

Abstract
Proline-rich tyrosine kinase 2 (Pyk2) is a member of the focal adhesion kinase (FAK) subfamily of cytoplasmic tyrosine kinases. The C-terminal Pyk2 focal adhesion-targeting (Pyk2-FAT) domain binds to paxillin, an adhesion molecule. Paxillin has five leucine-aspartate (LD) motifs (LD1–LD5). Here, we show that the second LD motif of paxillin, LD2, interacts with Pyk2-FAT, similar to the known Pyk2-FAT/LD4 interaction. Both LD motifs can target two ligand-binding sites on Pyk2-FAT. Interestingly, they also share similar binding affinity for Pyk2-FAT with preferential association to one site relative to the other. Nevertheless, the LD2–LD4 region of paxillin (paxillin133–290) binds to Pyk2-FAT as a 1:1 complex. However, our data suggests that the Pyk2-FAT and paxillin complex is dynamic and it appears to be a mixture of two distinct conformations of paxillin which almost equally compete for Pyk2-FAT binding. These studies provide insight into the underlying selectivity of paxillin for Pyk2 and FAK which may influence the differing behavior of these two closely-related kinases in focal adhesion sites.
Keywords: Cell adhesion and signaling, Focal adhesion kinase, Proline-rich tyrosine kinase 2, paxillin binding, NMR, dynamic complex
INTRODUCTION
Proline-rich tyrosine kinase 2 (Pyk2) is a focal adhesion kinase (FAK) related protein, also known as calcium-dependent tyrosine kinase (CADTK), cellular adhesion kinase β, related adhesion focal tyrosine kinase (RAFTK), or FAK2 [1–5]. Recently, Pyk2 has become a potentially important new therapeutic target or prognostic marker because overexpression of Pyk2 has been found in many human tumors [2, 3, 6–11]. Pyk2 and FAK function as scaffold proteins at focal adhesion sites, where they interact with cytoplasmic proteins and integrate different environmental signals [12–15] [16, 17]. Pyk2 interacts with a number of proteins, including Src [18], paxillin [19, 20] and p130cas [21], which also bind to FAK.
Although Pyk2 and FAK are similar, the activation mode of Pyk2 and FAK appears to be different; Pyk2 is activated by various stimuli, including elevated intracellular levels of calcium, protein kinase C activation, and exposure to stress factors [1, 22], whereas FAK is activated by integrin engagement [23, 24]. Nevertheless, Pyk2 can functionally replace FAK in biological processes where FAK is limited. For example, studies on endothelial cells (ECs) where FAK plays a critical role during the vascular development showed that they have an adaptive capacity to switch to Pyk2-dependent signaling after deletion of FAK [25]. In addition, Pyk2 and Src-family PTKs (Protein-tyrosine kinases) combine to promote fibronectin (FN) stimulated signaling events that promote activation of extracellular-signal-regulated kinase 2 (ERK2) in the absence of FAK, and Pyk2 tyrosine phosphorylation is enhanced by fibronectin (FN) stimulation of FAK– cells, but not in FAK+ cells [18]. It remains an open question why Pyk2 is capable but less effective than FAK in promoting focal adhesions (FAs) in these cases despite their structural similarity.
Both FAK and Pyk2 contain a large N-terminal FERM domain, a centrally located kinase domain, and a C-terminal focal adhesion targeting domain referred to as FAT domain in Pyk2 and FAK [13]. Similar to other FAT-like domains, the Pyk2-FAT domain forms a four-helix bundle [26]. In addition, the Pyk2-FAT and FAK-FAT domains are responsible for recruitment of Pyk2 and FAK, respectively, to FAs via their interaction with the N-terminus of paxillin, a multi-domain scaffolding protein [12, 26–32]. The N-terminus of paxillin has five leucine-rich LD (LDXLLXXL) motifs, LD1–LD5. Although this domain is largely unstructured, each LD motif is capable of forming an amphipathic alpha-helix which can interact with many other binding partners in FAs [33–36]. However, the binding mechanism by which different proteins interact with LD motif containing adaptor molecules is diverse. In FAK, for example, the binding of paxillin to FAT is mediated by LD2 and LD4 motifs [37, 38]. Specifically, LD2 motif peptide preferentially binds to the helices H1 and H4 interface (H1/H4) site and LD4 peptide binds to the H2/H3 site [39]. However, Schaller et al. showed that LD4 binding to the FAT H1/H4 is defective [40]. In contrast, studies of another FAT-like domain from G-protein-coupled receptor kinase-interacting protein 1 (GIT1) revealed that the binding site for both LD2 and LD4 peptides is located exclusively at the H1/H4 surface [41]. In addition, paxillin LD motifs also play important roles in localization of two closely related integrin-linked kinase (ILK) binding FA proteins, α and β- parvins, to FAs [42, 43].
The crystal structure of Pyk2-FAT in complex with a paxillin LD4 peptide has been reported, which also showed that it has two putative LD4 motif-binding sites located at helices H1/H4 and H2/H3 [26]. Since the FAT domain of FAK recognizes both LD2 and LD4 motifs of paxillin [39], we examined whether LD2 binds Pyk2-FAT as well. Different biophysical studies showed that indeed the LD2 motif of paxillin also binds to the Pyk2-FAT domain. In addition, to compare LD2 with LD4, we performed a series of biochemical and structural studies to determine the ability of LD2 and LD4 peptides to bind Pyk2-FAT in solution using various methods including Isothermal Titration Calorimetry (ITC), Analytical Ultracentrifugation (AUC) and Nuclear Magnetic Resonance (NMR). We also determined the crystal structure of LD2 bound to Pyk2-FAT. Finally, we addressed how these two LD motifs bind to Pyk2-FAT in the context of paxillin full-length mimic. Using a truncated paxillin construct comprising LD motifs 2, 3 and 4 (paxillin133–290), we determined the stoichiometry and binding affinity of this construct to Pyk2-FAT by NMR and various biophysical methods. Together, these structural and biochemical studies provide a better understanding of how key mechanistic differences in Pyk2 and FAK binding to paxillin may influence their ability to regulate FAs.
RESULTS
LD2 motif of paxillin binds the FAT domain of Pyk2
To investigate if the LD2 (residues 139–163 of chicken paxillin) motif binds to the Pyk2-FAT domain, interaction between LD2 peptide and Pyk2-FAT was first examined by ITC. For comparison, the LD4 (residues 262–278 of chicken paxillin) peptide was also examined (Supplemental Figure S1). Representative titrations are shown in Supplemental Figure S2, and the thermodynamic parameters of binding are listed in Table 1. The experimental data best fit a two-site sequential binding model for both LD2 and LD4 peptides. ITC data summarized in Table 1 shows that LD2 binds to the first site with a KD of ~ 7 μM and the second with a KD of ~ 35 μM. Similarly, the LD4 peptide binds to the first site with a KD of ~ 8 μM and the second site with a KD of ~ 46 μM. ITC results on LD2 and LD4 binding to the Pyk2-FAT domain establishes that these two sites are energetically different. Although LD3 has not been previously shown to associate with Pyk2-FAT, we measured LD3 peptide affinity for Pyk2-FAT by ITC. LD3 (residues 217–237 of chicken paxillin, Supplemental Figure S1) binding to Pyk2-FAT appears to be very weak compared to LD2 and LD4 peptides (Supplemental Figure S3). This relatively weak affinity suggests that LD3 does not compete with LD2 or LD4 for Pyk2-FAT binding.
Table 1.
Thermodynamic parameters for binding of Pyk2-FAT to paxillin LD2, LD3 and LD4 peptides and paxillin133–290 obtained by ITC
| Pyk2-FAT Domain | ||||
|---|---|---|---|---|
| Site (n)b | KD (μM)c | ΔHc | −T.ΔSc | |
| kcal/mol | ||||
| LD2a | First | 6.9±0.1 | −2.4±0.1 | −4.5 |
| Second | 35.2±1.8 | 0.3±0.1 | −6.2 | |
| LD3a | First | NA | NA | NA |
| Second | ||||
| LD4a | First | 8.0±0.6 | −2.8±0.2 | −4.1 |
| Second | 46.3±9.9 | 0.9±0.3 | −6.8 | |
| Paxillin133–290 | 1.2 | 1.8±0.1 | −5.0±0.1 | −2.8 |
The peptide sequences are shown in Figure. S1.
Binding stoichiometry.
KD is the dissociation constant. ΔH and ΔS are the change is enthalpy and entropy upon binding at T=298 K, respectively.
To confirm the results from the ITC studies, we also examined Pyk2-FAT/LD2 and Pyk2-FAT/LD4 interactions by AUC. The binding affinities were assessed from analysis of AUC sedimentation equilibrium (SE) experiments (Supplemental Figures S4–S6, Table 2A). Both 1:1 and 1:2 Pyk2-FAT to LD peptide experiments were performed. AUC-SE data fits very well to a single site hetero-association model (A+B ↔ AB) for 1:1 stoichiometry experiments giving equilibrium dissociation constants (KAB) of ~ 1 μM for LD2 and ~ 1μM for LD4. For 1:2 stoichiometry experiments, the data best fits a two-site hetero-association model (A+B+B ↔ ABB) for both peptides. LD2 binds Pyk2-FAT with a KAB of ~ 1 μM for the first site and ~ 5 μM for the second site KABB. Strikingly similar to LD2, LD4 binds Pyk2-FAT with a KAB of ~ 1 μM for the first site and ~ 5 μM for the second site KABB. The global free energy difference for the association between the first and second sites (ΔGAB and ΔGABB) is −0.76 kcal/mol for LD2 and −0.77 kcal/mol for LD4. This result indicates that both LD motifs show similar binding affinity for Pyk2-FAT with a preferential association to one-site relative to the other.
Table 2.
AUC analysis
| A. Sedimentation equilibrium-AUC analysis of Pyk2-FAT/LD2 and Pyk2-FAT/LD4 | ||||
|---|---|---|---|---|
| Model | KAB(μM)c | KABB (μM)d | Delta Ge (kcal/Mol) | Delta Gf (kcal/Mol) |
| Pyk2-FAT + LD2 ↔ Pyk2-FAT/LD2a | 1.00 ±0.01 | −7.61 | ||
|
Pyk2-FAT + 2(LD2) ↔ Pyk2-FAT/LD2 + LD2 ↔ Pyk2-FAT-(LD2)2b |
1.32±0.02 | 5.30±0.12 | −7.45 | −6.69 |
| Pyk2-FAT + LD4 ↔ Pyk2-FAT/LD4a | 1.17±0.02 | −7.52 | ||
|
Pyk2-FAT + 2(LD4) ↔ Pyk2-FAT/LD4 + LD4 ↔ Pyk2-FAT-(LD4)2b |
1.29±0.09 | 5.18±0.36 | −7.47 | −6.70 |
| B. Sedimentation equilibrium-AUC analysis of Pyk2-FAT-paxillin133–290 | ||
|---|---|---|
| Sample |
KAB (μM)b (hetero-association) |
r.m.s.d.c |
| Pyk2-FAT – paxillin133–290a | 0.4 | 0.0050 |
| C. Sedimentation velocity experiments of Pyk2-FAT and paxillin133–290 | |||
|---|---|---|---|
| Sample | s20 (Svedberg)b | M (Da)c | f/f0d |
| Pyk2-FAT free (0.15) | 1.54 (99%) | 15,600 (15,242) | 1.34 |
| paxillin133–290 free (0.01) | 1.32 (48%) | 15,000 (18,464) | 1.70 |
| Pyk2-FAT-paxillin133–290 1:2 (0.12) | 2.45 (70%) | 30,200 (33,706) | 1.40 |
Single site hetero-association model.
Two site hetero-association model.
First or single site dissociation equilibrium constant; Monte Carlo Error Analysis, 500 iterations confidence level 0.68.
Second equivalent two site dissociation equilibrium constant; Monte Carlo Error Analysis, 500 iterations, confidence level 0.68.
Delta G at T = 277.149994K, RT = 0.551kcal/mol for single or first binding site.
Delta G at T = 277.149994K, RT = 0.551kcal/mol for second binding site (both occupied).
Total loading concentration is 35 μM (Pyk2-FAT-paxillin133–290 1:1) in 20 MES, 50 mM NaCl, pH 6.2.
Dissociation equilibrium constant for reversible Pyk2-FAT (species A) and paxillin (species B) single site hetero-association model.
Root mean square deviation of the fit, units in absorbance.
Concentration of peak in mg/ml in parenthesis.
Sedimentation coefficient taken from the ordinate maximum of each peak in the best-fit c(s) distribution at 20 °C with percentage protein amount in parenthesis. Sedimentation coefficient (s-value) is a measure of the size and shape of a protein in a solution with a specific density and viscosity at a specific temperature.
Molar mass values taken from the c(s) distribution that was transformed to the c(M) distribution. The theoretical mass of the monomer is given in parenthesis.
Best-fit weight-average frictional ratio values (f/f0)w taken from the c(s) distribution.
Mapping the paxillin LD2 and LD4 binding sites on Pyk2-FAT by NMR
We next used NMR to further examine the interactions between the Pyk2-FAT domain and the two LD peptides in detail. NH backbone resonances of free and LD2 or LD4 bound Pyk2-FAT were assigned based on the three-dimensional HNCO, HNCA, CBCA(CO)NH, and HNCACB spectra. Chemical shift perturbations (CSPs) for paxillin LD2 peptides bound to Pyk2-FAT were measured by adding unlabeled LD2 and LD4 peptides to 15N-labeled Pyk2-FAT (Figure 1A and 1B). Examining a series of 1H-15N correlation spectra of Pyk2-FAT in the presence of different concentrations of LD peptides (Pyk2:LD peptide ratio of 1:0.2, 1:0.4, 1:0.8, 1:1.2 and 1:2.0), we observed that the binding sites for both LD2 and LD4 peptides are located to the central region of H2/H3 and H1/H4 solvent exposed surfaces. At a high concentration of peptide (Pyk2-FAT: LD2 ratio of 1:2), we observed two clusters of largely shifted peaks in the perturbation plot, indicating two peptides binds per molecule of Pyk2-FAT. This 1:2 stoichiometry of peptide binding is further supported by AUC sedimentation velocity data for Pyk2-FAT/LD2 and Pyk2-FAT/LD4 (Supplemental Figure S5, Supplemental Table S1), and is consistent with previously observed FAK-FAT/LD2 and FAK-FAT/LD4 interactions [44]. However, in NMR titration experiments at lower concentrations of LD2 or LD4 peptide, most of the perturbations were observed at H2/H3. On average, larger chemical shift perturbations were observed at the H2/H3 binding site compared to H1/H4, suggesting a stronger association of LD2 and LD4 peptides at this site (Figure 1A and 1B). The solvent-exposed residues Glu904, Val909, Lys911, Val909, Val913, Leu915, Asn947 and Ala951 from H2/H3 exhibited significant perturbations after binding to LD2 and LD4 peptides, whereas the residues from H1/H4 showed only moderate changes. Overall the residues from H2/H3 binding site exhibit intermediate slow exchange on NMR timescale. For example, residues Glu904, Val907, Val909, Lys911, Val913, Gly914, Leu915, Gly922, Asn947, Ala951 and Ile954 undergo intermediate slow exchange after adding LD2 or LD4 peptide. However, most of the residues at H1/H4 undergo fast exchange. In addition, we also observed a notable biphasic chemical shift behavior for H1/H4 residues during the titration. For example, Leu894 and Thr978 of H1/H4 show a biphasic behavior with a break at 1:1 ratio of protein to peptide (Figure 1C and 1D). In contrast, residues from H2/H3 display mostly a monophasic behavior in all titration experiments. The feature of biphasic peak-shift induced by the peptide binding suggests that both peptides undergo sequential binding characteristics, and that peptide binds first to H2/H3 and then H1/H4.
Figure 1. NMR analysis of the binding of the Pyk2-FAT to paxillin LD2 and LD4 peptides.
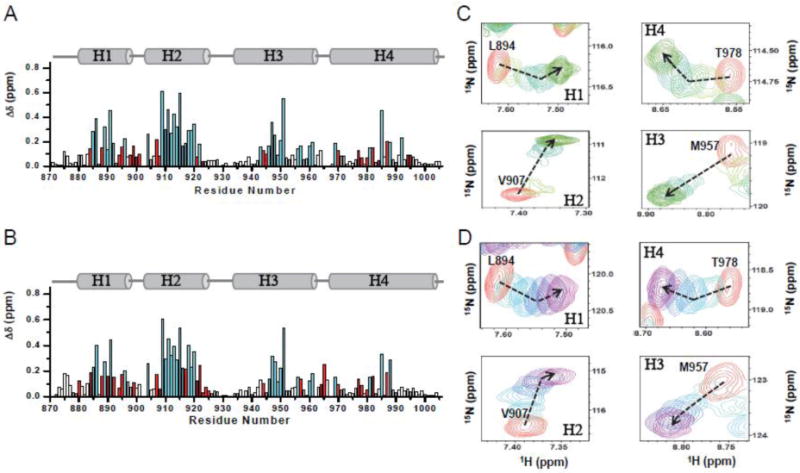
(A and B) Chemical shift changes (Δδ) are plotted as a function of the residue number for LD2 and LD4, respectively. Schematic indicating alpha helices 1–4 is shown above each plot. Residues in slow or intermediate exchange on the NMR time-scale upon LD2 and LD4 peptide binding are colored cyan. Residues that show biphasic curve behavior are colored in red. (C and D) Selected residues from 1H-15N-HSQC spectra of Pyk2-FAT (H2/H3 and H1/H4 region) showing biphasic and monophasic curve after titrating with unlabeled paxillin LD2 peptide (panel C) and LD4 peptide (panel D), at different molar ratios: 1:0 (free Pyk2-FAT, red), 1:0.2 (LD2-lime, LD4-Turquoise), 1:0.4 (LD2-bright green, LD4-sky blue), 1:1.0 (LD2-sea green, LD4-blue), 1:1.5 (LD2-forest, LD4-indigo), and 1:2.0 (LD2-dark green, LD4-magenta). Dashed arrows show the direction of perturbation.
To confirm the sequential binding, we used a paramagnetic relaxation enhancement (PRE) experiment to map the binding interface at lower concentrations of peptides (Pyk2-FAT:peptide ratio of 1:0.2). This method provides distance information from the spin center up to 20–25 Å [45]. 1H-15N HSQC spectra of Pyk2-FAT were acquired with methane thiosulphonate (MTSL)-derivatized LD2 and LD4 peptides (Figure 2A). The data clearly showed that residues from H2/H3 disappeared or showed reduced intensity after addition of LD2MTSL and LD4MTSL peptides at lower concentrations (Pyk2-FAT:peptide ratio of 1:0.2) (Figure 2B and 2C). For example, when spin-labeled LD2 (Q156C) or LD4 (S262C) were added, H2/H3 residues Ser899, Glu904, Gly905, Val907, Val908, Val909, Lys911 Asn912 and Val913 disappeared. Hence, both NMR and ITC data provide strong evidence for two site sequential binding model during Pyk2-FAT/LD2 and Pyk2-FAT/LD4 complex formation. Furthermore, H2/H3 is the stronger binding site for both LD2 and LD4 peptides as evidenced by NMR.
Figure 2. Ligand binding of Pyk2-FAT monitored by NMR spectroscopy.
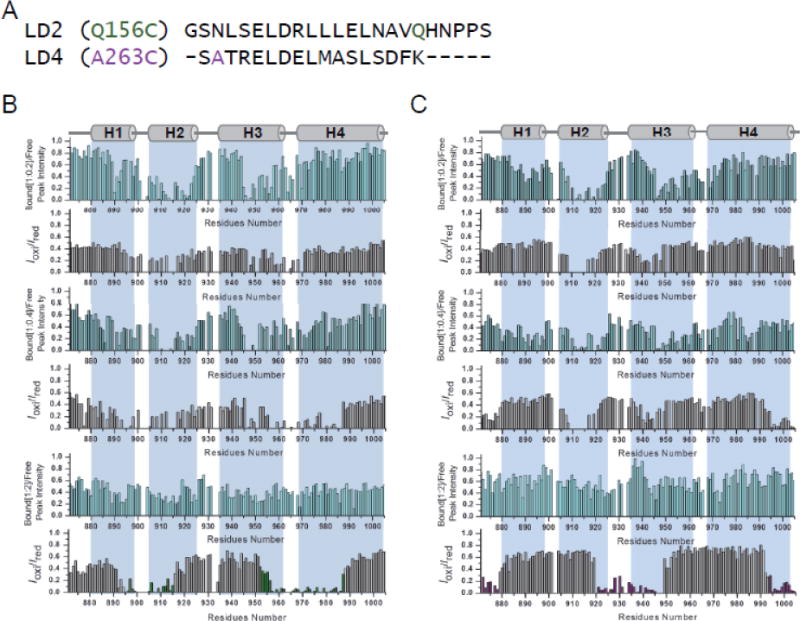
(A) Sequence of paramagnetically-labeled derivatives of paxillin LD2 and LD4 peptides. (B and C) Ratios of the HSQC peak intensity for protein alone to that of Pyk2-FAT bound to LD2 and LD4 peptide, respectively, are plotted versus residue numbers (upper graph; cyan). Ratios of Pyk2-FAT cross-peak intensities in the presence of oxidized or reduced (Iox/Ired) LD2 (Q156C-MTSL) and LD4 (A263C-MTSL) peptide versus residue number (lower graph; grey). Based on an Iox/Ired threshold of 0.4, Pyk2-FAT residues impacted by LD2 and LD4 peptides are colored in green and purple, respectively.
Crystallographic studies of paxillin LD peptides bound to Pyk2-FAT
Due to the limited NOEs available for LD2 bound to Pyk2-FAT, we were unable to determine the complex structure by NMR. To investigate the nature of these molecular interactions, we determined the co-crystal structure of Pyk2-FAT in complex with LD2 motif peptides. X-ray diffraction data exhibited strong anisotropy in reciprocal lattice dimension c*. Therefore, the 3.5 Å scaled data was submitted to the UCLA Diffraction Anisotropy Server [46] for anisotropy correction prior to refinement. Molecular replacement was performed using the previously published Pyk2-FAT crystal structure (PDB: 3GM3) [26]. As the data were relatively low resolution, refinement using DEN restraints during simulated annealing with CNS [47] produced improved maps for initial placement of LD2 peptides. Data collection and refinement statistics are provided in Table 3 and representative electron density is shown in Figure 3A and Supplemental Figure S7B.
Table 3.
Data collection and Refinement Statistics
| Pyk2-FAT/LD2 | Pyk2-FAT/LD4 | ||
|---|---|---|---|
| Data collection | |||
| Space group | P6222 | P212121 | |
| Cell dimensions | |||
| a, b, c (Å) | 183.0, 183.0, 52.9 | 83.5, 83.8, 170.6 | |
| α, β, γ (°) | 90, 90, 120 | 90, 90, 90 | |
| Resolution (Å) | 3.5 (3.62–3.50)a | Truncated* | 3.1 (3.22–3.10) |
| Rmerge | 10.4 (30.2) | 11.8 (27.2) | 6.0 (64.7) |
| I/σI | 25.3 (2.0) | 30.4 (6.9) | 25.8 (1.8) |
| Completeness (%) | 84.4 (31.9)b | 80.5 (5.4) | 97.9 (94.4) |
| Redundancy | 9.5 (2.9) | 10.4 (4.7) | 5.0 (3.9) |
| Refinement | |||
| Resolution (Å) | 30.0–3.5 | 30.0–3.1 | |
| No. reflections | 5,735 | 20,847 | |
| Rwork/Rfree | 22.1/24.0 | 21.8/26.6 | |
| No. atoms | |||
| Protein | 945 | 3,834 | |
| Peptide | 159 | 455 | |
| Water | 0 | 3 | |
| B-factors | |||
| Protein | 87.4 | 105.9 | |
| Peptide | 119.5 | 128.3 | |
| Water | 99.5 | ||
| R.m.s. deviations | |||
| Bond lengths(Å) | 0.007 | 0.008 | |
| Bond angles (°) | 0.97 | 1.1 |
Values in parenthesis are for the highest resolution shell.
Truncated values correspond to statistics after elliptical truncation prompted by strong anisotropy.
Pyk2-FAT/LD2 data completeness 100% to 4.2 Å before and after truncation.
Figure 3. Structures of Pyk2-FAT bound to paxillin LD2 and LD4 motif peptides.
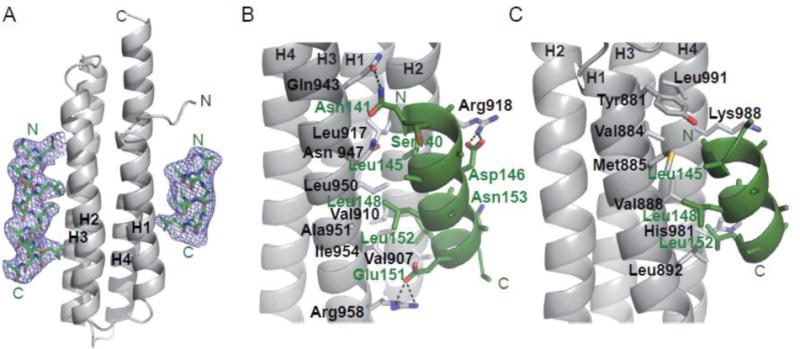
(A) Fo-Fc simulated annealing omit map for Pyk2-FAT/LD2 contoured at 2.5σ. Pyk2-FAT and paxillin LD2 peptide are colored in grey and green, respectively. Secondary structure elements of Pyk2-FAT are labeled. (B) The interface between the Pyk2-FAT H2/H3 binding site and paxillin LD2 peptide. Observed interacting residues from the LD2 peptide and Pyk2-FAT were shown in sticks with labels colored in green and black, respectively. Black dotted lines indicate hydrogen bonds. (C) The interface between Pyk2-FAT H1/H4 binding site and paxillin LD2 peptide. Due to weak electron density for the LD2 peptide at the H1/H site, the LD2 sequence registry was unclear and was therefore based on homology with previous crystal structures FAK-FAT/LD2 (1OW8) and Pyk2-FAT/LD4 (3GM1). Additionally, only side chains Leu148 and Leu152 were included in the final model.
We observed that two LD2 peptides bind to one molecule of Pyk2-FAT; one binds to H1/H4 and the other to H2/H3 (Figure 3). The solvent exposed LD2 peptides form an amphipathic helix with hydrophobic residues facing toward the hydrophobic cleft of Pyk2-FAT. At the H2/H3 interface, Leu145 of LD2 makes hydrophobic contact with Leu917, Ile921 and Leu950 of Pyk2-FAT and van der Waals interaction with Arg918 (Figure 3B). Leu148 makes hydrophobic interactions with Leu950 and Ile954, and van der Waals interaction with Ala951. Furthermore, Leu152 makes hydrophobic interactions with Val907 and Val910, and van der Waals interaction with Lys911 on H2/H3.
In addition to hydrophobic and van der Waals interactions, hydrogen bonds strengthen the association between LD2 peptide and the H2/H3 surface of Pyk2-FAT. The main chain carbonyl oxygen of Asn141 on LD2 makes a hydrogen bond with Asn947, whereas the side chain of Asn141 makes a hydrogen bond with Gln943. In addition, Asp146 makes a hydrogen bond with Arg918, and Glu151 is in hydrogen bonding proximity with Arg958 of Pyk2-FAT (Figure 3B). Although the electron density was not sufficient to determine the side chain positions for Pyk2-FAT residue Lys911, superposition of the Pyk2-FAT/LD2 structure with the previously published Pyk2-FAT/LD4 structure (PDB: 3GM1) indicates a hydrogen bond between Asn153 of LD2 (equivalent to Ser274 in LD4) and Lys911 is likely.
The electron density of the LD2 peptide bound to H1/H4 is relatively weak when compared to H2/H3. The polarity of the LD2 peptide bound to the H1/H4 site was apparent from the DEN electron density maps (Supplemental Figure S7A). However, as sidechain character was essentially absent for most of the peptide, (final 11 residue alpha-helical model includes Leu148 and Leu152 sidechains only) the registry of the peptide was not discernible from the electron density (Figure 3A, 3C, Supplemental Figure S7B). Chemical environment and packing at the hydrophobic interface provided clues to the sequence registry. Ultimately, the final sequence assignment was based on homology with previously reported crystal structures. These include the FAK-FAT in complex with LD2 peptide (1OW8; chains A/D) and the Pyk2-FAT in complex with LD4 peptide (3GM1; A/E and B/C) [26, 38]. However, superimposing our LD2 bound 15-residue helix from the H2/H3 site onto LD2 bound at H1/H4 suggests a registry shift of −3 would still allow many of the hydrophobic residues to face the H1/H4 hydrophobic site. For example, the shift would place Leu142 versus Leu145, Leu145 versus Leu148, and Leu149 versus Leu152 at the interface. This shift in registry, however, seems less likely as Asp 146 would replace Leu149 at the hydrophobic interface in proximity to Val 888. Additionally, Leu148 would replace solvent exposed Glu151.
In order to equitably address the difference between the LD2 and LD4 motifs of paxillin, based on sequence analysis (Supplemental Figure S1), the LD4 peptide used in this study is longer than the LD4 peptide used in the Pyk2-FAT/LD4 complex structure previously reported (Supplemental Figure S8A) [26]. To keep consistency, we decided to elucidate the crystal structure of the Pyk2-FAT domain in complex with the LD4 peptide used in this study. Using a condition similar to the study of Pyk2-FAT/LD2 complex structure, the Pyk2-FAT/LD4 complex (PDB: 3U3F) structure was determined at a resolution of 3.2 Å by molecular replacement. Data collection and refinement statistics are provided in Table 3. Representative electron density is shown in Supplemental Figure S8B. Comparing with the previously published Pyk2-FAT/LD4 complex structure (3GM1), most of the key interactions between LD4 peptide and the two binding sites of Pyk2-FAT are consistent (Supplemental Figure S8C and S8D). However, there are some clear differences between the two structures. The most striking difference we observed is that our relatively long LD4 peptide (residues 262–278 versus 263–275) forms an extended helix on the N-terminal side (Supplemental Figure S8E) when bound to H2/H3.
Conformational change of Pyk2-FAT induced by the binding of LD peptides
To further characterize the effects of LD peptides binding on Pyk2-FAT, we used H/D exchange experiment [48] to examine the conformational dynamics of Pyk2-FAT before and after peptide binding. For free Pyk2-FAT, fast 15N-HSQC spectra were collected immediately after dissolving the lyophilized 15N-labeled PAT in D2O. After the first 10 min, 40% of amide peaks had disappeared due to H/D exchange. All the protected peaks were in helical regions, whereas most peaks that vanished belonged to the residues in the loop regions and the termini (Supplemental Figure S9A). We then performed H/D exchange experiments for both Pyk2-FAT/LD2 and Pyk2-FAT/LD4. Data shows that all of the peaks within the core of the four-helix bundle were protected after 6 h. However, residues from H2/H3 and H1/H4 were protected even after 10 hrs (Supplemental Figure S9B and S9C), indicating extensively protected core regions at the H2/H3 and H1/H4 binding sites.
To further understand loop stabilization upon peptide binding, we also acquired steady-state 1H-15N heteronuclear NOE measurements, which offer information on motions [49, 50] and can be used to identify order induced binding or ordered conformations in partially folded ensembles. Typically, the value for the heteronuclear 15N [1H] NOE of folded residues is ~1–0.7, and the NOE for a flexible loop is <0.5. The study of free Pyk2-FAT showed that residues from loop 1 (899–903), loop 2 (926–934) and loop 3 (963–967) showed faster motions, with heteronuclear NOE values less than 0.7 (Supplemental Figure S10A). Upon ligand binding, steady-state NOE values of the amide resonances on Pyk2-FAT/LD2 and Pyk2-FAT/LD4 significantly increased relative to free Pyk2-FAT, indicating that the loop regions of Pyk2-FAT become stabilized (Supplemental Figure S10B and S10C).
Dynamic nature of LD2 peptide binding to Pyk2-FAT measured by NMR
In order to address the dynamic properties of LD2 peptide upon Pyk2-FAT binding, we have expressed and purified 15N-labeled chicken paxillin LD2 peptide (residues 139–163, Supplemental Methods). The 15N-1H HSQC spectra of LD2 peptide were assigned using standard triple resonance experiments. Chemical shift perturbations for paxillin LD2 peptides bound to Pyk2-FAT were measured by adding unlabeled Pyk2-FAT to 15N-labeled LD2 peptide. Examining a series of 1H-15N correlation spectra of LD2 peptide in the presence of different concentrations of Pyk2-FAT (Pyk2-FAT:LD2 ratio of 1:0.2, 1:0.4, 1:0.8, 1:1.2 and 1:2.0), we observed that most of the LD2 resonances were broadened beyond detection (Supplemental Figure S11A–S11C). Therefore, our binding data illustrates that LD2 peptide binds Pyk2-FAT as a dynamic complex.
Characterization of paxillin133–290 binding to Pyk2-FAT by ITC
Because both LD2 and LD4 peptides bind to the H2/H3 site of Pyk2-FAT 5-fold better than the H1/H4 site, it is unclear whether full-length paxillin utilizes both LD2 and LD4 motifs to bind Pyk2-FAT, or there is a preferential association of LD2 or LD4 for a particular site. To address this issue, we performed binding studies of Pyk2-FAT with a paxillin construct that comprises the LD2-LD3-LD4 region (paxillin133–290). Using ITC we determined the binding affinities between paxillin133–290 and Pyk2-FAT. Representative plot for each titration are shown in Supplemental Figure S12, and a summary of the thermodynamic parameters is given in Table 1. The data could be fitted to a single-site model assuming 1:1 stoichiometry with KD values of approximately 1.8 μM for the paxillin133–290/Pyk2-FAT complex. The binding affinity of paxillin133–290 is approximately 10-fold higher than for LD2 and LD4 peptides alone.
Analytical Ultracentrifugation of the paxillin133–290/Pyk2-FAT Complex
From sedimentation velocity and equilibrium experiments, it is evident that paxillin133–290 and Pyk2-FAT form a 1:1 complex in solution. The apparent molecular mass of the 1:1 complex from the velocity data is 30.20 kDa, reasonably close to the theoretical mass of the 1:1 complex (33,706 Da) (Table 2C). Interaction analysis from velocity data may not always yield accurate masses for the different species due to inter-conversions of species such as reversible association/dissociation on a timescale comparable with sedimentation data collection. Also, different species may have different shapes that could influence the calculation of the masses. The analysis of the sedimentation equilibrium data with a 1:1 hetero-association model yields a dissociation equilibrium constant of 0.41 μM (Supplemental Figure S6, Table 2B). The calculated frictional ratio value (f/f0 – value) for free Pyk2-FAT, 1.34, signifies a slightly elongated molecular shape. The unbound paxillin appears to form some soluble associated species (oligomers) of undetermined stoichiometry’s, but the main species appears to be a monomer with an f/f0 – value of 1.7, indicative of a highly extended, largely unfolded protein (Table 2C).
Chemical shift mapping of paxillin133–290 binding to Pyk2-FAT
We next used NMR chemical shift perturbation to study the interaction between paxillin133–290 and Pyk2-FAT. NH backbone resonances of paxillin133–290 were assigned based on the three-dimensional HNCO, HNCA, CBCA(CO)NH, and HNCACB spectra. We collected 1H-15N-transverse relaxation optimized spectroscopy (TROSY) [51] during the titration of unlabeled paxillin133–290 to 15N-labeled Pyk2-FAT (Pyk2-FAT: paxillin133–290 ratio of 1:0.2, 1:0.4, 1:0.6, 1:1, 1:2). During NMR titration of paxillin133–290 to Pyk2-FAT, most of the amide resonances from the H2/H3 binding site disappeared beyond detection (Supplemental Figure S13A). This peak disappearance is due to intermediate slow exchange regime, indicating tight peptide binding at H2/H3. However, addition of excess paxillin133–290 to Pyk2-FAT induces loss of signals from H1/H4 binding site as well (Supplemental Figure S13B). Such paxillin133–290 binding preference for H2/H3 is consistent with our LD2 and LD4 peptide studies, in which we observed that H2/H3 is the preferential binding site for both peptides. These amide resonances from the H2/H3 and H1/H4 binding sites of Pyk2-FAT did not reappear in the HSQC-TROSY spectra even after adding a 2-fold excess of paxillin133–290.
We also performed a reverse NMR titration by adding unlabeled Pyk2-FAT to 15N-labeled paxillin133–290 (paxillin133–290: Pyk2-FAT ratio of 1:0.2, 1:0.4, 1:0.6, 1:1, 1:2). The 1H-15N TROSY spectra of free paxillin133–290 showed dispersion of signals characteristic of an intrinsically unstructured protein (Supplemental Figure S14). During the titration of Pyk2-FAT to paxillin133–290, we observed that several peaks from the LD2 and LD4 regions disappeared beyond detection (Figure 4). Similar to the Pyk2-FAT titration experiment, amide peaks from the LD2 and LD4 regions of paxillin133–290 did not reappear in the spectra even after adding a 2-fold excess of Pyk2-FAT. This is most likely due to conformational switching between two competing conformations of Pyk2-FAT/paxillin133–290 complex; one conformation utilizes the LD2 motif to bind H2/H3 and the LD4 motif to bind H1/H4, and vice versa.
Figure 4. Mapping the paxillin133–290 and Pyk2-FAT interaction using NMR spectroscopy.
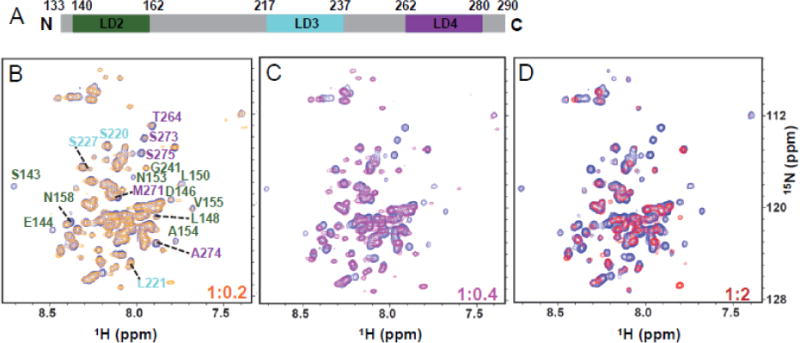
(A) Schematic of paxillinLD2-LD3-LD4 highlighting the distinct motifs used in NMR, x-ray crystallography and other biochemical experiments. LD2 motif is colored in green, LD3 motif is colored in cyan and LD4 motif is colored in purple. This same color scheme is used for residue labeling in panels B–D. (B–D) Superposition of 1H-15N TROSY-HSQC spectra of paxillin133–290 (residues 133–290) with added unlabeled Pyk2-FAT at different molar ratios were shown in blue (1:0), orange (1:0.2), magenta (1:0.4) and red (1:2).
To further address the role of LD3 in the Pyk2-FAT/paxillin interaction, two additional paxillin constructs, paxillin133–242 (LD2–LD3) and paxillin212–284 (LD3–LD4, Supplemental Figure S15A), were generated. Again, both NMR and ITC studies were used to examine the interactions between the two paxillin constructs and the Pyk2-FAT. In the NMR titration studies, at lower paxillin concentrations (Pyk2-FAT: paxillin ratio of 1:0.4 or less) we observed that the residues that had chemical shift perturbations induced by the binding of the two constructs respectively were those located in the H2/H3 (Supplemental Figure S15B and S16A). However, at higher concentrations (Pyk2-FAT: paxillin ratio of 1:1 or greater) we found that both paxillin133–242 and paxillin212–282 could occupy the H2/H3 and H1/H4 binding sites of Pyk2-FAT (Supplemental Figure S15C and S16B). Consistently, ITC experiments demonstrated a two-site sequential binding model for paxillin133–242 and paxillin212–282. Representative ITC titrations are shown in Supplemental Figure S17, and the thermodynamic parameters of the interactions summarized in Supplemental Table S2. Both NMR and ITC data clearly indicated that, in Pyk2-FAT interaction, paxillin133–242 and paxillin212–282 closely resemble LD2 and LD4 peptides respectively, suggesting that the LD3 segment in paxillin plays little role in the Pyk2-FAT/paxillin interaction.
DISCUSSION
The C-terminal FAT domain of Pyk2 is necessary for the recruitment of Pyk2 to focal adhesions by direct interaction with FA proteins such as paxillin. The Pyk2-FAT domain has a classical up-down-up-down four-helix bundle (H1–H4) structure, which is conserved among FAK related proteins [37, 41]. These homologs use either the H1/H4, and/or the H2/H3 binding surface to interact with adaptor proteins in localization to focal adhesion sites. From our ITC and AUC studies of paxillin LD2 and LD4 binding to Pyk2-FAT, it is evident that these two binding sites on the Pyk2-FAT surface are energetically different, with a 5-fold difference in their affinity for both LD2 and LD4 peptides. Indeed, both CSP and PRE NMR data support such sequential association, whereby peptide binds H2/H3 first, followed by H1/H4 (described as KAB and KABB in the AUC data, respectively). Moreover, in both Pyk2-FAT/LD2 and Pyk2-FAT/LD4 complex crystal structures we observed weak electron density for peptide bound at H1/H4 compared to H2/H3, which suggests a weaker association of peptide at the H1/H4 site. Comparison of the two paxillin binding surfaces of Pyk2-FAT mapped by CSP shows that residues on the H2/H3 surface create an extensive and well-defined binding pocket for both LD2 and LD4 peptides, whereas the H1/H4 binding surface is localized to only a few residues (Figure 5).
Figure 5. Chemical shift perturbation (CSP) induced in Pyk2-FAT amide resonances by binding to paxillin LD2 and LD4-derived peptides.
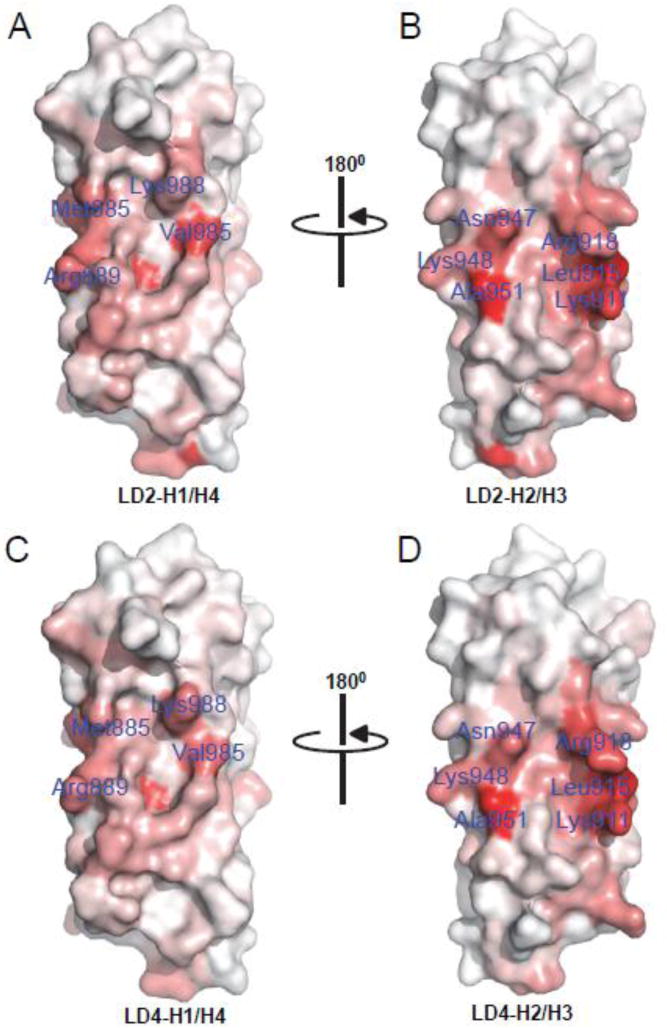
Chemical shift changes upon binding to paxillin LD2 and LD4 peptide were mapped to the surface of Pyk2-FAT. (A and B) LD2 peptide binding at H1/H4 and H2/H3 binding sites of Pyk2-FAT, respectively. (C and D) LD4 binding at H1/H4 and H2/H3 binding sites of Pyk2-FAT, respectively. Protein residues on the Pyk2-FAT surface were colored based on the degree of CSP; darker red color indicates high CSP. The orientation of the peptides corresponds to Figure 4A and 4D.
Comparison of Pyk2-FAT/LD2 and Pyk2-FAT/LD4 crystal complex structures reveals that both structures are very similar (Supplemental Figure S18). As their structural similarity and CSP mapping suggests, LD2 and LD4 bind to H2/H3 with approximately the same affinity (KD of 7 μM and 8 μM, respectively) and bind to H1/H4 with approximately the same affinity (KD of 35 μM and 46 μM, respectively). To further elucidate the binding mechanism of paxillin-Pyk2, we studied the interaction between paxillin133–290 (LD2–LD4 region) and Pyk2-FAT. Paxillin133–290 and Pyk2-FAT associate with a 1:1 stoichiometry as determined by ITC and AUC with an apparent KD of 1.8 μM and 0.4 μM, respectively. This binding affinity is approximately ten times higher than the affinity of paxillin fragments LD2 or LD4 for Pyk2-FAT. Indeed, FAT also exhibits a 10-fold greater binding affinity for a similar paxillin133–290 construct compared to either LD2 or LD4 peptides alone [35]. However, our NMR studies on Pyk2-FAT and paxillin133–290 binding suggests that this one-to-one complex has a unique dynamic feature. During the NMR titration of unlabeled Pyk2-FAT to labeled paxillin133–290, most of the resonances for LD2 and LD4 residues were severely broadened and disappeared with increasing Pyk2-FAT concentration (Figure 4). These LD2 and LD4 resonances did not reappear in the spectra even after adding excess of Pyk2-FAT, suggesting two competing conformations exist; one with LD2 occupying H2/H3 and LD4 occupying H1/H4 and vice versa. These findings are consistent with our peptide studies where we observed equal binding affinities of LD2 and LD4 for Pyk2-FAT, and a 5-fold preference for binding H2/H3 over H1/H4. Therefore, we propose a conformational affinity-switching model to describe the Pyk2 and paxillin association (Figure 6A). In contrast, FAK is proposed to bind paxillin in a concerted fashion with the LD2 motif binding H1/H4 of FAK-FAT and wrapping around to stably bind the LD4 motif at H2/H3 (Figure 6B) [39].
Figure 6. Proposed models of paxillin133–290 binding to the FAT domain of FAK and the FAT domain of Pyk2.
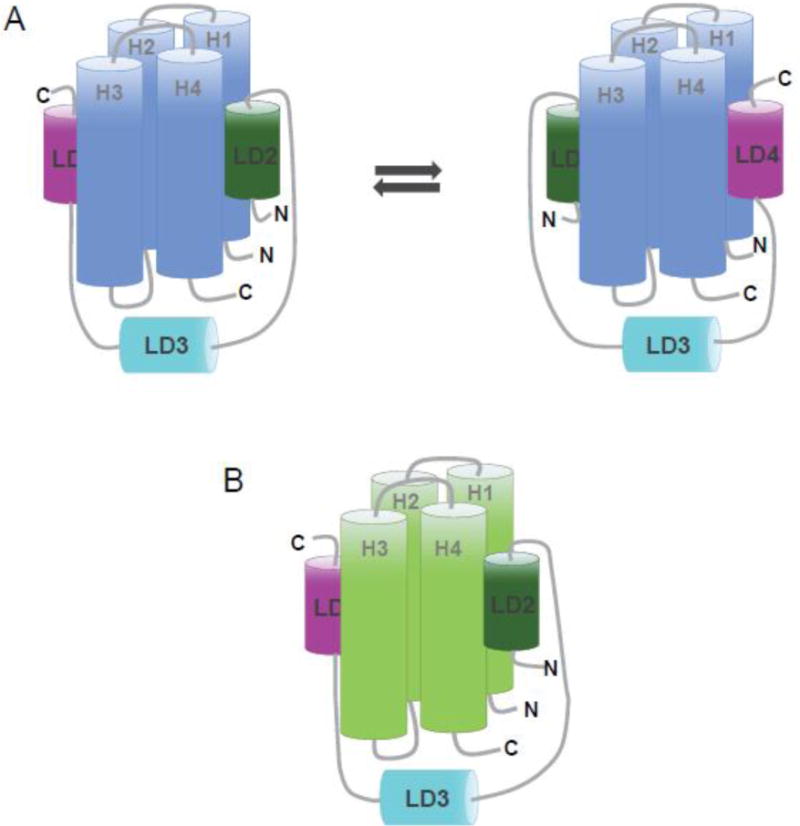
(A) In Pyk2-FAT, the LD2 and LD4 motifs of paxillin133–290 undergo a conformational switching mechanism in which two discrete conformations of paxillin compete for binding; one with LD2 occupying H1/H4 and LD4 occupying H2/H3, and the other conformation with LD4 at H1/H4 and LD2 at H2/H3. (B) In FAT, the LD2 and LD4 motifs of paxillin133–290 binds to opposite faces of the four-helix bundle (H2/H3 and H1/H4) and are oriented in the same direction.
The LD2 motif of paxillin plays a key role in distinguishing the two FAT domains. It is well established that the LD2 of paxillin specifically recognizes the H1/H4 site of FAK-FAT [39, 40]. On the other hand, LD2 binds to both H1/H4 and H2/H3 sites of Pyk2-FAT equally well. Such functional difference is likely due to the key sequence differences of the two FAT domains in the H3 region (marked in Figure 7A). In Pyk2-FAT/LD2 and Pyk2-FAT/LD4 crystal structures, we observed that Ala 951 forms hydrophobic interactions with Leu 148 and Leu 270 of LD2 and LD4 peptides, respectively (Figure 7B and Supplemental Figure S19). The interactions are further validated by NMR studies, where we observed that Ala 951 exhibits the highest chemical shift perturbations of any residue in H3 induced by the peptide bindings (Supplemental Figure S19C). However, in FAK-FAT, this residue is replaced by glycine in the equivalent position that effectively destructs the hydrophobic interactions. Moreover, in Pyk2-FAT, residue 937 is threonine and residue 941 is glycine, whereas the two residues at equivalent positions in FAK-FAT are arginine and methionine, respectively (Figure 7A). We speculate that these two bulky residues in FAK-FAT may play the key role in preventing LD2 to interact the H2/H3 sites in FAK/paxillin interaction. Indeed, the LD2 peptide used in the crystallography study of FAK-FAT/LD2 complex is very short [37, 38]. Based on the structure, we predict that N-terminal end of LD2 motif in full-length paxillin may not be able to interact with this site of FAK-FAT due to steric hindrance (Figure 7C). However, in Pyk2-FAT, the less bulky residues Thr937 and Gly941 allow N-terminal end of LD2 motif to exploit further interaction with extended core binding site of Pyk2-FAT at the H2/H3 site. Indeed, in Pyk2-FAT/LD2 structure, we observed that Asn141 of LD2 peptide makes hydrogen bond with Gln943 of Pyk2-FAT. Furthermore, in the NMR titration studies, we found that several residues in the N-terminal LD2 peptide interacting region of the H2/H3 site, such as Thr937 and Glu940, experienced chemical shift perturbations (Figure 1A and 1B).
Figure 7. Differential binding modes of Pyk2-FAT and FAK-FAT to Paxillin.
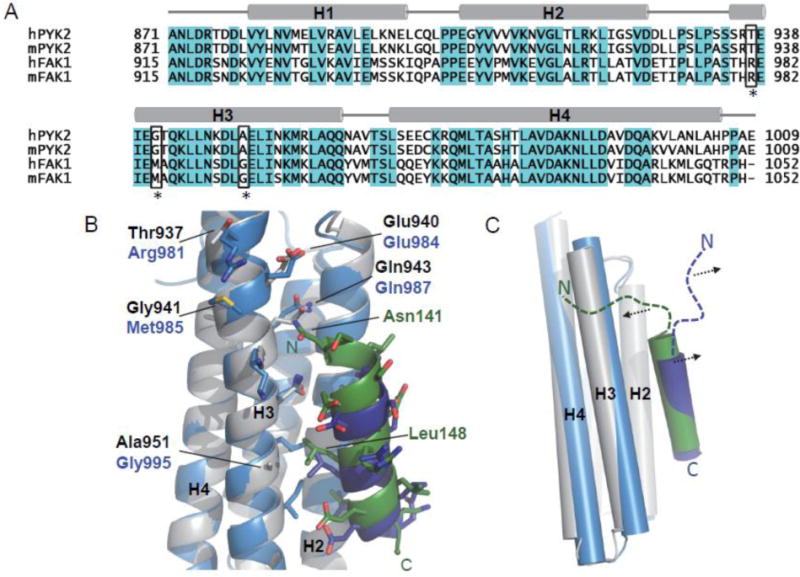
Sequence differences in H2/H3 may contribute to the differential binding mode of Pyk2-FAT and FAK-FAT to paxillin. (A) Structure-based sequence alignment of Pyk2-FAT with FAK-FAT. Human Pyk2 residues 871–1009 (SwissProt Q14289), mouse Pyk2 residues 871–1009 (SwissProt Q9QVP9), human FAK residues 915–1052 (SwissProt Q05397), and mouse FAK residues 915–1052 (SwissProt P34152) are shown. Conserved residues are highlighted in cyan. Key non-conserved residues in H3 are highlighted with black boxes and asterisks. These residues include Thr 937, Gly 941and Ala 951 of Pyk2-FAT, and equivalent residues Arg 981, Met 985 and Gly 995 of FAK-FAT. (B) Superposition of Pyk2-FAT/LD2 (Pyk2-FAT: Grey, LD2: Green; PDB: 4R32) with FAK-FAT/LD2 structure (FAT: Light Blue, LD2: Dark Blue; PDB: 1OW8). Residues from Pyk2-FAT and FAK-FAT are shown in sticks with labels colored in black and blue, respectively. Interacting residues from LD2 peptide are shown in green sticks. Black dotted lines indicate hydrogen bonds. (C) Predicted model showing that Arg 981 and Met 985 of FAK-FAT may induce steric hindrance and displace the N-terminal end of the LD2 motif of full-length paxillin (shown as a blue dashed line) away from H2/H3. However, in the Pyk2-FAT and full-length paxillin interaction, the less bulky residues Thr 937 and Gly 941 at equivalent position of Pyk2-FAT may allow the N-terminal end of the LD2 motif (shown as a green dashed line) to exploit further interaction at H2/H3. Pyk2-FAT is shown in grey and the LD2 motif in green. FAK-FAT is shown in light blue and the LD2 motif in deep blue.
The dynamic binding mode between Pyk2 and paxillin may indicate that in normal cells, paxillin may not be the primary target for Pyk2. However, even with such a dynamic binding mechanism, it appears that Pky2 binds to paxillin with a similar binding affinity as FAK. Therefore, overexpression of Pyk2 in tumor cells is likely to interfere with normal function(s) of FAK. Future studies will address whether other paxillin homologs known to bind Pyk2, such as leupaxin [52] and Hic-5 [53], bind Pyk2 in a similar fashion or more closely resemble paxillin-FAK binding. In the meantime, the identification of H2/H3 of Pyk2-FAT as the preferred paxillin binding site in this study may provide a novel target for structure-based anti-tumor drug discovery.
MATERIALS AND METHODS
Cloning, Expression and Purification of the FAT domain of Pyk2
The FAT domain of human Pyk2 (residues 871–1005) was amplified by PCR from a human cDNA library and cloned into a pET28 vector (Novagen) with an N-terminal His6-tag. The protein was expressed in E. coli BL21 (DE3) cells and purified by nickel-column affinity chromatography followed by gel filtration. The 15N-labeled and 13C/15N-labeled samples were prepared by growing the cells in MOPS-buffered media containing 15NH4Cl (1 g/liter) and [13C6] glucose (3 g/liter) [54]. The final protein buffer used for all NMR experiments, biochemical studies, and X-ray crystallization was 20mM MES, pH 6.2.
Cloning, Expression and purification of LD2–LD4 Paxillin (Paxillin133–290)
The coding sequence of paxillin133–290 (chicken paxillin; residues 133–290) was cloned into the pET-28 expression vector (Novagen). Chicken paxillin sequence (residues 133–290) is 90% identical to that of human paxillin. LD3 and LD4 motifs of human and chicken paxillin are same, whereas LD2 motif of chicken paxillin has an additional serine at the C-terminus (S162). However, S162 is outside of LDXLLXLL binding motif. Hence, we choose chicken paxillin133–290 construct for interaction studies with Pyk2-FAT. Protein was then expressed in E. coli Rosetta2 (DE3) pLysS cells (Novagen). Cells were initially grown at 37°C and induced with 1 mM IPTG at 18°C overnight. Protein was purified using nickel-column affinity chromatography followed by HPLC. In expression and purification of paxillin133–290, to avoid nonspecific proteolysis by thrombin, N-terminal His-tag was not cleaved during the purification. The protein was confirmed by SDS-PAGE (Supplemental Figure S20) and MALDI-TOF mass spectrometry (Supplemental Figure S21). The 15N-labeled and 13C/15N-labeled samples were prepared by growing the cells in MOPS-buffered media containing 15NH4Cl (1 g/liter) and [13C6] glucose (3 g/liter) [54]. All biochemical and structural studies of paxillin133–290 were performed in 20 mM MES buffer, pH 6.2.
Synthesis of paxillin peptide mimics LD2, LD3 and LD4
To keep the consistency with paxillin133–290 construct, we used chicken LD2, LD3 and LD4 sequence for the peptide synthesis. The length of LD2, LD3 and LD4 peptide was determined based on our previous paxillin binding studies with FAK [55] and GIT1 [41]. In addition, these peptide residues show NMR chemical shift perturbation during the titration of Pyk2-FAT to paxillin133–290. Paxillin derived peptides LD2 (chicken paxillin; residues 139–163), LD3 (chicken paxillin; residues 217–237) and LD4 (chicken paxillin; residues 262–278) were chemically synthesized and purified by high pressure liquid chromatography (HPLC) at the Hartwell Center of Bioinformatics and Biotechnology. All peptide stocks were prepared at a concentration of 5 mM in 20 mM MES, pH 6.2.
Isothermal Titration Calorimetry (ITC)
ITC experiments were performed using a Microcal ITC200 instrument (Microcal). Sample conditions for the ITC studies were the same as that for the NMR studies, in terms of buffer conditions. The sample cell of the calorimeter was loaded with 10 μM Pyk2-FAT in 20 mM MES, pH 6.2. The syringe was loaded with LD2, LD3 and LD4 peptides and paxillin133–290 (100 μM of LD2, LD3 and LD4, 50 μM of paxillin133–290) for titrations in the same buffer. All solutions were degassed for 10 min. Titrations were performed at 25°C with injection volumes of 2 μl and a spacing of 120 s. Control experiments were performed by injecting LD2, LD3 and LD4 peptide and paxillin 133–290 into the buffer solution in an identical manner and the resulting heat changes were subtracted from the measured heats of binding. The data were fit using a one-site binding model and two-site sequential binding model available in the Origin ITC data analysis software (v. 7.0). All ITC experiments were performed in duplicate. The results are shown in Table 1.
Analytical Ultracentrifugation (AUC)
AUC experiments were carried out in a ProteomeLab XL-I analytical ultracentrifuge (Beckman-Coulter, Palo Alto, CA). The buffer used was 20 mM MES, pH 6.2, with 50 mM NaCl and the same buffer without salt. The partial specific volumes of the proteins were calculated using the software SEDNTERP [56]. Sedimentation took place at 50,000 rpm at a temperature of 20°C. The loading volume was 400 μl and fringe displacement data at time intervals of 1.0 min were collected with the Rayleigh interference system for 10 hours and analysed with SEDFIT software using a sedimentation coefficient distribution model c(s) [57, 58]. Sedimentation equilibrium for Pyk2-FAT/LD2, Pyk2-FAT/LD4 and Pyk2-FAT/paxillin133–290 mixtures were attained at 30, 26 and 18 h at 4 °C at increasing speeds of 18.4, 26.9 and 46k rpm, respectively [59]. Protein concentrations between 35 and 96 μM (130 μL) were loaded into double-sector centrepieces and absorbance distributions recorded at 280 and 230 nm in 0.001 cm radial intervals with 20 replicates for each point. Global least squares modelling were performed at multiple rotor speeds with the software SEDPHAT (www.analyticalultracentrifugation.com). For 1:1 stoichiometry experiments, we used a single site hetero-association model (A+B↔AB, where Pyk2-FAT is the A species and LD2, LD4 or paxillin133–290 is the B species, respectively) [59]. For 1:2 stoichiometry experiments using either LD2 or LD4 peptide, the data best fit to a two site hetero-association model (A+B+B↔ ABB). However, for the Pyk2-FAT: paxillin133–290, 1:2 molar ratio sample, the data best fit a single site hetero-association model.
NMR spectroscopy and chemical shift perturbation (CSP) analysis
All NMR experiments were collected at 600 and 800 MHz on 13C/15N- and 15N-labeled samples of Pyk2-FAT and paxillin133–290 at 305K. NMR spectra were then processed with Topspin 3.1 and analyzed using CARA 1.8.4 [60]. All experiments of Pyk2-FAT and paxillin133–290 were carried out in 20 mM MES, pH 6.2 and the molar concentration of 400–500 μM was used. LD2, LD3 and LD4 peptide stocks were prepared in the Pyk2-FAT dialysis buffer, and their pH was readjusted to 6.2 before titration. Backbone assignments of Pyk2-FAT and paxillin133–290 were obtained based on CBCA(CO)NH, HNCACB, HNCA, and HN(CO)CA experiments. For the H/D exchange experiment, a series of FAST 1H-15N- HSQC spectra were collected after hydration of lyophilized samples in D2O at various time intervals up to 10 h. To ensure structural integrity of the lyophilized protein, HSQC-spectra were acquired in H2O before and after D2O exchange. Steady-state heteronuclear NOE measurements were performed at 600 MHz at 305 K using already-reported pulse sequences [61] on the free and LD2 and LD4 bound Pyk2-FAT. The heteronuclear NOE values were then obtained from the ratio of the peak intensity for 1H-saturated and unsaturated spectra.
For CSP analysis, NMR titration of LD2 and LD4 peptide to Pyk2-FAT was performed by adding the following protein: peptide ratios: 1:0.2, 1:0.4, 1:0.8, 1:1.2 and 1:2. Pyk2-FAT titration to paxillin133–290 was also performed by adding similar ratios. A series of 15N-HSQC spectra were collected on Bruker 600 and 800 MHz spectrometer and data was analyzed using CARA 1.8.4. 15N and 1H chemical shift values for the displaced peaks in the 15N-HSQC titration experiments were determined for each of the successive titration points by using CARA. To determine the per-residue chemical shift perturbation upon binding and account for differences in spectral widths between 15N and 1H resonances [62], weighted average chemical shift differences, Δav(HN) were calculated for the backbone amide 1H and 15N resonances using the equation: ∆av (NH) = [(∆H2+ (∆N/5)2)/2]1/2, where ΔH and ΔN are chemical-shift differences for 1H and 15N, respectively [63, 64].
Paramagnetic relaxation enhancement (PRE) NMR experiments
Samples for PRE NMR experiments contained uniformly 15N-labeled Pyk2-FAT prepared in 20 mM MES, pH 6.2. For spin labeling, LD2 and LD4 peptides were resuspended in 20 mM phosphate buffer pH 7.2 and reacted with a fivefold molar excess of spin label (MTSL) for 8 h at room temperature. The spin labeled peptide was then repurified by HPLC and redissolved in 20 mM MES, pH 6.2 after lyophilization. Initial NMR titration experiments were performed at low protein to peptide ratio (Pyk2-FAT: peptide ratio of 1:0.2). The reduced compound was generated by exposure to a five molar excess ascorbic acid for one hour. To evaluate the paramagnetic broadening effects of LD2MTSL and LD4MTSL, the calculated ratios from the peak intensities of the oxidized and reduced spectra (Iox/Ired) were determined. Measurements for proline or overlapped residues are absent from the intensity ratio plot.
Crystallization, structure determination and model quality
The FAT domain of Pyk2 was co-crystallized with LD2 peptide by sitting-drop vapor diffusion at 18° C. The 4 μl drop contained 2 μl protein-LD2 peptide mixture (20 mM MES, pH 6.2, 1mM protein, 2mM peptide), and 2 μl mother liquor (100 mM Tris pH 8.0, 2.0 M ammonium phosphate monobasic). The crystals were cryopreserved in 20 % (v/v) ethylene glycol/ 80% (v/v) mother liquor. Native data (1.0 Å) were collected at SER-CAT 22ID. Data were integrated and scaled to 3.5 Å using HKL2000 [65]. X-ray diffraction data exhibited strong anisotropy. Therefore, data were submitted to the Diffraction Anisotropy Server at UCLA [46] for automatic determination of resolution limits, ellipsoidal truncation and anisotropic scaling prior to refinement. Resolution limits of 1/3.8, 1/3.8, and 1/3.5 Å2 in a*, b*, c*, respectively, were applied. Of the 6054 reflections, 319 were discarded. The remaining 5,735 reflections had anisotropic scale factors and an isotropic B of −33.95 Å2 applied.
The 2.6 Å crystal structure of Pyk2-FAT (3GM3) served as the Phaser molecular replacement model [66]. The crystals belong to space group P6222 with one Pyk2-FAT and two LD2 peptides. Following molecular replacement, we observed electron density for the peptides at the 1/4 and 2/3 sites. However, due to the 80% solvent content of the crystals, the maps were extremely noisy. Therefore, we proceeded with a low resolution Deformable Elastic Network (DEN) protocol [47] in CNS [67] without incorporation of the solvent exposed peptides or prior refinement. The 3GM3 model served as the reference model. DEN refinement was instrumental in producing better quality maps for initial model building efforts. In addition, simulated annealing with DEN restraints greatly reduced the number of Ramachandran outliers that had to be fixed manually when compared to earlier efforts to perform simulated annealing in the absence of DEN restraints. The model with the lowest Rfree was used in subsequent model building and refinement. Electron density for the bound peptides exhibited characteristic alpha-helical quality, and the polarity of the helices was readily discerned in the DEN maps (refer to Figure S7). A 15-residue alpha-helical peptide was modeled at the 2/3 site, and an 11-residue alpha helical peptide at the 1/4 site. Although some side-chain character was present for the 2/3 site peptide, no attempts were made to initially determine the registry of either peptide. Rigid body, minimization, ADP, and TLS refinement were performed in Phenix [68]. As the model and maps improved, observable sidechains were added. The registry of the 2/3 site was uniquely determined by a combination of electron density features, along with considerations of chemical environment. In particular, several interaction partners at the interface were helpful in the assignment, as they were relatively well observed in the electron density, and fulfilled unique distance and chemical environment requirements in the context of the LD2 peptide sequence. These include Pyk2-FAT/LD2 interaction partners Gln 943/Asn 141 which are in hydrogen bonding proximity, several hydrophobic Pyk2-FAT residues packed with Leu145 and Leu148, as well as Arg918/Asp146 and Arg958/Glu 51 which are in hydrogen bonding proximity. The 1/4 site electron density quality was insufficient to ascertain the registry of the peptide. Chemical environment and packing at the interface were considered in the sequence assignment, and the resulting registry is ultimately based on homology with previously reported crystal structures of the FAK Focal Adhesion Targeting (FAT) domain in complex with LD2 motif peptide (1OW8; chains A/D) and the Pyk2 Focal Adhesion Targeting (FAT) domain in complex with LD4 motif peptide (3GM1; A/E and B/C) [26, 38].
Residues 871–872 from the N-terminus and residue 1005 from the C-terminus of Pyk2-FAT were not modeled due to insufficient electron density. Final simulated annealing omit maps for LD2 peptides are provided in Figure 3A and Supplemental Figure S7B. Ramachandran statistics show that 92.1% and 7.9% of the residues are in the preferred and allowed regions, respectively.
The Pyk2-FAT was co-crystallized with LD4 peptide by sitting-drop vapor diffusion at 18° C. The 4 μl drop contained 2 μl protein-LD4 peptide mixture (20 mM MES, pH 6.2, 1mM protein, 2mM peptide), and 2 μl ML (100 mM MES pH 6.3, 4.2 M NaCl, 2 % (v/v) glycerol). The crystals were cryo-preserved in 20 % (v/v) glycerol/ 80% (v/v) mother liquor. Native data (1.0 Å) were collected at SER-CAT 22ID to 3.1 Å. Molecular replacement proceeded as described for Pyk2-FAT/LD2. The crystals belong to space group P212121 with four Pyk2-FAT protomers (chains A, B, C, D) and six bound peptides (chains E, F, G, and H bound to H2/H3 site; chains I and J bound to H1/H4 site) in the asymmetric unit. All peptides were solvent exposed, and therefore not stabilized by crystal packing. Of the 4 FAT domain protomers A, B, C, and D, the 1/4 site for C and D was blocked by crystal packing. Residues 871–875 from the N-terminus and residue 1005 from the C-terminus of Pyk2-FAT were not modeled due to insufficient electron density. Ramachandran statistics show that 96.6% and 3.4% of the residues are in the preferred and allowed regions, respectively.
Model building was performed using COOT [69]. Structure refinement was performed with CNS [67] and Phenix [68], and 5% of the data was sequestered for the calculation of Rfree. The final structure statistics were calculated using MolProbity [70]. Structural figures were generated with PyMOL [71].
Supplementary Material
HIGHLIGHTS.
-
➢
Pyk2-FAT interacts with paxillin LD2 and LD4 motifs as observed for FAT of FAK.
-
➢
Paxillin LD2 and LD4 motifs preferentially dock at the helix 2/3 binding site of Pyk2-FAT.
-
➢
Pyk2-FAT binding to paxillin is dynamic and follows a conformational switching mechanism.
-
➢
Pyk2 and FAK exhibit differential binding behavior for paxillin association.
Acknowledgments
We thank Christy R. R. Grace for NMR assistance. Data were collected at Southeast Regional Collaborative Access Team (SER-CAT) 22-ID beamline at the Advanced Photon Source, Argonne National Laboratory. Supporting institutions may be found at www.ser-cat.org/members.html. Use of the Advanced Photon Source was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. W-31-109-Eng-38.
ABBREVIATIONS
- Pyk2
proline-rich tyrosine kinase 2
- FAK
focal adhesion kinase
- FAs
focal adhesions
- GIT1
G-protein-coupled receptor kinase-interacting protein 1
- ITC
isothermal titration calorimetry
- AUC
analytical ultracentrifugation
- NMR
nuclear magnetic resonance
- HSQC
heteronuclear single quantum coherence
- NOE
nuclear Overhauser enhancement
- TROSY
transverse relaxation optimized spectroscopy
- PDB
Protein Data Bank
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
ACCESSION NUMBERS
Atomic coordinates and structure factors have been deposited in the Protein Data Bank with accession code 4R32 for the Pyk2-FAT/LD2 complex and 3U3F for the Pyk2-FAT/LD4 complex.
AUTHOR CONTRIBUTIONS
M.V. designed and performed all NMR and biochemical experiments. D.J.M. designed and executed all X-ray crystallization analysis with crystallization assistance by M.V. C.G. made constructs and A.N. performed AUC. M.V., D.J.M. and J.J.Z. contributed in manuscript preparation. J.J.Z. conceived, designed and supervised the studies.
References
- 1.Lev S, Moreno H, Martinez R, Canoll P, Peles E, Musacchio JM, et al. Protein tyrosine kinase PYK2 involved in Ca(2+)-induced regulation of ion channel and MAP kinase functions. Nature. 1995;376:737–45. doi: 10.1038/376737a0. [DOI] [PubMed] [Google Scholar]
- 2.Sasaki H, Nagura K, Ishino M, Tobioka H, Kotani K, Sasaki T. Cloning and characterization of cell adhesion kinase beta, a novel protein-tyrosine kinase of the focal adhesion kinase subfamily. J Biol Chem. 1995;270:21206–19. doi: 10.1074/jbc.270.36.21206. [DOI] [PubMed] [Google Scholar]
- 3.Avraham S, London R, Fu Y, Ota S, Hiregowdara D, Li J, et al. Identification and characterization of a novel related adhesion focal tyrosine kinase (RAFTK) from megakaryocytes and brain. J Biol Chem. 1995;270:27742–51. doi: 10.1074/jbc.270.46.27742. [DOI] [PubMed] [Google Scholar]
- 4.Yu H, Li X, Marchetto GS, Dy R, Hunter D, Calvo B, et al. Activation of a novel calcium-dependent protein-tyrosine kinase. Correlation with c-Jun N-terminal kinase but not mitogen-activated protein kinase activation. J Biol Chem. 1996;271:29993–8. doi: 10.1074/jbc.271.47.29993. [DOI] [PubMed] [Google Scholar]
- 5.Herzog H, Nicholl J, Hort YJ, Sutherland GR, Shine J. Molecular cloning and assignment of FAK2, a novel human focal adhesion kinase, to 8p11.2–p22 by nonisotopic in situ hybridization. Genomics. 1996;32:484–6. doi: 10.1006/geno.1996.0149. [DOI] [PubMed] [Google Scholar]
- 6.Le HT, Maksumova L, Wang J, Pallen CJ. Reduced NMDA receptor tyrosine phosphorylation in PTPalpha-deficient mouse synaptosomes is accompanied by inhibition of four src family kinases and Pyk2: an upstream role for PTPalpha in NMDA receptor regulation. J Neurochem. 2006;98:1798–809. doi: 10.1111/j.1471-4159.2006.04075.x. [DOI] [PubMed] [Google Scholar]
- 7.Sun CK, Ng KT, Sun BS, Ho JW, Lee TK, Ng I, et al. The significance of proline-rich tyrosine kinase2 (Pyk2) on hepatocellular carcinoma progression and recurrence. Br J Cancer. 2007;97:50–7. doi: 10.1038/sj.bjc.6603827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Paulino VM, Yang Z, Kloss J, Ennis MJ, Armstrong BA, Loftus JC, et al. TROY (TNFRSF19) is overexpressed in advanced glial tumors and promotes glioblastoma cell invasion via Pyk2-Rac1 signaling. Mol Cancer Res. 2010;8:1558–67. doi: 10.1158/1541-7786.MCR-10-0334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Katsumi A, Kiyoi H, Abe A, Tanizaki R, Iwasaki T, Kobayashi M, et al. FLT3/ITD regulates leukaemia cell adhesion through alpha4beta1 integrin and Pyk2 signalling. Eur J Haematol. 2011;86:191–8. doi: 10.1111/j.1600-0609.2010.01556.x. [DOI] [PubMed] [Google Scholar]
- 10.Liebau MC, Hopker K, Muller RU, Schmedding I, Zank S, Schairer B, et al. Nephrocystin-4 regulates Pyk2-induced tyrosine phosphorylation of nephrocystin-1 to control targeting to monocilia. J Biol Chem. 2011;286:14237–45. doi: 10.1074/jbc.M110.165464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Riggs D, Yang Z, Kloss J, Loftus JC. The Pyk2 FERM regulates Pyk2 complex formation and phosphorylation. Cell Signal. 2011;23:288–96. doi: 10.1016/j.cellsig.2010.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mitra SK, Hanson DA, Schlaepfer DD. Focal adhesion kinase: in command and control of cell motility. Nat Rev Mol Cell Biol. 2005;6:56–68. doi: 10.1038/nrm1549. [DOI] [PubMed] [Google Scholar]
- 13.Parsons JT. Focal adhesion kinase: the first ten years. J Cell Sci. 2003;116:1409–16. doi: 10.1242/jcs.00373. [DOI] [PubMed] [Google Scholar]
- 14.Polte TR, Hanks SK. Interaction between focal adhesion kinase and Crk-associated tyrosine kinase substrate p130Cas. Proc Natl Acad Sci USA. 1995;92:10678–82. doi: 10.1073/pnas.92.23.10678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu Y, Loijens JC, Martin KH, Karginov AV, Parsons JT. The association of ASAP1, an ADP ribosylation factor-GTPase activating protein, with focal adhesion kinase contributes to the process of focal adhesion assembly. Mol Biol Cell. 2002;13:2147–56. doi: 10.1091/mbc.E02-01-0018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hildebrand JD, Taylor JM, Parsons JT. An SH3 domain-containing GTPase-activating protein for Rho and Cdc42 associates with focal adhesion kinase. Mol Cell Biol. 1996;16:3169–78. doi: 10.1128/mcb.16.6.3169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ren XR, Du QS, Huang YZ, Ao SZ, Mei L, Xiong WC. Regulation of CDC42 GTPase by proline-rich tyrosine kinase 2 interacting with PSGAP, a novel pleckstrin homology and Src homology 3 domain containing rhoGAP protein. J Cell Biol. 2001;152:971–84. doi: 10.1083/jcb.152.5.971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sieg DJ, Ilic D, Jones KC, Damsky CH, Hunter T, Schlaepfer DD. Pyk2 and Src-family protein-tyrosine kinases compensate for the loss of FAK in fibronectin-stimulated signaling events but Pyk2 does not fully function to enhance FAK-cell migration. EMBO J. 1998;17:5933–47. doi: 10.1093/emboj/17.20.5933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li X, Earp HS. Paxillin is tyrosine-phosphorylated by and preferentially associates with the calcium-dependent tyrosine kinase in rat liver epithelial cells. J Biol Chem. 1997;272:14341–8. doi: 10.1074/jbc.272.22.14341. [DOI] [PubMed] [Google Scholar]
- 20.Salgia R, Avraham S, Pisick E, Li JL, Raja S, Greenfield EA, et al. The related adhesion focal tyrosine kinase forms a complex with paxillin in hematopoietic cells. J Biol Chem. 1996;271:31222–6. doi: 10.1074/jbc.271.49.31222. [DOI] [PubMed] [Google Scholar]
- 21.Astier A, Avraham H, Manie SN, Groopman J, Canty T, Avraham S, et al. The related adhesion focal tyrosine kinase is tyrosine-phosphorylated after beta1-integrin stimulation in B cells and binds to p130cas. J Biol Chem. 1997;272:228–32. doi: 10.1074/jbc.272.1.228. [DOI] [PubMed] [Google Scholar]
- 22.Tokiwa G, Dikic I, Lev S, Schlessinger J. Activation of Pyk2 by stress signals and coupling with JNK signaling pathway. Science. 1996;273:792–4. doi: 10.1126/science.273.5276.792. [DOI] [PubMed] [Google Scholar]
- 23.Guan JL, Shalloway D. Regulation of focal adhesion-associated protein tyrosine kinase by both cellular adhesion and oncogenic transformation. Nature. 1992;358:690–2. doi: 10.1038/358690a0. [DOI] [PubMed] [Google Scholar]
- 24.Schaller MD, Borgman CA, Cobb BS, Vines RR, Reynolds AB, Parsons JT. pp125FAK a structurally distinctive protein-tyrosine kinase associated with focal adhesions. Proc Natl Acad Sci USA. 1992;89:5192–6. doi: 10.1073/pnas.89.11.5192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Weis SM, Lim ST, Lutu-Fuga KM, Barnes LA, Chen XL, Gothert JR, et al. Compensatory role for Pyk2 during angiogenesis in adult mice lacking endothelial cell FAK. The Journal of cell biology. 2008;181:43–50. doi: 10.1083/jcb.200710038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lulo J, Yuzawa S, Schlessinger J. Crystal structures of free and ligand-bound focal adhesion targeting domain of Pyk2. Biochem Biophys Res Commun. 2009;383:347–52. doi: 10.1016/j.bbrc.2009.04.011. [DOI] [PubMed] [Google Scholar]
- 27.Du QS, Ren XR, Xie Y, Wang Q, Mei L, Xiong WC. Inhibition of PYK2-induced actin cytoskeleton reorganization, PYK2 autophosphorylation and focal adhesion targeting by FAK. J Cell Sci. 2001;114:2977–87. doi: 10.1242/jcs.114.16.2977. [DOI] [PubMed] [Google Scholar]
- 28.Avraham H, Park SY, Schinkmann K, Avraham S. RAFTK/Pyk2-mediated cellular signalling. Cellular signalling. 2000;12:123–33. doi: 10.1016/s0898-6568(99)00076-5. [DOI] [PubMed] [Google Scholar]
- 29.Brown MC, Turner CE. Paxillin: adapting to change. Physiol Rev. 2004;84:1315–39. doi: 10.1152/physrev.00002.2004. [DOI] [PubMed] [Google Scholar]
- 30.Liu S, Thomas SM, Woodside DG, Rose DM, Kiosses WB, Pfaff M, et al. Binding of paxillin to alpha4 integrins modifies integrin-dependent biological responses. Nature. 1999;402:676–81. doi: 10.1038/45264. [DOI] [PubMed] [Google Scholar]
- 31.Yu JA, Deakin NO, Turner CE. Paxillin-kinase-linker tyrosine phosphorylation regulates directional cell migration. Mol Biol Cell. 2009;20:4706–19. doi: 10.1091/mbc.E09-07-0548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Deakin NO, Turner CE. Paxillin comes of age. J Cell Sci. 2008;121:2435–44. doi: 10.1242/jcs.018044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Turner CE, Brown MC, Perrotta JA, Riedy MC, Nikolopoulos SN, McDonald AR, et al. Paxillin LD4 motif binds PAK and PIX through a novel 95-kD ankyrin repeat, ARF-GAP protein: A role in cytoskeletal remodeling. J Cell Biol. 1999;145:851–63. doi: 10.1083/jcb.145.4.851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brown MC, Perrotta JA, Turner CE. Identification of LIM3 as the principal determinant of paxillin focal adhesion localization and characterization of a novel motif on paxillin directing vinculin and focal adhesion kinase binding. J Cell Biol. 1996;135:1109–23. doi: 10.1083/jcb.135.4.1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thomas JW, Cooley MA, Broome JM, Salgia R, Griffin JD, Lombardo CR, et al. The role of focal adhesion kinase binding in the regulation of tyrosine phosphorylation of paxillin. J Biol Chem. 1999;274:36684–92. doi: 10.1074/jbc.274.51.36684. [DOI] [PubMed] [Google Scholar]
- 36.Alam T, Alazmi M, Gao X, Arold ST. How to find a leucine in a haystack? Structure, ligand recognition and regulation of leucine-aspartic acid (LD) motifs. The Biochemical journal. 2014;460:317–29. doi: 10.1042/BJ20140298. [DOI] [PubMed] [Google Scholar]
- 37.Gao G, Prutzman KC, King ML, Scheswohl DM, DeRose EF, London RE, et al. NMR solution structure of the focal adhesion targeting domain of focal adhesion kinase in complex with a paxillin LD peptide: evidence for a two-site binding model. J Biol Chem. 2004;279:8441–51. doi: 10.1074/jbc.M309808200. [DOI] [PubMed] [Google Scholar]
- 38.Hoellerer MK, Noble ME, Labesse G, Campbell ID, Werner JM, Arold ST. Molecular recognition of paxillin LD motifs by the focal adhesion targeting domain. Structure. 2003;11:1207–17. doi: 10.1016/j.str.2003.08.010. [DOI] [PubMed] [Google Scholar]
- 39.Bertolucci CM, Guibao CD, Zheng J. Structural features of the focal adhesion kinase-paxillin complex give insight into the dynamics of focal adhesion assembly. Protein Sci. 2005;14:644–52. doi: 10.1110/ps.041107205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Scheswohl DM, Harrell JR, Rajfur Z, Gao G, Campbell SL, Schaller MD. Multiple paxillin binding sites regulate FAK function. J Mol Signal. 2008;3:1. doi: 10.1186/1750-2187-3-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang ZM, Simmerman JA, Guibao CD, Zheng JJ. GIT1 paxillin-binding domain is a four-helix bundle, and it binds to both paxillin LD2 and LD4 motifs. J Biol Chem. 2008;283:18685–93. doi: 10.1074/jbc.M801274200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lorenz S, Vakonakis I, Lowe ED, Campbell ID, Noble ME, Hoellerer MK. Structural analysis of the interactions between paxillin LD motifs and alpha-parvin. Structure. 2008;16:1521–31. doi: 10.1016/j.str.2008.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stiegler AL, Draheim KM, Li X, Chayen NE, Calderwood DA, Boggon TJ. Structural basis for paxillin binding and focal adhesion targeting of beta-parvin. J Biol Chem. 2012;287:32566–77. doi: 10.1074/jbc.M112.367342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Garron ML, Arthos J, Guichou JF, McNally J, Cicala C, Arold ST. Structural basis for the interaction between focal adhesion kinase and CD4. J Mol Biol. 2008;375:1320–8. doi: 10.1016/j.jmb.2007.11.040. [DOI] [PubMed] [Google Scholar]
- 45.Gillespie JR, Shortle D. Characterization of long-range structure in the denatured state of staphylococcal nuclease. I. Paramagnetic relaxation enhancement by nitroxide spin labels. J Mol Biol. 1997;268:158–69. doi: 10.1006/jmbi.1997.0954. [DOI] [PubMed] [Google Scholar]
- 46.Strong M, Sawaya MR, Wang S, Phillips M, Cascio D, Eisenberg D. Toward the structural genomics of complexes: crystal structure of a PE/PPE protein complex from Mycobacterium tuberculosis. Proc Natl Acad Sci USA. 2006;103:8060–5. doi: 10.1073/pnas.0602606103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schroder GF, Levitt M, Brunger AT. Super-resolution biomolecular crystallography with low-resolution data. Nature. 2010;464:1218–22. doi: 10.1038/nature08892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schanda P, Brutscher B. Very fast two-dimensional NMR spectroscopy for real-time investigation of dynamic events in proteins on the time scale of seconds. J Am Chem Soc. 2005;127:8014–5. doi: 10.1021/ja051306e. [DOI] [PubMed] [Google Scholar]
- 49.Barbar E, Hare M, Makokha M, Barany G, Woodward C. NMR-detected order in core residues of denatured bovine pancreatic trypsin inhibitor. Biochemistry. 2001;40:9734–42. doi: 10.1021/bi010483z. [DOI] [PubMed] [Google Scholar]
- 50.Green TB, Ganesh O, Perry K, Smith L, Phylip LH, Logan TM, et al. IA3, an aspartic proteinase inhibitor from Saccharomyces cerevisiae, is intrinsically unstructured in solution. Biochemistry. 2004;43:4071–81. doi: 10.1021/bi034823n. [DOI] [PubMed] [Google Scholar]
- 51.Pervushin K, Riek R, Wider G, Wuthrich K. Attenuated T2 relaxation by mutual cancellation of dipole-dipole coupling and chemical shift anisotropy indicates an avenue to NMR structures of very large biological macromolecules in solution. Proc Natl Acad Sci USA. 1997;94:12366–71. doi: 10.1073/pnas.94.23.12366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lipsky BP, Beals CR, Staunton DE. Leupaxin is a novel LIM domain protein that forms a complex with PYK2. J Biol Chem. 1998;273:11709–13. doi: 10.1074/jbc.273.19.11709. [DOI] [PubMed] [Google Scholar]
- 53.Ohsako M, To H, Nakamura C, Yamada M, Tsuzuki O, Tatsuo E, et al. Change in erythrocyte shape induced by cyclosporine administration. Biological & pharmaceutical bulletin. 1998;21:1236–9. doi: 10.1248/bpb.21.1236. [DOI] [PubMed] [Google Scholar]
- 54.Neidhardt FC, Bloch PL, Smith DF. Culture medium for enterobacteria. J Bacteriol. 1974;119:736–47. doi: 10.1128/jb.119.3.736-747.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liu G, Guibao CD, Zheng J. Structural insight into the mechanisms of targeting and signaling of focal adhesion kinase. Mol Cell Biol. 2002;22:2751–60. doi: 10.1128/MCB.22.8.2751-2760.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Laue TM, Shah BD, Ridgeway TM, Pelletier S. Computer-aided interpretation of analytical sedimentation data for proteins. In: Harding SE, et al., editors. Analytical ultracentrifugation in biochemistry and polymer science. The Royal Society of Chemistry; Cambridge, UK: 1992. pp. 90–125. [Google Scholar]
- 57.Schuck P. Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys J. 2000;78:1606–19. doi: 10.1016/S0006-3495(00)76713-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schuck P, Perugini MA, Gonzales NR, Howlett GJ, Schubert D. Size-distribution analysis of proteins by analytical ultracentrifugation: strategies and application to model systems. Biophys J. 2002;82:1096–111. doi: 10.1016/S0006-3495(02)75469-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Balbo A, Brown PH, Braswell EH, Schuck P. Measuring protein-protein interactions by equilibrium sedimentation. Curr Protoc Immunol. 2007 doi: 10.1002/0471142735.im1808s79. Chapter 18:Unit. [DOI] [PubMed] [Google Scholar]
- 60.Keller R. The Computer Aided Resonance Assignment Tutorial: Verlag. Cantina; Switzerland: 2004. [Google Scholar]
- 61.Farrow NA, Muhandiram R, Singer AU, Pascal SM, Kay CM, Gish G, et al. Backbone dynamics of a free and phosphopeptide-complexed Src homology 2 domain studied by 15N NMR relaxation. Biochemistry. 1994;33:5984–6003. doi: 10.1021/bi00185a040. [DOI] [PubMed] [Google Scholar]
- 62.Farmer BT, 2nd, Constantine KL, Goldfarb V, Friedrichs MS, Wittekind M, Yanchunas J, Jr, et al. Localizing the NADP+ binding site on the MurB enzyme by NMR. Nat Struct Biol. 1996;3:995–7. doi: 10.1038/nsb1296-995. [DOI] [PubMed] [Google Scholar]
- 63.Grzesiek S, Bax A, Clore GM, Gronenborn AM, Hu JS, Kaufman J, et al. The solution structure of HIV-1 Nef reveals an unexpected fold and permits delineation of the binding surface for the SH3 domain of Hck tyrosine protein kinase. Nat Struct Biol. 1996;3:340–5. doi: 10.1038/nsb0496-340. [DOI] [PubMed] [Google Scholar]
- 64.Garrett DS, Seok YJ, Peterkofsky A, Clore GM, Gronenborn AM. Identification by NMR of the binding surface for the histidine-containing phosphocarrier protein HPr on the N-terminal domain of enzyme I of the Escherichia coli phosphotransferase system. Biochemistry. 1997;36:4393–8. doi: 10.1021/bi970221q. [DOI] [PubMed] [Google Scholar]
- 65.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Method Enzymol. 1997;276:307–26. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 66.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–74. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, et al. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905–21. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 68.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–21. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–32. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 70.Chen VB, Arendall WB, 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta crystallographica Section D, Biological crystallography. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.DeLano WL. The PyMOL Molecular Graphics System. DeLano Scientific; San Carlos: 2002. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.