


Abstract
Non-coding RNAs (ncRNAs) are known to play important functional roles in the cell. However, their identification and recognition in genomic sequences remains challenging. In silico methods, such as classification tools, offer a fast and reliable way for such screening and multiple classifiers have already been developed to predict well-defined subfamilies of RNA. So far, however, out of all the ncRNAs, only tRNA, miRNA and snoRNA can be predicted with a satisfying sensitivity and specificity. We here present ptRNApred, a tool to detect and classify subclasses of non-coding RNA that are involved in the regulation of post-transcriptional modifications or DNA replication, which we here call post-transcriptional RNA (ptRNA). It (i) detects RNA sequences coding for post-transcriptional RNA from the genomic sequence with an overall sensitivity of 91% and a specificity of 94% and (ii) predicts ptRNA-subclasses that exist in eukaryotes: snRNA, snoRNA, RNase P, RNase MRP, Y RNA or telomerase RNA. AVAILABILITY: The ptRNApred software is open for public use on http://www.ptrnapred.org/.
INTRODUCTION
There are many different types of RNA with multiple functions in the cell. Some RNA molecules contribute to the translation of genetic information into protein and the regulation of genes. Others function enzymatically by catalyzing biological reactions. While the non-coding regions in the genome were first believed to be dispensable sequences, they have been shown to code for RNA families that play important roles in the eukaryotic cell. These so-called non-coding RNAs (ncRNAs) do not code for protein but are involved in many regulatory processes and can be divided into a tremendous variety of highly plethoric and versatile families that are essential for the cellular function (1). Hence, they form a vast and to a large extent unexplored reservoir of potentially valuable medical biomarkers (2,3). For their identification, modern techniques like next-generation-sequencing and microarray-technologies are being employed (4,5). These techniques provide an immense amount of data and offer ample opportunities to identify novel classes of non-coding RNA. However, the experimental analysis of new sequences is time-consuming and complex, indicating the need to find alternative approaches for their analysis. Promising and auspicious approaches are given by in silico methods. Due to phylogenetic relationships, sequences of non-coding RNA show similarities regarding their properties. They can be divided into subclasses based on their conserved properties, meaning sequence conservation and structural conservation (6). Computational methods, such as classification tools, offer a fast and reliable way to analyze and classify sequences by exploiting conserved properties among the sequences (6,7).
Various classification systems have been developed to predict different subsets of RNA, using machine learning and phylogenetic approaches (8–12). So far, tRNAs can be detected reliably using tRNAScan-SE (13). Furthermore, various approaches have been established to detect miRNA (14) and other small RNA subsets. Recently, snoReport was introduced, which is designed to recognize small nucleolar RNA (snoRNA) from the genome without using any target information (15). Most of these systems achieve a satisfying accuracy, however not every RNA family can be predicted. For example, to this point, there is no tool for the prediction of small nuclear RNA (snRNA), Ribonuclease P (RNase P), Ribonuclease MRP (RNase MRP), Y RNA and telomerase RNA. Facing the continuing increase in the number of human RNAs in databases like Rfam (16,17), it is necessary to extend the current possibilities of RNA prediction. SnRNA, RNase P, RNase MRP, Y RNA and telomerase RNA have in common, that, besides snoRNA, they are involved in post-transcriptional modification or DNA replication in eukaryotes (18–23).
The aim of this study was to develop a tool that can predict and differentiate among sequences coding for these RNA families, for which, for the sake of convenience, we here use the term ‘post-transcriptional RNA (ptRNA)’.
PtRNA-subclasses are not only distinct regarding their function, but also regarding their sequence. Therefore, we hypothesized that implementation of certain algorithms would make it possible to identify ptRNA from a genomic sequence. Machine learning algorithms, such as support vector machines (SVMs), have shown high accuracy in the development of classification systems (24–26). Recent advancement in small non-coding miRNA prediction has achieved high performance using machine learning approaches (27–30). Moreover, SVMs are also employed in the prediction of sequence based secondary structure of RNA (31,32). The available tools for designing the machine learning classifier, like SVM-Light (33) and LibSVM (34) are often used for the development of new algorithms for RNA prediction.
With the introduction of ‘ptRNApred’, this study offers a novel opportunity to detect and classify ptRNA without using any target information. ‘ptRNApred’ (i) detects RNA sequences coding for post-transcriptional RNA from the genomic sequence and (ii) predicts ptRNA-subclasses that exist in eukaryotes: snRNA, snoRNA, RNase P, RNase MRP, Y RNA or telomerase RNA.
MATERIALS AND METHODS
Dataset
2040 sequences of ncRNA were obtained from the NONCODE database (35), including 268 sequences of RNase P, 14 sequences of RNase MRP, 1443 sequences of snoRNA (1430 + 13 scaRNA), 46 sequences of telomerase RNA, 14 sequences of Y RNA and 255 sequences of snRNA (Table 1). These sequences were used as a dataset for the multiclass-classifier and as a positive set for our binary classifier. The negative set was made up of sequences of tRNA, 5s ribosomal RNA and miRNA that were derived from Rfam (17). Since our classifier focuses on eukaryotes, our selection of miRNA-sequences was restricted to sequences from the species Homo sapiens, Mus musculus and Drosophila melanogaster. The redundancy of the sequences within a set was removed using CD-Hit (36) at a threshold of 0.9 for the positive set and at 0.8 for the negative set of sequences. After removal of redundancy, the ratio of sequences in the negative to positive set was 3:2. Comparable ratios have been frequently used for the generation of SVMs (37,38).
Table 1. Total number of test and training sequences.
| Training sequences | Testing sequences | |
|---|---|---|
| RNase P | 178 | 90 |
| RNase MRP | 9 | 5 |
| snoRNA + scaRNA | 978 + 9 | 452 + 4 |
| telomerase RNA | 29 | 17 |
| Y RNA | 9 | 5 |
| snRNA | 170 | 85 |
The table displays the number of training and testing sequences of each ptRNA-subclass used for the SVM.
All sets of sequences were divided into two parts: two-third of each set of sequences were used for training the classifiers and one-third was used for testing the performance of the classifier. Table 1 gives an overview on the number of training and testing sequences of each ptRNA-subclass.
A detailed list of the sequences is provided on the classifier's website http://www.ptrnapred.org/. For testing its performance on RNase P, 329 RNase P sequences were downloaded from ‘The Ribonuclease P Database’ (48). Performance on coding RNA was tested using 10 000 randomly downloaded mRNA sequences from Ensembl (Release 72, June 2013).
Features for classification
Feature selection and SVM training was performed using two sets of input parameters: The first set was based on the primary sequence and the second set considered the secondary structure which was predicted with RNAfold (Version 2.0.7) (39). Primary sequence properties were mainly derived from dinucleotide properties employing DiProGB (40), using a sliding window approach (window size: 2 nucleotides). Some of the properties in DiProGB are highly correlating to each other. The use of highly correlating features for classification would not only be redundant in information, but would overfit the classifier. In order to determine which of the features that we derived from DiProGB were correlating, we determined the Pearson correlation coefficients among all possible dinucleotide properties. Two features were considered as highly correlating when the Pearson coefficient was >0.9. As an example, the dinucleotide property ‘stacking energy’ was highly correlated to the property ‘melting temperature’. Whenever one of two features were highly correlating, one of them was randomly discarded. In our example, we used ‘stacking energy’ as an input feature and discarded ‘melting temperature’ from consideration as a feature for classification. A table of the selected dinucleotide properties as well as their dinucleotide values (40) is provided in the supplement (Supplementary Table S1).
Secondary structures of every sequence were calculated via RNAfold (39), accessing the Vienna RNA Package (41). Fifty-two different properties were derived from the secondary structure, e.g. the number of loops, the number of bulges, the number of hairpins or the frequency of nucleotides involved in substructures.
Additionally, we included 32 triplet element properties employed by miPred, a triplet SVM for the classification of miRNA (42): MiPred considers the middle nucleotide among the triplet elements, resulting in 32 (4 × 8) possible combinations, which are denoted as ‘U(((’, ‘A((.’, etc.
Altogether, ptRNApred uses 91 features for classification. A detailed description of the feature selection is provided in the supplement (Supplementary Section S1).
Classification system
To create optimal conditions for classification, we compared the outcome of two different algorithms.
On the one hand, we employed a Random Forest according to Breiman (43) as a sophisticated classification method. Random Forests operate by constructing a multitude of decision trees at training time and outputting the class that is the mode of the classes output by individual trees.
On the other hand, we employed LibSVM (34), a library of SVM, which serves as an interface to train and build SVMs based on certain vectors. LibSVM was a superior machine learning algorithm for training our classifiers, since it gave us a better accuracy than Random Forest prediction. Further information is provided in the ‘Validation of the algorithm’ part of the ‘Results’ section.
Implementation of LibSVM
The SVM algorithm defines a hyperplane in the feature space with maximum margin distinguishing positive instances from negative (44). LibSVM provides a python script to optimize the grid parameters C and gamma (γ). C stands for cost function, i.e. penalty for the misclassification in the training set and γ is a free parameter deciding upon the impact of each training vector. The parameters are thus used for designing the classifier from a training set. Therefore, we used a (Gaussian) Radial Basis Function (RBF) (45) kernel for classification.
In order to decide the parameters C and γ, LibSVM obtains cross validation (CV) accuracy for each possible parameter setting. Regarding our binary classifier, the highest CV accuracy was achieved when C was set to 32768 and γ was set to 0.008 (Figure 1a). These parameters were used to train the whole training set and to generate the final model.
Figure 1.
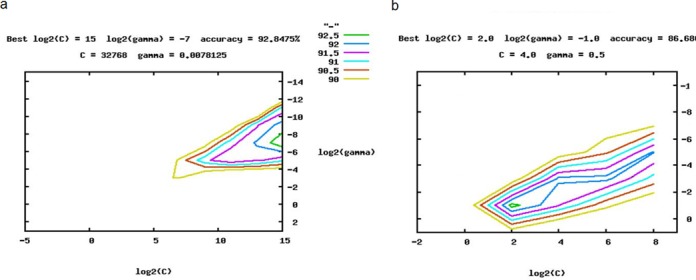
C and γ determination and 5-fold cross validation using LibSVM. The figure shows graphs for different values of the parameters C (a trade-off for misclassification) and γ (inverse width of RBF kernel) on a logarithmic X and Y axis. The ranges of the axes describe the different values that were tested, searching the optimal C and γ values in the grid space. The different colors in the diagram display the different accuracies obtained while optimizing C and γ values. We chose the C and γ values according to the green graphs, respectively, representing the C and γ value with the highest accuracy. (a) C and γ determination and 5-fold cross validation of the two-class SVM. The green graph represents the optimal values for C and gamma. In this case, the highest 5-fold cross validation accuracy (92.89%) is achieved when C = 32768 and γ = 0.008. (b) C and γ determination and 5-fold cross validation of the multi-class SVM. The green graph represents the optimal values for C and gamma. In this case, the highest 5-fold cross validation accuracy (86.69%) is achieved when C = 4 and γ = 0.5.
For multi-class classification, under a given (C, γ), LibSVM uses the one-against-one, one-against-all and sparse method to build hyper planes and to obtain the CV accuracy. Hence, the parameter selection tool suggests the same (C, γ) for all k(k−1)/2 decision functions. Yuan et al. (46,47) discuss issues of using the same or different parameters for the k(k−1)/2 two-class problems. In our case, C = 4 and γ = 0.5 as well as usage of the sparse method gave us the best CV accuracy for the multi-class classification (Figure 1b).
Work flow and output of ptRNApred
The web server implementation accepts sequences in a FASTA-format as an input that can be either uploaded as a file or pasted into the text box (Supplementary Figure S1). By checking ‘Post-Transcriptional RNA’, an in-built Perl script calculates input vectors for the pre-trained model to predict whether or not the input sequence belongs to the group of post-transcriptional RNA. Additionally selecting ‘RNA family’, the server also predicts the RNA-subclass.
Altogether, the output includes the prediction for ptRNA as well as the classification of the RNA class within the ptRNA. Additionally, it displays the minimum free energy using RNA-fold (39) as well as the secondary structure, using VARNA (Version 3.1) (48) (Supplementary Figure S2). The output can directly be downloaded.
RESULTS
We created a two-step classifier to distinguish sequences of ptRNA and non-ptRNA in a binary classification, and for the prior separate six classes of post-transcriptional RNA (snRNA, snoRNA, RNase P, RNase MRP, Y RNA or telomerase RNA) in a multi-class classification. Since the binary and multi-class classifiers were trained with separate data and thus function independently, the accuracy was calculated for each individual classifier.
In a 5-fold cross validation, using balanced amounts of randomly selected sequences throughout the positive and negative sets, the binary classifier yields an accuracy of 93% within the training set (Figure 1a) and the multi-class classifier yields a 5-fold cross validation accuracy of 87% (Figure 1b).
When testing the classifiers with the test set of sequences, the binary classifier showed an accuracy of 93%, with a sensitivity of 91%, a specificity of 94% and an overall precision of 90%. The multi-class classifier showed an accuracy of 91%.
The fact that the accuracy in the test set is higher than the 5-fold cross validation accuracy in the training set suggests that an increase in the number of sequences leads to a more accurate prediction. Regarding this matter, we observed an increase of the accuracy of the multi-class classification when adding more sequences to the training set.
Validation of the method
In order to validate our tool by comparing it to existing tools that can predict ptRNA, we found snoReport (15) as an advanced tool for the prediction of snoRNA. snoReport can predict orphan snoRNA without using target information and is therefore similar to our approach. To compare our tool to snoReport, we derived snoRNA sequences from a mouse genome from Ensembl (49) and used them as an independent set. In total, we used an input of 1603 sequences of snoRNA. As a result, snoReport identified 733 sequences correctly. In contrast, our classifier identified 1589 sequences correctly. Furthermore, we abstracted a human dataset with 1641 sequences of snoRNA from Ensembl. While snoReport identified 852 of the snoRNA-sequences correctly, our tool identified 1611 (Table 2).
Table 2. Comparison between snoReport and ptRNApred.
| Organism | RNA class | Total number of sequencesa | Number of sequences identified by snoReport (% of total number of sequences) | Number of sequences identified by ptRNApred (% of total number of sequences) |
|---|---|---|---|---|
| M. musculus | snoRNA | 1603 | 737(46%) | 1589(99%) |
| H. sapiens | snoRNA | 1641 | 852(52%) | 1611(98%) |
| snoU13b | 245b | 0(0%)b | 245(100%)b |
A murine and a human dataset of snoRNA was abstracted from Ensembl (49) and performance of ptRNApred was compared to snoReport as a well-established tool for snoRNA prediction. ptRNApred achieved higher sensitivity than snoReport (99 versus 46% on the murine and 98 versus 52% on the human set of sequences). Regarding snoU13, a member of the snoRNAs, there is an even larger difference in the sensitivity (100 versus 0%).
aTotal number of snoRNA-sequences downloaded from Ensembl (49).
bsnoU13 among the human snoRNA sequences.
In order to analyze the low sensitivity of snoReport we inspected the sequences that it fails to classify. We found that snoReport was not able to detect a major snoRNA-subclass snoU13. snoU13 was identified in 1989 (50). It has been well characterized in 35 species by both functional assay and prediction. It is involved in the nucleolytic cleavage at the 3′ end of 18S rRNA where it works as a trans-acting factor (51).
snoReport was unable to assign any of 245 snoU13-sequences in a human cohort to snoRNA. Our tool ptRNApred however identified all of them correctly (Table 2). Current approaches to identify different RNA families heavily rely on their secondary structure conservation. Consequently, these approaches are accurate as long as the RNA families show high secondary structure conservation. However, as soon as a RNA-family lacks a secondary structure because it e.g. has no complementary sequences within itself, it will be misclassified. This explains why snoReport failed to identify snoU13 and possibly also the remaining ∼550 snoRNA sequences: snoU13 does not form any secondary structure conservation as it forms a loop.
Validation of the algorithm
As mentioned in the ‘Materials and Methods’ section, we compared the algorithm implemented in our tool to Random Forest classification. As a result, implementation of Random Forest yielded an overall accuracy of 82%. In comparison, using LibSVM, our multi-class classifier developed yields an accuracy of 91%. Detailed results of the performance of Random Forest based prediction of the ptRNA-subclasses can be found in the Supplement (Supplementary Table S2).
Variable importance
Classification features have an individual impact on the differentiation of RNA-classes. To determine the importance of each of the 91 features for classification of ptRNA, an F-score was calculated for each feature, using LibSVM. F-scores can be interpreted as a weighted average of the precision and recall, where an F-score reaches its best value at 1 and worst score at 0. Supplementary Table S3 depicts the F-score corresponding to every feature.
Additionally, even though Random Forests did not contribute to ptRNApred predictions, those provide useful information on feature importance. One of the measures of variable importance in Random Forest is the mean decrease in accuracy, calculated using the out-of-bag sample. The difference between the prediction accuracy on the untouched out-of-bag sample and that on the out-of-bag sample permuted on one predictor variable is averaged over all trees in the forest and normalized by the standard error. This gives the mean decrease in accuracy of that particular predictor variable which has been permuted. Thus, the importance of the predictor variables can be ranked by their mean decrease in accuracy. Supplementary Table S3 depicts the Gini-Index corresponding to every feature.
Interestingly, comparing the 25 most discriminative feature variables according to F-score and Gini-Index (Supplementary Table S4), dinucleotide properties achieve high ranks: 9 of the 10 most discriminative features according to the F-score are composed of dinucleotide properties. Furthermore, all of the 15 dinucleotide properties can be found among the 25 most discriminative properties. According to the Gini-Index, 12 properties can be found among the 25 most discriminative properties, whereas only three of them can be found among the top 10, indicating the importance of the secondary structure.
Validation of the feature number
As mentioned in the ‘Materials and Methods’ section, a general concern for all machine learning approaches is that one has too many features, i.e. that one trains on features that are not relevant—referred to as overfitting. This was excluded by the above mentioned cross validation test. On the other hand, too few features would lead to loss of (overall) accuracy. In order to confirm that using less features would lead to loss of accuracy, we selected the 78 most discriminative features based on Random Forest prediction, using the R package ‘Boruta’. When using these 78 features instead of 91 features, the 5-fold cross validation accuracy decreased from 92.89 to 74.46% (Supplementary Figure S3).
Performances on a non-eukaryotic system
Even though ptRNApred is designed to primarily predict eukaryotic sequences, ptRNApred was tested for performance on RNase P sequences, using 329 RNase P sequences from ‘The Ribonuclease P Database’ (52). RNAse P has not only been described in eukaryotic systems (53), but rather distributes among different organisms (20). Interestingly, our tool predicted the RNAse P sequences with an accuracy of 97.3%.
Performances on coding RNA
Over the last few years, several tools have been developed to distinguish coding from non-coding RNA (54–57). Even though our aim was to develop a novel tool that can differentiate between subclasses of non-coding RNAs and not to distinguish between coding and non-coding RNAs, ptRNApred was tested for performance on mRNA. Therefore, ptRNApred was challenged by 10 000 mRNA randomly downloaded sequences from Ensembl. Surprisingly, only 15 of the sequences were misclassified as ptRNA. Therefore, the accuracy of separating out mRNA is 99.85%.
DISCUSSION
RNA classes that are involved in post-transcriptional modification or DNA replication in eukaryotes not only have functional similarities, but rather form a distinct group of ncRNA with sequence and conformational similarities, that make it possible to accurately distinguish them from other RNA classes.
We here present a novel user-friendly tool that employs discriminative properties to (i) distinguish what we here call ‘post-transcriptional RNA’ from other classes of ncRNA and (ii) discriminate between the different types of ptRNA.
An advantage of the tool is its highly accurate and therefore reliable output. This is based on its working principle: More than 90 features that are derived from the primary sequence and secondary structure are used to define properties for characterization and differentiation between the subclasses. Analyzing the most discriminative feature variables according to F-score and Gini-Index, the most important features are not only based on the secondary structure, but even more importantly on dinucleotide properties. This might be due to the fact that many nucleic acid properties such as nucleic acid stability, for example, seem to depend primarily on the identity of nearest-neighbor nucleotides (58). Furthermore, the corresponding nearest-neighbor model is also the basis for RNA secondary structure prediction by free-energy minimization (59). It has long been known that also thermodynamic but also conformational nucleotide properties may play a role. It has been shown, for example, that promoter locations can be predicted adopting dinucleotide stiffness parameters derived from molecular dynamic simulations (60). Our tool underscores the value of these properties.
Recently, a focus has been on the characterization of snoRNA. However, there has been no classifier that could predict snRNA, RNase P or RNase MRP, even though these subclasses have conserved secondary structures. Identification of those RNA classes has as yet been dependent on sequence alignment. This technique frequently leads to misidentification, especially if the particular homologous sequence is not present in any database.
Furthermore, ptRNApred can be used to elucidate unknown relations and derivation of RNA classes. Based on the assumption that evolutionarily close RNA families have similar sequence properties, one may speculate that tools like ptRNApred will falsely arrange evolutionarily close RNA families into the same group.
A deficiency of this tool is that its accuracy is dependent on the amount of published ptRNA sequences. Some classes of ptRNA, for example Y RNA, are to this point just rarely available in the NONCODE database, making it hard to define discriminative sequence properties and setting limits to the accuracy. In the current era of high-throughput next generation sequencing, where a large amount of genomic data is generated each day, ptRNA sequences that will be added to the database in the future can be used to increase training and test set, setting a base to improve the classifier. On the other hand, discovery of new candidates for ptRNA requires a method which can classify them rapidly and reliably. Our tool offers a solution to this problem. Also, facing the huge amount of new sequences that are found in Next Generation Sequencing (NGS) or RNA-seq data (61), it is important to include such algorithms into NGS pipelines. For such purposes, we provide a standalone version.
We implemented our method as a web-based server for free public use. Transparent and user-friendly design makes it possible for everyone to understand and employ the tool. Data and scripts for the development of the tool can be downloaded, allowing anyone to acquire the working principle and improve ptRNApred.
Collectively, our tool offers a fast and reliable way to analyze cDNA and RNA sequences and outperforms the existing classifiers. Furthermore, the tool provides comprehensive annotations. Therefore, ptRNApred introduces different opportunities to identify and classify new and un-annotated RNA sequences.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Footnotes
The authors wish it to be known that, in their opinion, the first two authors should be regarded as Joint First Authors.
FUNDING
Deutsche Forschungsgemeinschaft: the research training programs Modulation of Autoimmunity [GRK1727/1]; Genes, Environment and Inflammation [GRK 1743/1]; Excellence Cluster Inflammation at Interfaces [EXC 306/2]. Funding for open access charge: Deutsche Forschungsgemeinschaft: the research training programs Modulation of Autoimmunity [GRK1727/1]; Genes, Environment and Inflammation [GRK 1743/1]; Excellence Cluster Inflammation at Interfaces [EXC 306/2].
Conflict of interest statement. None declared.
REFERENCES
- 1.Carninci P., Kasukawa T., Katayama S., Gough J., Frith M.C., Maeda N., Oyama R., Ravasi T., Lenhard B., Wells C., et al. The transcriptional landscape of the mammalian genome. Science. 2005;309:1559–1563. doi: 10.1126/science.1112014. [DOI] [PubMed] [Google Scholar]
- 2.Mallardo M., Poltronieri P., D'Urso O. Non-protein coding RNA biomarkers and differential expression in cancers: a review. J. Exp. Clin. Cancer Res. 2008;27:19. doi: 10.1186/1756-9966-27-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kim T., Reitmair A. Non-coding RNAs: functional aspects and diagnostic utility in oncology. Int. J. Mol. Sci. 2013;14:4934–4968. doi: 10.3390/ijms14034934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beck D., Ayers S., Wen J., Brandl M.B., Pham T.D., Webb P., Chang C.-C., Zhou X. Integrative analysis of next generation sequencing for small non-coding RNAs and transcriptional regulation in Myelodysplastic Syndromes. BMC Med. Genomics. 2011;4:19. doi: 10.1186/1755-8794-4-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jung C.-H., Hansen M.A., Makunin I.V., Korbie D.J., Mattick J.S. Identification of novel non-coding RNAs using profiles of short sequence reads from next generation sequencing data. BMC Genomics. 2010;11:77. doi: 10.1186/1471-2164-11-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bompfunewerer A. Evolutionary patterns of non-coding RNAs. Theory Biosci. 2005;123:301–369. doi: 10.1016/j.thbio.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 7.Lu Z.J., Yip K.Y., Wang G., Shou C., Hillier L.W., Khurana E., Agarwal A., Auerbach R., Rozowsky J., Cheng C., et al. Prediction and characterization of noncoding RNAs in C. elegans by integrating conservation, secondary structure, and high-throughput sequencing and array data. Genome Res. 2011;21:276–285. doi: 10.1101/gr.110189.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Artzi S., Kiezun A., Shomron N. miRNAminer: a tool for homologous microRNA gene search. BMC Bioinformatics. 2008;9:39. doi: 10.1186/1471-2105-9-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hertel J., Stadler P.F. Hairpins in a Haystack: recognizing microRNA precursors in comparative genomics data. Bioinformatics. 2006;22:e197–e202. doi: 10.1093/bioinformatics/btl257. [DOI] [PubMed] [Google Scholar]
- 10.Laslett D., Canback B. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 2004;32:11–16. doi: 10.1093/nar/gkh152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lagesen K., Hallin P., Rødland E.A., Staerfeldt H.-H., Rognes T., Ussery D.W. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007;35:3100–3108. doi: 10.1093/nar/gkm160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lowe T.M., Eddy S.R. A computational screen for methylation guide snoRNAs in yeast. Science. 1999;283:1168–1171. doi: 10.1126/science.283.5405.1168. [DOI] [PubMed] [Google Scholar]
- 13.Lowe T.M., Eddy S.R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–964. doi: 10.1093/nar/25.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yoon S., De Micheli G. Prediction and Analysis of Human microRNA Regulatory Modules. Conf. Proc. IEEE Eng. Med. Biol. Soc. 2005;5:4799–4802. doi: 10.1109/IEMBS.2005.1615545. [DOI] [PubMed] [Google Scholar]
- 15.Hertel J., Hofacker I.L., Stadler P.F. SnoReport: computational identification of snoRNAs with unknown targets. Bioinformatics. 2008;24:158–164. doi: 10.1093/bioinformatics/btm464. [DOI] [PubMed] [Google Scholar]
- 16.Griffiths-Jones S., Bateman A., Marshall M., Khanna A., Eddy S.R. Rfam: an RNA family database. Nucleic Acids Res. 2003;31:439–441. doi: 10.1093/nar/gkg006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Burge S.W., Daub J., Eberhardt R., Tate J., Barquist L., Nawrocki E.P., Eddy S.R., Gardner P.P., Bateman A. Rfam 11.0: 10 years of RNA families. Nucleic Acids Res. 2013;41:D226–D232. doi: 10.1093/nar/gks1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thore S., Mayer C., Sauter C., Weeks S., Suck D. Crystal structures of the Pyrococcus abyssi Sm Core and its complex with RNA: common features of binding in archaea and eukarya. J. Biol. Chem. 2003;278:1239–1247. doi: 10.1074/jbc.M207685200. [DOI] [PubMed] [Google Scholar]
- 19.Kiss T. Small nucleolar RNA-guided post-transcriptional modification of cellular RNAs. EMBO J. 2001;20:3617–3622. doi: 10.1093/emboj/20.14.3617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pannucci J.A., Haas E.S., Hall T.A., Harris J.K., Brown J.W. RNase P RNAs from some archaea are catalytically active. Proc. Natl. Acad. Sci. U.S.A. 1999;96:7803–7808. doi: 10.1073/pnas.96.14.7803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Woodhams M.D., Stadler P.F., Penny D., Collins L.J. RNase MRP and the RNA processing cascade in the eukaryotic ancestor. BMC Evol. Biol. 2007;7(Suppl. 1):S13. doi: 10.1186/1471-2148-7-S1-S13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Perreault J., Perreault J.-P., Boire G. Ro-associated Y RNAs in metazoans: evolution and diversification. Mol. Biol. Evol. 2007;24:1678–1689. doi: 10.1093/molbev/msm084. [DOI] [PubMed] [Google Scholar]
- 23.Lustig A.J. Crisis intervention: the role of telomerase. Proc. Natl. Acad. Sci. U.S.A. 1999;96:3339–3341. doi: 10.1073/pnas.96.7.3339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang C., Ding C., Meraz R.F., Holbrook S.R. PSoL: a positive sample only learning algorithm for finding non-coding RNA genes. Bioinformatics. 2006;22:2590–2596. doi: 10.1093/bioinformatics/btl441. [DOI] [PubMed] [Google Scholar]
- 25.Arrial R.T., Togawa R.C., Brigido M. Screening non-coding RNAs in transcriptomes from neglected species using PORTRAIT: case study of the pathogenic fungus Paracoccidioides brasiliensis. BMC Bioinformatics. 2009;10:239. doi: 10.1186/1471-2105-10-239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Song Y., Liu C., Qu J., National Science Foundation . Learning parameters for non-coding RNA sequence-structure alignment. Washington DC: IEEE; 2009. 2009 IEEE INTERNATIONAL CONFERENCE ON BIOINFORMATICS AND BIOMEDICINE WORKSHOPS; pp. 73–77. [Google Scholar]
- 27.Williams P.H., Eyles R., Weiller G. Plant MicroRNA prediction by supervised machine learning using C5.0 Decision Trees. J. Nucleic Acids. 2012;2012:1–10. doi: 10.1155/2012/652979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sturm M., Hackenberg M., Langenberger D., Frishman D. TargetSpy: a supervised machine learning approach for microRNA target prediction. BMC Bioinformatics. 2010;11:292. doi: 10.1186/1471-2105-11-292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jha A., Shankar R. Employing machine learning for reliable miRNA target identification in plants. BMC Genomics. 2011;12:636. doi: 10.1186/1471-2164-12-636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ding J., Zhou S., Guan J. MiRenSVM: towards better prediction of microRNA precursors using an ensemble SVM classifier with multi-loop features. BMC Bioinformatics. 2010;11:S11. doi: 10.1186/1471-2105-11-S11-S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhao Y., Wang Z. RNA secondary structure prediction based on support vector machine classification. Sheng Wu Gong Cheng Xue Bao. 2008;24:1140–1148. [PubMed] [Google Scholar]
- 32.Zhao Y., Wang Z. Consensus RNA secondary structure prediction based on support vector machine classification. Chin. J. Biotechnol. 2008;24:1140–1148. [PubMed] [Google Scholar]
- 33.Joachims T. Making large-Scale SVM Learning Practical. Advances in Kernel Methods - Support Vector Learning. MIT-Press. 1999;11:41–56. [Google Scholar]
- 34.Chang C.-C., Lin C.-J. LIBSVM: a library for support vector machines. ACM Trans. Intell. Syst. Technol. 2011;2:1–27. [Google Scholar]
- 35.Bu D., Yu K., Sun S., Xie C., Skogerbo G., Miao R., Xiao H., Liao Q., Luo H., Zhao G., et al. NONCODE v3.0: integrative annotation of long noncoding RNAs. Nucleic Acids Res. 2012;40:D210–D215. doi: 10.1093/nar/gkr1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li W., Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22:1658–1659. doi: 10.1093/bioinformatics/btl158. [DOI] [PubMed] [Google Scholar]
- 37.Han L.Y. Prediction of RNA-binding proteins from primary sequence by a support vector machine approach. RNA. 2004;10:355–368. doi: 10.1261/rna.5890304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bhasin M., Raghava G.P.S. GPCRpred: an SVM-based method for prediction of families and subfamilies of G-protein coupled receptors. Nucleic Acids Res. 2004;32:W383–W389. doi: 10.1093/nar/gkh416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ding Y. Statistical and Bayesian approaches to RNA secondary structure prediction. RNA. 2006;12:323–331. doi: 10.1261/rna.2274106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Friedel M., Nikolajewa S., Sühnel J., Wilhelm T. DiProGB: the dinucleotide properties genome browser. Bioinformatics. 2009;25:2603–2604. doi: 10.1093/bioinformatics/btp436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lorenz R., Bernhart S.H., Höner zu Siederdissen C., Tafer H., Flamm C., Stadler P.F., Hofacker I.L. ViennaRNA Package 2.0. Algorithms Mol. Biol. 2011;6:26. doi: 10.1186/1748-7188-6-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xue C., Li F., He T., Liu G.-P., Li Y., Zhang X. Classification of real and pseudo microRNA precursors using local structure-sequence features and support vector machine. BMC Bioinformatics. 2005;6:310. doi: 10.1186/1471-2105-6-310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Breiman L. Bagging Predictors. Machine Learning. 1996;24:123–140. [Google Scholar]
- 44.Vapnik V.N. An overview of statistical learning theory. IEEE Trans Neural Netw. 1999;10:988–999. doi: 10.1109/72.788640. [DOI] [PubMed] [Google Scholar]
- 45.Kaminski W., Strumillo P. Kernel orthonormalization in radial basis function neural networks. IEEE Trans Neural Netw. 1997;8:1177–1183. doi: 10.1109/72.623218. [DOI] [PubMed] [Google Scholar]
- 46.Yuan G.-X., Ho C.-H., Lin C.-J. Recent advances of large-scale linear classification. Proceedings of the IEEE. 2012;100 [Google Scholar]
- 47.Chen Y., Lu B.-L., Zhao H. The 2012 International Joint Conference on Neural Networks (IJCNN). Brisbane: IEEE; 2012. Parallel learning of large-scale multi-label classification problems with min-max modular LIBLINEAR; pp. 1–7. [Google Scholar]
- 48.Darty K., Denise A., Ponty Y. VARNA: Interactive drawing and editing of the RNA secondary structure. Bioinformatics. 2009;25:1974–1975. doi: 10.1093/bioinformatics/btp250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Flicek P., Ahmed I., Amode M.R., Barrell D., Beal K., Brent S., Carvalho-Silva D., Clapham P., Coates G., Fairley S., et al. Ensembl 2013. Nucleic Acids Res. 2013;41:D48–D55. doi: 10.1093/nar/gks1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tyc K., Steitz J.A. U3, U8 and U13 comprise a new class of mammalian snRNPs localized in the cell nucleolus. EMBO J. 1989;8:3113–3119. doi: 10.1002/j.1460-2075.1989.tb08463.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cavaillé J., Hadjiolov A.A., Bachellerie J.P. Processing of mammalian rRNA precursors at the 3’ end of 18S rRNA. Identification of cis-acting signals suggests the involvement of U13 small nucleolar RNA. Eur. J. Biochem. FEBS. 1996;242:206–213. doi: 10.1111/j.1432-1033.1996.0206r.x. [DOI] [PubMed] [Google Scholar]
- 52.Brown J.W. The Ribonuclease P Database. Nucleic Acids Res. 1999;27:314. doi: 10.1093/nar/27.1.314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jarrous N., Reiner R. Human RNase P: a tRNA-processing enzyme and transcription factor. Nucleic Acids Res. 2007;35:3519–3524. doi: 10.1093/nar/gkm071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Badger J.H., Olsen G.J. CRITICA: coding region identification tool invoking comparative analysis. Mol. Biol. Evol. 1999;16:512–524. doi: 10.1093/oxfordjournals.molbev.a026133. [DOI] [PubMed] [Google Scholar]
- 55.Liu J., Gough J., Rost B. Distinguishing protein-coding from non-coding RNAs through support vector machines. PLoS Genet. 2006;2:e29. doi: 10.1371/journal.pgen.0020029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kong L., Zhang Y., Ye Z.-Q., Liu X.-Q., Zhao S.-Q., Wei L., Gao G. CPC: assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007;35:W345–W349. doi: 10.1093/nar/gkm391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gaspar P., Moura G., Santos M.A.S., Oliveira J.L. mRNA secondary structure optimization using a correlated stem-loop prediction. Nucleic Acids Res. 2013;41:e73. doi: 10.1093/nar/gks1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.SantaLucia J., Jr A unified view of polymer, dumbbell, and oligonucleotide DNA nearest-neighbor thermodynamics. Proc. Natl. Acad. Sci. U.S.A. 1998;95:1460–1465. doi: 10.1073/pnas.95.4.1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mathews D.H., Turner D.H. Prediction of RNA secondary structure by free energy minimization. Curr. Opin. Struct. Biol. 2006;16:270–278. doi: 10.1016/j.sbi.2006.05.010. [DOI] [PubMed] [Google Scholar]
- 60.Goñi J.R., Pérez A., Torrents D., Orozco M. Determining promoter location based on DNA structure first-principles calculations. Genome Biol. 2007;8:R263. doi: 10.1186/gb-2007-8-12-r263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Morin R., Bainbridge M., Fejes A., Hirst M., Krzywinski M., Pugh T., McDonald H., Varhol R., Jones S., Marra M. Profiling the HeLa S3 transcriptome using randomly primed cDNA and massively parallel short-read sequencing. Biotechniques. 2008;45:81–94. doi: 10.2144/000112900. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.