilizumab8; therefore, tocilizumab was incorporated into the management of severe cytokine-release syndrome in this study. Nine patients received tocilizumab, which resulted in rapid defervescence and stabilization of blood pressure, with improvement (weaning from vasopressor support) over a period of 1 to 3 days. Six patients also received short courses of glucocorticoids, and four patients received a second dose of tocilizumab for recrudescence of the cytokine-release syndrome after transient improvement with the first dose. All the patients recovered fully, and there was a complete reversal of symptoms and a normalization of laboratory results. Relapses occurred in two of the nine patients who received immunosuppressive therapy for the cytokine-release syndrome.
ENCEPHALOPATHY
Thirteen patients had neurologic toxic effects, which ranged from delirium during the period of high temperatures to global encephalopathy with one or more of the following: aphasia, confusion, delirium, and hallucinations. Six patients had delayed encephalopathy that occurred after high temperatures had resolved and was independent of the severity of the cytokine-release syndrome and whether the patient had received prior tocilizumab therapy. Symptoms were self-limiting (lasting 2 to 3 days and resolving over 2 to 3 days), and they resolved fully without further intervention or apparent long-term sequelae. One patient with encephalopathy had two seizures that may have been caused by concomitant electrolyte abnormalities. Several patients had normal computed tomographic or magnetic resonance imaging of the head and lumbar puncture that was negative for infection or leukemia.
B-CELL APLASIA
We performed flow cytometry to detect CD19-positive B cells in order to monitor patients for the development of B-cell aplasia, which can be used as a pharmacodynamic measure of CTL019 function (Fig. 3). B-cell aplasia occurred in all the patients who had a response and persisted for up to 1 year after CTL019 cells were no longer detectable by means of flow cytometry. Patients with B-cell aplasia received immunoglobulin replacement to maintain IgG levels greater than 500 mg per deciliter.
DISCUSSION
The engineering of T lymphocytes to express chimeric antibodies that target tumor antigens has been studied for more than 20 years.10,11 Clinical progress had been limited by poor in vivo expansion of engineered T cells and failure of these cells to persist after infusion.12–16 We and others have documented a high level of proliferation, activity against bulk disease, and long-term persistence of chimeric antigen receptor T cells.4–8,17,18
In this study, we found a 90% rate of complete remission among 30 children and adults who received CTL019 for ALL that was relapsed or refractory. With a follow-up period of 2 to 24 months, sustained remissions were observed in 19 patients (15 of whom received no further therapy) and were associated with the persistence of CTL019 and B-cell aplasia that continued beyond 2 to 3 months, suggesting continued effector function. Three patients who were in remission subsequently underwent allogeneic stem-cell transplantation. However, for several reasons (e.g., the lack of a suitable donor, prior stem-cell transplantation, or family choice), most patients did not undergo stem-cell transplantation; this allowed for longer follow-up. The rate of event-free survival at the median follow-up of 6 months was 67% in this heavily pretreated population. Several patients with relapse received salvage therapy, and the rate of overall survival at 6 months was 78%. In comparison, with the use of clofarabine, nelarabine, and liposomal-encapsulated vincristine — the most recently approved drugs for relapsed ALL19–21 — the Food and Drug Administration label indicates rates of complete remission of less than 25%, with a median documented duration of response of 4 to 9 weeks.
Davila et al. recently reported results from an expanded cohort of 16 adult patients with B-cell ALL.22 The short-term results were similar to those in our study; the overall complete remission rate was 88% with the use of a CD19 chimeric antigen receptor with the CD28 costimulatory domain. However, the only long-term remissions that were reported occurred in patients who crossed over to allogeneic stem-cell transplantation. The median duration of persistence of 19–28z chimeric antigen receptor T cells was 30 days (range, 0 to 120). As a bridge to transplantation, high levels of proliferation with a short duration of persistence is sufficient to achieve remission and eligibility for stem-cell transplantation. However, in patients who are ineligible for stem-cell transplantation, short persistence of chimeric antigen receptor T cells is unlikely to produce long-term remission. We observed prolonged persistence of CTL019 cells and B-cell aplasia for as long as 2 years in this cohort and for more than 3 years in patients with CLL.4
There was no discernible effect of the density of CD19 antigen or cell dose on either efficacy or toxicity (Table S2 in the Supplementary Appendix), although this study was not powered to investigate these correlations. Both complete remissions and severe cytokine-release syndrome were observed at the lower doses of cells administered. Although there was generally good concordance of CTL019 proliferation as measured by means of flow cytometry and quantitative PCR, in the cases of the three patients who did not have a response, the degree of proliferation of CLT019 cells determined by means of quantitative PCR was matched neither by the detection of cells expressing surface chimeric antigen receptor nor by the induction of B-cell aplasia. The mechanisms underlying this observation, including possible gene silencing of CTL019, are unknown. B-cell aplasia can be used as a pharmacodynamic measure of the persistence of functional CD19-targeted T cells. Continued B-cell aplasia was seen in all patients who had a sustained remission, and none of the patients with B-cell aplasia had a CD19-positive relapse.
Relapses were associated with either lack of CTL019 persistence or CD19 escape variants. In two cases of loss of CTL019, relapse was heralded by recovery of normal B cells, suggesting that monitoring for the return of normal B cells will be important to identify patients at the highest risk for relapse and potentially provide a window for repeat treatment or stem-cell transplantation. Selection of CD19-negative clones or down-regulation of CD19 expression are concerns associated with CD19-directed therapies. Data are lacking to determine the mechanism of CD19-negative relapses.
The most prominent and serious toxic effect of chimeric antigen receptor T-cell therapy is the cytokine-release syndrome, an inflammatory process marked by dramatic elevations of cytokine levels.23 Because this cytokine release probably both results from and contributes to T-cell activation, some degree of cytokine-release syndrome is probably necessary for efficacy. In the majority of our patients, the cytokine-release syndrome was self-limited, with high temperatures and myalgias that resolved spontaneously in a few days. However, in approximately 30% of the patients in our study, hypotension and respiratory insufficiency associated with this syndrome required treatment in the intensive care unit. The cytokine-release syndrome has been described by several groups in association with CD19 chimeric antigen receptor T-cell therapy,8,22 but until recently, management was nonspecific, glucocorticoid-based, and directed primarily toward supportive care. In severely affected patients, glucocorticoids may have a limited effect on toxicity but a substantial negative effect on T-cell proliferation and persistence.4,7,8,22
We previously observed marked elevation of interleukin-6 levels in a patient with severe cytokine-release syndrome after infusion of CTL019, and our study showed the benefit of cytokine-directed therapy.8 Tocilizumab, an interleukin-6–receptor blocking antibody, produced a rapid and profound improvement in severe manifestations of the cytokine-release syndrome without an apparent impact on the efficacy of CTL019. We have now incorporated tocilizumab as a first-line agent in the management of severe cytokine-release syndrome. Tocilizumab could blunt maximal T-cell proliferation by interfering with a cytokine feedback loop; therefore, we have limited its use to patients with grade 4, or life-threatening, toxic effects. Despite this theoretical concern, in this study, all the patients who received tocilizumab had robust proliferation of CTL019 cells, a complete remission, and the continued persistence of CTL019 cells. Future studies are needed to determine whether tocilizumab can be used preemptively without compromising efficacy.
We observed that many findings in patients with the cytokine-release syndrome were similar to those in patients with the macrophage activation syndrome or hemophagocytic lymphohistiocytosis.8,24,25 Hyperferritinemia is a hallmark of these two conditions, and we observed marked elevations in ferritin levels during the period of maximal T-cell proliferation. The degree of elevation in ferritin and C-reactive protein levels was significantly higher in patients with severe cytokine-release syndrome.
Neurologic toxic effects were seen in a subgroup of patients. Encephalopathy typically occurred after symptoms of the cytokine-release syndrome peaked and was self-limited. CNS toxic effects have been associated with other T-cell–activating therapies, including other chimeric antigen receptor–modified T-cell therapies and blinatumomab.22,26,27 The pathophysiology of encephalopathy after the use of CTL019 is not known. The mechanism could involve direct T-cell–mediated toxicity or it could be cytokine-mediated. We have observed no clear predictors of neurologic toxic effects. CTL019-modified T cells were detected in the cerebrospinal fluid of 17 of 19 patients with specimens that could be evaluated. This may have implications for successful surveillance of CNS disease, yet only a fraction of the patients had encephalopathy. The 2 patients (of the 25 patients in the pediatric trial) in whom cerebrospinal fluid leukemic blasts were detected (CNS-2 disease) did not have encephalopathy. Neurologic toxic effects did not obviously correlate with the severity of the cytokine-release syndrome and was not prevented by tocilizumab. Further studies are needed to determine the potential predictors, mechanism, and treatment of neurologic toxicity.
Relapsed ALL is a considerable therapeutic challenge, particularly in patients who do not have a second complete remission or have a relapse after stem-cell transplantation.1–3,28,29 Salvage therapy induces remissions in only 40% of children who have had two or more bone marrow relapses, and long-term survival is quite poor.3 This study included patients with multiple relapses or with disease that was refractory to two or more attempts at reinduction, and 18 patients (60%) had had a relapse after allogeneic stem-cell transplantation. Two complete remissions were achieved in 3 patients with disease that was refractory to blinatumomab, a promising CD19-directed bispecific antibody for relapsed ALL,30 suggesting that a lack of response to CD19-directed therapy may not preclude successful therapy with CTL019. The complete remission rate of 90% and sustained remissions of up to 2 years that were seen in this study are encouraging.
Supplementary Material
Acknowledgments
Supported in part by a grant from Novartis (to Dr. June), by grants from the National Institutes of Health (1R01CA165206, to Dr. June; and R01CA102646 and R01CA116660, to Dr. Grupp), the Leukemia and Lymphoma Society, the Jeffrey Jay Weinberg Memorial Foundation, and the Children’s Hospital of Philadelphia Hematologic Malignancy Research Fund, a Stand Up to Cancer–St. Baldrick’s Pediatric Dream Team translational research grant (SU2C-AACR-DT1113), a St. Baldrick’s Foundation Scholar Award (to Dr. Maude), and a Research Scholar Grant from the American Cancer Society (RSG-14-022-01-CDD, to Dr. Teachey).
We thank Jeff Finklestein, Farzana Nazimuddin, Harit Parakandi, and Marina Bogush for sample processing and flow-cytometric analysis; Irina Kulikovskaya and Minnal Gupta for the quantitative polymerase-chain-reaction assay; Fang Chen for the Luminex assay; Marybeth Helfrich, Gerald Wertheim, and Michele Paessler for assistance with interpretation of the marrow findings; Alexey Bersenev, Anne Lamontagne, Alexander Malykhin, Neel Manvar, Matthew O’Rourke, Megan Suhoski-Davis, and members of the Clinical Cell and Vaccine Production Facility for cell manufacturing and testing; Colleen Callahan, Christine Strait, Brittany Girard, Margaret Tartaglione, Trish Hankins, Brandon Loudon, Christopher Grupp, Maya Mudambi, Laura Motley, Elizabeth Veloso, Lester Lledo, Joan Gilmore, Holly McConville, Christina Jemison, and James Capobianchi for clinical research assistance; Wei-Ting Hwang for statistical support; Michele Sharr and Patricia Wood at Novartis for discussions and protocol support; Children’s Hospital of Philadelphia Vector Core (Frasier Wright and Katherine High) for clinical-grade vector production; Kenneth Cornetta and the National Gene Vector Biorepository at the Indiana University School of Medicine for postinfusion testing of the product for replication-competent lentivirus; Bipulendu Jena and Laurence Cooper for provision of the chimeric antigen receptor anti-idiotype detection reagent; the data and safety monitoring board; our hematology–oncology and critical care faculty for providing clinical support; and the nurses, residents, and fellows at our two institutions.
Footnotes
Disclosure forms provided by the authors are available with the full text of this article at NEJM.org.
References
- 1.Bhojwani D, Pui C-H. Relapsed childhood acute lymphoblastic leukaemia. Lancet Oncol. 2013;14(6):e205–e217. doi: 10.1016/S1470-2045(12)70580-6. [DOI] [PubMed] [Google Scholar]
- 2.Forman SJ, Rowe JM. The myth of the second remission of acute leukemia in the adult. Blood. 2013;121:1077–82. doi: 10.1182/blood-2012-08-234492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Raetz EA, Bhatla T. Where do we stand in the treatment of relapsed acute lymphoblastic leukemia? Hematology Am Soc Hematol Educ Program. 2012;2012:129–36. doi: 10.1182/asheducation-2012.1.129. [DOI] [PubMed] [Google Scholar]
- 4.Kalos M, Levine BL, Porter DL, et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med. 2011;3:95ra73. doi: 10.1126/scitranslmed.3002842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;365:725–33. doi: 10.1056/NEJMoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kochenderfer JN, Wilson WH, Janik JE, et al. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood. 2010;116:4099–102. doi: 10.1182/blood-2010-04-281931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brentjens RJ, Davila ML, Riviere I, et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Science Transl Med. 2013;5:177ra38. doi: 10.1126/scitranslmed.3005930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grupp SA, Kalos M, Barrett D, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. 2013;368:1509–18. doi: 10.1056/NEJMoa1215134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Milone MC, Fish JD, Carpenito C, et al. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol Ther. 2009;17:1453–64. doi: 10.1038/mt.2009.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Irving BA, Weiss A. The cytoplasmic domain of the T cell receptor zeta chain is sufficient to couple to receptor-associated signal transduction pathways. Cell. 1991;64:891–901. doi: 10.1016/0092-8674(91)90314-o. [DOI] [PubMed] [Google Scholar]
- 11.Eshhar Z, Waks T, Gross G, Schindler DG. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc Natl Acad Sci U S A. 1993;90:720–4. doi: 10.1073/pnas.90.2.720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kalos M, June CH. Adoptive T cell transfer for cancer immunotherapy in the era of synthetic biology. Immunity. 2013;39:49–60. doi: 10.1016/j.immuni.2013.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kershaw MH, Westwood JA, Parker LL, et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin Cancer Res. 2006;12:6106–15. doi: 10.1158/1078-0432.CCR-06-1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lamers CH, Sleijfer S, Vulto AG, et al. Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: first clinical experience. J Clin Oncol. 2006;24(13):e20–e22. doi: 10.1200/JCO.2006.05.9964. [DOI] [PubMed] [Google Scholar]
- 15.Till BG, Jensen MC, Wang J, et al. Adoptive immunotherapy for indolent non-Hodgkin lymphoma and mantle cell lymphoma using genetically modified autologous CD20-specific T cells. Blood. 2008;112:2261–71. doi: 10.1182/blood-2007-12-128843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Savoldo B, Ramos CA, Liu E, et al. CD28 costimulation improves expansion and persistence of chimeric antigen receptor-modified T cells in lymphoma patients. J Clin Invest. 2011;121:1822–6. doi: 10.1172/JCI46110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brentjens RJ, Rivière I, Park JH, et al. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood. 2011;118:4817–28. doi: 10.1182/blood-2011-04-348540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kochenderfer JN, Dudley ME, Carpenter RO, et al. Donor-derived CD19-targeted T cells cause regression of malignancy persisting after allogeneic hematopoietic stem cell transplantation. Blood. 2013;122:4129–39. doi: 10.1182/blood-2013-08-519413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.O’Brien S, Schiller G, Lister J, et al. High-dose vincristine sulfate liposome injection for advanced, relapsed, and refractory adult Philadelphia chromosome-negative acute lymphoblastic leukemia. J Clin Oncol. 2013;31:676–83. doi: 10.1200/JCO.2012.46.2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jeha S, Gandhi V, Chan KW, et al. Clofarabine, a novel nucleoside analog, is active in pediatric patients with advanced leukemia. Blood. 2004;103:784–9. doi: 10.1182/blood-2003-06-2122. [DOI] [PubMed] [Google Scholar]
- 21.Berg SL, Blaney SM, Devidas M, et al. Phase II study of nelarabine (compound 506U78) in children and young adults with refractory T-cell malignancies: a report from the Children’s Oncology Group. J Clin Oncol. 2005;23:3376–82. doi: 10.1200/JCO.2005.03.426. [DOI] [PubMed] [Google Scholar]
- 22.Davila ML, Riviere I, Wang X, et al. Efficacy and toxicity management of 19-.28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Science Transl Med. 2014;6:224ra25. doi: 10.1126/scitranslmed.3008226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Maude SL, Barrett D, Teachey Grupp SA. Managing cytokine release syndrome associated with novel T cell-engaging therapies. Cancer J. 2014;20:119–22. doi: 10.1097/PPO.0000000000000035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Teachey DT, Rheingold SR, Maude, et al. Cytokine release syndrome after blinatumomab treatment related to abnormal macrophage activation and ameliorated with cytokine-directed therapy. Blood. 2013;121:5154–7. doi: 10.1182/blood-2013-02-485623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Risma K, Jordan MB. Hemophagocytic lymphohistiocytosis: updates and evolving concepts. Curr Opin Pediatr. 2012;24:9–15. doi: 10.1097/MOP.0b013e32834ec9c1. [DOI] [PubMed] [Google Scholar]
- 26.Schlegel P, Lang P, Zugmaier G, et al. Pediatric posttransplant relapsed/refractory B-precursor acute lymphoblastic leukemia shows durable remission by therapy with the T-cell engaging bispecific antibody blinatumomab. Haematologica. 2014;99:1212–9. doi: 10.3324/haematol.2013.100073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Topp MS, Kufer P, Gökbuget N, et al. Targeted therapy with the T-cell-engaging antibody blinatumomab of chemotherapy-refractory minimal residual disease in B-lineage acute lymphoblastic leukemia patients results in high response rate and prolonged leukemia-free survival. J Clin Oncol. 2011;29:2493–8. doi: 10.1200/JCO.2010.32.7270. [DOI] [PubMed] [Google Scholar]
- 28.Nguyen K, Devidas M, Cheng SC, et al. Factors influencing survival after relapse from acute lymphoblastic leukemia: a Children’s Oncology Group study. Leukemia. 2008;22:2142–50. doi: 10.1038/leu.2008.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Poon LM, Hamdi A, Saliba R, et al. Outcomes of adults with acute lymphoblastic leukemia relapsing after allogeneic hematopoietic stem cell transplantation. Biol Blood Marrow Transplant. 2013;19:1059–64. doi: 10.1016/j.bbmt.2013.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Topp MS, Gökbuget N, Zugmaier G, et al. Long-term follow-up of hematologic relapse-free survival in a phase 2 study of blinatumomab in patients with MRD in B-lineage ALL. Blood. 2012;120:5185–7. doi: 10.1182/blood-2012-07-441030. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.