

Summary
Immunity to many intracellular pathogens requires the proliferation, differentiation, and function of CD8+ cytotoxic T lymphocytes (CTLs). While the majority of effector CTLs die upon clearance of the pathogen, a small proportion of them survive to become long-lived memory CTLs. Memory CTLs can provide protective immunity against re-exposure to the same pathogen and are the principle motivation behind T-cell- based vaccine design. While a large body of cellular immunologic research has proven invaluable to define effector and memory CTLs by their different phenotypes and functions, an emerging focus in the field has been to understand how environmental cues regulate CTL differentiation on a genomic level. Genome-wide studies to profile transcriptional and epigenetic changes during infection have revealed that dynamic changes in DNA methylation patterns and histone modifications accompany transcriptional signatures that define and regulate CTL differentiation states. In this review, we emphasize the importance of epigenetic regulation of CD8+ T-cell differentiation and the likely role that transcription factors play in this process.
Keywords: epigenetic, transcription, CD8, differentiation, effector, memory
Introduction
Immunity to many intracellular pathogens, both viral and bacterial, requires the proliferation, differentiation, and function of cytotoxic CD8+ cytotoxic T lymphocytes (CTLs) (1). Upon clearance of the pathogen, the majority of effector CTLs die, while just a small proportion of them survive to become long-lived memory CTLs. The cardinal role of memory CTLs is to rapidly clear a previously encountered pathogen on secondary exposure without the need for another primary adaptive immune response. This rapid response prevents full-blown re-infection and is the principle motivation behind T-cell-based vaccine design. While a large body of cellular immunologic research has proven invaluable to define effector and memory CTLs by their different phenotypes and functions, an emerging focus in the field has been to understand how environmental cues regulate CTL differentiation on a genomic level (2).
From a developmental standpoint, T-cell differentiation is a useful model system to study how complex epigenetic processes are regulated by environmental cues and changes in transcriptional networks to affect gene expression. Genome- wide studies to profile transcriptional and epigenetic changes during infection have revealed that dynamic changes in DNA methylation patterns and histone modifications accompany transcriptional signatures that define and regulate CTL differentiation states (3–6). In this review, we emphasize the importance of epigenetic regulation of CD8+ T-cell differentiation and the likely role that transcription factors play in this process.
Effector and memory CD8+ T-cell heterogeneity
Kinetics of CD8+ T-cell differentiation
When naive CD8+ T cells encounter their cognate antigen, they rapidly become activated, undergo clonal expansion, and differentiate into killer CTLs that can secrete effector molecules such as IFNγ, perforin and granzyme to provide sterilizing immunity against the invading microbe (reviewed in (1). While the majority of effector CTLs die soon after the pathogen is cleared during a phase of programmed contraction, a small proportion of these cells will survive and form a population of long-lived memory CTLs. Although the pinnacle of effector CTL expansion and maturation occurs approximately 8 days post infection, the development of long-lived memory CTLs is a much slower progression (reviewed in 1, 2). Immunologic memory is typically defined as those CTLs persisting at least 1–2 months after infection, the time at which memory CD8+ T cells become endowed with the unique ability to homeostatically proliferate in response to IL-7/IL-15 cytokines and robustly respond to secondary infection (reviewed in 1). Memory CTLs are exceptionally long-lived, often persisting the life of the host animal and providing lifelong immunity. Throughout the course of infection, there is considerable heterogeneity in the populations of effector and memory CTLs that form (discussed in more detail below). This heterogeneity is driven by differences in antigen-specificity, precursor frequency, the duration of antigenic stimulation, exposure to inflammation, among other signals, that leads to a spectrum of differentiation fates (reviewed in 2).
To better understand the factors that contribute to the heterogeneity of effector and memory CD8+ T-cell differentiation states, a number of groups have taken a reductionist cellular approach. Single-cell adoptive transfer experiments have elegantly demonstrated that effector and memory CTLs can arise from a single daughter cell, and that cues from the environment greatly influence their heterogeneity (7–10). Although T-cell receptor (TCR) affinity for peptide-MHC can greatly influence the diversity of antigen- specific CTLs that from during infection, these studies suggest that TCR avidity (strength of interaction between the TCR and peptide/MHC) does not appear to play as important a role (8, 9). Instead, these single-cell transfer experiments have demonstrated that the summation of all the individual clonal responses is equivalent to the endogenous polyclonal response to any given pathogen, suggesting that the breadth of the polyclonal T-cell response ensures that both short-lived effector and long-lived memory CTLs form (7, 9). The important conclusion from these studies is that a great deal of heterogeneity in the qualities of effector and memory progeny may arise from a single common naive T-cell precursor, and thus heterogeneity is an inevitable consequence of T-cell differentiation. Herein, we review the literature in CD8+ T cells that has shaped our understanding of the transcriptional and epigenetic regulatory events that generate diverse CD8+ T-cell populations with different functional properties and long-term fates following acute infection.
Gene expression profiling of effector and memory CD8+ T-cell development
Our understanding of the gene expression profiles associated with CD8+ T-cell differentiation stems from several important profiling studies of CTLs as they differentiate from a naive to an effector to a memory state. Work by Kaech et al., (3) that was later expanded upon by Best et al. (4), has helped immensely to further our understanding of the genome- wide transcriptional changes that occur in CTLs as they differentiate following acute infection in mice. Several interesting patterns of gene expression emerged from these analyses that can be used to infer their potential role in regulating CTL differentiation. For example, Best et al. (4) showed that within hours after activation, a number of important genes involved in T-cell metabolism and cell cycle progression are rapidly upregulated and represent a core signature of recently activated CD8+ T cells. Both studies noted that many genes are differentially upregulated or downregulated as CTLs transition from naive to effector to memory CTLs. Of some of the more interesting patterns in global gene expression, however, were genes that were (i) increased at the peak of the effector response (i.e. down in naive, up in effector, down in memory), (ii) increased during memory (i.e. down in naive, down in effector, up in memory), (iii) enriched in all activated CTLs (down in naive, up in effector, up in memory), or (iv) enriched in ‘quiescent’ CTLs (up in naive, down in effector, up in memory). Importantly, the timing of these changes in global gene expression is indicative, and perhaps predictive, of their importance during CD8+ T-cell differentiation. This information can be used to extrapolate how different transcription factors may regulate these transcriptional programs to promote or suppress gene expression (4). In a recent review by Weng et al. (11), drawing on data from multiple gene expression profiling studies, the reviewers noted that approximately 95% of genes that were highly expressed in memory CD8+ T cells are shared with naive CD8+ T cells. Similarly, Luckey et al. (12) also found that for a handful of genes that were coordinately regulated in memory CTL and B cells (up or down) virtually all of these were shared with hematopoietic stem cells, suggesting that this gene program may represent common features of long-lived cells that are capable of self-renewal. In addition, such studies are a useful frame of reference for understanding how gene expression in CTLs changes under physiological or pathophysiological states. For example, comparing gene expression profiles of CTLs that develop in the setting of an acute or chronic viral infection have demonstrated marked differences in global gene expression and transcriptional networks (13, 14). Similarly, by examining gene expression data of memory CTLs after secondary, tertiary, and quaternary recall, Wirth et al. demonstrated that repetitive antigenic stimulation of CD8+ T cells, a clinically relevant strategy used to expand rare population of CTLs, and their exposure to inflammation drives their progressive loss of various cardinal features of memory, including long-term homeostasis, tissue distribution, and function, but not their ‘exhaustion’ (15, 16). Thus, genes that were progressively downregulated or upregulated during this process were representative of memory or effector CTL signatures, respectively.
Defining and refining the subsets
A long-standing question in the field has been: how do long-lived memory CTLs form following acute infection? Numerous studies have outlined the intrinsic heterogeneity in long-term fates of various subsets of effector CD8+ T cells (reviewed in 2). Building on the seminal work of Schluns et al. (17) demonstrating the importance of IL-7 and IL-7R expression on CTLs for the homeostasis and survival of memory CTLs, Kaech et al.(18) went on to show how IL-7Rhi CTLs at the peak of the effector response were the direct precursors of central memory CD8+ T cells that are capable of self-renewal. Later, Joshi et al. (19) and Sarkar et al. (20) demonstrated that effector cells with higher expression of KLRG-1 and lower expression of IL-7R can identify CTLs with potent effector functions, but shortened lifespans compared to those that express the converse pattern of markers. It is now well appreciated that at the peak of the effector response following a number of different infections, the differentiation of KLRG-1hi IL-7Rlo short-lived effector and KLRG-1lo IL-7Rhi memory precursor CTL subsets form to varying degrees, further illustrating the heterogeneity of effector CTL responses and how they can vary according to different infectious environments. Further work demonstrated that the use of additional surface markers can help distinguish CTLs with enhanced memory potential and function (21).
Broadly speaking, memory CD8+ T cells can be divided into three groups based not only on their phenotype but also on their tissue distribution including: central, effector, and tissue resident memory (reviewed in 2). The ability to distinguish effector and memory CTL subsets based on phenotype has allowed us to study the underlying mechanisms regulating effector and memory CTL differentiation at the molecular level in greater detail, and the discovery of a number of transcription factors including, T-bet (19, 22), Blimp-1 (23), and Id-2 (24), that are highly expressed in effector cells and FoxO1 (25–28), Eomes (22, 29), and TCF-1 (30) that are highly expressed in naïve and memory CTLs in controlling the differentiation of short-lived and long-lived memory CTLs, respectively (reviewed in 2). Expanding on the earlier article of Chang et al. (31–33) regarding the role of asymmetric cell division in establishing effector and memory CTL fates, Arsenio et al. (34) have now used single-cell high-throughput quantitative PCR analysis to show that the transcriptome of the proximal (closest to the APC) and distal (furthest from the APC) daughter CTLs are predictive of a effector and memory CTL fates, respectively. Mechanistically, they suggest that asymmetric partitioning of IL-2Rα, a signal that is known to influence early commitment between effector and memory CTL lineages, may intrinsically bias these cells toward one fate or the other even prior to the first cell division. Conceptually, these studies and others have helped to further our understanding of how environmental cues can shape competing transcriptional programs to regulate the effector and memory CTL differentiation. In addition, this study serves as the basis for how epigenetics, or changes to the DNA and chromatin, impact CD8+ T-cell differentiation, the topic of this review.
Epigenetic regulation of CD8+ T-cell differentiation
Differentiation is undoubtedly a complex process involving the interpretation of environmental signals with, often significant, changes in gene expression, re-organization of transcriptional networks, and commitment. As part of this, unique gene expression programs that accompany effector and memory CTL transitions (discussed above) must be initiated, matured, and stabilized over time (Fig. 1, left). Epigenetic modifications to the chromatin in the form of alterations to the DNA and/or histones are essential to allow access of transcriptional machinery to the underlying genome to enable changes in gene expression and differentiation. Transcription factors that function to integrate environmental signals with changes in gene expression are likely to be involved in virtually every facet of this process, from initiating modifications to the epigenetic landscape to establishing lineage commitment (Fig. 1). In the following sections, we will discuss how DNA methylation and histone modifications are involved in shaping the differentiation and function of CD8+ T cells, and define the role of lineage-defining transcription factors in controlling this epigenetic regulation.
Fig. 1. Model for epigenetic and transcriptional control of T-cell differentiation.
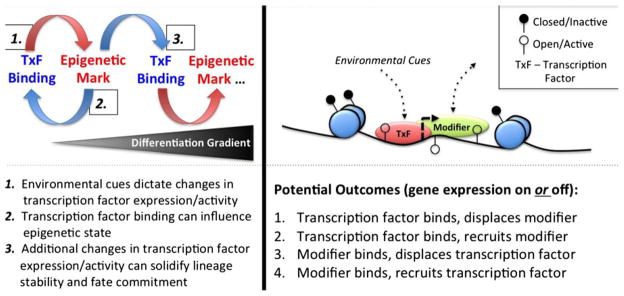
Epigenetic modifications play an important role in the development and differentiation of CD4 and CD8 lineages in vivo. Methylation of the DNA and modifications to histones through acetylation and methylation can promote or suppress gene transcription. Environmental cues (often in the form of antigen and inflammation) play an important role in shaping T-cell differentiation following infection. Transcription factors that lie downstream of these signals instruct T-cell differentiation fate and lineage stability through their regulation of gene expression that is achieved, in part, through their ability to influence chromatin structure. (Left) Effector T cells develop along a differentiation gradient. Cells that are the least committed often have the lowest expression of a particular lineage-defining transcription factor, while those with the highest expression are often terminally differentiated (i.e. fate committed). An example of this is T-bet, that controls the differentiation of short-lived effector CTLs during acute infection. Herein, binding of a transcription factor can have a profound influence on the chromatin structure leading to further changes in transcription factor and gene expression. These changes can ultimately be reinforced and stabilized by modifications to the epigenetic landscape to regulate cell fate and lineage commitment. (Right) A model for how environmental cues can influence the activity of a transcription factor (TxF) that can directly bind to the DNA and regulate the recruitment or displacement of epigenetic enzymes, other transcription factors or RNA polymerase II (modifiers), and visa versa. For example, to enhance gene expression, a transcription factor can bind to the DNA to facilitate the recruitment of a modifier that can enhance gene transcription; whereas to suppress gene expression, a transcription factor can displace a modifier that would otherwise enhance gene expression, and so on. The most likely relationships between transcription factor and modifier that can either turn gene transcription on or off are listed.
Lessons learned from CD4+ T cells
Helper CD4+ T cells (Th) play an important role in shaping the immune response to virtually every type of infection and come in a variety of flavors, including Th1, Th2, Th17 and Treg (reviewed in 35, 36). The importance of chromatin epigenetic modifications in T-cell differentiation and function and the role of transcription factors in controlling chromatin remodeling were first described in the CD4+ T-cell field (reviewed in 37). A number of pioneering studies have established that epigenetic regulation through DNA methylation and histone modifications is important in controlling the development, differentiation, and function of CD4+ T cells (reviewed in 11, 38). In 1998, Bird et al. (39) were the first to clearly show that epigenetic modifications regulate T-cell differentiation by demonstrating that modulation of epigenetics controls Th1 versus Th2 differentiation. Th1 cells are characterized predominantly by the expression of IFNγ and lack of IL-4 expression, while Th2 cells have the opposite cytokine profile. CD4+ T cells polarized in Th1 conditions have an IFNγ locus that is epigenetically open, while the IL-4 locus is closed, suggesting that helper T-cell gene programs are mutually exclusive (39, 40). The study by Bird et al. (39) provided one of the first examples of how changes at the epigenetic level could regulate mutually exclusive differentiation programs in Th1 and Th2 cells. Treatment of Th1 cells with the epigenetic modifying drugs sodium butyrate (an HDAC inhibitor) or 5-Aza-2-deoxycytidine (Aza, a cytosine methylation inhibitor) relieved suppressive marks at the Il4 locus, thereby allowing these Th1 cells to start to make the proto-typical Th2 cytokine, IL-4 (39). These experiments indicate that alternative programs are being actively suppressed by epigenetic modifications to the chromatin or DNA, and highlight the importance of epigenetic regulation in T-cell differentiation and function.
CD4+ T-helper subsets are defined not only by their phenotype and function but also perhaps more specifically by the transcription factors that control their differentiation for example: T-bet in Th1, GATA-3 in Th2, RORγT in Th17, Foxp3 in Treg, and so on. The role for many of these and other transcription factors in controlling epigenetics to establish and maintain their identity was first established in CD4+ T cells (reviewed in 37). For example, in Th1 CD4+ T cells, the promoter and distal upstream regulatory regions of the Ifng gene are H4 acetylated (permissive) (41, 42); however, while the majority of these require Th1 polarizing cytokine IL-12 and STAT-4 activity, only some appear to be T-bet dependent (43, 44). Mechanistically, T-bet has been shown to displace the histone deacteylase, Sin3a, to facilitate permissive H4 marks that enforce IFNγ expression and the differentiation of Th1 cells (45). Most recently, article by Vahedi et al. (46) provided a detailed, genome-wide view of the epigenetic regulation of CD4+ T-cell differentiation (and reviewed in 47). This study showed that certain STAT proteins that have previously been shown to regulate T-helper identity bind to enhancer regions in CD4+ T cells to open the chromatin, acting as pioneers to allow access for lineage-defining transcription factors to bind to regulate gene expression (46) (Fig 1, right). Thus, in response to IL-12 signals, STAT-4 activation facilitates chromatin remodeling to the at the enhancer regions of Th1 genes that allows for the subsequent recruitment of T-bet and commitment to the Th1 lineage. Similarly, Th2 commitment requires the stepwise activities of STAT-6 and GATA-3 in response to IL-4 stimulation.
In addition to establishing CD4+ T-helper lineage differentiation, transcription factor control of epigenetic modifications also confers stability in maintaining these differentiated states (reviewed in 37). It is now well appreciated that CD4+ T-helper lineages exhibit a certain degree of developmental plasticity that can be attributed to the co-expression and functional interplay between some of these transcription factors under certain circumstances (reviewed in 37). Genome-wide profiling of histone modifications in polarized T cells demonstrated that loci encoding lineage-defining transcription factors that regulate alternative T-cell fates exist in a bivalent state, containing both permissive and repressive (48). These data suggest that while commitment to a particular lineage is typically under the regulation of a single ‘master’ transcription factor, other lineage-defining transcription factors, and alternative fates, although repressed at the epigenetic level, remain in a poised state perhaps to allow for a certain degree of developmental plasticity. This may be explained to a large degree by the specific activity of the Enhancer of Zeste Homolog 2, EZH2, which is the enzymatically active part of the histone methylation polycomb repressor complex, PRC2, which lays repressive H3K27me3 marks to suppress gene expression. Notably, CD4+ T cells deficient in EZH2 fail to commit exclusively to either the TH1 or TH2 lineage under polarizing conditions, instead remaining plastic, thereby demonstrating that epigenetic histone modifications maintain lineage stability, and commitment (49, 50). In TH9 cells, Smad proteins that are activated in response to TGF-β signaling function to displace EZH2 from the Il9 locus (51). Finally, in Treg cells, the lineage-defining transcription factor FoxP3 stabilizes and maintains this lineage by recruiting EZH2 to repress its target genes (52). Based on this body of literature from the CD4+ T-cell field, transcription factors control of epigenetics is clearly involved in both the establishment and maintenance of T-cell differentiation states. Therefore, transcription factors not only promote T-cell differentiation but also function to secure commitment through their ability to broadly influence the epigenetic states and gene expression programs that define a particular lineage.
Although lesser advanced than our knowledge on CD4+ T-cell differentiation, for the remainder of this review, we focus on how epigenetic mechanisms in CD8+ T cells, specifically DNA methylation and histone modifications, contribute to the formation and function of terminally differentiated effector and long-lived memory CD8+ T cells. We discuss evidence supporting a role for transcription factors in both establishing and maintaining CD8+ T-cell differentiation and lineage commitment through control of epigenetic regulation.
DNA methylation in the control of CD8+ T-cell differentiation
DNA methylation on cytosine residues of CpG dinucleotides is an epigenetic modification associated with gene silencing that has been shown to play an important role in the differentiation and function of CD8+ T cells. DNA methylation is deposited de novo and maintained by the DNA methyltransfe- rases: DNMT1, DNMT3A, and DNMT3B (52, 53). De novo methylation is canonically attributed to DNMT3A and DNMT3B, while maintenance is mostly accomplished by DNMT1 with support from DNMT3A and DNMT3B (53–56). DNMT1 is important for thymocyte development, where it is critical for survival of double negative cells and differentiation of double positive cells (57). In response to viral infection DNMT1 is required for the normal clonal expansion, survival, and polyfunctionality of CD8+ T cells (57). These studies in DNMT1-deficient CD8+ T cells provide broad evidence that DNA methylation is important in T-cell survival and function, but fall short of mechanistically elucidating how this happens. Additionally, although de novo DNA methylation is undoubtedly important in effector and memory CD8+ T-cell differentiation and function, the roles of DNMT3A and DNMT3B have not been investigated. While DNMT deficiency studies have been informative in showing the necessity of these enzymes, a more detailed understanding of the regulation of DNA methylation in naïve and effector CD8+ T cells has come from recent genome-wide studies.
The first genome-wide evaluation of DNA methylation during CD8+ T-cell differentiation by Scharer et al. (6) has revealed that DNA methylation changes dynamically during infection and correlates inversely with gene expression. Effector genes, such as Gzmb (Granzyme B) and Ifng (IFNγ), have markedly increased expression and decreased promoter methylation in effector CD8+ T cells relative to naive cells, while homeostasis genes, such as Tcf7, expressed highly in naïve and memory cells have reduced expression and increased promoter methylation in effector relative to naive CD8+ T cells (6). These findings support the concept that gene silencing by DNA methylation is associated with the acquisition of differential gene expression profiles unique to effector CD8+ T cells. While this global profiling study provides a rich dataset and correlative support for the hypothesis that DNA methylation is important in CD8+ T-cell differentiation, there are many unanswered questions. First, do terminal effector and memory precursor CD8+ T cells have differential DNA methylation patterns? Second, does differential DNA methylation drive effector versus memory lineage formation in CD8+ T cells, or is it a secondary consequence of otherwise determined fates? Third, does DNA methylation have an important role in stabilizing/maintaining differentiation status? And finally, how is DNA methylation regulated in response to environmental cues, such as inflammation or antigen re-exposure, known to shape CD8+ T-cell differentiation? The answer to the final question has been investigated in relation to antigen re-exposure in perhaps the most intriguing and illuminating studies on DNA methylation in CD8+ T cells.
A fundamental feature of memory CD8+ T cells is their ability to rapidly re-acquire effector function and massively proliferate upon cognate antigen encounter. Why memory CD8+ T cells are capable of this unique rapid response to antigen relative to naïve cells is poorly understood. Epigenetic remodeling of effector gene loci by altering DNA methylation may be an important molecular mechanism underlying this process. Although DNA methylation in CD8+ T cells is dynamic during infection, DNA methylation patterns of effector gene loci in memory cells actually closely resemble those in naive cells (58). At the IFNγ locus, effector CD8+ T cells lose the high levels of repressive methylation seen in naive cells, while memory CD8+ T cells reacquire significant methylation almost to the level of naïve cells (58). For DNA methylation, therefore, permanent remodeling and removal of silencing methylation on effector gene loci does not account for the rapid recall ability of a memory CD8+ T cell. Instead, memory CD8+ T cells have the exclusive ability to rapidly and completely demethylate effector gene loci following antigen exposure, while naïve cells remain methylated in the same time frame (58). Permanent remodeling of DNA methylation patterns does not, therefore, account for the ability of memory cells to rapidly acquire effector gene expression upon recall. Rather, memory cells are uniquely capable of quickly removing repressive DNA methylation at effector gene loci. The mechanism that underlies rapid removal of repressive DNA methylation is of profound interest and importance. One possibility is that memory cells express a unique enzyme or protein, absent in naive cells, that promotes demethylation. This factor may be a transcription factor, perhaps T-bet that guides demethylation machinery to the appropriate loci upon antigen stimulation (59). Another possibility is that activated CD8+ T cells undergo permanent remodeling of their chromatin structure at the histone level, which in turn influences rapid removal of DNA methylation upon antigen stimulation. In support of this idea, there is a growing body of literature that links DNA methylation and histone modifications (60). Indeed, histone modifying proteins, such as G9a, are reported to bind to DNMT3; and DNMT3L, the DNMT3 binding partner, is able to bind unmethylated but not methylated H3K4 residues (60).
Although much work is still required, DNA methylation is clearly important in CD8+ T-cell differentiation and function. This importance is demonstrated by the develop- mental defects of DNMT deficient CD8+ T cells and by the unique capacity of memory CD8+ T cells to rapidly demethylate effector gene loci on antigen re-exposure. The possible link between DNA methylation and histone modifications is intriguing and worthy of future investigation, especially considering the importance of histone modifications in CD8+ T-cell differentiation, discussed in the next section.
Dynamic changes in histone modifications underlie CD8+ T-cell differentiation
Epigenetic-mediated transcription factor control of differentiation can be accomplished through alterations to the chromatin structure by covalent modifications to histones. Indeed, modulation of histone modifications, which can be permissive or repressive in nature (Fig. 2), appears to influence expression of memory and effector genes in CTLs. By examining H3K9 acetylation at effector gene loci in naïve and memory human CD8+ T cells, Araki et al. and Fann et al. have demonstrated that histone hyper-acetylation aids rapid recall response of memory CTLs by encoding ‘chromatin memory’ that allows rapid and robust expression of hyperacetylated genes in memory CTLs (61, 62). Permissive H3K9Ac marks are enriched at the proximal promoter and first exon of the Eomes gene in memory CD8+ T cells relative to naive cells (61). Following in vitro stimulation, Eomes mRNA expression is induced in memory CD8+ T cells; however, pharmacological hypo-acetylation of the Eomes locus prevents this induction. Induction of Prf1 (Perforin) and Gzmb (GranzymeB) mRNA is similarly dependent on H3K9 hyperacetylation at these gene loci (61). Enhanced chromatin accessibility through histone hyper-acetylation may, therefore, be an important mechanism for rapid re-expression of effector genes, and the unique capacity for rapid recall may be epigenetically permitted in memory CD8+ T cells due to the combined effects of hyper-acetylation of effector gene loci and rapid DNA demethylation upon antigen exposure.
Fig. 2. Histone modifications associated with chromatin states.

As the fundamental structural unit of chromatin, the nucleosome controls DNA accessibility as a function of the combined modifications of its histone subunits. Histone modifications are made to residues within the globular core and on the N-terminal tail of all four histone subunits with the H3 tail being the most prominent and extensively modified domain (93, 94). Modifications to the globular histone core are thought to regulate the chromatin state by altering nucleosome structure, while N-terminal tail modifications are believed to primarily regulate the binding of non-histone proteins to the chromatin (95). Histone tail modifications therefore serve as recruitment signals for effector proteins capable of reading and interpreting the histone modification code. Analysis of genome-wide epigenetic marks has identified distinct chromatin states defined by recurrent and spatially related patterns of chromatin modifications that encode functionally distinct roles (96, 97). Study by Ernst et al. (96, 97) has identified chromatin states for multiple functional genetic elements, including promoters, enhancers, insulators, transcribed regions, polycomb repressed regions, and heterochromatin/inactive regions. The chromatin state of a functional genetic element is uniquely identified by its signature of histone marks and spatial relationship with putative gene targets (96, 97). The figure depicts a simplified representation of this histone modification map. Histone acetylation is associated with active chromatin elements. Histone methylation is more complicated with some marks associated with repression (H3K27me3) while others are associated with activation (H3K4me1/2/3). CTCF is a zinc finger protein which may act as an insulator through enhancer-blocking (98).
Activating histone modifications, such as H3K9Ac, are generally associated with active effector and memory gene expression. Indeed, Dispirito et al. have shown that di-acetylated histone H3 (diAcH3) is enriched in activated effector CTLs and remains enriched in cells that acquire a central memory phenotype (64). Remarkably, CD4+ T-cell help, known to be important for normal differentiation of memory CTLs, is also required for the maintenance of the diAcH3 mark in memory CD8+ T cells. One of the functions of CD4+ T-cell help may, therefore, be to promote global epigenetic remodeling of effector and memory CD8+ T cells. The precise mechanism by which CD4+ T cells may regulate the epigenetic state of CD8+ T cells is unclear.
In memory CD8+ T cells, histone acetylation serves not only to ensure robust active expression of effector and cytolytic genes following antigen exposure but also helps to maintain stable expression of memory defining genes. Chandele et al. (64) have demonstrated that reciprocal action of the transcription factors Gfi-1 and GABPα regulates IL7Rα expression in CTLs through modulation of Il7rα gene acetylation. Gfi-1 recruits HDAC1 to the Il7ra locus in terminal effector CD8+ T cells, driving deacetylation and repression of the Il7rα locus, while GABPα antagonized Gfi-1 in memory precursor cells to promote maintenance of Il7rα acetylation and expression. HDAC7 has been similarly shown to differentially regulate genes important for effector function and migration (65). These studies have demonstrated that CTL subsets have differential histone modifications at certain ‘pro-effector’ or ‘pro-memory’ genes, and that these differences are functionally important. This study highlighted the need to perform genome-wide studies on CTL subsets to better understand how differential histone modification of certain genes impacts CD8+ T-cell differentiation.
The pioneering study of Araki et al. was the first to examine genome-wide differences in epigenetic marks in CD8+ T cells and the functional relevance of these marks to differential gene expression and T-cell differentiation. Araki et al. studied histone methylation states in naïve and memory human CD8+ T cells, showing an association of histone methylation patterns with differential gene expression in memory CD8+ T cells (5). Their study utilized genome-wide ChIP-Seq to study two methylation marks: H3K4me3, which positively correlates with gene expression, and H3K27me3, which negatively correlates with gene expression, in naive (Tn), effector memory (Tem), and central memory (Tcm) CD8+ T cells. Globally, Tem and Tcm cells have more genes with high levels of H3K4me3 marks than naive cells, indicating global deposition of active histone marks following T-cell activation (5). By comparing mRNA expression with genome-wide H3K27me3 and H3K4me3 profiles of naive and memory CD8+ T cells, Araki et al. (5) identified four modes of association between gene expression and histone methylation: repressed, active, poised, and bivalent genes. Repressed genes have low mRNA expression in resting memory cells, high H3K27me3, and low H3K4me3. Active genes have high mRNA expression in resting memory cells, low H3K27me3, and high H3K4me3, and include such genes included PRDM1 (Blimp1) and KLRG1. Poised genes have low mRNA expression in naïve and resting memory cells, markedly increased expression in activated memory cells, and histone methylation patterns similar to those of active genes (5). Similar to poised genes, bivalent genes have low mRNA expression in naive and resting memory cells and markedly increased expression in activated memory cells, however, their methylation patterns have high H3K27me3 and high H3K4me3 (5). Intriguingly, Arkai et al. found that central memory CD8+ T cells contain a greater number of poised genes than effector memory subsets. One such example is Id2, a gene that has been shown to be necessary for optimal CD8+ T-cell expansion after infection (66). This seminal study by Araki et al. has established that genes known to regulate CTL differentiation are differentially marked by histone modifications, yet several questions remains. First, which histone modifications are most important for promoting differentiation to effector or memory fates? Second, are certain histone marks especially important for memory cell survival, and correspondingly, how do memory cells maintain their epigenetic identity as they self-renew and homeostatically proliferate? Answering these questions will be a significant contribution to our field. Nonetheless, with our current knowledge, we may conclude that the epigenetic state of CD8+ T cells changes dynamically during an immune response to infection, and that resting memory CTLs posses a hybrid epigenetic state in which many effector genes are not actively expressed but remain poised for rapid re-expression. Lineage- defining transcription factors play an important role in shaping and controlling this epigenetic state in CTLs, which we will address in the next section.
Transcriptional control of epigenetics
Transcriptional and epigenetic regulation of short-lived effector CTL differentiation by T-bet
The role of T-bet in the epigenetic control of T-cell differentiation in CD4+ and CD8+ T cells has been fairly well characterized for a number of genes whose expression are T-bet dependent. Although, much of this study stems from the study of CD4+ T-cell differentiation, in effector CD8+ T cells T-bet binds to the promoters of the IFNγ, granzyme B, and perforin genes to directly regulate their expression (67, authors’ unpublished data). As previously discussed, within hours of TCR stimulation, the IFNγ gene becomes active due to rapid DNA demethylation, histone acetylation, and recruitment of T-bet (58, 67, 68). T-bet is both necessary and sufficient for the induction of permissive histone modifications at the IFNγ locus (69). Remarkably, while deletion of T-bet greatly compromises the expression of IFNγ, treatment of T-bet deficient T cells with the HDAC inhibitor, trichostatin-A (TSA), can fully restore IFNγ expression (45). This epigenetic rescue suggests that T-bet functions, at least partially, by affecting histone modifying enzymes to promote maximal gene expression at the modified locus. In 2007, Joshi et al. demonstrated the importance of inflammation driven T-bet expression in the control of effector CTL differentiation. Gene expression profiling of antigen-specific CTLs in the absence of T-bet revealed its critical importance to direct the transcriptional program that regulates the differentiation of this subset. Future studies are necessary to determine if to what extent T-bet regulates epigenetic modifications, and how these are conserved between CD4+ and CD8+ T cells.
Epigenetic regulation by Blimp-1 re-enforces the differentiation of short-lived effector CTLs
Like T-bet, increased expression of Blimp-1 promotes the terminal differentiation of short-lived effector CTLs and Blimp-1 deficiency enhances the differentiation of memory CTLs (23, 70). In the absence of T-bet, expression of Blimp-1 is reduced, suggesting it potential dependence on T-bet for its expression (23). Blimp-1 too has a clear role in regulating the epigenetic state of effector CD8+ T cells. Recently, Shin et al. (71) demonstrated that Blimp-1 functions as a transcriptional repressor in CD8+ T cells by recruiting the histone methyltransferase G9a and deacetylase HDAC2, but not Ezh2, to mediate epigenetic closing of the Il2ra and Cd27 loci. Similarly, Blimp-1 has been shown to mediate the repression of the inhibitory receptor PD-1 on activated CD8+ T cells by competing with NFATc1, a known inducer of PD-1 expression (72, 73). Blimp-1 directly bound to the Pdcd1 locus where it enhanced deposition of repressive H3K27me3 marks, although the role for Ezh2 at this locus was not examined. Given that PD-1 has also been shown to be repressed by DNA methylation, the details of how DNA methylation and histone modifications are coordinated to suppress the expression of the same gene and if Blimp-1 is involved in this process remains to be determined. Recently, it was demonstrated that prolonged exposure to IL-2 promotes short-lived effector CTL differentiation at the expense of CTL memory (74, 75). In the study by Pipkin et al. IL-2 signaling initiated the recruitment of STAT5, Eomes, and RNA Polymerase II to the Ifng, Il2ra and Gzmb loci. In addition, IL-2 promoted the expression of Blimp-1, while suppressing the expression of the pro-memory transcription factor, Bcl-6. Furthermore, exposure of CTLs to high doses of IL-2 prevented these cells from downregulating the effector gene program and inhibited their ability to upregulate memory genes, which may suggest a potential role for epigenetics and Blimp-1 in this process. Intriguingly, Blimp-1 that is induced by IL-2 can also mediate repression of Il2ra (71), suggesting that in addition to its role in promoting effector CTL differentiation, Blimp-1 can temper IL-2 signaling, although the relevance of this negative feedback pathway on effector CTL differentiation has not yet been explored. Nevertheless, considering the important role of Blimp-1 in the transcriptional program and the differentiation of short-lived effector CTLs and, furthermore, its conserved function as a transcriptional repressor, it will be important in the future to determine the extent to which Blimp-1 mediates global epigenetic silencing to regulate fate commitment of this subset.
Transcriptional and, potential, epigenetic regulation of memory CTL differentiation by FoxO1
While short-lived effector CTL fate depends on T-bet, the differentiation of memory CTLs has been shown to be under the direct regulation of the transcription factor FoxO1 including the dependence of a number of other transcription factors that are known to regulate memory CTL differentiation and survival (25–28). Although, it can undergo a plethora of different posttranslational modifications to regulate its function and specificity (reviewed in 76), notably for this review, FoxO transcription factors have been shown to be directly acetylated by p300/CBP and de-acetylated by class IIa HDACs and Sirt1, a NAD-dependent class III HDAC (77–79). For example, a Sirt1-FoxO1 axis has been noted to promote longevity in response to caloric restriction in multiple cell types and organisms (reviewed in 80), and HDAC4 de-acetylation of FoxO1 can promote FoxO1-dependent metabolism (81, 82). These data support the notion that FoxO1 and HDACs not only interact but may also cooperate to enhance or repress FoxO1-dependent gene transcription. Intriguingly, similar to the phosphorylation of FoxO1 by AKT that promotes its nuclear export, phosphorylation of class IIa HDACs by PKC also promotes their binding to 14- 3-3 adapter proteins results in their nuclear exclusion (reviewed in 83). Likewise, the phosphatase PP2A, which de-phosphorylates FoxO1 allowing it to dissociate from 14- 3-3 and return to the nucleus, can act in an analogous fashion to de-phosphorylate and class IIa HDACs to enhance their activity (84–87). These data suggest that FoxO1 and class IIa HDACs activities could in fact be linked or at the very least regulated by similar mechanisms. In support of this, salt-inducible kinases that have recently been implicated in the differentiation of Th17 CD4+-helper T cells have been shown to regulate both the phosphorylation and activities of FoxO1 and class II HDACs (82, 88, 89).
The seemingly opposing functions of T-bet and FoxO1 in short-lived effector and long-lived memory CTL fates suggest that these transcription factors could oppose each other’s functions at the transcriptional and epigenetic level. In support of this idea, in the absence of FoxO1, the expression of T-bet and T-bet target genes KLRG-1, IFNγ, granzyme B, and perforin are markedly increased, while the opposite is true in the absence of T-bet for a number of genes that are FoxO1 targets (19, 25–28, authors’ unpublished data). Intriguingly, AKT signaling that inhibits memory CTL differentiation results in the phosphorylation FoxO1 and inhibition of its transcriptional activity, while mTORC1 signaling (downstream of AKT) has been shown to promote T-bet expression (28, 90, 91). Thus, FoxO1 and T-bet that are differentially regulated by AKT/mTOR signaling, control opposing transcriptional programs and CTL differentiation fates. However, while T-bet has been shown to directly regulate modifications at the chromatin level, a similar role for FoxO1 in the regulation of memory CTLs remains to be explored. Furthermore, whether there is relationship between class IIa HDAC activity and FoxO1-dependent gene expression in regulating the epigenetic state of CTLs has not been tested, but represents a potential area future research.
Conclusions and future directions
In this review, we discuss the evidence supporting a role for epigenetics in the regulation of CD8+ T-cell differentiation following an acute infection. Following T-cell activation, epigenetic changes occur at many effector loci to regulate gene expression. Chromatin remodeling and DNA methylation are likely dependent on transcription factors whose activities are regulated by TCR and cytokine signaling. These initial changes in the chromatin structure likely allow expression of additional transcription factors that can bind to the open regions and either enforce or suppress gene expression by inducing further epigenetic modifications or recruitment of transcriptional machinery. It seems likely, then, that the various transcription factors that help to define and maintain lineage fates in CD8+ T cells are critical components of the epigenetic program that underlies both effector and memory differentiation. In the future, it will be instrumental to dissect the relative contribution of certain fate determining transcription factors (e.g. T-bet in short-lived differentiation, and FoxO1 in memory CTL differentiation) as well as their temporal requirements in the epigenetic regulation of CD8+ T-cell differentiation. It will be important to link changes in DNA and chromatin modifications to environmental cues [e.g. IL-2 in short-lived differentiation (74, 75), and IL-10/IL-21 and STAT-3 in memory CTL differentiation (92)], identify how global epigenetic changes are influenced by the absence of CD4+ T-cell help (63), and examine how the epigenome is remodeled following secondary recall (67). It is likely that epigenome remodeling during secondary recall differs greatly from those changes that are associated with activation of naive T cells. Considering the central importance of the PI3K/AKT/mTOR axis in the regulation of terminal effector and memory cell fate decisions, it seems likely that this pathway may also directly influences the epigenetic state of CD8+ T cells. How these signals are related to changes in transcription factor expression and/or activities and how environmental signals can influence these epigenetic changes warrant further investigation.
Acknowledgments
We thank the members of the Kaech laboratory for helpful comments and discussions. This work was supported by grants to S.M.K. from the US National Institutes of Health (grants R01AI074699, R37AI066232, and R21AI097767) and from the Howard Hughes Medical Institute. S.M.G. is supported by the Yale MD/PhD Program (Grant NIH MSTP TG T32GM07205).
Footnotes
The authors have no conflicts of interest to declare.
References
- 1.Wherry EJ, Ahmed R. Memory CD8 T-cell differentiation during viral infection. J Virol. 2004;78:5535–5545. doi: 10.1128/JVI.78.11.5535-5545.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kaech SM, Cui W. Transcriptional control of effector and memory CD8+ T cell differentiation. Nat Rev Immunol. 2012;12:749–761. doi: 10.1038/nri3307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kaech SM, Hemby S, Kersh E, Ahmed R. Molecular and functional profiling of memory CD8 T cell differentiation. Cell. 2002;111:837–851. doi: 10.1016/s0092-8674(02)01139-x. [DOI] [PubMed] [Google Scholar]
- 4.Best JA, et al. Transcriptional insights into the CD8(+) T cell response to infection and memory T cell formation. Nat Immunol. 2013;14:404–412. doi: 10.1038/ni.2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Araki Y, et al. Genome-wide analysis of histone methylation reveals chromatin state-based regulation of gene transcription and function of memory CD8+ T cells. Immunity. 2009;30:912–925. doi: 10.1016/j.immuni.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Scharer CD, Barwick BG, Youngblood BA, Ahmed R, Boss JM. Global DNA methylation remodeling accompanies CD8 T cell effector function. J Immunol. 2013;191:3419–3429. doi: 10.4049/jimmunol.1301395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gerlach C, et al. Heterogeneous differentiation patterns of individual CD8+ T cells. Science. 2013;340:635–639. doi: 10.1126/science.1235487. [DOI] [PubMed] [Google Scholar]
- 8.Gerlach C, et al. One naive T cell, multiple fates in CD8+ T cell differentiation. J Exp Med. 2010;207:1235–1246. doi: 10.1084/jem.20091175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Plumlee CR, Sheridan BS, Cicek BB, Lefrancois L. Environmental cues dictate the fate of individual CD8+ T cells responding to infection. Immunity. 2013;39:347–356. doi: 10.1016/j.immuni.2013.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Buchholz VR, et al. Disparate individual fates compose robust CD8+ T cell immunity. Science. 2013;340:630–635. doi: 10.1126/science.1235454. [DOI] [PubMed] [Google Scholar]
- 11.Weng NP, Araki Y, Subedi K. The molecular basis of the memory T cell response: differential gene expression and its epigenetic regulation. Nat Rev Immunol. 2012;12:306–315. doi: 10.1038/nri3173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Luckey CJ, et al. Memory T and memory B cells share a transcriptional program of self-renewal with long-term hematopoietic stem cells. Proc Natl Acad Sci USA. 2006;103:3304–3309. doi: 10.1073/pnas.0511137103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Doering TA, et al. Network analysis reveals centrally connected genes and pathways involved in CD8+ T cell exhaustion versus memory. Immunity. 2012;37:1130–1144. doi: 10.1016/j.immuni.2012.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wherry EJ, et al. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity. 2007;27:670–684. doi: 10.1016/j.immuni.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 15.Wirth TC, Martin MD, Starbeck-Miller G, Harty JT, Badovinac VP. Secondary CD8+ T-cell responses are controlled by systemic inflammation. Eur J Immunol. 2011;41:1321–1333. doi: 10.1002/eji.201040730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wirth TC, et al. Repetitive antigen stimulation induces stepwise transcriptome diversification but preserves a core signature of memory CD8(+) T cell differentiation. Immunity. 2010;33:128–140. doi: 10.1016/j.immuni.2010.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schluns KS, Kieper WC, Jameson SC, Lefrancois L. Interleukin-7 mediates the homeostasis of naive and memory CD8 T cells in vivo. Nat Immunol. 2000;1:426–432. doi: 10.1038/80868. [DOI] [PubMed] [Google Scholar]
- 18.Kaech SM, et al. Selective expression of the interleukin 7 receptor identifies effector CD8 T cells that give rise to long-lived memory cells. Nat Immunol. 2003;4:1191–1198. doi: 10.1038/ni1009. [DOI] [PubMed] [Google Scholar]
- 19.Joshi NS, et al. Inflammation directs memory precursor and short-lived effector CD8(+) T cell fates via the graded expression of T-bet transcription factor. Immunity. 2007;27:281–295. doi: 10.1016/j.immuni.2007.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sarkar S, et al. Functional and genomic profiling of effector CD8 T cell subsets with distinct memory fates. J Exp Med. 2008;205:625–640. doi: 10.1084/jem.20071641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Joshi NS, et al. Increased numbers of preexisting memory CD8 T cells and decreased T-bet expression can restrain terminal differentiation of secondary effector and memory CD8 T cells. J Immunol. 2011;187:4068–4076. doi: 10.4049/jimmunol.1002145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Intlekofer AM, et al. Effector and memory CD8+ T cell fate coupled by T-bet and eomesodermin. Nat Immunol. 2005;6:1236–1244. doi: 10.1038/ni1268. [DOI] [PubMed] [Google Scholar]
- 23.Rutishauser RL, et al. Transcriptional repressor Blimp-1 promotes CD8(+) T cell terminal differentiation and represses the acquisition of central memory T cell properties. Immunity. 2009;31:296–308. doi: 10.1016/j.immuni.2009.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Masson F, et al. Id2-mediated inhibition of E2A represses memory CD8+ T cell differentiation. J Immunol. 2013;190:4585–4594. doi: 10.4049/jimmunol.1300099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Michelini RH, Doedens AL, Goldrath AW, Hedrick SM. Differentiation of CD8 memory T cells depends on Foxo1. J Exp Med. 2013;210:1189–1200. doi: 10.1084/jem.20130392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim MV, Ouyang W, Liao W, Zhang MQ, Li MO. The transcription factor foxo1 controls central-memory CD8(+) T cell responses to infection. Immunity. 2013;39:286–297. doi: 10.1016/j.immuni.2013.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tejera MM, Kim EH, Sullivan JA, Plisch EH, Suresh M. FoxO1 controls effector-to-memory transition and maintenance of functional CD8 T cell memory. J Immunol. 2013;191:187–199. doi: 10.4049/jimmunol.1300331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rao RR, Li Q, Gubbels Bupp MR, Shrikant PA. Transcription factor Foxo1 represses T-bet-mediated effector functions and promotes memory CD8(+) T cell differentiation. Immunity. 2012;36:374–387. doi: 10.1016/j.immuni.2012.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pearce EL, et al. Control of effector CD8+ T cell function by the transcription factor Eomesodermin. Science. 2003;302:1041–1043. doi: 10.1126/science.1090148. [DOI] [PubMed] [Google Scholar]
- 30.Zhou X, et al. Differentiation and persistence of memory CD8(+) T cells depend on T cell factor 1. Immunity. 2010;33:229–240. doi: 10.1016/j.immuni.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ciocca ML, Barnett BE, Burkhardt JK, Chang JT, Reiner SL. Cutting edge: asymmetric memory T cell division in response to rechallenge. J Immunol. 2012;188:4145–4148. doi: 10.4049/jimmunol.1200176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chang JT, et al. Asymmetric proteasome segregation as a mechanism for unequal partitioning of the transcription factor T-bet during T lymphocyte division. Immunity. 2011;34:492–504. doi: 10.1016/j.immuni.2011.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chang JT, et al. Asymmetric T lymphocyte division in the initiation of adaptive immune responses. Science. 2007;315:1687–1691. doi: 10.1126/science.1139393. [DOI] [PubMed] [Google Scholar]
- 34.Arsenio J, et al. Early specification of CD8+ T lymphocyte fates during adaptive immunity revealed by single-cell gene-expression analyses. Nat Immunol. 2014;15:365–372. doi: 10.1038/ni.2842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhu J, Yamane H, Paul WE. Differentiation of effector CD4 T cell populations (*) Annu Rev Immunol. 2010;28:445–489. doi: 10.1146/annurev-immunol-030409-101212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhu J, Paul WE. Peripheral CD4+ T-cell differentiation regulated by networks of cytokines and transcription factors. Immunol Rev. 2010;238:247–262. doi: 10.1111/j.1600-065X.2010.00951.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kanno Y, Vahedi G, Hirahara K, Singleton K, O’Shea JJ. Transcriptional and epigenetic control of T helper cell specification: molecular mechanisms underlying commitment and plasticity. Annu Rev Immunol. 2012;30:707–731. doi: 10.1146/annurev-immunol-020711-075058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wilson CB, Rowell E, Sekimata M. Epigenetic control of T-helper-cell differentiation. Nat Rev Immunol. 2009;9:91–105. doi: 10.1038/nri2487. [DOI] [PubMed] [Google Scholar]
- 39.Bird JJ, et al. Helper T cell differentiation is controlled by the cell cycle. Immunity. 1998;9:229–237. doi: 10.1016/s1074-7613(00)80605-6. [DOI] [PubMed] [Google Scholar]
- 40.Agarwal P, et al. Gene regulation and chromatin remodeling by IL-12 and type I IFN in programming for CD8 T cell effector function and memory. J Immunol. 2009;183:1695–1704. doi: 10.4049/jimmunol.0900592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fields PE, Kim ST, Flavell RA. Cutting edge: changes in histone acetylation at the IL-4 and IFN-gamma loci accompany Th1/Th2 differentiation. J Immunol. 2002;169:647–650. doi: 10.4049/jimmunol.169.2.647. [DOI] [PubMed] [Google Scholar]
- 42.Zhou W, Chang S, Aune TM. Long-range histone acetylation of the Ifng gene is an essential feature of T cell differentiation. Proc Natl Acad Sci USA. 2004;101:2440–2445. doi: 10.1073/pnas.0306002101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chang S, Aune TM. Dynamic changes in histone-methylation ‘marks’ across the locus encoding interferon-gamma during the differentiation of T helper type 2 cells. Nat Immunol. 2007;8:723–731. doi: 10.1038/ni1473. [DOI] [PubMed] [Google Scholar]
- 44.Hatton RD, et al. A distal conserved sequence element controls Ifng gene expression by T cells and NK cells. Immunity. 2006;25:717–729. doi: 10.1016/j.immuni.2006.09.007. [DOI] [PubMed] [Google Scholar]
- 45.Chang S, Collins PL, Aune TM. T-bet dependent removal of Sin3A-histone deacetylase complexes at the Ifng locus drives Th1 differentiation. J Immunol. 2008;181:8372–8381. doi: 10.4049/jimmunol.181.12.8372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vahedi G, et al. STATs shape the active enhancer landscape of T cell populations. Cell. 2012;151:981–993. doi: 10.1016/j.cell.2012.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.O’Shea JJ, Lahesmaa R, Vahedi G, Laurence A, Kanno Y. Genomic views of STAT function in CD4+ T helper cell differentiation. Nat Rev Immunol. 2011;11:239–250. doi: 10.1038/nri2958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wei G, et al. Global mapping of H3K4me3 and H3K27me3 reveals specificity and plasticity in lineage fate determination of differentiating CD4+ T cells. Immunity. 2009;30:155–167. doi: 10.1016/j.immuni.2008.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tumes DJ, et al. The polycomb protein Ezh2 regulates differentiation and plasticity of CD4(+) T helper type 1 and type 2 cells. Immunity. 2013;39:819–832. doi: 10.1016/j.immuni.2013.09.012. [DOI] [PubMed] [Google Scholar]
- 50.Wang A, et al. Cutting edge: Smad2 and Smad4 regulate TGF-beta-mediated Il9 gene expression via EZH2 displacement. J Immunol. 2013;191:4908–4912. doi: 10.4049/jimmunol.1300433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Arvey A, et al. Inflammation-induced repression of chromatin bound by the transcription factor Foxp3 in regulatory T cells. Nat Immunol. 2014;15:580–587. doi: 10.1038/ni.2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012;13:484–492. doi: 10.1038/nrg3230. [DOI] [PubMed] [Google Scholar]
- 53.Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99:247–257. doi: 10.1016/s0092-8674(00)81656-6. [DOI] [PubMed] [Google Scholar]
- 54.Jones PA, Liang G. Rethinking how DNA methylation patterns are maintained. Nat Rev Genet. 2009;10:805–811. doi: 10.1038/nrg2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen T, Ueda Y, Dodge JE, Wang Z, Li E. Establishment and maintenance of genomic methylation patterns in mouse embryonic stem cells by Dnmt3a and Dnmt3b. Mol Cell Biol. 2003;23:5594–5605. doi: 10.1128/MCB.23.16.5594-5605.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lee PP, et al. A critical role for Dnmt1 and DNA methylation in T cell development, function, and survival. Immunity. 2001;15:763–774. doi: 10.1016/s1074-7613(01)00227-8. [DOI] [PubMed] [Google Scholar]
- 57.Chappell C, Beard C, Altman J, Jaenisch R, Jacob J. DNA methylation by DNA methyltransferase 1 is critical for effector CD8 T cell expansion. J Immunol. 2006;176:4562–4572. doi: 10.4049/jimmunol.176.8.4562. [DOI] [PubMed] [Google Scholar]
- 58.Kersh EN, et al. Rapid demethylation of the IFN-gamma gene occurs in memory but not naive CD8 T cells. J Immunol. 2006;176:4083–4093. doi: 10.4049/jimmunol.176.7.4083. [DOI] [PubMed] [Google Scholar]
- 59.Sullivan BM, Juedes A, Szabo SJ, von Herrath M, Glimcher LH. Antigen-driven effector CD8 T cell function regulated by T-bet. Proc Natl Acad Sci USA. 2003;100:15818–15823. doi: 10.1073/pnas.2636938100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cedar H, Bergman Y. Linking DNA methylation and histone modification: patterns and paradigms. Nat Rev Genet. 2009;10:295–304. doi: 10.1038/nrg2540. [DOI] [PubMed] [Google Scholar]
- 61.Araki Y, Fann M, Wersto R, Weng N-P. Histone acetylation facilitates rapid and robust memory CD8 T cell response through differential expression of effector molecules (eomesodermin and its targets: perforin and granzyme B) J Immunol. 2008;180:8102–8108. doi: 10.4049/jimmunol.180.12.8102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fann M, et al. Histone acetylation is associated with differential gene expression in the rapid and robust memory CD8+ T-cell response. Blood. 2006;108:3363–3370. doi: 10.1182/blood-2006-02-005520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dispirito JR, Shen H. Histone acetylation at the single-cell level: a marker of memory CD8+ T cell differentiation and functionality. J Immunol. 2010;184:4631–4636. doi: 10.4049/jimmunol.0903830. [DOI] [PubMed] [Google Scholar]
- 64.Chandele A, et al. Formation of IL-7Rαhigh and IL-7Rαlow CD8 T cells during infection is regulated by the opposing functions of GABPα and Gfi-1. J Immunol. 2008;180:5309–5319. doi: 10.4049/jimmunol.180.8.5309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Navarro MN, Goebel J, Feijoo-Carnero C, Morrice N, Cantrell DA. Phosphoproteomic analysis reveals an intrinsic pathway for the regulation of histone deacetylase 7 that controls the function of cytotoxic T lymphocytes. Nat Immunol. 2011;12:352–361. doi: 10.1038/ni.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cannarile MA, et al. Transcriptional regulator Id2 mediates CD8+ T cell immunity. Nat Immunol. 2006;7:1317–1325. doi: 10.1038/ni1403. [DOI] [PubMed] [Google Scholar]
- 67.Northrop JK, Wells AD, Shen H. Cutting edge: chromatin remodeling as a molecular basis for the enhanced functionality of memory CD8 T cells. J Immunol. 2008;181:865–868. doi: 10.4049/jimmunol.181.2.865. [DOI] [PubMed] [Google Scholar]
- 68.Northrop JK, Thomas RM, Wells AD, Shen H. Epigenetic remodeling of the IL-2 and IFN-gamma loci in memory CD8 T cells is influenced by CD4 T cells. J Immunol. 2006;177:1062–1069. doi: 10.4049/jimmunol.177.2.1062. [DOI] [PubMed] [Google Scholar]
- 69.Lewis MD, Miller SA, Miazgowicz MM, Beima KM, Weinmann AS. T-bet’s ability to regulate individual target genes requires the conserved T-box domain to recruit histone methyltransferase activity and a separate family member-specific transactivation domain. Mol Cell Biol. 2007;27:8510–8521. doi: 10.1128/MCB.01615-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kallies A, Xin A, Belz GT, Nutt SL. Blimp-1 transcription factor is required for the differentiation of effector CD8(+) T cells and memory responses. Immunity. 2009;31:283–295. doi: 10.1016/j.immuni.2009.06.021. [DOI] [PubMed] [Google Scholar]
- 71.Shin HM, et al. Epigenetic modifications induced by Blimp-1 Regulate CD8(+) T cell memory progression during acute virus infection. Immunity. 2013;39:661–675. doi: 10.1016/j.immuni.2013.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lu P, et al. Blimp-1 represses CD8 T cell expression of PD-1 using a feed-forward transcriptional circuit during acute viral infection. J Exp Med. 2014;211:515–527. doi: 10.1084/jem.20130208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Oestreich KJ, Yoon H, Ahmed R, Boss JM. NFATc1 regulates PD-1 expression upon T cell activation. J Immunol. 2008;181:4832–4839. doi: 10.4049/jimmunol.181.7.4832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kalia V, et al. Prolonged interleukin-2Ralpha expression on virus-specific CD8+ T cells favors terminal-effector differentiation in vivo. Immunity. 2010;32:91–103. doi: 10.1016/j.immuni.2009.11.010. [DOI] [PubMed] [Google Scholar]
- 75.Pipkin ME, et al. Interleukin-2 and inflammation induce distinct transcriptional programs that promote the differentiation of effector cytolytic T cells. Immunity. 2010;32:79–90. doi: 10.1016/j.immuni.2009.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Calnan DR, Brunet A. The FoxO code. Oncogene. 2008;27:2276–2288. doi: 10.1038/onc.2008.21. [DOI] [PubMed] [Google Scholar]
- 77.Perrot V, Rechler MM. The coactivator p300 directly acetylates the forkhead transcription factor Foxo1 and stimulates Foxo1-induced transcription. Mol Endocrinol. 2005;19:2283–2298. doi: 10.1210/me.2004-0292. [DOI] [PubMed] [Google Scholar]
- 78.Frescas D, Valenti L, Accili D. Nuclear trapping of the forkhead transcription factor FoxO1 via Sirt-dependent deacetylation promotes expression of glucogenetic genes. J Biol Chem. 2005;280:20589–20595. doi: 10.1074/jbc.M412357200. [DOI] [PubMed] [Google Scholar]
- 79.Brunet A, et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science. 2004;303:2011–2015. doi: 10.1126/science.1094637. [DOI] [PubMed] [Google Scholar]
- 80.Giannakou ME, Partridge L. The interaction between FOXO and SIRT1: tipping the balance towards survival. Trends Cell Biol. 2004;14:408–412. doi: 10.1016/j.tcb.2004.07.006. [DOI] [PubMed] [Google Scholar]
- 81.Mihaylova MM, et al. Class IIa histone deacetylases are hormone-activated regulators of FOXO and mammalian glucose homeostasis. Cell. 2011;145:607–621. doi: 10.1016/j.cell.2011.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wang B, et al. A hormone-dependent module regulating energy balance. Cell. 2011;145:596–606. doi: 10.1016/j.cell.2011.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Finlay D, Cantrell D. The coordination of T-cell function by serine/threonine kinases. Cold Spring Harbor perspectives in biology. 2011;3:a002261. doi: 10.1101/cshperspect.a002261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Martin M, et al. Protein phosphatase 2A controls the activity of histone deacetylase 7 during T cell apoptosis and angiogenesis. Proc Natl Acad Sci USA. 2008;105:4727–4732. doi: 10.1073/pnas.0708455105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Paroni G, et al. PP2A regulates HDAC4 nuclear import. Mol Biol Cell. 2008;19:655–667. doi: 10.1091/mbc.E07-06-0623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Singh A, et al. Protein phosphatase 2A reactivates FOXO3a through a dynamic interplay with 14-3-3 and AKT. Mol Biol Cell. 2010;21:1140–1152. doi: 10.1091/mbc.E09-09-0795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Yan L, et al. PP2A regulates the pro-apoptotic activity of FOXO1. J Biol Chem. 2008;283:7411–7420. doi: 10.1074/jbc.M708083200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kleinewietfeld M, et al. Sodium chloride drives autoimmune disease by the induction of pathogenic TH17 cells. Nature. 2013;496:518–522. doi: 10.1038/nature11868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wu C, et al. Induction of pathogenic TH17 cells by inducible salt-sensing kinase SGK1. Nature. 2013;496:513–517. doi: 10.1038/nature11984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kim EH, et al. Signal integration by Akt regulates CD8 T cell effector and memory differentiation. J Immunol. 2012;188:4305–4314. doi: 10.4049/jimmunol.1103568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hand TW, et al. Differential effects of STAT5 and PI3K/AKT signaling on effector and memory CD8 T-cell survival. Proc Natl Acad Sci USA. 2010;107:16601–16606. doi: 10.1073/pnas.1003457107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Cui W, Liu Y, Weinstein JS, Craft J, Kaech SM. An interleukin-21-interleukin-10-STAT3 pathway is critical for functional maturation of memory CD8+ T cells. Immunity. 2011;35:792–805. doi: 10.1016/j.immuni.2011.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res. 2011;21:381–395. doi: 10.1038/cr.2011.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.van Leeuwen F, Gafken PR, Gottschling DE. Dot1p modulates silencing in yeast by methylation of the nucleosome core. Cell. 2002;109:745–756. doi: 10.1016/s0092-8674(02)00759-6. [DOI] [PubMed] [Google Scholar]
- 95.Mersfelder EL, Parthun MR. The tale beyond the tail: histone core domain modifications and the regulation of chromatin structure. Nucleic Acids Res. 2006;34:2653–2662. doi: 10.1093/nar/gkl338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ernst J, Kellis M. Discovery and characterization of chromatin states for systematic annotation of the human genome. Nat Biotechnol. 2010;28:817–825. doi: 10.1038/nbt.1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ernst J, et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature. 2011;473:43–49. doi: 10.1038/nature09906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Phillips JE, Corces VG. CTCF: master weaver of the genome. Cell. 2009;137:1194–1211. doi: 10.1016/j.cell.2009.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]