


Abstract
Efficient transcription of the HIV-1 genome is regulated by Tat, which recruits P-TEFb from the 7SK small nuclear ribonucleoprotein (snRNP) and other nucleoplasmic complexes to phosphorylate RNA polymerase II and other factors associated with the transcription complex. Although Tat activity is dependent on its binding to the viral TAR sequence, little is known about the cellular factors that might also assemble onto this region of the viral transcript. Here, we report that the splicing factor SRSF1 (SF2/ASF) and Tat recognize overlapping sequences within TAR and the 7SK RNA. SRSF1 expression can inhibit Tat transactivation by directly competing for its binding to TAR. Additionally, we provide evidence that SRSF1 can increase the basal level of viral transcription in the absence of Tat. We propose that SRSF1 activates transcription in the early stages of viral infection by recruiting P-TEFb to TAR from the 7SK snRNP. Whereas in the later stages, Tat substitutes for SRSF1 by promoting release of the stalled polymerase and more efficient transcriptional elongation.
INTRODUCTION
After integration into the host genome, the HIV-1 provirus is transcribed into a single pre-mRNA from a promoter located within the 5′ long terminal repeat (LTR) of the viral genome. Binding of cellular factors, including NF-kB, Sp1 and the TATA box binding protein, to the viral promoter stimulates the RNA polymerase II (RNAPII) complex to assemble and initiate transcription (1). However, in the absence of the viral factor Tat, RNAPII processivity is dramatically reduced, resulting in transcription pausing and the prevalent synthesis of short viral transcripts (2).
Tat binds to an RNA stem-loop structure, called TAR, located at the 5′ end of the nascent viral transcripts. Tat's binding triggers efficient elongation of the viral transcripts via the recruitment of the P-TEFb complex, which is composed of cellular cyclin T1 (CycT1) and the cyclin-dependent kinase 9 (CDK9). P-TEFb activates viral transcription through several mechanisms: (i) it triggers the release of the paused transcription-elongation complexes by phosphorylating components of the Negative Elongation Factor (NELF), composed of fours subunits (NELF-A, -B, C or -D and -E), and 5,6-Dichloro-1-β-D-ribofuranosylbenzimidazole (DRB) sensitivity inducing factor (DSIF), composed of hSPT4 and hSTP5 (3), which assembles onto the transcription complex (4); (ii) it phosphorylates the C-terminal domain (CTD) of RNAPII to increase the polymerase processivity (5) and (iii) it stimulates the assembly of new transcription complexes by directing the recruitment of TATA box binding protein to the LTR promoter (6). Tat's role in viral transcription is not limited to the recruitment of P-TEFb; it also promotes the recruitment of chromatin-modifying enzymes with histone acetyl transferase (HAT) activity, which modify chromatin conformation, relieving the repression exerted on the LTR promoter by nucleosomes (7). Additionally, the phosphorylation of transcription factors, including Sp1, CREB, the alpha subunit of eukaryotic initiation factor 2 (eIF2a) and NF-kB, has also been reported to be triggered by Tat and increases viral transcription (8).
The cellular machineries regulating the transcription and processing of eukaryotic RNAs are intimately coupled. Trans-acting factors required for capping, splicing and polyadenylation are found within the RNAPII complex, modulate the processing of the nascent pre-mRNA and, in some cases, processing feeds back to regulate the activity of the transcription complex (9,10).
Our understanding of the association between viral transcription and RNA processing is still in its infancy. Nevertheless, several factors associate with both the Tat-dependent transcription complex and components of the cellular splicing machinery. TATSF1, TCERG1 and SKIP modulate transcription and interact with spliceosomal components (11–13). The Tat-dependent transcription complex modulates splicing through multiple mechanisms (14,15) and conversely, splicing factors can regulate viral transcription (16).
In the past two decades, biochemical and cell-based approaches have been utilized to isolate the components of the Tat-dependent transcription complex and to modulate its activity. Studies have been mostly aimed at the characterization of known regulators of the RNAPII complex but little effort has been devoted to characterize the role played by RNA-processing factors. Given the emerging role of RNA-processing factors in the modulation of the RNAPII transcription complex (17), we sought to determine if cellular factors that regulate mRNA processing, export and stability could also modulate Tat transactivation of viral transcription. In this work, we utilized a HIV-1-derived minigene to screen an RNA-binding protein (RBP) expression library for activity in Tat transactivation. We identified a series of cellular RBPs that can significantly regulate viral transactivation. In addition, we addressed the mechanism by which one of those proteins, SRSF1, inhibits Tat activity.
MATERIALS AND METHODS
Plasmids and cells
The pLTR-Xm-LR reporter construct was obtained by cloning the HCV internal ribosomal entry site (IRES) (provided by Dr. T. Tellinghuisen, SCRIPPS Florida) and the modified luciferase gene Ppy R8 (provided by Dr. B.R. Branchini, Connecticut College) downstream the Rev responsive element (RRE) sequence in the pLTR-S1Xm-R (15). pLTR-XTm-LR was obtained by deleting TAR from pLTR-Xm-LR. pLTR-Luc was previously described (19). The SRSF1 deletion mutants have been previously described (18), the enhanced green fluorescent protein (EGFP)-tagged Tat expression plasmid (pTat-GFP) was obtained by cloning the Tat coding sequence in pEGFP-N1 (Clontech). RBPs expression plasmids were obtained from various sources (Supplementary Table S1). HEK293 were transfected in 24-well plates, for RNA extraction, and 96-well plates, for luciferase assays, with Lipofectamine 2000 (Invitrogen), a carrier plasmid was added to specific reaction to normalize the amount of total DNA added. Anti SRSF1 siRNAs (Qiagen, Cat # SI02655086, SI02655093) and control siRNA (cat. # 1022076) were transfected at a final concentration of 40 pM. A Rev expression vector (pCMV-Rev) was co-transfected with the reporter pLTR-Xm-LR to optimize the expression of unspliced mRNAs. A minimum of three independent assays were carried out for each experiment. Each luciferase assay was performed in triplicates giving a minimum of nine data points. The Transactivation Change (TrC) measures the change in transactivation efficiency and is defined as log2[(LTBP/LBP)/(LTE/LE)]. LTBP: luciferase expression in cells transfected with a given RBP in the presence of Tat. LBP: luciferase expression in cells transfected with a given RBP in the absence of Tat. LTE: luciferase expression values in cells transfected with the EGFP control in the presence of Tat. LE: luciferase expression values in cells transfected with the EGFP control in the absence of Tat. Samples were analyzed utilizing a BMG FluoStar Omega reader and the BMG MARS software. Data are represented as means ± SEM. Cellular viability was measured utilizing the CellTiterGlo (Promega) adenosine triphosphate (ATP) production assay according to the manufacturer's instructions. RNA isolation and quantitative polymerase chain reaction (qPCR) assays were performed as indicated in the supplemental experimental procedures. HLM1 cells (obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH: HLM30 Cells from Dr. Reza Sadaie) were transfected by electroporation utilizing a GenePulserII (BioRad).
RNA isolation and transcripts quantification
Total RNA was extracted 48 after transfection, unless differently indicated, with the Total RNA Isolation Kit (Agilent) and DNase treated with Turbo DNase (Ambion). RNA was reverse transcribed utilizing a random pd(N)6 primer and Superscript II RT (LifeTechnologies). qPCR analysis of pLTR-Xm-LR was performed with primers RRE1 (TTGAGGCGCAACAGCATCTG) and RRE2 (TCCAAGGCACAGCAGTGGTG) and as previously described for the pMtat(-) viral clone (19). Each sample was normalized for the relative expression of the housekeeping gene glyceraldehyde 3-phosphate dehydrogenase (GAPDH). qPCR was performed utilizing a Stratagene Mx3005P system and analyzed with MxPro V3.0 software. Data are represented as means ± SEM.
RNA-affinity chromatography (RAC) and purified proteins
Substrate RNAs were synthesized in vitro using T7 RNAP and DNA oligonucleotide templates containing the sequences depicted in Figures 4 and 6. The RNA was bound to adipic acid-agarose beads as previously described (20) and incubated in a reaction mixture containing 400 μl of buffer B (20 mM HEPES-KOH pH 7.9, 5% Glycerol, 0.1 M KCl, 0.2 mM ethylenediaminetetraacetic acid, 0.5 mM DTT, 4 mM ATP, 4 mM MgCl2), SRSF1 purified from HeLa cells as previously described (21) and recombinant Tat (NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH: HIV-1 Tat protein). The proteins specifically bound to the immobilized RNA were eluted, separated on polyacrylamide sodium dodecyl sulphate gels and probed with antibodies anti-SRSF1 (provided by Dr. A. R. Krainer, Cold Spring Harbor Laboratories) and Tat (obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH: Antiserum to HIV-1 Tat from Dr. Bryan Cullen).
Figure 4.
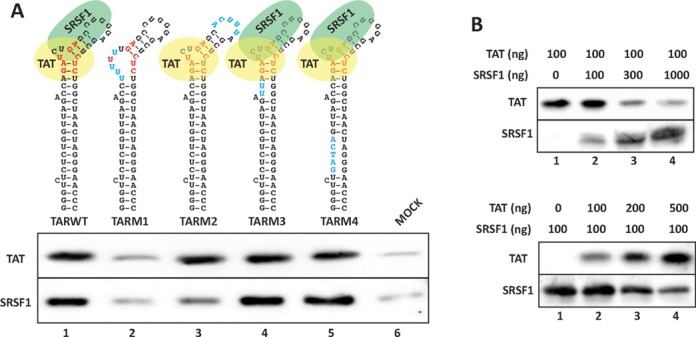
SRSF1 and Tat bind overlapping sequences within TAR. (A) RAC assays were set up with bait RNAs containing the wild-type (TARWT) and mutated (TARM) TAR sequences. The RNA substrates were incubated with 100 ng of recombinant Tat or purified SRSF1 in separate reactions. (B) Tat and SRSF1 compete for binding onto TAR. RAC assays were set up with the wild-type TAR sequence as bait, and either 100 ng of recombinant Tat and increasing amounts of purified SRSF1 (upper panel) or increasing amounts of Tat and 100 ng of SRSF1 (lower panel).
Figure 6.
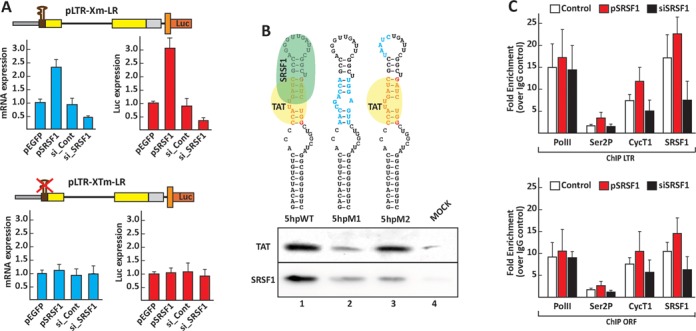
SRSF1 binds to the 7SK RNA and activates transcription of the viral promoter. (A) SRSF1 transcription activation is dependent on TAR. HEK293 cells were transfected with the reporter constructs pLTR-Xm-LR and pLTR-XTm-LR, which carries a deletion of the TAR sequence, the control pEGFP, the SRSF1 expression constructs, control or SRSF1-specific siRNAs I the absence of Tat. Luciferase activity and mRNA expression were assayed 48 h after transfection. (B) SRSF1 and Tat bind overlapping sequences within the 5′ hairpin of the 7SK RNA. RAC assays were set up with bait RNAs containing the wild-type (5hpWT) and mutated (5hpM) sequences of the 5′ hairpin of the 7SK RNA. The RNA substrates were then incubated with 100 ng of recombinant Tat or purified SRSF1 in separate reactions. (C) HEK293 cells were transfected with the viral clone pMtat(–), a control plasmid, the SRSF1 expression clone or SRSF1 siRNA. ChIP assays were performed 48 h after transfection, with antibodies for the indicated proteins and the GFP-tagged Tat. Values represent the relative enrichment in ChIP signal to the TAR and ORF relative to the control IgG, and are the average of three independent PCRs from two independent ChIP experiments.
Chromatin immunoprecipitation (ChIP) assay
HEK293 cells transfected with the indicated plasmids. At 48 h post-transfection cells were cross-linked in 1% formaldehyde and ChIP was performed as previously described (22) utilizing the following antibodies RNAPII (Millipore, clone CTD4H8), RNAPII Phospho Ser2 (AbCam, #ab24758), GFP (Santa Cruz Biotechnology, #sc-9996), SRSF1, Cyclin T1 (Santa Cruz Biotechnology, sc-10750) or normal rabbit immunoglobulin G (IgG) (Cell Signaling Technologies, #2729). The green fluorescent protein (GFP) antibody was utilized to precipitate the product of the pTat-GFP construct. The data collected were derived from three independent experiments and quantified by qPCR assays carried out with primer pairs specific for the LTR promoter (TAR_Fb: GGAACCCACTGCTTAAGCCT; TAR_Rb: GGATCTCTAGTTACCAGAGT) and the open reading frame (ORF) (RF21.21: TTCTTCAGAGCAGACCAGAGC; RF20.22: GCTGCCAAAGAGTGATCTGA) utilizing a Stratagene Mx3005P and analyzed with MxPro V3.0 software. The data sets were normalized to input values and results expressed as fold enrichment over the IgG control. Data are represented as means ± SEM.
RESULTS
RBPs modulate Tat transactivation
To study the effects of cellular RBPs on Tat-dependent transactivation, we screened a human RBP cDNA expression library utilizing an HIV-1-derived reporter minigene. Minimal constructs containing a reporter gene driven by the viral promoter have been widely utilized for the past two decades. Nevertheless, given the role played in transcription regulation by factors and mechanisms regulating different aspects of RNA biogenesis, the activity of some cellular factors differs in reporter constructs containing a minimal LTR sequence and lacking downstream sequences that regulate the processing and stability of the viral mRNA (15,23).
In previous work, we showed that an HIV-1-derived minigene containing the LTR promoter, downstream sequences with several splicing regulatory sequences and the RRE, which allows the stabilization and efficient export of unspliced mRNAs, can reliably reproduce the regulation of viral transcription, splicing and nuclear export (15,24). A luciferase gene was inserted at the 3′ end of the minigene under the control of an IRES to facilitate the analysis of the viral promoter activity in a multiple-well plate format (Figure 1A). Tat expression activated transcription of the pLTR-XmLR reporter up to 60-fold in a dose-dependent manner, which was quantified by both luciferase activity and qPCR with comparable results (Figure 1B). Although, it is not well understood why expression of higher amounts of Tat cause a decrease in transactivation it is plausible that at higher concentration this factor might bind and sequester components of the transcription complex.
Figure 1.
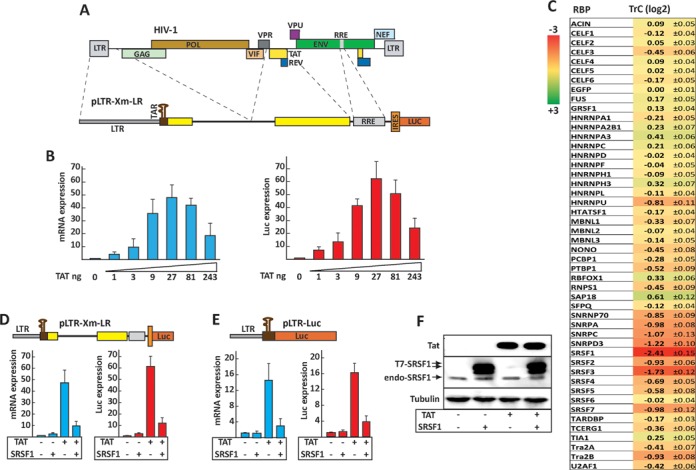
Screening of the RBP expression library reveals novel cellular factors regulating Tat transactivation. (A) Schematic map of the pLTR-Xm-LR reporter minigene. Dashed lines indicate the viral sequences utilized to construct the minigenes in relation to the HIV-1 genome (top). (B) Transactivation of the pLTR-Xm-LR reporter. HEK293 cells were transfected with the pLTR-Xm-LR reporter, a Rev expression vector and increasing amounts of Tat expression vector. Expression of the reporter mRNA and gene product were assayed by qPCR (left panel) and an enzymatic luciferase assay (right panel). (C) Screening of the RBP expression library. HEK293 cells were transfected with each RBP and the control EGFP expression clones in the presence or absence of Tat. The TrC measures the change in transactivation efficiency and is defined as log2[(LTBP/LBP)/(LTE/LE)], where LTBP and LBP are luciferase expression values in cells transfected with a given RBP in the presence or absence of Tat, respectively, whereas LTE and LE are luciferase expression values in cells transfected with the EGFP control in the presence or absence of Tat, respectively. SRSF1 inhibits Tat-dependent transactivation of the HIV-1 promoter. HEK293 cells were transfected with either pLTR-Xm-LR (D) or pLTR-Luc (E) and the expression clones for Tat and SRSF1, as indicated. (F) Tat and SRSF1 expression. The SRSF1 expressed from the pSRSF1 vector is tagged with a T7 epitope (T7-SRSF1) and migrates slower than the endogenous protein (endo-SRSF1).
HEK293 cells were co-transfected with the pLTR-XmLR reporter and each RBP expressing clone in the absence or presence of the Tat coding plasmid. The fold change in Tat activation caused by the RBP versus the control was defined as the TrC (Figure 1C). A minimum of nine independent experimental data points were obtained for each assay, and TrC|1| with P < 0.05 were considered significant (Figure 1C). Among the RBPs screened, none increased overall transactivation by over 2-fold and only one increased it by more than 50% (SAP18). Five among the RBPs that caused the strongest decrease in transactivation (SRSF1, SRSF2, SRSF3, SRSF7 and TRA2β) belong to the SR protein or SR-like families of splicing regulators. Members of the SR family regulate the assembly of the splicing machinery (25), integrate multiple steps in RNA metabolism (26) and have been recently shown to modulate RNAPII activity (27,28).
The two strongest repressors of Tat activity (SRSF1 and SRSF3) decreased Tat activation from 62- to 12-fold (TrC-2.4) and 19-fold (TrC-1.7), respectively. SRSF1 (formerly SF2/ASF) and SRSF3 (formerly SRp20) share a high degree of homology and act by binding specific RNA sequences and activating splicing to nearby splice sites (29). Both proteins associate with the RNAPII complex (30) and have been shown to enhance co-transcriptional pre-mRNA processing by facilitating the recruitment of the spliceosome on nascent transcripts (31,32), associate with interphase chromatin (33) and regulate translation (34).
SRSF1 inhibition of Tat transactivation is dependent on the LTR promoter
Having established that SRSF1 acts as a strong downregulator of Tat transactivation and given the growing evidence for reverse coupling mechanisms between the splicing and transcription machineries, we sought to determine if SRSF1 binding to sequences downstream from the LTR promoter or its interaction with components of the splicing complex is required for the inhibition of transactivation. To this end, we deleted the viral sequences between TAR and the intronless Luciferase gene. Tat expression increased luciferase expression of the LTR-Luc reporter by roughly 17-fold, whereas SRSF1 expression reduced it to 4-fold (Figure 1E); this was comparable to the reduction observed utilizing the reporter pLTR-Xm-LR (Figure 1D) (TrC-2.1 versus −2.6). These data suggest that viral sequences downstream TAR are required for optimal transactivation (pLTR-Luc 17-fold versus pLTR-Xm-LR 62-fold), thus validating the use of a complex reporter minigene. Nevertheless, SRSF1 activity is solely dependent on the viral promoter sequences.
We also observed that Tat expression levels were not altered upon SRSF1 overexpression (Figure 1F). Furthermore, the decrease in transactivation was not due to a change in the basal level of transcription or translation, as evidenced by similar mRNA and luciferase expression levels in both reporter constructs tested. Taken together, these results demonstrate that SRSF1 inhibits transactivation by interfering with the Tat-dependent transcription complex.
Although, expression of cellular factors outside physiological ranges might trigger changes in cellular viability and other unwanted secondary effects we observed that the marked increase in SRSF1 (over 10-fold) expression (Figure 1F) is consistent with physiological changes in this protein levels in different tissues and under different physiological conditions (35). Furthermore, expression of SRSF1 did not significantly alter cell viability in our experimental system (Supplementary Figure S1).
Tat function is directly affected by SRSF1 expression
Although SRSF1 interacts with the RNAPolII complex, it is also an important splicing regulator. As such, it is plausible that SRSF1 may alter the processing of one of the components of the Tat-dependent transcriptional unit, instead of directly interfering with Tat's functions. If SRSF1 exerts its function through a secondary target, there should be a time lag between its overexpression and the suppression of Tat activity. To test this hypothesis, we set up a time course and monitored expression of Tat, SRSF1 and the reporter pLTR-Xm-LR (Figure 2). A clear increase in the mRNA expressed from the LTR promoter was observed as early as 12 h from transfection of the Tat-coding plasmids (Figure 2A). Simultaneously, we also detected inhibition of transactivation, which peaked 24 h after transfection. Similar results, although delayed by roughly 6 h, were observed by measuring luciferase activity (Figure 2B). The appearance of Tat and SRSF1 in the transduced cells correlates with transactivation and its inhibition (Figure 2C). The absence of a time lag between SRSF1 expression and the inhibition of transactivation suggest that SRSF1 affects Tat-transactivation directly, instead of altering the expression of secondary factors, which should require a longer time between SRSF1 expression and inhibition of Tat activity.
Figure 2.
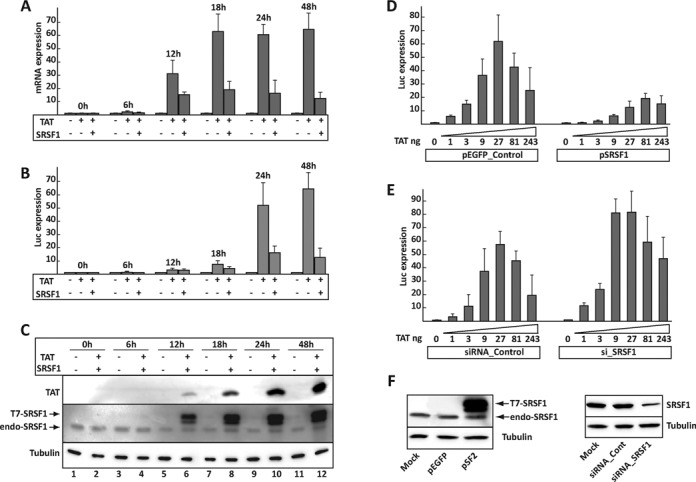
Tat transactivation is a primary target for SRSF1. (A) HEK293 cells were co-transfected with the pLTR-Xm-LR reporter, Tat alone or in combination with SRSF1. Cells were harvested during a 48-h time course. Transcripts generated by the reporter construct were quantified by qPCR. (B) Luciferase activity of the cells transfected during the time course. (C) Tat and SRSF1 expression during the time course; expression of SRSF1 does not interfere with expression of Tat (Figure 1F). Dose-response curves in HEK293 cells transfected with the pLTR-Xm-LR reporter, increasing amounts of the Tat expression construct and either a control (pEGFP), the SRSF1 expression construct or control (D) or SRSF1-specific siRNAs (E). (F) SRSF1 expression following transfection of the SRSF1 expression clone (left panel) or the anti SRSF1 siRNAs (right panel).
Since Tat's transcriptional effects are dose-dependent, we speculated that if SRSF1 directly inhibits Tat activity, the optimal amount of Tat required for efficient transactivation would be either increased or decreased in response to a higher or lower intracellular concentration of SRSF1. To address this hypothesis, we generated a Tat dose–response curve in cells in which SRSF1 was either up- or downregulated (Figure 2D and E). While optimal transactivation of 62-fold was achieved by transfection of 27 ng of the Tat expression plasmid, SRSF1 overexpression reduced it to 12-fold and required 81 ng of Tat expression plasmid. Accordingly, SRSF1 downregulation increased optimal transactivation to over 80-fold and required only 9 ng of the Tat expression plasmid. Taken together, these results indicate that Tat transcriptional activity is directly dependent on the relative intracellular SRSF1 concentration.
The SR2SF1 RNA recognition motifs (RRMs) are required and sufficient to inhibit Tat transactivation
Structurally, SRSF1 is composed of two RRM RNA-binding domains, which interact with specific RNA sequences, and an arginine/serine-rich (RS) carboxy-terminal domain, which functions as a protein-interaction domains. The RS domain contributes to the splicing function of SRSF1, but does not appear to affect the RNA binding specificity of the protein (29). We set out to determine what domains are required for SRSF1 inhibition of Tat transactivation, utilizing a series of deletion clones (Figure 3). Deletion of the RS domain indicated that SRSF1 activity was solely dependent on the RRMs. We observed that the clone carrying the deletion of the RS domain inhibited transactivation more efficiently than the wild-type protein (from 62- to 7-fold versus 12-fold). This may be due to the role played by the RS domain in promoting the association of SRSF1 with nuclear speckles and spliceosomal complexes (18). The protein carrying the RS domain deletion is unlikely to be sequestered within these nucleoplasmic complexes, and may be more available to be recruited to the Tat-dependent transcription complex.
Figure 3.

The SRSF1 RRM2 is required and sufficient to abolish Tat transactivation. (A) Schematic representation of the SRSF1 deletion clones. (B) Luciferase activity in HEK293 cells transfected with the pLTR-Xm-LR reporter, the Tat expression vector, the control pEGFP and the SRSF1 expression constructs. (C) Relative expression level of the T7-tagged SRSF1 deletion clones.
Although both RRMs contribute to the inhibition of transactivation, RRM2 alone is sufficient to repress Tat transactivation (from 62- to 25-fold), whereas expression of RRM1 alone did not induce significant changes. The SRSF1 RRM2 has been described as a pseudo-RRM, because it lacks the set of conserved aromatic residues usually involved in RNA binding found in canonical RRMs (36), but it has been shown to autonomously bind to RNA sequences characterized by the GGA motif and regulate the alternative splicing of some cellular pre-mRNAs by competing with other splicing factors for the binding to specific regulatory RNA sequences (37). These and our present findings suggest a mechanism by which Tat and SRSF1 compete for overlapping sequences within TAR.
SRSF1 and Tat compete for binding to overlapping sequences within TAR
Binding of Tat to a highly conserved bulge in the TAR hairpin is the initial step in the series of events that terminate with the increase in RNAPolII processivity and efficient elongation. To determine whether SRSF1 and Tat compete for overlapping binding sites on TAR, we utilized an RAC assay we had previously optimized to isolate proteins that bind to short RNA sequences under native conditions (20). Short RNAs containing the wild-type or mutated TAR sequence were covalently linked to agarose beads and incubated with recombinant Tat and SRSF1 purified from HeLa cells. Proteins bound to the bead-conjugated RNA were isolated and analyzed (Figure 4). Although this technique is often used to determine the complete assortment of weak and transient RNA–protein interactions within cell nuclear extracts, here we utilized purified proteins to avoid possible bridging effects caused by other components of the Tat-TAR complex. The binding profiles of SRSF1 and Tat indicated that both proteins bind the wild-type TAR sequence. Furthermore, whereas mutations affecting the bulge sequence, which is required for the recruitment of Tat, also abolished binding of SRSF1 (Figure 4A, cf. TARWT and TARM1), mutation of the apical loop disrupted binding of SRSF1 but not Tat (TARM2) indicating that the sequences recognized by the two proteins overlap but are not identical. Other control mutations within the TAR stem (TARM3, TARM4) did not interfere with the binding of either protein. A separate set of binding assays utilizing nuclear extracts confirmed these results (data not shown). The sequence recognized by SRSF1 contains the GGA motif, which is recognized by the SRSF1 RRM2 (37). Nevertheless, it does not overlap with a putative SRSF1 binding sites within the stem as predicted by the ESEfinder 3.0 (Supplementary Figure S2A) (38), a SR protein functional binding site prediction matrix. This could be due to the algorithm's inability to properly predict binding to a highly structured sequence, such as TAR.
Next, we wished to confirm that Tat and SRSF1 compete for binding to the same sequence within the apical region of TAR. We set up a RAC assay utilizing the wild-type TAR sequence and either an SRSF1 dose–response curve with a constant amount of Tat, or a Tat dose–response curve with a constant amount of SRSF1. Increasing the amount of SRSF1 in the reaction resulted in a progressive decrease in the amount of bound Tat recovered (Figure 4B, top), whereas increasing the amount of Tat reduced the amount of bound SRSF1 (Figure 4B, bottom). This result confirmed the hypothesis that Tat and SRSF1 compete for binding to overlapping sequences within TAR, indicating that SRSF1 downregulates transactivation by inhibiting Tat binding to TAR.
SRSF1 regulates transactivation and associates with the promoter in the context of the full-length viral genome
We established above that SRSF1 inhibits Tat activity and assembly onto TAR utilizing reporter minigenes and biochemical assays. Next, we set out to study SRSF1 association with the active RNAPII complex in the context of the full-length virus. Previous studies showed that SRSF1 associates with the RNAPII complex in an RNA-dependent manner (39) and is a key component of the promoter associated 7SK particle (27). To determine the contribution of Tat and SRSF1 to the formation of the RNAPII transcription complex, we examined Tat, SRSF1, RNAPII and CyclinT1 occupancy at the HIV-1 promoter and ORF by ChIP. HEK293 cells were transiently transfected with the proviral clone pMtat(–), which contains the full-length viral genome, but does not express Tat (40), in the presence or absence of the Tat and SRSF1 expression vectors. The ChIP signal for Tat showed a high signal-to-noise ratio at the promoter (Figure 5B) and the downstream ORF (Figure 5C), which is consistent with previous reports showing that Tat travels with the elongating RNAPII (41,42). RNAPII occupancy at the promoter increased by a factor of 2 in the presence of Tat, which is also consistent with previous data (42). Tat expression increased recruitment of the CycT1 component of P-TEFb to the promoter by 5- to 6-fold and triggered a marked increase in the presence of the elongating form of RNAPII (10-fold), as detected with antibodies to the phosphorylated Ser2 of the RNAPII CTD domain (Ser2P). SRSF1 was found associated at the promoter with great efficiency in mock-transfected cells. Following Tat expression, SRSF1 occupancy was reduced by a factor of ∼35. Conversely, overexpression of SRSF1 reduced Tat's presence at the promoter by a factor of 5, and consistent with this result, RNAPII, Ser2P and CycT1 were also reduced, whereas SRSF1 occupancy at the promoter was partially restored. These results are in agreement with the above RAC data (Figure 4) and indicate functional and spatial competition between Tat and SRSF1.
Figure 5.
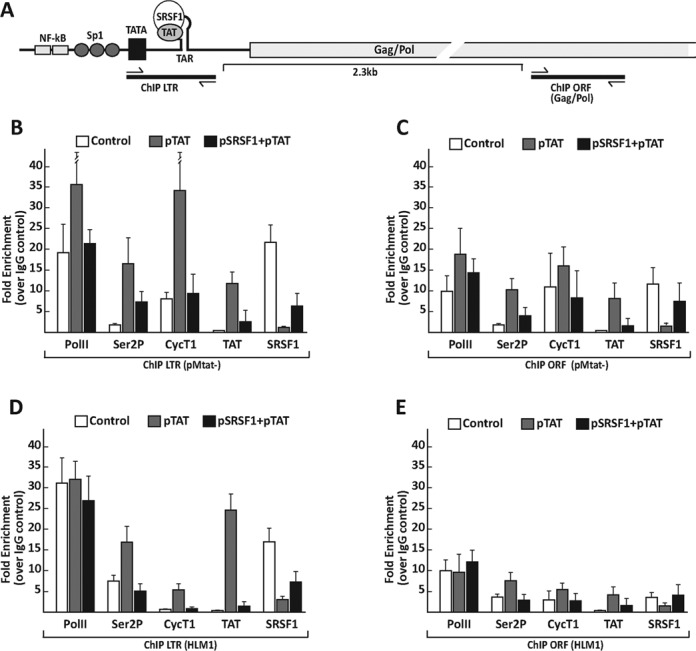
ChIP analysis of transcription and splicing factors at the HIV-1 promoter of transiently transfected and integrated proviruses. (A) The schematic diagram shows the HIV-1 promoter and downstream sequences, indicating the location of the primer sets used for ChIP of the TAR and ORF. (B and C) HEK293 cells were transfected with the viral clone pMtat(–), a control plasmid, the Tat expression vector alone or in combination with SRSF1. (D and E) HLM1 cells were transfected with a control plasmid, the Tat expression vector alone or in combination with SRSF1. ChIP assays were performed 48 h after transfection, with antibodies for the indicated proteins and the GFP-tagged Tat. Values represent the relative enrichment in ChIP signal to the TAR (B and D) and ORF (C and E) relative to the control IgG, and are the average of three independent PCRs from two independent ChIP experiments.
Next, we performed a ChIP assay in HLM1 cells, a stable cell line carrying a single copy of the integrated pMtat(–) (Figure 5D and E). The results confirmed the data obtained with the transient transfection of pMtat(–) with a more pronounced reduction of Tat occupancy at the promoter upon SRSF1 overexpression, which could be caused by the diverse stoichiometry of the SRSF1, Tat and TAR ratios in cells carrying a single copy of the viral genome versus the transfection assays, which delivers multiple copies per cell.
The interplay between Tat and SRSF1 was also observed by analysis of the factors associated with the downstream Gag-Pol ORF (Figure 5C), although the enrichment for most factors associated with the transcription complex was substantially reduced when compared to the promoter.
Tat and SRSF1 bind overlapping sequences in the 7SK RNA
Having confirmed the association of SRSF1 with the active transcription complex, we asked whether its role in viral transcription might extend beyond inhibiting Tat activity. The association of SRSF1 with the RNAPII transcription complex has been recently studied by Ji et al. (27). SRSF1 was found to be a component of the 7SK snRNP particle, a nuclear ribonucleoparticle containing the 7SK RNA that recruits and inhibits the activity of P-TEFb through the action of the RBPs HEXIM1 and HEXIM2 (43,44). In a subset of cellular promoters SRSF1 mediates the release and activation of P-TEFb from the 7SK particle and this process appears to require the presence of an SRSF1-binding sequence within the nascent transcript, in proximity to the transcription start site (TSS). Tat has been shown to mobilize the inactive 7SK-bound P-TEFb through a similar mechanism (42), directly binding the 7SK RNA and facilitating the release of P-TEFb from the 7SK snRNP and its transfer to TAR (45–47).
Given SRSF1 and Tat parallel activities in promoting the mobilization of P-TEFb from the 7SK snRNP we sought to determine if SRSF1 might promote viral transcription in the absence of Tat. To this end, we compared the basal transcription level of the viral promoter following overexpression or downregulation of SRSF1. The pLTR-Xm-LR reporter was transfected into HEK293 cells in combination with the SRSF1 expression vector, a control vector, an anti-SRSF1 siRNA or a control siRNA (Figure 6A). SRSF1 overexpression increased the basal transcription level by over 2-fold, whereas its downregulation decreased transcription to roughly one-third of the control. Mutation of the TAR sequence abolished the transcriptional activity of SRSF1. Our data are consistent with a model for SRSF1 transcriptional activation that depends upon the transfer of P-TEFb from the 7SK particle to the active RNAPII complex (27) and requires the presence of an SRSF1-binding site in the synthesized transcript, in proximity to the TSS.
Although Tat binds at the 5′-hairpin of the 7SK RNA and displaces HEXIM1, which otherwise sequesters P-TEFb into the transcriptionally inactive 7SK/HEXIM/P-TEFb snRNP (45), little is known about the mechanism by which SRSF1 promotes P-TEFb release from the 7SK particle. The 7SK RNA 5′-hairpin and the TAR sequences share remarkable structural similarities (cf. Figures 4A and 6B). As the Tat and SRSF1 binding sequences within TAR appear to partially overlap, we explored the possibility that the same arrangement of binding sequences for the two proteins is present within the 7SK RNA 5′-hairpin. We set up a RAC assay similar to the one we utilized to determine the SRSF1-binding site within TAR (Figure 6). Short RNAs containing the wild-type or mutated 7SK RNA 5′-hairpin sequence were covalently linked to agarose beads and incubated with recombinant Tat and purified SRSF1 proteins (Figure 6B). The binding profiles of SRSF1 and Tat on the wild-type sequence showed that both proteins bind the 5′-hairpin. Mutation of the region bound by Tat (45) reduced the binding of both Tat and SRSF1, whereas mutation within the apical loop of the 5′-hairpin reduced SRSF1 binding but not Tat binding. The sequence recognized by SRSF1 within the 7SK RNA 5′-hairpin co-localizes with a SRSF1 binding site predicted by the ESEfinder 3.0 (Supplementary Figure S2B).
Association of SRSF1 with the promoter and activation of the RNAPolII complex were confirmed carrying out a ChIP assay. Overexpression of SRSF1 induced an increased occupancy at the promoter of the elongating hyperphosphorylated form of RNAPII and CycT1 while downregulation of SRSF1 had the opposite effect on the assembly of the transcription complex (Figure 6C).
These results complement previous findings (27,42) and indicate that both, Tat and SRSF1, activate basal viral transcription through a mechanism that requires the recruitment of the 7SK particle to the RNAPII initiation complex and the presence of a Tat/SRSF1 binding site within the TAR sequence.
DISCUSSION
The mechanism controlling HIV-1 transcription has been widely studied to gain insights into the regulation of transcription of eukaryotic genes and to develop novel therapeutic targets to inhibit viral replication. Proteins that regulate cellular and viral RNA biogenesis are an integral component of a transcription-RNA processing super-complex with the ability to modulate the activity of the RNAPII holoenzyme. In this work, we demonstrate that SRSF1, an ubiquitously expressed cellular splicing factor, and the viral transactivator Tat recognize overlapping sequences within the TAR and 7SK RNA and play parallel roles in the activation of the viral promoter, although they do so with markedly different efficiencies.
The results we presented, together with data recently obtained by other groups (27,42), suggest that in the early stages of the viral infection, when little or no Tat has been synthesized, SRSF1 can enter the transcription complex at the promoter as part of the 7SK snRNP (Figure 7A). RNAPII starts transcription and clears the promoter, but the cellular factors DSIF and NELF cooperatively arrest transcription soon after the synthesis of TAR. Next, SRSF1 is transferred from the 7SK RNA to a binding site within the nascent TAR. This causes the release of the inactive P-TEFb from the 7SK particle and its repositioning onto TAR. At the same time the 7SK particle is ejected from the transcription complex. As a result, a transcriptionally active SRSF1:P-TEFb:TAR complex phosphorylates the RNAPII CTD on Ser2, DSIF and NELF, resulting in the transition from RNAPII pausing to elongation.
Figure 7.
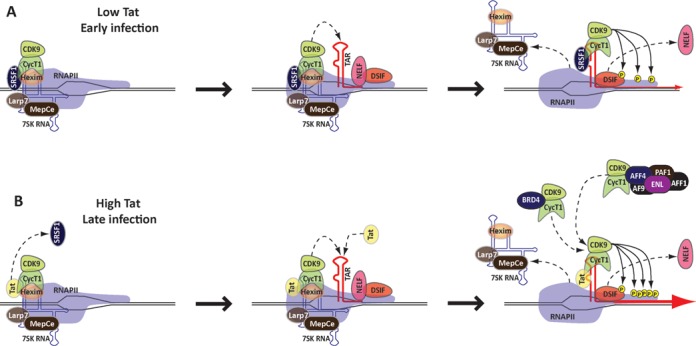
SRSF1 regulates HIV-1 transcription. (A) In the early stage of viral infection, SRSF1 associates with gene promoters, as part of the 7SK snRNP. Synthesis of TAR triggers the switch of SRSF1 from the 7SK RNA to the viral transcript, and the simultaneous release of P-TEFb from the 7SK complex and its recruitment onto the nascent RNA. The activated P-TEFb phosphorylates RNAPII and the components of the pausing complex NELF and DSIF, which result in transcription-pause release. (B) During the later stages of the viral infection, Tat is present at higher concentrations and is found in a complex with P-TEFb within the 7SK particle. Tat binding to TAR induces relocation of P-TEFb from the 7SK complex and other nucleoplasmic complexes to the nascent transcript within the paused RNAPII complex. This triggers activation of the P-TEFb kinase and hyper-phosphorylation of the RNAPII CTD and the NELF/DSIF complex. The difference in the efficiency of transactivation between Tat and SRSF1 might be due to the ability of Tat to recruit P-TEFb from nucleoplasmic complexes other than 7SK (dashed arrows).
In the later stages of the viral infection, when Tat is readily available, the viral protein enters the pre-initiation complex (PIC) as part of the 7SK particle (Figure 7B). Tat displaces Hexim1 from the inactive P-TEFb complex (46,47) possibly by simultaneously binding to a stem-loop near the 5′ end of the 7SK RNA (45), and interacting with the CycT1 component of P-TEFb. Once RNAPII clears the promoter, TAR captures Tat and P-TEFb resulting in the ejection of the 7SK snRNP components and productive transcription elongation by activating the CDK9 kinase component of P-TEFb, which leads to RNAPII CTD phosphorylation.
This interpretation of our findings is consistent with the strong increase in SRSF1 mRNA expression we observed in CD4+ T cells following activation with anti-CD3/28 antibodies and IL2 (5- to 8-fold) (Supplementary Figure S3), and might contribute to the mechanisms that regulate the activation of viral replication in latency. Future studies are warranted to determine the precise mechanism by which SRSF1 dislodges P-TEFb from the 7SK snRNP and positions it onto the active transcription complex. Does SRSF1 utilize a mechanism similar to Tat to displace the inhibitory factor HEXIM from its position within the 7SK snRNP and form a complex with P-TEFb, or does SRSF1 utilize a different strategy to mobilize P-TEFb from its HEXIM1 bound state?
Although SRSF1 and Tat can release the paused RNAPII complex through seemingly parallel mechanisms, it is unclear why the amplitude of transcription activation is drastically different between the two factors (3- versus 60-fold). It is plausible that Tat may have a higher affinity for sequences on the 7SK RNA and TAR; alternatively, it may be more efficient in displacing HEXIM1 or in better positioning P-TEFb within the active transcription complex. The extent of the contribution of P-TEFb captured from the 7SK snRNP to the overall Tat-dependent transactivation is unclear. It is plausible that Tat might be more efficient than SRSF1 in recruiting P-TEFb from cellular complexes other than the 7SK, such as the Brd4:P-TEFb (48) and the super elongation complex (49,50), which contains the cofactors ELL2, AFF4, ENL and AF9, or other pools of soluble P-TEFb. Furthermore, Tat promotes the recruitment of HAT, which acetylates histones to increase the accessibility of chromatin to the transcription complex (7) and triggers the phosphorylation of several transcription factors (8); these are functions that SRSF1 presumably lacks.
In previous work, we showed that Tat regulates viral splicing by recruiting SRSF1 to a downstream splicing enhancer through the Tat-associated factors Tat-SF1 and TCERG1 (15). Thus, although Tat and SRSF1 compete for binding onto TAR and the 7SK RNA, Tat appears to mediate the recruitment of this splicing factor to the nascent transcript. It is unclear if SRSF1 is transferred to the Tat:Tat-SF1:TCERG1 complex from the 7SK snRNP, or if it is recruited ex novo from a different snRNP complex or an unbound soluble pool of the protein. Future studies will be required to determine if SRSF1 is transferred to different compartments within the transcription complex and associates with different factors following PIC formation, initiation and elongation.
Our findings have implications for the development of therapeutics with novel mechanisms of action. SRSF1 can directly compete with Tat for its binding onto TAR, and expression of deletion mutants containing both RRMs or RRM2 alone can downregulate Tat activity with efficiency similar to the full-length protein. The possibility to downregulate Tat activity utilizing a single RNA-binding domain has potential for the development of novel therapeutic approaches utilizing truncated proteins or small compounds that mimic the key structural features of the SRSF1 RRMs.
We have shown that other members of the SR protein family or a related protein (SRSF2, SRSF3, SRSF7 and TRA2β) can also significantly downregulate Tat activity, although with lower efficiency than SRSF1. This is consistent with previous findings showing that members of the SR protein family share structural and functional similarities (29). Furthermore, since many of these splicing factors have been shown to bind regulatory elements throughout the transcript it is plausible that a larger regulatory complex might provide a feedback mechanism and modulate both transcription and processing of the viral messengers.
Given the presence of roughly 500 RBP coding genes in the human genome, our analysis with a small-scale, RBP expression library suggests that several other RBPs could similarly regulate Tat activity. Thus, the overall expression level of multiple RBPs might modulate viral replication in specific cell types and under different physiological conditions, contributing to the mechanisms underlying viral latency and activation.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We would like to thank Dr. Heiner Schaal, Emanuele Buratti, Thomas Cooper, Javier Caceres and Kristen Lynch for providing many of the clones utilized in the RBP expression library. We would also like to thank Dr. Mike Lu for his helpful suggestions and support. The BMG FluoStar Omega reader was acquired with funds provided from the Charles E. Schmidt Foundation.
FUNDING
NIH NIAID [R15AI093229 to M.C.]. Funding for open access charge: Personal funds [M.C.] and Department of Biomedical Science, FAU.
Conflict of interest statement. None declared.
REFERENCES
- 1.Karn J., Stoltzfus C.M. Transcriptional and posttranscriptional regulation of HIV-1 gene expression. Cold Spring Harb. Persp. Med. 2012;2:a006916. doi: 10.1101/cshperspect.a006916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kao S.Y., Calman A.F., Luciw P.A., Peterlin B.M. Anti-termination of transcription within the long terminal repeat of HIV-1 by tat gene product. Nature. 1987;330:489–493. doi: 10.1038/330489a0. [DOI] [PubMed] [Google Scholar]
- 3.Fujinaga K., Irwin D., Huang Y., Taube R., Kurosu T., Peterlin B.M. Dynamics of human immunodeficiency virus transcription: P-TEFb phosphorylates RD and dissociates negative effectors from the transactivation response element. Mol. Cell. Biol. 2004;24:787–795. doi: 10.1128/MCB.24.2.787-795.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wada T., Takagi T., Yamaguchi Y., Watanabe D., Handa H. Evidence that P-TEFb alleviates the negative effect of DSIF on RNA polymerase II-dependent transcription in vitro. EMBO J. 1998;17:7395–7403. doi: 10.1093/emboj/17.24.7395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Parada C.A., Roeder R.G. Enhanced processivity of RNA polymerase II triggered by Tat-induced phosphorylation of its carboxy-terminal domain. Nature. 1996;384:375–378. doi: 10.1038/384375a0. [DOI] [PubMed] [Google Scholar]
- 6.Raha T., Cheng S.W., Green M.R. HIV-1 Tat stimulates transcription complex assembly through recruitment of TBP in the absence of TAFs. PLoS Biol. 2005;3:e44. doi: 10.1371/journal.pbio.0030044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Easley R., Van Duyne R., Coley W., Guendel I., Dadgar S., Kehn-Hall K., Kashanchi F. Chromatin dynamics associated with HIV-1 Tat-activated transcription. Biochim. et Biophys. Acta. 2010;1799:275–285. doi: 10.1016/j.bbagrm.2009.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Romani B., Engelbrecht S., Glashoff R.H. Functions of Tat: the versatile protein of human immunodeficiency virus type 1. J. Gen. Virol. 2010;91:1–12. doi: 10.1099/vir.0.016303-0. [DOI] [PubMed] [Google Scholar]
- 9.Fong N., Bentley D.L. Capping, splicing, and 3′ processing are independently stimulated by RNA polymerase II: different functions for different segments of the CTD. Genes Dev. 2001;15:1783–1795. doi: 10.1101/gad.889101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pandit R.A., Svasti S., Sripichai O., Munkongdee T., Triwitayakorn K., Winichagoon P., Fucharoen S., Peerapittayamongkol C. Association of SNP in exon 1 of HBS1L with hemoglobin F level in beta0-thalassemia/hemoglobin E. Int. J. Hematol. 2008;88:357–361. doi: 10.1007/s12185-008-0167-3. [DOI] [PubMed] [Google Scholar]
- 11.Bres V., Gomes N., Pickle L., Jones K.A. A human splicing factor, SKIP, associates with P-TEFb and enhances transcription elongation by HIV-1 Tat. Genes Dev. 2005;19:1211–1226. doi: 10.1101/gad.1291705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carty S.M., Goldstrohm A.C., Sune C., Garcia-Blanco M.A., Greenleaf A.L. Protein-interaction modules that organize nuclear function: FF domains of CA150 bind the phosphoCTD of RNA polymerase II; Proc. Natl. Acad. Sci. U.S.A.; 2000. pp. 9015–9020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fong Y.W., Zhou Q. Stimulatory effect of splicing factors on transcriptional elongation. Nature. 2001;414:929–933. doi: 10.1038/414929a. [DOI] [PubMed] [Google Scholar]
- 14.Berro R., Kehn K., de la Fuente C., Pumfery A., Adair R., Wade J., Colberg-Poley A.M., Hiscott J., Kashanchi F. Acetylated Tat regulates human immunodeficiency virus type 1 splicing through its interaction with the splicing regulator p32. J. Virol. 2006;80:3189–3204. doi: 10.1128/JVI.80.7.3189-3204.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jablonski J.A., Amelio A.L., Giacca M., Caputi M. The transcriptional transactivator Tat selectively regulates viral splicing. Nucleic Acids Res. 2010;38:1249–1260. doi: 10.1093/nar/gkp1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Alexander M.R., Wheatley A.K., Center R.J., Purcell D.F. Efficient transcription through an intron requires the binding of an Sm-type U1 snRNP with intact stem loop II to the splice donor. Nucleic Aacids Res. 2010;38:3041–3053. doi: 10.1093/nar/gkp1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Braunschweig U., Gueroussov S., Plocik A.M., Graveley B.R., Blencowe B.J. Dynamic integration of splicing within gene regulatory pathways. Cell. 2013;152:1252–1269. doi: 10.1016/j.cell.2013.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Caceres J.F., Misteli T., Screaton G.R., Spector D.L., Krainer A.R. Role of the modular domains of SR proteins in subnuclear localization and alternative splicing specificity. J. Cell Biol. 1997;138:225–238. doi: 10.1083/jcb.138.2.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jablonski J.A., Caputi M. Role of cellular RNA processing factors in human immunodeficiency virus type 1 mRNA metabolism, replication, and infectivity. J. Virol. 2009;83:981–992. doi: 10.1128/JVI.01801-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Caputi M., Mayeda A., Krainer A.R., Zahler A.M. hnRNP A/B proteins are required for inhibition of HIV-1 pre-mRNA splicing. EMBO J. 1999;18:4060–4067. doi: 10.1093/emboj/18.14.4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zahler A.M. Purification of SR protein splicing factors. Methods Mol. Biol. 1999;118:419–432. doi: 10.1385/1-59259-676-2:419. [DOI] [PubMed] [Google Scholar]
- 22.Johnson D.S., Mortazavi A., Myers R.M., Wold B. Genome-wide mapping of in vivo protein-DNA interactions. Science. 2007;316:1497–1502. doi: 10.1126/science.1141319. [DOI] [PubMed] [Google Scholar]
- 23.Coiras M., Montes M., Montanuy I., Lopez-Huertas M.R., Mateos E., Le Sommer C., Garcia-Blanco M.A., Hernandez-Munain C., Alcami J., Sune C. Transcription elongation regulator 1 (TCERG1) regulates competent RNA polymerase II-mediated elongation of HIV-1 transcription and facilitates efficient viral replication. Retrovirology. 2012;10:124–141. doi: 10.1186/1742-4690-10-124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jean-Philippe J., Paz S., Lu M.L., Caputi M. A truncated hnRNP A1 isoform, lacking the RGG-box RNA binding domain, can efficiently regulate HIV-1 splicing and replication. BBA-Gene Regul. Mech. 2014;1839:251–258. doi: 10.1016/j.bbagrm.2014.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lin S., Fu X.D. SR proteins and related factors in alternative splicing. Adv. Exp. Med. Biol. 2007;623:107–122. doi: 10.1007/978-0-387-77374-2_7. [DOI] [PubMed] [Google Scholar]
- 26.Zhong X.Y., Wang P., Han J., Rosenfeld M.G., Fu X.D. SR proteins in vertical integration of gene expression from transcription to RNA processing to translation. Mol. Cell. 2009;35:1–10. doi: 10.1016/j.molcel.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ji X., Zhou Y., Pandit S., Huang J., Li H., Lin C.Y., Xiao R., Burge C.B., Fu X.D. SR proteins collaborate with 7SK and promoter-associated nascent RNA to release paused polymerase. Cell. 2013;153:855–868. doi: 10.1016/j.cell.2013.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lin S., Coutinho-Mansfield G., Wang D., Pandit S., Fu X.D. The splicing factor SC35 has an active role in transcriptional elongation. Nat. Struct. Mol. Biol. 2008;15:819–826. doi: 10.1038/nsmb.1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Long J.C., Caceres J.F. The SR protein family of splicing factors: master regulators of gene expression. Biochem. J. 2009;417:15–27. doi: 10.1042/BJ20081501. [DOI] [PubMed] [Google Scholar]
- 30.Kim E., Du L., Bregman D.B., Warren S.L. Splicing factors associate with hyperphosphorylated RNA polymerase II in the absence of pre-mRNA. J. Cell Biol. 1997;136:19–28. doi: 10.1083/jcb.136.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Das R., Yu J., Zhang Z., Gygi M.P., Krainer A.R., Gygi S.P., Reed R. SR proteins function in coupling RNAP II transcription to pre-mRNA splicing. Mol. Cell. 2007;26:867–881. doi: 10.1016/j.molcel.2007.05.036. [DOI] [PubMed] [Google Scholar]
- 32.de la Mata M., Kornblihtt A.R. RNA polymerase II C-terminal domain mediates regulation of alternative splicing by SRp20. Nat. Struct. Mol. Biol. 2006;13:973–980. doi: 10.1038/nsmb1155. [DOI] [PubMed] [Google Scholar]
- 33.Loomis R.J., Naoe Y., Parker J.B., Savic V., Bozovsky M.R., Macfarlan T., Manley J.L., Chakravarti D. Chromatin binding of SRp20 and ASF/SF2 and dissociation from mitotic chromosomes is modulated by histone H3 serine 10 phosphorylation. Mol. Cell. 2009;33:450–461. doi: 10.1016/j.molcel.2009.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sanford J.R., Gray N.K., Beckmann K., Caceres J.F. A novel role for shuttling SR proteins in mRNA translation. Genes Dev. 2004;18:755–768. doi: 10.1101/gad.286404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hanamura A., Cáceres J.F., Mayeda A., Franza B.R., Jr., Krainer A.R. Regulated tissue-specific expression of antagonistic pre-mRNA splicing factors. RNA. 1998;4:430–444. [PMC free article] [PubMed] [Google Scholar]
- 36.Clery A., Blatter M., Allain F.H. RNA recognition motifs: boring? Not quite. Curr. Opin. Struct. Biol. 2008;18:290–298. doi: 10.1016/j.sbi.2008.04.002. [DOI] [PubMed] [Google Scholar]
- 37.Clery A., Sinha R., Anczukow O., Corrionero A., Moursy A., Daubner G.M., Valcarcel J., Krainer A.R., Allain F.H. Isolated pseudo-RNA-recognition motifs of SR proteins can regulate splicing using a noncanonical mode of RNA recognition. Proc. Natl. Acad. Sci. U.S.A. 2013;110:E2802–E2811. doi: 10.1073/pnas.1303445110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cartegni L., Wang J., Zhu Z., Zhang M.Q., Krainer A.R. ESEfinder: a web resource to identify exonic splicing enhancers. Nucleic Acids Res. 2003;31:3568–3571. doi: 10.1093/nar/gkg616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Misteli T., Spector D.L. RNA polymerase II targets pre-mRNA splicing factors to transcription sites in vivo. Mol. Cell. 1999;3:697–705. doi: 10.1016/s1097-2765(01)80002-2. [DOI] [PubMed] [Google Scholar]
- 40.Sadaie M.R., Benter T., Wong-Staal F. Site-directed mutagenesis of two trans-regulatory genes (tat-III,trs) of HIV-1. Science. 1988;239:910–913. doi: 10.1126/science.3277284. [DOI] [PubMed] [Google Scholar]
- 41.Keen N.J., Gait M.J., Karn J. Human immunodeficiency virus type-1 Tat is an integral component of the activated transcription-elongation complex. Proc. Natl. Acad. Sci. U.S.A. 1996;93:2505–2510. doi: 10.1073/pnas.93.6.2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.D'Orso I., Frankel A.D. RNA-mediated displacement of an inhibitory snRNP complex activates transcription elongation. Nat. Struct. Mol. Biol. 2010;17:815–821. doi: 10.1038/nsmb.1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Byers S.A., Price J.P., Cooper J.J., Li Q., Price D.H. HEXIM2, a HEXIM1-related protein, regulates positive transcription elongation factor b through association with 7SK. J. Biol. Chem. 2005;280:16360–16367. doi: 10.1074/jbc.M500424200. [DOI] [PubMed] [Google Scholar]
- 44.Yik J.H., Chen R., Nishimura R., Jennings J.L., Link A.J., Zhou Q. Inhibition of P-TEFb (CDK9/Cyclin T) kinase and RNA polymerase II transcription by the coordinated actions of HEXIM1 and 7SK snRNA. Mol. Cell. 2003;12:971–982. doi: 10.1016/s1097-2765(03)00388-5. [DOI] [PubMed] [Google Scholar]
- 45.Muniz L., Egloff S., Ughy B., Jady B.E., Kiss T. Controlling cellular P-TEFb activity by the HIV-1 transcriptional transactivator Tat. PLoS Pathog. 2010;6:e1001152. doi: 10.1371/journal.ppat.1001152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sedore S.C., Byers S.A., Biglione S., Price J.P., Maury W.J., Price D.H. Manipulation of P-TEFb control machinery by HIV: recruitment of P-TEFb from the large form by Tat and binding of HEXIM1 to TAR. Nucleic Acids Res. 2007;35:4347–4358. doi: 10.1093/nar/gkm443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Barboric M., Yik J.H., Czudnochowski N., Yang Z., Chen R., Contreras X., Geyer M., Matija Peterlin B., Zhou Q. Tat competes with HEXIM1 to increase the active pool of P-TEFb for HIV-1 transcription. Nucleic Acids Res. 2007;35:2003–2012. doi: 10.1093/nar/gkm063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yang Z., Yik J.H., Chen R., He N., Jang M.K., Ozato K., Zhou Q. Recruitment of P-TEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4. Mol. Cell. 2005;19:535–545. doi: 10.1016/j.molcel.2005.06.029. [DOI] [PubMed] [Google Scholar]
- 49.Sobhian B., Laguette N., Yatim A., Nakamura M., Levy Y., Kiernan R., Benkirane M. HIV-1 Tat assembles a multifunctional transcription elongation complex and stably associates with the 7SK snRNP. Mol. Cell. 2010;38:439–451. doi: 10.1016/j.molcel.2010.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.He N., Liu M., Hsu J., Xue Y., Chou S., Burlingame A., Krogan N.J., Alber T., Zhou Q. HIV-1 Tat and host AFF4 recruit two transcription elongation factors into a bifunctional complex for coordinated activation of HIV-1 transcription. Mol. Cell. 2010;38:428–438. doi: 10.1016/j.molcel.2010.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.