


Abstract
In all organisms, the large ribosomal subunit contains multiple copies of a flexible protein, the so-called ‘stalk’. The C-terminal domain (CTD) of the stalk interacts directly with the translational GTPase factors, and this interaction is required for factor-dependent activity on the ribosome. Here we have determined the structure of a complex of the CTD of the archaeal stalk protein aP1 and the GDP-bound archaeal elongation factor aEF1α at 2.3 Å resolution. The structure showed that the CTD of aP1 formed a long extended α-helix, which bound to a cleft between domains 1 and 3 of aEF1α, and bridged these domains. This binding between the CTD of aP1 and the aEF1α•GDP complex was formed mainly by hydrophobic interactions. The docking analysis showed that the CTD of aP1 can bind to aEF1α•GDP located on the ribosome. An additional biochemical assay demonstrated that the CTD of aP1 also bound to the aEF1α•GTP•aminoacyl-tRNA complex. These results suggest that the CTD of aP1 interacts with aEF1α at various stages in translation. Furthermore, phylogenetic perspectives and functional analyses suggested that the eukaryotic stalk protein also interacts directly with domains 1 and 3 of eEF1α, in a manner similar to the interaction of archaeal aP1 with aEF1α.
INTRODUCTION
Protein synthesis on the ribosome is promoted by the action of several translational GTPase factors (1–3). For example, during the peptide elongation cycle, two translational GTPase factors function as elongation factors. One elongation factor, named aEF1α in archaea, eEF1α in eukaryotes and EF-Tu in bacteria, delivers aminoacyl-tRNA to the ribosome in its GTP-bound form. After codon recognition and GTP hydrolysis, aEF1α/eEF1α/EF-Tu is released from the ribosome in the GDP-bound form. Subsequently, the GTP-bound form of the second elongation factor, named aEF2 in archaea, eEF2 in eukaryotes and EF-G in bacteria, binds to the ribosome and then catalyzes the translocation of the tRNA accompanied by GTP hydrolysis. Multiple copies of an acidic ribosomal protein, the so-called stalk protein, play a crucial role in the recruitment of translational GTPase factors to the ribosome, GTP hydrolysis and associated factor-dependent events on the ribosome (4–6).
Ribosomes in all organisms contain the stalk protein. The comparison of amino acid sequences and biochemical analyses indicate that the eukaryotic stalk protein P1/P2 and the archaeal stalk protein aP1 are related closely to each other, but not to the bacterial stalk protein L12 (7,8). However, the archaeal/eukaryotic and bacterial stalk proteins share a similar domain organization. Namely, all the stalk proteins are composed of an N-terminal and a C-terminal domain (hereafter NTD and CTD, respectively), and a flexible hinge region that connects these domains (8–11). The stalk proteins form a homo- or heterodimer [(L12)2 homodimer in bacteria; P1•P2 heterodimer in eukaryotes; (aP1)2 homodimer in archaea] via the NTD (10,12–15). Through their dimerized NTDs, multiple stalk protein dimers bind to an anchor protein (L10 in bacteria; P0 in eukaryotes; aP0 in archaea) (13,14,16–19). The CTD of the archaeal/eukaryotic stalk is composed of approximately 20 amino acids and seems to be unstructured (8,11,20), in contrast to the large globular bacterial CTD, which is composed of approximately 70 amino acid residues that form three α-helices and a three-stranded β-sheet (12,21). It is interesting that, despite the absence of structural similarity in the CTDs between archaeal/eukaryotic and bacterial stalks, they share common functionalities, namely, the CTDs play crucial roles in the recruitment of translational GTPase factors to the ribosome, stimulation of GTP hydrolysis, and the associated actions of individual translational GTPase factors on the ribosome (13,14,19,22–26). Moreover, direct binding of the CTD to several translational GTPase factors has been demonstrated in both archaea and bacteria (26,27).
To understand the molecular mechanisms involved in the functional interactions between the CTD of the stalk protein and translational GTPase factors, nuclear magnetic resonance (NMR) chemical shift perturbation analysis using isolated bacterial stalk protein L12 was performed (27). This analysis identified the amino acid residues of the L12 CTD that contribute to the interactions with EF-G, EF-Tu, initiation factor 2 (IF-2) and release factor 3 (RF-3). However, no information was obtained about the amino acid residues in the translational GTPase factors that are involved in the interaction with L12 (27). On the other hand, the structures of the ribosome•EF-G complex that were determined by X-ray crystallography and cryo-electron microscopy (cryo-EM) showed that the L12 CTD interacts directly with EF-G through the G′ domain, a subdomain of domain 1 (28–31). However, EF-Tu has no domain that corresponds to the G’ domain of EF-G, and thus, it is of interest how the stalk recognizes EF-Tu. The cryo-EM structural studies also showed that the L12 CTD interacts with domain 1 of EF-Tu on the ribosome (32). However, owing to the low resolution and ambiguous electron density, which is presumably due to the weak interaction and high flexibility of L12, detailed structures of the interactions of L12 with these factors are not available. Furthermore, as yet no structural information has been obtained for the eukaryotic and archaeal proteins.
Recently, using proteins from a hyperthermophilic archaeon, Pyrococcus horikoshii, we showed that the CTD of aP1, which shares common characteristics with the eukaryotic stalk, forms stable complexes with several translational GTPase factors (26). In the present study, we determined the structure of a complex between the CTD of aP1 and the GDP-bound form of aEF1α from P. horikoshii at 2.3 Å resolution. This structure revealed unambiguously how aP1 recognized the aEF1α•GDP complex at atomic resolution. In addition, we showed that the CTD of aP1 also bound to aEF1α in the aEF1α•GTP•aminoacyl-tRNA ternary complex by biochemical analysis. Moreover, from the results of a functional assay using the eukaryotic system, we suggest that the mode of interaction between stalk proteins and aEF1α/eEF1α is conserved between archaea and eukaryotes. The functional significance of the interaction of the stalk protein with EF1α in translational efficiency is also discussed.
MATERIALS AND METHODS
Plasmid constructs
The gene that encodes P. horikoshii aEF1α was cloned into the pET-22b vector (Novagen) with a sequence that codes for a C-terminal His-tag. The plasmids for P. horikoshii aP1 and its mutants were constructed as described previously (26,33). The plasmids for silkworm P0ΔC55, P1 and P2 were constructed as described previously (17). The plasmids for the mutants of aEF1α, N-terminal FLAG-tagged aP1 and its CTD-truncated mutant, and the mutants of silkworm P1 and P2 were generated using the aforementioned respective vectors and a QuikChange Site-Directed Mutagenesis Kit (Stratagene). The plasmids for the expression of E. coli tRNALys and LysRS were gifts from Dr Takashi Yokogawa (Gifu University, Japan) (34).
Preparation of proteins, the aP1 peptide and aminoacyl-tRNA
aEF1α and its mutants were expressed in the Escherichia coli strain BL21-CodonPlus(DE3)-RIL (Stratagene). aEF1α for crystallography was purified by heat treatment, followed by chromatography using HisTrap HP and Resource S columns (GE Healthcare). For gel mobility shift assays, aEF1α and its mutants were purified by heat treatment and passage through a Ni-NTA Agarose column (Qiagen). aP1 and its mutants were prepared as described previously (33). N-terminal FLAG-tagged aP1 and its CTD-truncated mutant were expressed in the E. coli strain BL21-CodonPlus(DE3)-RIL (Stratagene). N-terminal FLAG-tagged aP1 was purified by heat treatment, followed by chromatography through HiTrap Q XL and Resource Q columns (GE Healthcare). N-terminal FLAG-tagged aP1 lacking the CTD was purified by heat treatment, followed by chromatography through HiTrap Q XL and HiLoad 16/60 Superdex 75 pg columns (GE Healthcare). A peptide that consisted of the C-terminal 32 amino acid residues of P. horikoshii aP1 [residues 77–108, hereafter aP1(CTD)] was synthesized and purified by Hokkaido System Science Co., Ltd. Silkworm P0ΔC55, P1, P2 and their mutants were prepared as described previously (35). E. coli tRNALys was expressed in the E. coli strain BL21(DE3), extracted using water-saturated phenol and purified with a HiTrap Q HP column (GE Healthcare). E. coli LysRS was prepared as described previously (36). The aminoacylation of tRNALys was performed under the same conditions as described previously (37) except that tRNALys, LysRS and lysine were used instead of tRNAAla, AlaRS and alanine.
Crystallization and data collection
Purified aEF1α was dialyzed against buffer A (20 mM Hepes-KOH, pH 7.6, 50 mM KCl, 7 mM β-mercaptoethanol, 10 mM MgCl2 and 10 μM GDP) and concentrated by ultrafiltration. Crystals of the aEF1α•GDP complex were prepared by the sitting drop vapor diffusion method at 4ºC by mixing 1 μl of protein solution (5.9 mg/ml aEF1α in buffer A) and an equal volume of reservoir solution [100 mM Hepes-NaOH, pH 7.5, 10% (w/v) PEG6000 and 5% (v/v) 2-methyl-2,4-pentanediol]. Crystals of the aP1(CTD)•aEF1α•GDP complex were prepared by the sitting drop vapor diffusion method at 20ºC by mixing 1 μl of sample solution [5.1 mg/ml aEF1α and 2.0 mg/ml aP1(CTD) peptide in buffer A that contained 10% (v/v) DMSO] and an equal volume of reservoir solution [200 mM NaCl, 100 mM phosphate-citrate, pH 4.2 and 20% (w/v) PEG8000]. Prior to data collection, 5 μl of reservoir solution that contained 25% (v/v) glycerol was added to the drops that contained crystals for cryoprotection. All diffraction data were collected with a wavelength of 1.000 Å at 95 K on the BL-5A of the Photon Factory (Tsukuba, Japan) using an ADSC Quantum 315r CCD detector. Diffraction data were processed with the program HKL2000 (HKL Research).
Structure determination
The initial model of the aEF1α•GDP complex was obtained by the molecular replacement method using the program BALBES (38). The best solution was obtained using the structure of the aEF1α•GDP complex from Sulfolobus solfataricus (PDB ID: 1JNY) (39) as a search model. The model was improved by auto model building with the programs BUCCANEER (40) and ARP/wARP (41). The initial model of the aP1(CTD)•aEF1α•GDP complex was obtained by the molecular replacement method using the program MOLREP (42) and the structure of P. horikoshii aEF1α•GDP as a search model. The model was improved subsequently by rigid body refinement with the program REFMAC5 (43), followed by building of the model for aP1(CTD). The models of the aEF1α•GDP and aP1(CTD)•aEF1α•GDP complexes were improved by iterative cycles of manual model building with the program COOT (44) and maximum likelihood refinement with the program REFMAC5.
Thin-layer chromatography
An aliquot of 4 nmol of purified aEF1α from E. coli was treated with phenol/chloroform to extract the bound nucleotide. The aqueous phase of the sample was recovered, concentrated and spotted onto a polyethylenimine cellulose TLC plate (Millipore). Aliquots of 4 nmol of standard GTP, GDP and GMP were also spotted. The plate was developed with a solvent system that consisted of 0.8 M LiCl and 0.8 M acetic acid. Nucleotides were detected under UV light (260 nm).
Removal of guanosine nucleotide from aEFlα
To prepare the GTP-bound form of aEF1α, first, tightly bound GDP was removed from purified aEF1α from E. coli (45). Bacterial alkaline phosphatase (1 U per mg of aEF1α, Takara) was added to a solution that contained 30 mM Tris-HCl, pH 8.0, 200 mM (NH4)2SO4, 1.5 M NaCl, 7 mM β-mercaptoethanol and 1 mg/ml aEF1α, and the sample solution was incubated for 5 h at 60ºC. To remove the released nucleotide and bacterial alkaline phosphatase, the sample solution was loaded onto a Ni-NTA Agarose column (Qiagen) that had been equilibrated with buffer B (20 mM Hepes-KOH, pH 7.6, 1 M NH4Cl, 5% (v/v) glycerol and 7 mM β-mercaptoethanol). Subsequently, the column was washed with buffer B that contained 20 mM imidazole, and the nucleotide-free form of aEFlα was eluted with buffer B that contained 250 mM imidazole.
Nitrocellulose-filter binding assay
The binding of GTP to aEF1α was checked by a nitrocellulose-filter binding assay as described previously with slight modifications (46). The binding reaction was performed on ice for 30 min in 100 μl of buffer C (20 mM Hepes-KOH pH 7.6, 50 mM KCl, 10 mM MgCl2 and 7 mM β-mercaptoethanol) that contained 1 μM nucleotide-free aEF1α and 10 μM [γ-32P]GTP. The reaction solutions were applied onto nitrocellulose filters (Millipore), and then the filters were washed with 1 ml of cold buffer C. The amount of GTP bound to aEF1α was determined from the radioactivity retained on the nitrocellulose filters. Assays were performed in triplicate.
Gel mobility shift assay
To analyze the binding between aP1 and the aEF1α•GDP complex, 200 pmol of wild-type or mutant aP1 and an equal molar amount of wild-type or mutant aEF1α•GDP were mixed in 10 μl of buffer D (20 mM Tris-HCl, pH 6.5, 10 mM MgCl2 and 20 mM KCl). To analyze the binding between the aEF1α•GTP complex and Lys-tRNALys or tRNALys, 400 pmol of aEF1α•GTP and an equal molar amount of Lys-tRNALys or tRNALys were mixed in 10 μl of buffer D. After incubation at 37ºC or 70ºC for 10 min, each sample was mixed with 2 μl of solution that contained 120 mM Tris-HCl, pH 6.5, 60 mM MgCl2, 120 mM KCl, 40% (v/v) glycerol and 0.12% (w/v) bromophenol blue, and was then subjected to gel electrophoresis as described previously (26).
Pull-down assay
An aliquot of 20 μl of Anti DYKDDDDK tag Antibody Beads (Wako) was incubated with 500 pmol of N-terminal FLAG-tagged aP1 in 50 μl of phosphate buffered saline (PBS) for 30 min at 4ºC, and then washed with PBS. Subsequently, 20 μl of the aP1-bound beads were incubated with 500 pmol of [14C]Lys-tRNALys (specific activity, 290 cpm/pmol) in the presence or absence of 500 pmol of the aEF1α•GTP complex in 50 μl of PBS for 30 min at 4ºC. After pull-down, the beads were washed with PBS and mixed with 500 μl of Insta-Gel Plus (PerkinElmer). The 14C radioactivity was measured by scintillation counting. The radioactivity derived from the nonspecific binding of [14C]Lys-tRNALys to the beads was also measured and subtracted as a background value. Assays were performed in triplicate.
Eukaryotic eEF1α-dependent GTPase activity assay
The silkworm stalk complex was reconstituted as described previously (17). The hybrid 50 S subunits were prepared as described previously (47). An aliquot of 30 μl of the reaction mixture containing 10 pmol of hybrid 50 S subunits, 30 pmol of 30 S subunits, 300 pmol of [γ-32P]GTP (270 cpm/pmol), 10 μg of poly(U), 20 pmol of Phe-tRNA, 5 mM MgCl2, 50 mM NH4Cl, 20 mM Tris-HCl, pH 7.5 and 0.2 μg of eEF1α was incubated at 37ºC for 20 min. The GTPase activity was measured as described previously (47). Assays were performed in triplicate.
RESULTS
Structure determination
We showed previously that the P. horikoshii ribosomal stalk protein aP1 directly bound to aEF1α and the binding was almost completely prevented by the addition of a synthetic peptide that comprised the C-terminal 18 amino acids of aP1 (26). From the results we concluded that the aP1 binding to aEF1α occurs via its C-terminal region. However, it was not clear whether the aEF1α used in the earlier study was bound to GTP, GDP or no nucleotide. To address this question, first we performed thin layer chromatography and found that the aEF1α used in the binding assay was predominantly in the GDP-bound form (Supplementary Figure S1). Consequently, to understand the mechanistic details of the interaction between aP1 and aEF1α, as a first step, we crystallized the GDP-bound form of aEF1α from P. horikoshii with a 32-mer peptide that included all C-terminal amino acid residues for the putative CTD of P. horikoshii aP1 [hereafter aP1(CTD)] and determined the structure of this complex at 2.30 Å resolution (Figure 1A and B). In addition, to compare this structure with that of the aP1(CTD)-free form, we also determined the structure of the GDP-bound form of aEF1α from P. horikoshii at 2.35 Å resolution (Figure 1C and D). The data collection and structure refinement statistics are summarized in Table 1.
Figure 1.
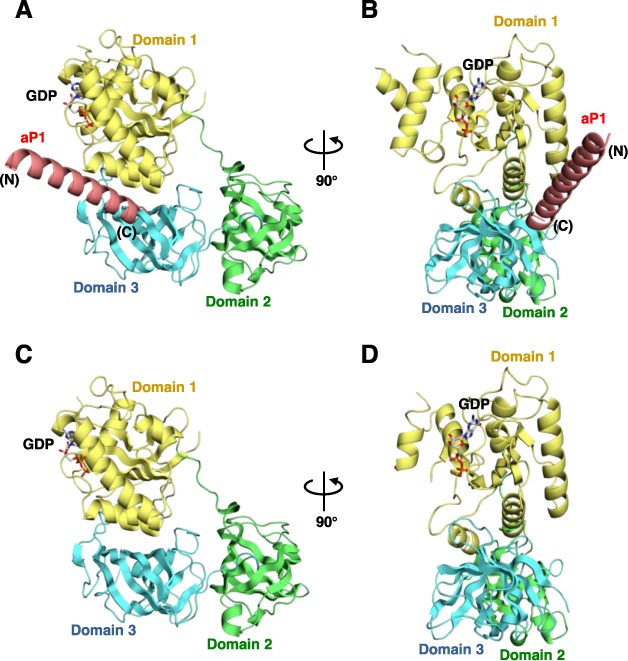
Overall structures of the aP1(CTD)•aEF1α•GDP complex and the aEF1α•GDP complex. (A) Overall structure of the aP1(CTD)•aEF1α•GDP complex from P. horikoshii. aP1(CTD) and aEF1α are represented by ribbon models. aP1(CTD) and domains 1, 2 and 3 of aEF1α are colored pink, yellow, green and light blue, respectively. The N- and C-termini of aP1(CTD) are indicated as ‘(N)’ and ‘(C)’, respectively. The bound GDP molecule is shown by a stick model. (B) An orthogonal view of (A). (C) Overall structure of the aEF1α•GDP complex from P. horikoshii. The figure is depicted in a manner similar to (A). (D) An orthogonal view of (C). All graphical figures in this paper were prepared using the program PyMol (http://www.pymol.org).
Table 1. Data collection and refinement statistics.
| aP1(CTD)•aEF1α•GDP | aEF1α•GDP | |
|---|---|---|
| Data collection | ||
| Space group | C2 | P212121 |
| Cell dimensions | ||
| a, b, c (Å) | 157.26, 87.42, 82.46 | 47.68, 78.28, 138.17 |
| α, β, γ (º) | 90.0, 92.3, 90.0 | 90.0, 90.0, 90.0 |
| Resolution range (Å) | 50.0–2.30 (2.34–2.30)a | 50.0–2.35 (2.39–2.35) |
| No. of measured reflections | 186879 | 100656 |
| No. of unique reflections | 49622 | 20974 |
| Completeness (%) | 100.0 (99.9) | 95.0 (95.0) |
| Redundancy | 3.8 (3.7) | 4.8 (4.1) |
| Average I/σ(I) | 34.5 (7.1) | 32.2 (5.6) |
| Rmergeb (%) | 5.8 (22.7) | 8.4 (36.0) |
| Refinement | ||
| Rwork/Rfreec | 19.4/23.8 | 19.5/24.3 |
| No. of complexes | 2 | 1 |
| Assigned residues/Chain | 5–43, 51–427/A, B | 5–35, 53–427/A |
| 83–108/C, D | ||
| No. of atoms | ||
| Protein | 6912 | 3087 |
| Ligand | 56 | 28 |
| Solvent | 136 | 76 |
| Average B factors (Å2) | ||
| Protein | 42.3 | 38.4 |
| Ligand | 31.0 | 32.7 |
| Solvent | 35.7 | 37.7 |
| RMS deviations | ||
| Bond length (Å) | 0.011 | 0.007 |
| Bond angle (º) | 1.512 | 1.239 |
| Ramachandran plot | ||
| Favored region (%) | 96.9 | 98.7 |
| Allowed region (%) | 2.8 | 1.0 |
| Outlier region (%) | 0.3 | 0.3 |
aValues in parentheses are for the highest resolution shell.
bRmerge = ΣhklΣi |Ii(hkl) – <I(hkl)>|/ΣhklΣi Ii(hkl), where Ii(hkl) is the i-th intensity measurement of reflection hkl, including symmetry-related reflections, and <I(hkl)> is its average.
cRfree was calculated by using 5% of randomly selected reflections that were excluded from the refinement.
Overall and domain structures
aEF1α consists of three domains (domains 1, 2 and 3). Domain 1 of aEF1α in the aP1(CTD)•aEF1α•GDP complex binds the GDP molecule and shares structural similarity with small GTPases (Supplementary Figure S2A). The structure of the aP1(CTD)•aEF1α•GDP complex revealed that the C-terminal 27 residues of aP1(CTD) formed a long extended α-helix, whereas the N-terminal 5 residues were disordered (Figure 1A and B). The structure also revealed that aP1(CTD) bound to a space between domains 1 and 3 of aEF1α and bridged these domains (Figures 1A and B and 2A). This binding site was ∼20 Å away from the GTP/GDP-binding site.
Figure 2.
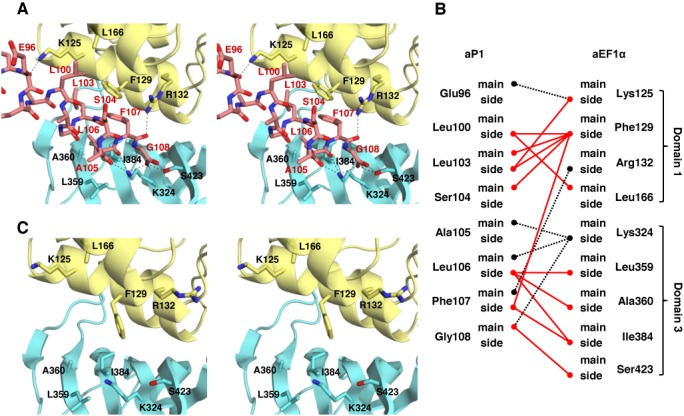
Interaction between aP1 and aEF1α. (A) Stereo view of the structure of the interaction interface between aP1 and aEF1α in the aP1(CTD)•aEF1α•GDP complex. aEF1α is represented by a ribbon model, and aP1 and the amino acid residues of aEF1α that participate in the interaction are represented by stick models. The color coding is the same as in Figure 1. Hydrogen bonds are represented by dashed lines. (B) Schematic representation of the interaction between aP1 and aEF1α. Hydrogen bonds and hydrophobic interactions are represented by black dashed lines and red lines, respectively. (C) Stereo view of the structure of the same region as that shown in (A) in the aEF1α•GDP complex.
Comparison between the structures of aP1(CTD)-bound and free aEF1α•GDP revealed that, although the orientation of two amino acid side chains in the aP1 binding site differed remarkably (see below), the overall structures of the two forms were highly similar (RMS deviation of 0.74 Å for equivalent Cα atoms) (Figure 1A–D). It is worth noting that the conformations of the key regions for GTP hydrolysis, namely the P-loop and switch I and II regions, were also similar between the two structures (Supplementary Figure S2A and S2B). These facts indicate that the binding of aP1 did not substantially affect the structure of the GDP-bound form of aEF1α.
Interaction between aP1 and aEF1α
The structure of the aP1(CTD)•aEF1α•GDP complex revealed that the C-terminal α-helix of aP1 interacted with domains 1 and 3 of aEF1α by an extensive hydrophobic interaction network, six hydrogen bonds and one salt bridge (Figure 2A and B, Supplementary Table SI). Specifically, on domain 1 of aEF1α, the main chains of Glu96 and Phe107 of aP1 formed hydrogen bonds with the side chains of Lys125 and Arg132 of aEF1α, respectively. The side chain of Phe129 on domain 1 of aEF1α interacted hydrophobically with the concave surface of aP1 that was formed by Leu100, Leu103, Ser104 and Phe107. This interaction included π-stacking between Phe129 of aEF1α and Phe107 of aP1. Furthermore, on domain 1 of aEF1α, the main chain of Leu166 and the side chain of Lys125 formed hydrophobic interactions with the side chains of Leu100 and Leu103 of aP1, respectively. On domain 3 of aEF1α, the side chain of Lys324 formed hydrogen bonds with the main chains of Ala105 and Leu106 of aP1. In addition, the side chain of Lys324 of aEF1α formed a salt bridge with the carboxyl group of the C-terminal Gly108 of aP1. In the vicinity of this salt bridge, the side chain of Ser423 of aEF1α formed a bifurcated hydrogen bond with the carboxyl group of the C-terminal Gly108 of aP1. In addition, a hydrophobic surface of domain 3 of aEF1α, which was formed by Leu359, Ala360 and Ile384, formed a hydrophobic interaction with the side chains of Leu106 and Phe107 of aP1.
As described in the previous section, the overall structures of aEF1α•GDP in the aP1(CTD)-bound and free forms were highly similar. However, there were marked differences in the side chains of two amino acids of aEF1α at the aP1 binding site. Compared to the structure of aP1(CTD)-free aEF1α•GDP, the side chain of Phe129 in the aP1(CTD)•aEF1α•GDP complex flipped toward Lys125 of aEF1α (Figure 2A and C). This residue fitted into the hydrophobic surface of aP1 formed by Leu100, Leu103, Ser104 and Phe107 in the aP1(CTD)•aEF1α•GDP complex (Figure 2A). Furthermore, the side chain of Arg132 flipped toward aP1 in the aP1(CTD)•aEF1α•GDP complex and formed a hydrogen bond with the main chain oxygen of Phe107 of aP1 (Figure 2A and C). These movements suggest an induced-fit mechanism for the binding between aP1 and aEF1α.
Effect of amino acid substitutions on the interaction between aP1 and aEF1α
The structure of the aP1(CTD)•aEF1α•GDP complex revealed the amino acid residues involved in the interaction between aP1 and GDP-bound aEF1α. To analyze the roles of these residues in the interaction, we performed a gel mobility shift assay using mutants of aP1 (Figure 3A) or aEF1α (Figure 3B). The amino acid residues that participated in the interaction through their side chains were selected as the targets of mutation, and hydrophobic and hydrophilic residues were replaced with serine and leucine, respectively. Wild-type aP1 exhibited a high mobility in the gel (Figure 3A and B, lane 1), whereas aEF1α alone hardly entered the gel because of its high basicity (Figure 3A, lane 7 and Figure 3B, lane 12). The incubation of wild-type aP1 with wild-type aEF1α led to a decrease in the band that corresponded to free aP1, and the appearance of a lower mobility band (Figure 3A and B, lane 2). We confirmed previously that this lower mobility band is a complex of aP1 and aEF1α (26). When wild-type aEF1α was incubated with the aP1 mutants L103S, L106S and F107S, no complex was formed (Figure 3A, lanes 4–6). In addition, a moderate disruption was detected with the L100S mutant (Figure 3A, lane 3). Our previous mutagenesis study revealed that Leu103, Leu106 and Phe107 of aP1 are also responsible for the interaction with aEF2 (26), which suggests that these hydrophobic amino acids participate in the binding of both aEF1α and aEF2. In the case of the aEF1α mutants, complete disruption of binding was observed with the F129S, K324L and I384S mutants (Figure 3B, lanes 4, 7 and 10). The R132L, L166S, L359S, A360S and S423L mutants also had a moderate effect on complex formation (Figure 3B, lanes 5, 6, 8, 9 and 11). Only the K125L mutant had little effect on complex formation (Figure 3B, lane 3). The substitution of K125 might be compensated for by other interactions. These results indicate that the interactions observed in the present crystal structure are responsible for the binding between aP1 and aEF1α.
Figure 3.
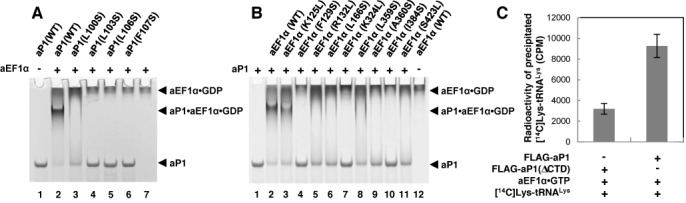
Analysis of the interaction between aP1 and aEF1α. (A) Analysis of the binding of aP1 mutants to the GDP-bound form of wild-type aEF1α by gel mobility shift assay. Wild-type aP1 (lane 2) or each of the aP1 mutants (lanes 3–6) was incubated with wild-type aEF1α and subjected to a gel mobility shift assay as described under the Materials and Methods section. Wild-type aP1 (lane 1) and wild-type aEF1α (lane 7) were also incubated alone and subjected to the gel mobility shift assay. (B) Analysis of the binding of wild-type aP1 to the GDP-bound form of the aEF1α mutants by gel mobility shift assay. Wild-type aP1 was incubated with wild-type aEF1α (lane 2) or each of the aEF1α mutants (lanes 3–11), and subjected to the gel mobility shift assay as in (A). Wild-type aP1 (lane 1) or wild-type aEF1α (lane 12) were also incubated alone and subjected to the gel mobility shift assay. (C) Analysis of the binding of the CTD of aP1 to the aEF1α•GTP•aminoacyl-tRNA ternary complex by a pull-down assay. Anti-FLAG beads that were prebound with N-terminal FLAG-tagged aP1 or N-terminal FLAG-tagged aP1 lacking the CTD were mixed with the aEF1α•GTP complex and [14C]Lys-tRNALys. The beads were precipitated and washed, and the 14C radioactivity on the beads was measured (n = 3, mean ± SE), as described under the Materials and Methods section.
Binding of the CTD of aP1 to the aEF1α•GTP•aminoacyl-tRNA ternary complex
To examine the involvement of the CTD of aP1 in the recruitment of aEF1α to the ribosome, we evaluated the capability of the CTD of aP1 to bind to the aEF1α•GTP•aminoacyl-tRNA ternary complex. First, to prepare the aEF1α•GTP binary complex, purified aEF1α expressed in E. coli was treated with bacterial alkaline phosphatase to remove associated GDP (45) and then incubated with GTP. A nitrocellulose-filter binding assay showed that 93.6 ± 2.5% (n = 3, mean ± SE) of the aEF1α was bound to GTP. Next, we prepared the aEF1α•GTP•aminoacyl-tRNA ternary complex by mixing aEF1α•GTP with [14C]Lys-tRNALys, and confirmed the association of aEF1α with [14C]Lys-tRNALys by a gel mobility shift assay (Supplementary Figure S3). Then, we examined the binding of the CTD of aP1 to the aEF1α•GTP•[14C]Lys-tRNALys complex by a pull-down assay. The results showed that a significant amount of [14C]Lys-tRNALys coprecipitated with anti-FLAG resin in the presence of N-terminal FLAG-tagged aP1 and aEF1α•GTP (Figure 3C). In contrast, only a small amount of [14C]Lys-tRNALys coprecipitated when N-terminal FLAG-tagged aP1 that lacked the CTD was used instead of full-length N-terminal FLAG-tagged aP1 (Figure 3C). These results indicate that aP1 forms a complex with the aEF1α•GTP•aminoacyl-tRNA ternary complex via its CTD and suggests the involvement of the CTD of aP1 in the recruitment of the ternary complex to the ribosome.
Functional contributions of the C-terminal residues in the eukaryotic stalk
The structure of the aP1(CTD)•aEF1α•GDP complex and the results of the gel mobility shift assays revealed the amino acid residues of the archaeal stalk CTD that are responsible for the interaction with aEF1α. Among these amino acid residues, Leu106 and Phe107 are strictly conserved from archaea to eukaryotes (Figure 4A) (48,49). Thus, to investigate the functional contribution of these amino acid residues in the eukaryotic stalk complex, we examined the effects of the substitution of these amino acid residues in the eukaryotic stalk P1/P2 on eukaryotic eEF1α-dependent GTPase activity by using a hybrid ribosome system (47) as follows. First, we mutated Leu110 or Phe111 in both silkworm P1 and P2 to serine. In both silkworm P1 and P2, Leu110 and Phe111 correspond to Leu106 and Phe107 of P. horikoshii aP1, respectively. Second, we reconstituted the stalk complex in vitro by mixing the P1 and P2 variants with the silkworm anchor protein P0 (Supplementary Figure S4A). In this reconstitution, to focus on the roles of the P1 and P2 variants, we used a truncation mutant of P0 that lacked the C-terminal 55 amino acid residues, whose sequence is very similar to that of the C-termini of P1 and P2. Third, we substituted the variants of the stalk complexes for the L10•L12 stalk complex on the E. coli 50S subunit in vitro (Supplementary Figure S4B), and then measured the eEF1α-dependent GTPase activity. For both the L110S and F111S substitutions, the GTPase activity was reduced to one-third of that obtained with the wild-type stalk protein (Figure 4B). These results indicate that, as in archaea, the strictly conserved leucine and phenylalanine in the CTD of eukaryotic stalk protein P1/P2 are involved in functional interaction with eEF1α.
Figure 4.
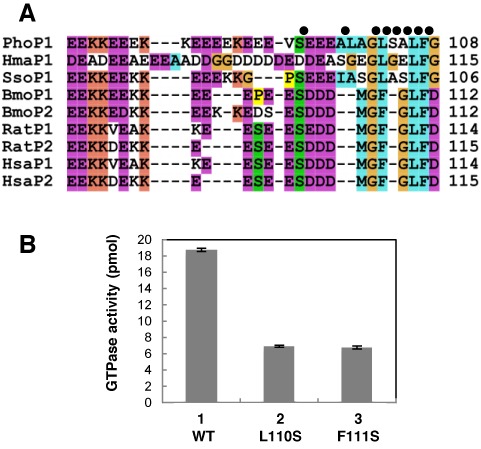
Effects of the substitution of amino acid residues in the CTD of the eukaryotic stalk protein P1/P2. (A) Amino acid sequence alignment of the CTDs of various stalk proteins. The sequences for archaeal aP1 from Pyrococcus horikoshii (Pho), Haloarcula marismortui (Hma) and Sulfolobus solfataricus (Sso), and eukaryotic P1/P2 from Bombyx mori (Bmo), Rattus norvegicus (Rat) and Homo sapiens (Hsa) were aligned using the program T-Coffee (48). The program Clustal X (49) was used to prepare the figure. The black dots above the sequences indicate the residues that participated in the interaction with aEF1α in P. horikoshii. (B) Eukaryotic eEF1α-dependent GTPase activity was measured (n = 3, mean ± SE) using hybrid ribosomes composed of the E. coli ribosome core and the following variants of the silkworm stalk complex: P0ΔC55•[P1(WT)•P2(WT)]2 (lane 1, WT), P0ΔC55•[P1(L110S)•P2(L110S)]2 (lane 2, L110S) and P0ΔC55•[P1(F111S)•P2(F111S)]2 (lane 3, F111S), as described under the Materials and Methods section.
DISCUSSION
A functional role for the CTDs of stalk proteins in the interaction between EF-Tu/aEF1α/eEF1α and ribosomes has long been suggested in prokaryotes, archaea and eukaryotes, even though little detailed structural information on the role of the CTD has been available. In the present study, we solved the structure of a complex composed of the CTD of archaeal stalk aP1, aEF1α and GDP. This is the first unambiguous structure of a complex of a stalk protein with a translational factor at atomic resolution. The structure clearly showed that the C-terminal peptide formed an α-helix and that it unexpectedly bound to a cleft between domains 1 and 3 of aEF1α (Figures 1A and B and 2A). The structure was stabilized by many hydrophobic interactions between aP1(CTD) and aEF1α (Figure 2A and B). The crystal structure data on the interactions were evaluated by gel mobility shift assay using mutant proteins. This confirmed that the interactions observed in the crystal structure were essential for the binding between aP1 and aEF1α•GDP (Figure 3A and B).
The present structural data indicated that most of the residues in the small CTD that are characteristic of archaeal P1 (or eukaryotic P1/P2) formed a single α-helix (Figures 1A and B and 2A), unlike the globular CTD in the bacterial stalk, at least when aP1 was bound to aEF1α•GDP. The structural features of the eukaryotic stalk CTD were previously reported. Kessenbrock et al. showed that the C-terminal six residues (GFGLFD) of the human P1 CTD form an α-helix (50). Lee et al. reported that the CTDs of human P1 and P2 are flexible and unfolded (11). Too et al. showed that four residues (GFGL) in the CTD of human P1/P2 adopt a type II β-turn in the complex with trichosanthin, a ribosome-inactivating protein (51). On the other hand, the present structural data revealed that the majority of the CTD residues of archaeal aP1 form a long extended α-helix in the complex with aEF1α•GDP (Figures 1A and B and 2A). It is interesting that, although the binding modes of human P1/P2•trichosanthin and archaeal aP1•aEF1α are different, the strictly conserved LF motifs in the CTDs of the two stalk proteins (L110-F111 in human and L106-F107 in P. horikoshii) both participate in the binding by forming many hydrophobic interactions.
To evaluate the structure and binding orientation of the archaeal stalk CTD, we attempted to construct a docking model of the aP1(CTD)•aEF1α•GDP complex bound to the ribosomal factor binding center. No atomic-resolution structure of the archaeal 70S ribosome in complex with aEF1α is available at present. However, a docking model could be constructed on the basis of the following facts: (i) a structure of a complex of the bacterial ribosome, EF-Tu•GDP, and aminoacyl-tRNA is available (52); (ii) the core structure and function of the ribosome is essentially conserved between archaea and bacteria (33,53); (iii) the amino acid sequences as well as the structures of aEF1α and EF-Tu are conserved (39) (Supplementary Figure S5); (iv) the structure of the P. horikoshii stalk complex aP0(aP1)2(aP1)2(aP1)2 (PDB ID: 3A1Y) is available (14); (v) the rRNA-binding domains of the archaeal and bacterial stalk complexes share a similar structure (14). The resultant docking model showed that the stalk CTD could access aEF1α•GDP on the ribosome through an unstructured hinge region without any steric hindrance and without changing the binding orientation of the C-terminal α-helix of aP1 (Figure 5A). Therefore, it appears that the structure of the aP1(CTD)•aEF1α•GDP complex (Figures 1A and B and 2A) represents the mode of binding of the CTD of aP1 to aEF1α•GDP on the ribosome, although the CTD of aP1 probably also binds to aEF1α•GDP released from the ribosome in the same manner.
Figure 5.
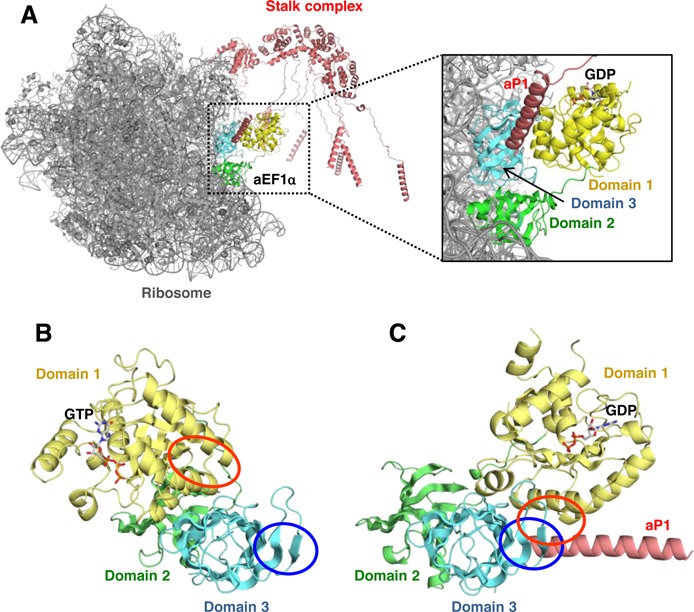
Implications for the functional significance of the interaction of the stalk protein with EF1α. (A) The docking model of the aP1(CTD)•aEF1α•GDP complex to the ribosome. The core of the ribosome and the stalk complex is represented by gray and pink, respectively. aEF1α is represented by the same color coding as in Figure 1. In the docking model, domains 2 and 3 of aEF1α in the aP1(CTD)•aEF1α•GDP complex were superposed onto those of EF-Tu on the bacterial ribosome. The rRNA-binding domain of the P. horikoshii stalk complex was superposed onto that of the bacterial ribosome. The CTDs of aP1 bound to aEF1α and the NTD of one of the multiple copies of aP1 are connected by a flexible hinge. Other hinge regions and C-terminal regions of aP1 are modeled arbitrarily. (B) and (C) Comparison of the structures of the GTP- and GDP-bound forms of aEF1α. aEF1α•GTP in the aEF1α•GTP•Pelota complex (B) (PDB ID: 3AGJ) (54) and aEF1α•GDP in the aP1(CTD)•aEF1α•GDP complex (C) are represented in a manner similar to Figure 1. Panels (B) and (C) are shown from the same direction with respect to domains 2 and 3. The aP1-binding region on domain 1 of aEF1α•GDP and the equivalent region on aEF1α•GTP are indicated by red circles. The aP1-binding region on domain 3 of aEF1α•GDP and the equivalent region on aEF1α•GTP are indicated by blue circles.
In the present study, biochemical analyses also showed that aP1 could bind directly to the aEF1α•GTP•aminoacyl-tRNA ternary complex via its CTD (Figure 3C). This result indicates that the CTD of aP1 participates in the recruitment of the aEF1α•GTP•aminoacyl-tRNA ternary complex to the ribosome. However, we have not yet obtained crystals of the aP1(CTD)•aEF1α•GTP•aminoacyl-tRNA complex. We think that the explanation for this is as follows. Upon GTP hydrolysis, aEF1α is known to undergo a substantial conformational change from the GTP-bound form to the GDP-bound form, in which domain 1 shifts its orientation by approximately 90º with respect to domains 2 and 3 (Figure 5B and C) (54). Due to this conformational change, aP1 seems to bind weakly to the GTP-bound form of aEF1α because the aP1-binding regions on domains 1 and 3 are separated in this state (Figure 5B). We think that this weak binding affects the crystallization. In contrast, aP1 seems to bind tightly to the GDP-bound form of aEF1α through both domain 1 and 3, as shown by the present crystal structure (Figures 1A and B, 2A and 5C). Taking these points into consideration, we propose an additional role for the ribosomal stalk, namely that the stalk protein promotes the conformational change in aEF1α that is associated with GTP hydrolysis through stabilizing the GDP-bound form of aEF1α, by bridging domains 1 and 3 on the ribosome, as presented in Figure 5A. This action of the stalk protein would facilitate the function of aEF1α on the ribosome, and thus support translation elongation. Further analyses are needed to validate this hypothesis.
In our previous research, we showed that the binding of aP1 to aEF2 was independent of the nucleotide state on aEF2 (26). This is in contrast with the present model for aEF1α, in which aP1 presumably binds weakly in the GTP-bound form, as compared with the GDP-bound form, as discussed above. This difference in the nucleotide dependency for the binding of aP1 to aEF-2 and to aEF1α may be interpreted as follows. In the case of aEF1α, domains 1 and 3 are responsible for binding to aP1, as demonstrated in the present research. On the other hand, in the case of aEF2, aP1 could bind to aEF2 only via domain 1 (26). From these results, we infer that the binding modes of aP1 to aEF2 and aEF1α are different.
The result on α-helix structure of the CTD of aP1 in the present study is consistent with previous NMR data on the C-terminal part of the human stalk P2 (50). Modes of dimerization of stalks and their binding to the anchor protein P0/aP0 are also similar between archaea and eukaryotes (11,14,55). Given this structural similarity between the archaeal and eukaryotic stalk proteins, and between aEF1α and eEF1α (39), and also the functional compatibility between the archaeal and eukaryotic translational GTPase factors (14,33), it seems to be likely that the mechanism by which the stalk binds to aEF1α/eEF1α and the above-mentioned role of the stalk protein is conserved between archaea and eukaryotes. Our present results support this view, that is, we identified several amino acid residues involved in the interaction between aP1 and aEF1α, and sequence alignment showed that these residues were conserved between archaea and eukaryotes. Specifically, Glu96, Leu103, Leu106 and Phe107 of aP1 were conserved between aP1 and eukaryotic P1/P2 (Figure 4A). For eEF1α/aEF1α, Lys125 of aEF1α was conserved as a basic residue in eEF1α, whereas Phe129, Leu166 and Ile384 of aEF1α were conserved as hydrophobic residues in eEF1α (Supplementary Figure S5). Indeed, the conserved amino acid residues were present at equivalent positions in aEF1α and eEF1α (Supplementary Figure S6). In addition to the sequence similarities, the results of the present functional study indicated that mutation of Leu110 and Phe111 of the silkworm P1/P2, which are strictly conserved between archaea and eukaryotes (Figure 4A), caused a significant reduction in eEF1α-dependent GTPase activity (Figure 4B). Furthermore, in a previous study, a monoclonal antibody against the conserved CTD of the eukaryotic stalk also seriously compromised eEF1α-dependent GTPase activity (22). On the basis of these facts, we infer that eukaryotic P1/P2 also interacts directly with domains 1 and 3 of eEF1α via Leu110 and Phe111, in a manner similar to the interactions of Leu106 and Phe107 in archaeal aP1 with aEF1α.
ACCESSION NUMBERS
Coordinates and structure factors have been deposited to RCSB Protein Data Bank with the accession codes 3WY9 for the aP1(CTD)•aEF1α•GDP complex and 3WYA for the aEF1α•GDP complex.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We thank Kentaro Baba, Hirotatsu Imai and Yuji Uehara (Niigata University) for sample preparation, and the beamline staff of the PF and PF-AR (KEK, Tsukuba) for technical assistance during data collection.
FUNDING
Grant-in-Aid for Young Scientists (B) from the Japan Society for the Promotion of Science [21770108 to K.I.]; Uchida Energy Science Promotion Foundation [22-1-10 to K.I.]; Grant-in-Aid for Scientific Research (B) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan [24370073 to T.U.]. Funding for open access charge: Grant-in-Aid for Scientific Research (B) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan [24370073 to T.U.].
Conflict of interest statement. None declared.
REFERENCES
- 1.Kubarenko A.V., Sergiev P.V., Rodnina M.V. GTPases of translational apparatus. Mol. Biol. (Mosk) 2005;39:746–761. [PubMed] [Google Scholar]
- 2.Hauryliuk V.V. GTPases of prokaryotic translation apparatus. Mol. Biol. (Mosk) 2006;40:769–783. [PubMed] [Google Scholar]
- 3.Liljas A., Ehrenberg M. Structural Aspects of Protein Synthesis. 2nd edn. Singapore: World Scientific; 2013. The catalysts—translation factors; pp. 149–228. [Google Scholar]
- 4.Wahl M.C., Möller W. Structure and function of the acidic ribosomal stalk proteins. Curr. Protein Pept. Sci. 2002;3:93–106. doi: 10.2174/1389203023380756. [DOI] [PubMed] [Google Scholar]
- 5.Gonzalo P., Reboud J.P. The puzzling lateral flexible stalk of the ribosome. Biol. Cell. 2003;95:179–193. doi: 10.1016/s0248-4900(03)00034-0. [DOI] [PubMed] [Google Scholar]
- 6.Wilson D.N., Nierhaus K.H. Ribosomal proteins in the spotlight. Crit. Rev. Biochem. Mol. Biol. 2005;40:243–267. doi: 10.1080/10409230500256523. [DOI] [PubMed] [Google Scholar]
- 7.Shimmin L.C., Ramirez C., Matheson A.T., Dennis P.P. Sequence alignment and evolutionary comparison of the L10 equivalent and L12 equivalent ribosomal proteins from archaebacteria, eubacteria, and eucaryotes. J. Mol. Evol. 1989;29:448–462. doi: 10.1007/BF02602915. [DOI] [PubMed] [Google Scholar]
- 8.Grela P., Bernadó P., Svergun D., Kwiatowski J., Abramczyk D., Grankowski N., Tchórzewski M. Structural relationships among the ribosomal stalk proteins from the three domains of life. J. Mol. Evol. 2008;67:154–167. doi: 10.1007/s00239-008-9132-2. [DOI] [PubMed] [Google Scholar]
- 9.Liljas A., Gudkov A.T. The structure and dynamics of ribosomal protein L12. Biochimie. 1987;69:1043–1047. doi: 10.1016/0300-9084(87)90004-6. [DOI] [PubMed] [Google Scholar]
- 10.Bocharov E.V., Sobol A.G., Pavlov K.V., Korzhnev D.M., Jaravine V.A., Gudkov A.T., Arseniev A.S. From structure and dynamics of protein L7/L12 to molecular switching in ribosome. J. Biol. Chem. 2004;279:17697–17706. doi: 10.1074/jbc.M313384200. [DOI] [PubMed] [Google Scholar]
- 11.Lee K.M., Yusa K., Chu L.O., Yu C.W., Oono M., Miyoshi T., Ito K., Shaw P.C., Wong K.B., Uchiumi T. Solution structure of human P1•P2 heterodimer provides insights into the role of eukaryotic stalk in recruiting the ribosome-inactivating protein trichosanthin to the ribosome. Nucleic Acids Res. 2013;41:8776–8787. doi: 10.1093/nar/gkt636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wahl M.C., Bourenkov G.P., Bartunik H.D., Huber R. Flexibility, conformational diversity and two dimerization modes in complexes of ribosomal protein L12. EMBO J. 2000;19:174–186. doi: 10.1093/emboj/19.2.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Diaconu M., Kothe U., Schlünzen F., Fischer N., Harms J.M., Tonevitsky A.G., Stark H., Rodnina M.V., Wahl M.C. Structural basis for the function of the ribosomal L7/12 stalk in factor binding and GTPase activation. Cell. 2005;121:991–1004. doi: 10.1016/j.cell.2005.04.015. [DOI] [PubMed] [Google Scholar]
- 14.Naganuma T., Nomura N., Yao M., Mochizuki M., Uchiumi T., Tanaka I. Structural basis for translation factor recruitment to the eukaryotic/archaeal ribosomes. J. Biol. Chem. 2010;285:4747–4756. doi: 10.1074/jbc.M109.068098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee K.M., Yu C.W., Chiu T.Y., Sze K.H., Shaw P.C., Wong K.B. Solution structure of the dimerization domain of the eukaryotic stalk P1/P2 complex reveals the structural organization of eukaryotic stalk complex. Nucleic Acids Res. 2012;40:3172–3182. doi: 10.1093/nar/gkr1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lalioti V.S., Pérez-Fernández J., Remacha M., Ballesta J.P. Characterization of interaction sites in the Saccharomyces cerevisiae ribosomal stalk components. Mol. Microbiol. 2002;46:719–729. doi: 10.1046/j.1365-2958.2002.03179.x. [DOI] [PubMed] [Google Scholar]
- 17.Hagiya A., Naganuma T., Maki Y., Ohta J., Tohkairin Y., Shimizu T., Nomura T., Hachimori A., Uchiumi T. A mode of assembly of P0, P1, and P2 proteins at the GTPase-associated center in animal ribosome: in vitro analyses with P0 truncation mutants. J. Biol. Chem. 2005;280:39193–39199. doi: 10.1074/jbc.M506050200. [DOI] [PubMed] [Google Scholar]
- 18.Krokowski D., Boguszewska A., Abramczyk D., Liljas A., Tchórzewski M., Grankowski N. Yeast ribosomal P0 protein has two separate binding sites for P1/P2 proteins. Mol. Microbiol. 2006;60:386–400. doi: 10.1111/j.1365-2958.2006.05117.x. [DOI] [PubMed] [Google Scholar]
- 19.Naganuma T., Shiogama K., Uchiumi T. The N-terminal regions of eukaryotic acidic phosphoproteins P1 and P2 are crucial for heterodimerization and assembly into the ribosomal GTPase-associated center. Genes Cells. 2007;12:501–510. doi: 10.1111/j.1365-2443.2007.01067.x. [DOI] [PubMed] [Google Scholar]
- 20.Soares M.R., Bisch P.M, Campos de Carvalho A.C., Valente A.P., Almeida F.C. Correlation between conformation and antibody binding: NMR structure of cross-reactive peptides from T. cruzi, human and L. braziliensis. FEBS Lett. 2004;560:134–140. doi: 10.1016/S0014-5793(04)00088-2. [DOI] [PubMed] [Google Scholar]
- 21.Leijonmarch M., Liljas A. Structure of the C-terminal domain of the ribosomal protein L7/L12 from Escherichia coli at 1.7 Å. J. Mol. Biol. 1987;195:555–578. doi: 10.1016/0022-2836(87)90183-5. [DOI] [PubMed] [Google Scholar]
- 22.Uchiumi T., Traut R.R., Kominami R. Monoclonal antibodies against acidic phosphoproteins P0, P1, and P2 of eukaryotic ribosomes as functional probes. J. Biol. Chem. 1990;265:89–95. [PubMed] [Google Scholar]
- 23.Santos C., Ballesta J.P. The highly conserved protein P0 carboxyl end is essential for ribosome activity only in the absence of proteins P1 and P2. J. Biol. Chem. 1995;270:20608–20614. doi: 10.1074/jbc.270.35.20608. [DOI] [PubMed] [Google Scholar]
- 24.Mohr D., Wintermeyer W., Rodnina M.V. GTPase activation of elongation factors Tu and G on the ribosome. Biochemistry. 2002;41:12520–12528. doi: 10.1021/bi026301y. [DOI] [PubMed] [Google Scholar]
- 25.Kothe U., Wieden H.J., Mohr D., Rodnina M.V. Interaction of helix D of elongation factor Tu with helices 4 and 5 of protein L7/12 on the ribosome. J. Mol. Biol. 2004;336:1011–1021. doi: 10.1016/j.jmb.2003.12.080. [DOI] [PubMed] [Google Scholar]
- 26.Nomura N., Honda T., Baba K., Naganuma T., Tanzawa T., Arisaka F., Noda M., Uchiyama S., Tanaka I., Yao M., et al. Archaeal ribosomal stalk protein interacts with translation factors in a nucleotide-independent manner via its conserved C terminus. Proc. Natl. Acad. Sci. U.S.A. 2012;109:3748–3753. doi: 10.1073/pnas.1112934109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Helgstrand M., Mandava C.S., Mulder F.A., Liljas A., Sanyal S., Akke M. The ribosomal stalk binds to translation factors IF2, EF-Tu, EF-G and RF3 via a conserved region of the L12 C-terminal domain. J. Mol. Biol. 2007;365:468–479. doi: 10.1016/j.jmb.2006.10.025. [DOI] [PubMed] [Google Scholar]
- 28.Datta P.P., Sharma M.R, Qi L., Frank J., Agrawal R.K. Interaction of the G’ domain of elongation factor G and the C-terminal domain of ribosomal protein L7/L12 during translocation as revealed by cryo-EM. Mol. Cell. 2005;20:723–731. doi: 10.1016/j.molcel.2005.10.028. [DOI] [PubMed] [Google Scholar]
- 29.Connell S.R., Takemoto C., Wilson D.N., Wang H., Murayama K., Terada T., Shirouzu M., Rost M., Schüler M., Giesebrecht J., et al. Structural basis for interaction of the ribosome with the switch regions of GTP-bound elongation factors. Mol. Cell. 2007;25:751–764. doi: 10.1016/j.molcel.2007.01.027. [DOI] [PubMed] [Google Scholar]
- 30.Gao Y.G., Selmer M., Dunham C.M., Weixlbaumer A., Kelley A.C., Ramakrishnan V. The structure of the ribosome with elongation factor G trapped in the posttranslocational state. Science. 2009;326:694–699. doi: 10.1126/science.1179709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tourigny D.S., Fernández I.S., Kelley A.C., Ramakrishnan V. Elongation factor G bound to the ribosome in an intermediate state of translocation. Science. 2013;340:1235490. doi: 10.1126/science.1235490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stark H., Rodnina M.V., Rinke-Appel J., Brimacombe R., Wintermeyer W., van Heel M. Visualization of elongation factor Tu on the Escherichia coli ribosome. Nature. 1997;389:403–406. doi: 10.1038/38770. [DOI] [PubMed] [Google Scholar]
- 33.Nomura T., Nakano K., Maki Y., Naganuma T., Nakashima T., Tanaka I., Kimura M., Hachimori A., Uchiumi T. In vitro reconstitution of the GTPase-associated centre of the archaebacterial ribosome: the functional features observed in a hybrid form with Escherichia coli 50S subunits. Biochem. J. 2006;396:565–571. doi: 10.1042/BJ20060038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fukunaga J., Ohno S., Nishikawa K., Yokogawa T. A base pair at the bottom of the anticodon stem is reciprocally preferred for discrimination of cognate tRNAs by Escherichia coli lysyl- and glutaminyl-tRNA synthetases. Nucleic Acids Res. 2006;34:3181–3188. doi: 10.1093/nar/gkl414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shimizu T., Nakagaki M., Nishi Y., Kobayashi Y., Hachimori A., Uchiumi T. Interaction among silkworm ribosomal proteins P1, P2 and P0 required for functional protein binding to the GTPase-associated domain of 28S rRNA. Nucleic Acids Res. 2002;30:2620–2627. doi: 10.1093/nar/gkf379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shimizu Y., Inoue A., Tomari Y., Suzuki T., Yokogawa T., Nishikawa K., Ueda T. Cell-free translation reconstituted with purified components. Nat. Biotechnol. 2001;19:751–755. doi: 10.1038/90802. [DOI] [PubMed] [Google Scholar]
- 37.Ito K., Murakami R., Mochizuki M., Qi H., Shimizu Y., Miura K., Ueda T., Uchiumi T. Structural basis for the substrate recognition and catalysis of peptidyl-tRNA hydrolase. Nucleic Acids Res. 2012;40:10521–10531. doi: 10.1093/nar/gks790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Long F., Vagin A.A., Young P., Murshudov G.N. BALBES: a molecular-replacement pipeline. Acta Crystallogr. D Biol. Crystallogr. 2008;64:125–132. doi: 10.1107/S0907444907050172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vitagliano L., Masullo M., Sica F., Zagari A., Bocchini V. The crystal structure of Sulfolobus solfataricus elongation factor 1α in complex with GDP reveals novel features in nucleotide binding and exchange. EMBO J. 2001;20:5305–5311. doi: 10.1093/emboj/20.19.5305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cowtan K. The Buccaneer software for automated model building. 1. Tracing protein chains. Acta Crystallogr. D Biol. Crystallogr. 2006;62:1002–1011. doi: 10.1107/S0907444906022116. [DOI] [PubMed] [Google Scholar]
- 41.Perrakis A., Morris R., Lamzin V.S. Automated protein model building combined with iterative structure refinement. Nat. Struct. Biol. 1999;6:458–463. doi: 10.1038/8263. [DOI] [PubMed] [Google Scholar]
- 42.Vagin A., Teplyakov A. MOLREP: an automated program for molecular replacement. J. Appl. Crystallogr. 1997;30:1022–1025. [Google Scholar]
- 43.Murshudov G.N., Vagin A.A., Dodson E.J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 44.Emsley P., Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 45.Raimo G., Masullo M., Bocchini V. The interaction between the archaeal elongation factor 1α and its nucleotide exchange factor 1β. FEBS Lett. 1999;451:109–112. doi: 10.1016/s0014-5793(99)00560-8. [DOI] [PubMed] [Google Scholar]
- 46.Masullo M., Raimo G., Parente A., Gambacorta A., De Rosa M., Bocchini V. Properties of the elongation factor 1α in the thermoacidophilic archaebacterium Sulfolobus solfataricus. Eur. J. Biochem. 1991;199:529–537. doi: 10.1111/j.1432-1033.1991.tb16151.x. [DOI] [PubMed] [Google Scholar]
- 47.Uchiumi T., Honma S., Nomura T., Dabbs E.R., Hachimori A. Translation elongation by a hybrid ribosome in which proteins at the GTPase center of the Escherichia coli ribosome are replaced with rat counterparts. J. Biol. Chem. 2002;277:3857–3862. doi: 10.1074/jbc.M107730200. [DOI] [PubMed] [Google Scholar]
- 48.Notredame C., Higgins D.G., Heringa J. T-Coffee: a novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 2000;302:205–217. doi: 10.1006/jmbi.2000.4042. [DOI] [PubMed] [Google Scholar]
- 49.Larkin M.A., Blackshields G., Brown N.P., Chenna R., McGettigan P.A., McWilliam H., Valentin F., Wallace I.M., Wilm A., Lopez R., et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 50.Kessenbrock K., Fritzler M.J., Groves M., Eissfeller P., von Müheln C.A., Höpfl P., Mahler M. Diverse humoral autoimmunity to the ribosomal P proteins in systemic lupus erythematosus and hepatitis C virus infection. J. Mol. Med. 2007;85:953–959. doi: 10.1007/s00109-007-0239-5. [DOI] [PubMed] [Google Scholar]
- 51.Too P.H., Ma M.K., Mak A.N., Wong Y.T., Tung C.K., Zhu G., Au S.W., Wong K.B., Shaw P.C. The C-terminal fragment of the ribosomal P protein complexed to trichosanthin reveals the interaction between the ribosome-inactivating protein and the ribosome. Nucleic Acids Res. 2009;37:602–610. doi: 10.1093/nar/gkn922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schmeing T.M., Voorhees R.M., Kelley A.C., Gao Y.G., Murph F.V, Weir J.R., Ramakrishnan V. The crystal structure of the ribosome bound to EF-Tu and aminoacyl-tRNA. Science. 2009;326:688–694. doi: 10.1126/science.1179700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ban N., Nissen P., Hansen J., Moore P.B., Steitz T.A. The complete atomic structure of the large ribosomal subunit at 2.4 Å resolution. Science. 2000;289:905–920. doi: 10.1126/science.289.5481.905. [DOI] [PubMed] [Google Scholar]
- 54.Kobayashi K., Kikuno I., Kuroha K., Saito K., Ito K., Ishitani R., Inada T., Nureki O. Structural basis for mRNA surveillance by archaeal Pelota and GTP-bound EF1α complex. Proc. Natl. Acad. Sci. U.S.A. 2010;107:17575–17579. doi: 10.1073/pnas.1009598107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Baba K., Tumuraya K., Tanaka I., Yao M., Uchiumi T. Molecular dissection of the silkworm ribosomal stalk complex: the role of multiple copies of the stalk proteins. Nucleic Acids Res. 2013;41:3635–3643. doi: 10.1093/nar/gkt044. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.