

Abstract
Members of the bone morphogenetic protein (BMP) superfamily, including transforming growth factor-betas (TGFβ), regulate multiple aspects of chondrogenesis. Smad7 is an intracellular inhibitor of BMP and TGFβ signaling. Studies in which Smad7 was overexpressed in chondrocytes demonstrated that Smad7 can impact chondrogenesis by inhibiting BMP signaling. However, whether Smad7 is actually required for endochondral ossification in vivo is unclear. Moreover, whether Smad7 regulates TGFβ in addition to BMP signaling in developing cartilage is unknown. In this study, we found that Smad7 is required for both axial and appendicular skeletal development. Loss of Smad7 led to impairment of the cell cycle in chondrocytes and to defects in terminal maturation. This phenotype was attributed to upregulation of both BMP and TGFβ signaling in Smad7 mutant growth plates. Moreover, Smad7−/− mice develop hypocellular cores in the medial growth plates, associated with elevated HIF1α levels, cell death, and intracellular retention of types II and X collagen. Thus, Smad7 may be required to mediate cell stress responses in the growth plate during development.
Keywords: BMP, bone morphogenetic protein, TGFβ, transforming growth factor β, Smad, Mouse, Chondrocytes, Cartilage, Growth plate, Hypoxia, ER stress
Introduction
The majority of the vertebrate skeleton is formed via endochondral ossification. This process begins with the condensation of mesenchymal cells, which then differentiate into chondrocytes that proliferate and secrete extracellular matrix (ECM) proteins. Chondrocytes in the centers of these cartilage condensations exit the cell cycle and mature into prehypertrophic, and then hypertrophic chondrocytes, which eventually undergo apoptosis. Meanwhile, the cartilage ECM is invaded by blood vessels, osteoblasts, and osteoclasts, which degrade the ECM and replace cartilage with bone.
Endochondral ossification is regulated by local paracrine factors, including bone morphogenetic proteins (BMPs) and transforming growth factor βs (TGFβ). Cellular responses to BMP and TGFβ are mediated by canonical (Smad) and noncanonical (non-Smad) pathways (Derynck and Zhang, 2003; Massague, 1998), all of which are initiated via activation of the type I receptors. In the canonical pathway, activated type I receptors phosphorylate regulatory Smads (R-Smads1/5/8 for BMP signaling; R-Smads2/3 for TGFβ signaling). R-Smads complex with Smad4 and translocate to the nucleus to regulate target gene expression. In noncanonical pathways, BMPs and TGFβs transduce their signals via MAP kinases (ERK, JNK, p38) through TGFβ-activating kinase (TAK1) (Moriguchi et al., 1996; Yamaguchi et al., 1995).
Downregulation of BMP and TGFβ signaling is mediated extracellularly by ligand antagonists, and intracellularly by attenuation of R-Smad activity, in part by inhibitory Smads (I-Smad) 6 and 7. I-Smads recruit E3 ubiquitin ligases to type I receptors, leading to their degradation (Inoue and Imamura, 2008; Murakami et al., 2003). In addition, I-Smads can interfere with R-Smad phosphorylation (Hayashi et al., 1997; Imamura et al., 1997; Nakao et al., 1997). While Smad6 specifically inhibits the BMP pathway, Smad7 can inhibit both BMP and TGFβ pathways (Massague et al., 2005).
Genetic analyses in mice demonstrate that the majority of BMP signaling in cartilage development occurs via the canonical pathway, as evidenced by severe chondrodysplasia in mice lacking R-Smads1/5/8 (Retting et al., 2009). The role of TGFβ signaling in endochondral ossification is less clear, as variable phenotypes have been reported in mice. For example, mice with cartilage-specific deletion of the type II TGFβ receptor exhibited defects in axial, but not appendicular development (Baffi et al., 2004). Moreover, appendicular bones were apparently normal in Smad3−/− mice up to one month after birth (Yang et al., 2001). On the other hand, axial and appendicular skeletal development was impaired in mice with conditional deletion of the type I TGFβ receptor (ALK5) in skeletal progenitor cells (Matsunobu et al., 2009). Taken together, these findings suggest that TGFβ pathways are important at condensation stages, but may be less important in committed chondrocytes.
Gain- and loss-of function studies in vivo (Estrada et al., 2011; Horiki et al., 2004) and in vitro (Li et al., 2003; Scharstuhl et al., 2003) reveal important roles for Smad6 in chondrocytes. Studies in which Smad7 was overexpressed in chondrocytes showed that Smad7 has the capacity to limit BMP signaling (Iwai et al., 2008), but did not define the physiological role of Smad7 in developing cartilage. Moreover, whether Smad7 regulates TGFβ signaling during cartilage development remained unknown. Here, we show that decreased levels of Smad7 lead to skeletal defects resulting from increased BMP and TGFβ activity in chondrocytes.
Materials and methods
Generation of Smad7 knockout mice
Smad7 knockout (Smad7−/−) mice were generated as described (Li et al., 2006a). Embryos and mice were on a mixed C57BL/6J/CD1 background and were genotyped by PCR using the primers 5′-CCCTCCTGCTGTGCAAAGTGTTC-3′ and anti-sense primer 5′-GCATGTC-TATTCAGTAGAAGGATAAG-3′ to detect the wild-type allele, and 5′-GCTTCCTCGTGCTTTACGGTATC-3′ and the above anti-sense primer to detect the mutant allele.
Skeletal preparation and histology
Skeletal preparations were performed as in Estrada et al. (2011). For histology, embryos were fixed with 10% buffered formalin (Fisher Scientific) overnight at 4 °C, decalcified with Immunocal (Decal Chemical Corp., Tallman, NY, USA) overnight at 4 °C, embedded in paraffin, and cut at a thickness of 5–7 μm. For Alcian blue staining, sections were stained as in Estrada et al. (2011). Safranin-O staining was performed as described (Rosenberg, 1971). Heights of proliferative and hypertrophic zones were measured directly from images (n = 5) taken from each of five mice per genotype and significance was established using Student’s t-test.
Immunohistochemical and immunoflourescence staining
Antigen retrieval was performed by boiling in citrate buffer pH 6.0 for 15 min at 95 °C or incubating in 1 mg/ml hyaluronidase (Sigma-Aldrich) in PBS (Mediatech, Inc., Manassas, VA, USA) for 1 h at 37 °C.
For detection of GRP78/BiP (Cell Signaling, 3177), HIF1α (Santa Cruz Biotechnologies, sc10790), Ihh (Santa Cruz Biotechnologies, sc1196), MMP-13 (Abcam, ab84594), osteopontin (Thermo Scientific, RB-9097-P0), phospho-Smad1/5/8 (Cell Signaling, 9511), phospho-Smad2 (Cell Signaling, 3108), phospho-TAK1 (Cell Signaling, 4508), and Ptc1 (Novus Biologicals, NB100-91923), sections were quenched in 3% H2O2 in methanol, blocked with 0.5% blocking reagent (TSA™ Biotin System, Perkin Elmer, NEL700A) in TBS (100 mM Tris pH 7.5, 150 mM NaCl), and incubated with primary antibody overnight at 4 °C. Detection of binding was performed using the TSA™ Biotin System according to the manufacturer’s protocol. Fluorescent detection was conducted using Streptavidin-AlexaFluor-555 (Invitrogen) secondary antibodies; sections were counterstained with DAPI (Invitrogen, D1306).
For detection of Type II Collagen (Abcam, ab21291) and Type X Collagen (Abcam, ab58632 and Abcam ab140230), sections were blocked and incubated with primary antibody as above, incubated with AlexaFluor-488 or -555 (Invitrogen) secondary antibodies for 30 min at room temperature, and then counterstained with DAPI. For detection of Smad7 (Thermo Scientific, PA1-41506), sections were blocked, incubated with primary antibody, and quenched in 3% H2O2 in methanol, as above. Sections were then incubated with biotin-XX anti-rabbit (Invitrogen, B2770) and Streptavidin-HRP (Perkin Elmer) secondary antibodies. Chromogenic detection was performed with the DAB Peroxidase Substrate Kit (Vector Laboratories, SK-4100) followed by counterstaining with Hematoxylin QS (Vector Laboratories, H-3404).
In situ hybridization
Radioactive in situ hybridization was performed as described previously (Retting et al., 2009; Wang et al., 2009) using the following probes: Ihh (St-Jacques et al., 1999), Ptc1 (Milenkovic et al., 1999), Col10a1 (Jacenko et al., 1993).
Cell proliferation and TUNEL labeling
For detection of cell proliferation in vivo, immunofluorescence staining was performed using anti-PCNA (Zymed, 13-3900), anti-phospho-histone H3 (Ser10) primary antibodies (Cell Signaling, 9701), and p57 (Santa Cruz, sc1037), and biotin-XX anti-mouse (Invitrogen) and Streptavidin-AlexaFluor-555 (Invitrogen) secondary antibodies. For TUNEL labeling, the fluorescein In Situ Cell Death Detection Kit (Roche Applied Sciences) was used according to the manufacturer’s instructions. Quantitation of phosphoH3-positive, PCNA-positive and p57-positive cells was performed as described previously; at least three sections from five mice per genotype were examined by individuals blinded to genotype (Estrada et al., 2011).
Cell culture
Primary chondrocytes were isolated from costal cartilage (Estrada et al., 2011), and seeded at 3 × 106 cells/well in 12-well plates. For quantitative real-time PCR, cells were maintained for 2–8.5 days in chondrogenic media (DMEM supplemented with 10% FBS, 1% antibiotic–antimycotic (Invitrogen) and 50 μg/ml ascorbic acid). Each experiment was independently repeated twice. For Western analyses, cells were maintained for 3 days in chondrogenic media. Cells were serum-starved overnight with DMEM supplemented with 1% antibiotic–antimycotic, then stimulated with 50 ng/ml BMP2 (R&D Systems, 355-BM) or 5 ng/ml TGFβ1 (R&D Systems, 240-B) for 2, 4, 8, 12 or 24 h or with equal volume of DMSO. This experiment was performed independently three times.
ATDC5 cells were maintained in DMEM:F12 (1:1) supplemented with 5% FBS, 1% antibiotic–antimycotic, 10 μg/ml transferrin, and 3 × 10−8 M sodium selenite. Cells were serum-starved overnight, and then pretreated with DMSO or 5 ng/ml TGFβ1 in the presence or absence of the ALK5 inhibitor (SB431542, 10 μM) for 20 h at 20% O2. The media was then replaced with media containing DMSO/agonist/inhibitors and the cultures were maintained in 20% O2 or 2% O2 for 24 h. The experiment was performed independently twice.
Quantitative real-time PCR (qRT-PCR) and Western blot analyses
RNA was extracted using the RNeasy Kit (Qiagen). Synthesis of cDNA was performed with the First Strand cDNA Synthesis Kit (Fermentas). qRT-PCR reactions were performed with a SYBR Green Master Mix (Fermentas) using the Mx3005P QPCR System (Stratagene). Primer sequences were as follows: β-actin: forward 5′-CTGAACCCTAAGGCCAACCG-3′, reverse 5′-GTCACGCACGATTTCCCTCTC-3′; MMP-13 and Col10a1 (Li et al., 2006b) and Col2a1 as described (Clark et al., 2009).
For Western analysis, cells were lysed in RIPA buffer (25 mM Tris pH 7.4, 150 mM NaCl, 1% NP-40, 1% Na-deoxycholate, 0.1% SDS) supplemented with protease (complete Mini Tablets, Roche Applied Science) and phosphatase inhibitors (Sigma-Aldrich, P5726). Whole cell lysates were run on 8 or 10% SDS-polyacrylamide gels and transferred semidry onto PVDF membranes. The membranes were blocked with 5% milk in TBS-tween (30 mM Tris pH 7.4, 300 mM NaCl, 0.2% tween-20), incubated with primary antibody (from Cell Signaling: phospho-p38 [9215], p38 [9212], phospho-Smad1/5/8 [9511], phospho-Smad2 [3108], Smad2 [3122] or Smad5 [9517]; Sigma-Aldrich: β-actin [A5316] or tubulin [T6793]; Abcam: HIF1α [ab65979])) diluted in blocking buffer overnight at 4 °C, and then incubated with secondary antibody diluted in blocking buffer for 1 h at room temperature. Binding was detected using the ECL Plus kit (GE Healthcare, Piscataway, NJ, USA) and images were captured using the GE Healthcare Typhoon 9400 Imager and quantified as described (Hall-Glenn et al., 2012).
Results
Smad7 localization
Immunohistochemistry was performed to evaluate the spatial and temporal expression of Smad7 in developing appendicular elements. Smad7 was highly expressed in the lower proliferative, prehypertrophic and hypertrophic zones at E15.0 and P0, and at lower levels in the reserve and upper proliferative zones (Fig. 1A and B). These results suggest that Smad7 may play a role in regulating the onset of hypertrophic differentiation, as well as terminal maturation of chondrocytes in vivo.
Fig. 1.

Skeletal defects in Smad7−/− mice at P0. Immunohistochemical staining for Smad7 (brown color) in (A) WT femurs at E15.0 and (B) proximal tibial growth plates at P0 demonstrates expression in prehypertrophic and hypertrophic chondrocytes. Higher magnification of the prehypertrophic and hypertrophic zones at (A′) E15.0 and (B′) P0 shows that Smad7 is localized in hypertrophic cells; low levels of expression are evident in the (B″) reserve and proliferative zones at P0. R, reserve zone; P, proliferative zone; PH, prehypertrophic zone; H, hypertrophic zone. (C) Whole mount skeletal preparations of P0 WT and Smad7−/− mice. (D) Top panel, anterior view of the skull. Red brackets highlight wider frontal and sagittal sutures in mutants. f, Frontal bone; p, parietal bone. Lateral view of cervical vertebrae. Second panel, red asterisk highlights rib anlagen indicative of posterior transformation of the seventh cervical vertebra (C7) in mutants. Third panel, ventral view of the sternum shows fused ST4 and ST5 in mutants. Bottom panel, dorsal view of lumbar vertebrae. Red arrowheads highlight sacro-iliac joints at L6 in mutants.
Skeletal defects in Smad7−/− mice
The Smad7 mutant allele employed in this study encodes a severe hypomorphic variant of Smad7 (Li et al., 2006a). We chose to use this allele because it is well characterized structurally and phenotypically (e.g. (Li et al., 2006a; Liu et al., 2013; Wang et al., 2013). Smad7−/− embryos were recovered in Mendelian ratios up to E18.5. At weaning (21 days after birth), Smad7−/− mice represented only 5% of heterozygous intercross progeny (n = 134). As early as E12.5, Smad7−/− embryos were smaller than wild-type (WT) littermates (not shown) and dwarfism was maintained postnatally (Fig. 1C). In addition, Smad7−/− mice that survived to weaning appeared malnourished and hunched, and were thus euthanized. These findings are consistent with those of Li and colleagues (Li et al., 2006a). The difference in the percentages of homozygous mice we found (5%) vs. Li et al. (15%) may reflect differences in background strain or vivarium conditions.
Examination of skeletal preparations at postnatal day 0 (P0) revealed defects in ossification of calvarial bones in mutants (Fig. 1D), raising the possibility that Smad7 plays a direct role in ossification. With respect to chondrogenesis, no defects in the chondrocranium were detected (not shown). Loss of Smad7 also led to defects in axial patterning. Smad7−/− P0 mice exhibited posterior transformation of the seventh cervical vertebra (C7), evidenced by the presence of a small rib rudiment at C7 (Fig. 1D). Similarly, WT mice had six ossified sternebrae separated by cartilaginous intersternebrae, while Smad7−/− mice exhibited a fusion of the fourth (ST4) and fifth (ST5) sternebrae (Fig. 1D). Lastly, posterior transformation of the sixth lumbar vertebra (L6) was seen in mutants (Fig. 1D), as evidenced by the formation of sacro-iliac joints, normally found in the first sacral vertebra, at L6 in Smad7−/− mice (Fig. 1D).
Hypocellularity and shortened hypertrophic zones in Smad7−/− growth plates
Histological examination did not reveal any differences in the sizes or morphologies of skeletal condensations at E12.5 or E14.5 in Smad7 mutants and WT littermates (Supplemental Fig. S1 and data not shown). Alcian blue staining revealed no differences in growth plate organization in WT vs. Smad7−/− embryos at E15.5 (Fig. 2A). In WT embryos, vessels from the perichondrium had invaded the hypertrophic zone to facilitate replacement of cartilage by bone (Fig. 2A); however, delayed vascular invasion was observed in Smad7−/− mutants, accompanied by a shorter hypertrophic zone (Fig. 2A). By E17.5, vascular invasion of the hypertrophic zone was observed in mutants, but the hypertrophic zone remained shorter than in WT littermates (Fig. 2B). Immunostaining for PECAM was performed at E16.5 to investigate whether the delayed vascular invasion in mutants is a consequence of a defect in the vasculature as opposed to a delay in chondrocyte differentiation (Supplemental Fig. S2). While a primary defect in vascularization might make a contribution to the Smad7 growth plate phenotype, the indistinguishable presence of blood vessels adjacent to the prospective zone of invasion in the growth plates in mutants and WT littermates is more supportive of the hypothesis that the delay in chondrocyte differentiation is a direct consequence of Smad7 action in chondrocytes.
Fig. 2.
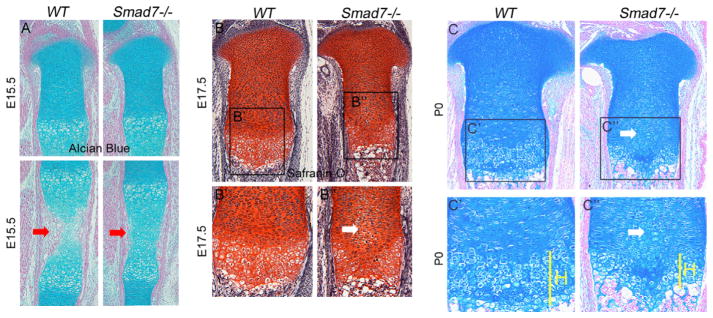
Hypocellularity and shortened hypertrophic zones in Smad7−/− growth plates. (A) Alcian blue/nuclear fast red staining of WT and Smad7−/− tibiae at E15.5. Red arrows highlight delayed vascular invasion in mutant growth plates. (B) Safranin-O, Fast Green, and hematoxylin staining of WT and Smad7−/− tibial growth plates at E17.5. Higher magnification of (B′) WT and (B″) Smad7−/− growth plates at E17.5. White arrow highlights enlarged cells in the proliferative zone of mutant growth plates. (C) Alcian blue and nuclear fast red staining of WT and Smad7−/− tibiae at P0. Higher magnification of (C′) WT and (C″) Smad7−/− growth plates at P0, highlighting shortened hypertrophic zones in mutant growth plates. H, hypertrophic zone.
Chondrocytes in the proliferative zone are normally flattened and organized into well-defined columns. However, beginning at E17.5, enlarged atypical cells were observed in the medial growth plate in Smad7−/− mice (Fig. 2B″). This aberrant morphology persisted through P0, leading to hypocellularity in the medial growth plates of mutants (Fig. 2C). Additionally, the hypertrophic zones remained shorter in P0 Smad7−/− growth plates (Fig. 2C″). At this stage, quantitation of WT and Smad7 mutant littermates confirmed that the hypertrophic zones were shorter in mutants (27±2% reduction, p<.005), but revealed no alteration in the height of the proliferative zones (n = 5; p = 0.9). At postnatal stage (P)7, the hypocellular core diminished, but the shorter hypertrophic zones persisted in mutants (Fig. 3A). By P17, both the proliferative and hypertrophic zones were shorter, and an accumulation of cells exhibiting the morphology of prehypertrophic chondrocytes, and a paucity of enlarged hypertrophic cells, was observed in mutants (Fig. 3B and B″). Overall, these results show that loss of Smad7 significantly impacts chondrocyte proliferation and/or differentiation in the growth plate.
Fig. 3.

Expanded prehypertrophic zones in postnatal Smad7−/− growth plates. (A) Alcian blue and nuclear fast red staining of WT and Smad7−/− tibiae at P7. (B) Safranin-O, Fast Green, and hematoxylin staining of WT and Smad7−/− tibial growth plates at P17, highlighting shortened proliferative zones, as well as expanded prehypertrophic and shortened hypertrophic zones in mutant growth plates. P, proliferative zone; PH, prehypertrophic zone; H, hypertrophic zone. Higher magnification of (B′) WT and (B″) Smad7−/− growth plates.
Smad7 deficiency leads to impaired cell cycle progression and survival in the growth plate
The shorter hypertrophic zones and hypocellular cores in Smad7−/− mice may reflect impaired chondrocyte proliferation. Immunofluorescence staining for phospho-histone H3, a marker for M-phase, revealed no differences in the numbers of cells entering the cell cycle between genotypes in the resting zone at E16.5 (WT = 1.4%±0.25%, mutant = 0.94±0.1%, p = 0.058) but a decrease was seen in the proliferative zone in mutants (WT = 4.74%±0.11%, mutant = 3.14%±0.57%, p = 0.05) (Fig. 4A). Staining for PCNA, a marker for late G1 through S-phase, demonstrated, as reported previously, that PCNA expression was most abundant in lower proliferative/upper prehypertrophic chondrocytes in WT mice (Arikawa-Hirasawa et al., 1999); however, this zone was expanded in mutants (Fig. 4B and C). Analysis of expression of p57, a cell cycle inhibitor required for hypertrophic differentiation (Supplemental Fig. S3) revealed an increased percentage of p57-positive cells was seen at E16.5 in the proliferative zones of mutants (WT = 37.65%±1.48%, mutant = 47.53%, p<0.05; n = 5). This increase persisted in mutants at P0. Taken together, these results indicate that loss of Smad7 leads to defects in chondrocyte proliferation.
Fig. 4.
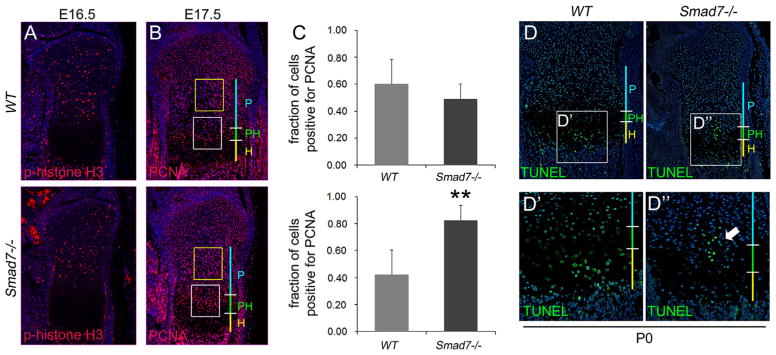
Impaired cell cycle and increased cell death in medial growth plates of Smad7−/− mice. (A) Immunofluorescence staining of E16.5 proximal tibial growth plates for Phospho-histone H3. (B) Immunofluorescence staining of E17.5 tibial growth plates for the proliferation marker PCNA. Yellow box demarcates upper proliferative zone. White box demarcates lower proliferative/prehypertrophic zone. (C) Quantification of the number of cells positive for PCNA in upper proliferative (top) and lower proliferative/prehypertrophic (bottom) zones. Student’s t-test; **p < 0.001. (D) TUNEL staining of P0 proximal tibiae. Higher magnification of TUNEL staining in (D′) WT and (D″) Smad7−/− growth plates. Arrow highlights increased cell death in medial growth plates in mutant mice. P, proliferative zone; PH, prehypertrophic zone; H, hypertrophic zone.
Apoptotic cells, normally restricted to the lower hypertrophic zone, were found in the medial region of the proliferative zone in Smad7−/− growth plates (Fig. 4D), suggesting that the hypocellularity seen in mutants is partly a consequence of reduced cell survival.
Smad7 deficiency leads to defects in terminal chondrocyte maturation
Immunofluorescence staining for type II collagen, normally expressed by proliferating chondrocytes, was observed throughout the proliferative zones in WT and mutant littermates. However, intracellular retention of type II collagen was observed in the medial growth plates of mutants (Fig. 5A). Type X collagen is normally expressed in the early phase of hypertrophic differentiation, and is retained in terminal hypertrophic chondrocytes. In Smad7−/− P0 growth plates, the domain of type X collagen expression was shorter, and protein was retained intracellularly (Fig. 5A). A second antibody against type X collagen revealed some extracellular deposition in lower hypertrophic chondrocytes in mutants, but at reduced levels, and confirmed the intracellular retention of type X collagen in mutants (Supplemental Fig. S4A). Reduced levels of extracellular type X collagen were also seen at E16.5 (Supplemental Fig. S4B). We performed in situ hybridization to obtain additional information. This analysis confirmed that the domain of Col10a1 expression was shorter in both E16.5 and P0 mutant growth plates (Supplemental Fig. S4C and D). These results suggest that mutant chondrocytes initiate hypertrophic differentiation, but may not be able to complete the process.
Fig. 5.

Defects in terminal maturation of chondrocytes in Smad7−/− growth plates at P0. (A) Alcian blue/nuclear fast red staining of P0 proximal tibiae and immunofluorescence staining of proximal tibial growth plates for types II and X collagen. Type X collagen immunostaining was performed using ab58632. Higher magnification of Col2 staining in (A′) WT and (A″) Smad7−/− growth plates. (B) Real-time PCR analysis of Col2a1 and Col10a1 mRNA levels in WT and Smad7−/− primary chondrocytes cultured in chondrogenic media for 2, 4, or 8.5 days. The data represent averages from triplicate reactions and are expressed as fold change±s.d. Student’s t-test; *p < 0.05. (C) Safranin-O, Fast Green, and hematoxylin staining of of P0 proximal tibiae and immunofluorescence staining of tibial growth plates for osteopontin (Opn) and MMP-13. (D) Real-time PCR analysis of Mmp13 mRNA levels in WT and Smad7−/− primary chondrocytes cultured in chondrogenic media for 2, 4, or 8.5 days. The data represent averages from triplicate reactions and are expressed as fold change±s.d. Student’s t-test; *p < 0.05.
Given that Smad7−/− mice are global knockouts, impaired secretion of types II and X collagen may be secondary to defective growth plate angiogenesis. Thus, direct effects of loss of Smad7 on Col2a1 and Col10a1 mRNA expression were investigated in primary chondrocytes. Both Col2a1 and Col10a1 levels were elevated in Smad7-deficient chondrocytes at all time points (Fig. 5B). Thus, loss of Smad7 has direct effects on expression of these genes. The increased transcript levels could be a compensatory response to, or a cause of, increased intracellular retention and decreased extracellular localization of types II and X collagen.
As discussed above, mutant chondrocytes exhibit impaired entry and exit from the cell cycle (Fig. 4B and C). This may contribute to the shortened hypertrophic zone in mutants. It is also possible that the shortened hypertrophic zone is due to accelerated chondrocyte maturation. If so, increased expression of terminal maturation markers osteopontin (OPN) and MMP-13 would be expected. However, OPN and MMP-13 levels were reduced in Smad7−/− mice (Fig. 5C). qRT-PCR analyses of Mmp13 levels in primary chondrocytes confirmed reduced levels in mutant chondrocytes (Fig. 5D). The data thus suggest that the shorter hypertrophic zone in mutant growth plates may be due in part to defects in proliferation and completion of terminal maturation.
Smad7 deficiency does not severely impact Ihh signaling in the growth plate
As discussed above, chondrocytes appear to accumulate in the lower proliferative/prehypertrophic zones in mutants (Fig. 3B). We therefore assessed whether the defect in progression through the cell cycle in Smad7−/− growth plates results from aberrant Ihh signaling. Ihh and PTHrP interact in a negative feedback loop to promote proliferation and limit hypertrophic differentiation (Kronenberg, 2003). In situ hybridization and immunostaining for Ihh revealed no apparent differences in the expression domains in WT vs. Smad7−/− growth plates at E16.5 and P0 (Supplemental Fig. S5A–C). Ihh activity was evaluated via examination of its direct target, Patched 1 (Ptc1). The domain of Ptc1 mRNA expression was reduced in Smad7 mutants at E16.5 and P0 (Supplemental Fig. S5D and E). Immunostaining at P0 further demonstrated that while Ptc1 levels were similar in mutants and WT littermates in the upper resting zone, Ptc1 localization was less pronounced in the lower proliferative zone in Smad7 mutants (Supplemental Fig. S5F). Therefore, impaired entry and exit from the cell cycle in Smad7 mutants may be due in part to decreased Ihh pathway activity.
TGFβ and BMP pathways are elevated in Smad7−/− growth plates and isolated primary chondrocytes
Smad7 is a key intracellular antagonist of both BMP and TGFβ pathways (Hanyu et al., 2001; Ishisaki et al., 1999; Mochizuki et al., 2004). Thus, we examined the effect of loss of Smad7 on activation of canonical and noncanonical BMP and TGFβ pathways in cartilage. Increased pSmad2 staining was seen in the proliferative zone of Smad7−/− growth plates (Fig. 6A). Staining for pSmad1/5/8 revealed increased levels in the proliferative, prehypertrophic, and hypertrophic zones of mutant growth plates (Fig. 6B). By P17, the pSmad2 expression domain was expanded in mutants, particularly in the enlarged prehypertrophic zone (Fig. 6A). In contrast, pSmad1/5/8 expression was expanded throughout both the prehypertrophic and hypertrophic zones (Fig. 6B).
Fig. 6.

TGFβ and BMP pathways are elevated in Smad7−/− growth plates and isolated primary chondrocytes. (A) Immunostaining of proximal tibial growth plates for phospho-Smad2 (pSmad2) at E17.5 and P17. Asterisk highlights elevated pSmad2 levels in the proliferative zone in mutants. (B) Immunostaining of proximal tibial growth plates for phospho-Smad1/5/8 (pSmad1/5/8) at E17.5 and P17. (C) Immunostaining of P0 proximal tibial growth plates for phospho-TAK1 (pTAK1 (D) Western blot analysis shows elevated levels of pSmad2 and phospho-p38 in lysates isolated from WT and Smad7−/− primary chondrocytes treated with TGFβ1 (5 ng/ml) for 0, 2, 4, 8, 12 h. Western analysis shows elevated levels of pSmad1/5/8 in lysates isolated from WT and Smad7−/− primary chondrocytes treated with BMP2 (50 ng/ml) for 0, 4, 8, 12 h.
The upstream regulator of noncanonical BMP and TGFβ signaling, TAK1, is widely expressed in developing cartilage, and then becomes restricted to the prehypertrophic and hypertrophic zones postnatally (Shim et al., 2009). By P0, activated TAK1 (phospho-TAK1) was localized primarily in the prehypertrophic zones of WT growth plates, and this expression domain was expanded in Smad7−/− growth plates (Fig. 6C). These results indicate that both canonical and noncanonical BMP and TGFβ signaling are elevated in Smad7−/− growth plates.
The direct effects of loss of Smad7 on BMP and TGFβ responsiveness were examined in primary chondrocytes. The extent and duration of canonical and noncanonical TGFβ signaling were then evaluated via treatment with TGFβ1. Levels of pSmad2 were greater in Smad7-deficient chondrocytes compared to WT cells at 2 h, and for up to 24 h post-stimulation (Fig. 6D). Likewise, phospho-p38, a downstream target of pTAK1, levels were elevated under basal conditions and for up to 24 h post-stimulation in mutant chondrocytes (Fig. 6E). Canonical BMP signaling was also evaluated. Basal levels of pSmad1/5/8 were elevated in mutants at all time points (Fig. 6D). Thus, loss of Smad7 leads to upregulation of both canonical and noncanonical BMP/TGFβ pathways in vivo, likely due to increased responsiveness to TGFβ and BMP.
Loss of Smad7 leads to increased cell stress responses in the growth plate
The growth plate functions at a lower oxygen tension than most tissues. Schipani et al. (2001) have shown that the hypoxic area of the growth plate is localized to the medial region of the reserve and proliferative zones, the same area where hypocellularity and increased cell death occurred in Smad7−/− growth plates. Thus, the Smad7 growth plate phenotype may result from defective adaption to the hypoxic environment. Expression of HIF1α, the main activator of the hypoxic stress response, was therefore evaluated. Increased HIF1α levels were found throughout the proliferative zones of Smad7−/− embryos, with the highest levels in the lower proliferative/upper prehypertrophic zones (Fig. 7A). These results indicate Smad7 impacts the hypoxic stress response via HIF1α in developing cartilage.
Fig. 7.
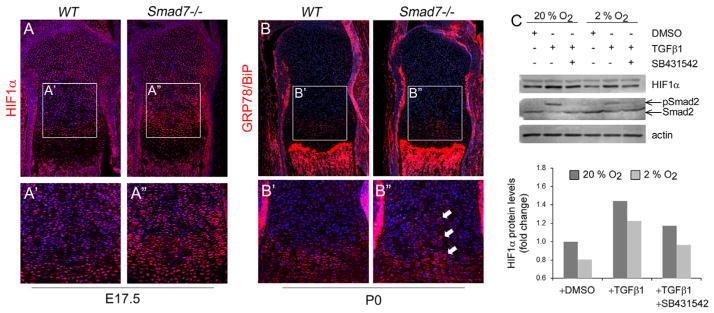
Loss of Smad7 leads to increased cell stress responses in the growth plate. (A) Immunostaining of proximal tibial growth plates for HIF1α at E17.5. Higher magnification of HIF1α staining in (A′) WT and (A″) Smad7−/− growth plates. (B) Immunostaining of tibial growth plates for the ER stress marker, GRP78/BiP, at P0. Higher magnification of GRP78/BiP staining in (B′) WT and (B″) Smad7−/− growth plates. White arrow highlights increased levels in the medial region of the proliferative zones in mutant growth plates. (C) Top: Representative Western blot analysis shows increased stability of HIF1α upon treatment of ATDC5 cells with 5 ng/ml TGFβ1 at both normoxic (20% O2) and hypoxic (2% O2) conditions. Addition of 10 μM of the ALK5 inhibitor, SB431542, attenuates TGFβ-mediated stabilization of HIF1α. Bottom: Densitometric analysis of HIF1α levels was performed using ImageJ software on four independent Western blots as described (Hall-Glenn et al., 2012). The quantitation was done for the major (lower) band and for both bands. The relative intensities were identical in both analyses. A representative image is shown along with its quantitation.
Hypoxia can activate the unfolded protein response as a consequence of endoplasmic reticulum (ER) stress (Wouters and Koritzinsky, 2008). To evaluate whether loss of Smad7 might impact the ER stress response, we performed immunostaining for the master regulator of ER stress, GRP78/BiP. Increased GRP78/BiP levels were evident in the medial region of the proliferative zones in mutants (Fig. 7B). While this finding is consistent with elevated ER stress, it is not definitive. However, taken together, the elevated expression of HIF1α and BiP, hypocellularity and increased cell death indicate abnormal stress responses in mutant growth plates.
A functional connection between TGFβ signaling and the hypoxic stress response has been demonstrated in tumor cells, where TGFβ1 leads to HIF1α stabilization in normoxic conditions (McMahon et al., 2006). It is unknown whether TGFβ signaling impacts HIF1α stability in chondrocytes. Treatment with TGFβ1 led to increased stabilization of HIF1α in chondrocytic ATDC5 cells in both normoxic and hypoxic conditions (Fig. 7C) that was attenuated by treatment with the ALK5-selective inhibitor, SB431542. These results suggest that TGFβ signaling via ALK5 promotes HIF1α stabilization in chondrocytes, and thus, the elevated HIF1α levels in vivo may result from increased responsiveness of Smad7-deficient chondrocytes to TGFβ.
Discussion
Overexpression studies demonstrated that Smad7 has the potential to regulate BMP signaling in cartilage (Iwai et al., 2008). However, given that overexpression studies results in non-physiological expression levels and most often do not recapitulate actual expression patterns, it is still unknown whether Smad7 is actually required for endochondral bone formation. In this study, we show that Smad7 regulates axial and appendicular skeletal development. Smad7−/− mice exhibit defects in anterior/posterior (A/P) patterning, as evidenced by posterior transformation of cervical and lumbar vertebrae. In appendicular bones, loss of Smad7 results in the retention of chondrocytes in a lower proliferative/prehypertrophic state, accompanied by a shorter hypertrophic zone. These defects are attributed, at least in part, to elevated BMP and TGFβ signaling in Smad7-deficient chondrocytes. Thus, our results provide in vivo evidence that Smad7 plays an essential role in limiting both BMP and TGFβ signaling during endochondral bone formation.
Smad7 and Smad6 have overlapping functions in anterior/posterior patterning
Smad7−/− mice exhibit posterior vertebral transformations. Smad6−/− mice also exhibit posterior transformation of the seventh cervical vertebra (Estrada et al., 2011); lumbar patterning, however, was normal in Smad6−/− mice. Thus, Smad7 and Smad6 appear to have overlapping functions in A/P patterning of cervical vertebrae, but Smad7 has a unique function in patterning lumbar vertebrae.
BMP and TGFβ signaling differentially affect the expression pattern and activity of members of the Hox family of transcription factors, which regulate vertebral axial patterning. For example, mice lacking ALK5 in skeletal progenitor cells exhibited normal levels of Hoxc8, but the expression of Hoxc10, was absent (Andersson et al., 2006). Smad1, which transduces BMP signals, can interact with Hoxc8 to regulate osteopontin gene expression (Shi et al., 1999). The difference in A/P patterning of the axial skeleton in Smad6−/− and Smad7−/− mice may be due to differences in the signaling pathways mediated by either Smad6 or Smad7. That is, Smad6−/− mice may exhibit patterning defects due to alterations in BMP signaling, while defects in Smad7−/− mice may reflect alterations in both BMP and TGFβ signaling. Further studies are warranted to determine the mechanisms by which Smad6 and Smad7 regulate the expression and activity of Hox transcription factors.
Smad7 limits BMP and TGFβ signaling during cartilage development
Smad7 deficiency led to an expanded prehypertrophic zone and a reduced hypertrophic zone. Our results, reflecting the consequences of gain of TGFβ function, are consistent with in vivo studies showing that loss of TGFβ pathway activity leads to the opposite result. For example, loss of TGFβ activity in mice due to expression of a kinase-dead TGFβ type II receptor (Serra et al., 1997) or deficiency in Smad3 (Yang et al., 2001) results in accelerated hypertrophic differentiation associated with expanded domains of type X collagen expression. Moreover, in vitro studies have shown that Smad3 plays important roles in suppressing chondrocyte maturation (Li et al., 2006b). These findings suggest that Smad7 mediates its effects on chondrocyte maturation at least in part through antagonizing TGFβ pathways in the growth plate.
However, our findings suggest that increased TGFβ signaling is not the only factor contributing to the mutant phenotype. We observed increased pSmad1/5/8 activity and expression of Col10a1 mRNA in Smad7−/− chondrocytes. BMP signaling promotes hypertrophic differentiation in part by stimulating Col10a1 expression (Grimsrud et al., 1999, 2001; Minina et al., 2001; Valcourt et al., 2002). Given that Smad7-deficient chondrocytes were found to be more responsive to BMP, increased Col10a1 mRNA levels in Smad7−/− chondrocytes could result from upregulation of BMP signaling. We showed previously that loss of Smad6 leads to elevated BMP signaling in the growth plate (Estrada et al., 2011). It is thus likely that Smad6 and Smad7 exert compensatory function in the regulation of BMP pathway activity in the growth plate.
Smad7 mediates stress responses in the growth plate
A striking aspect of the Smad7−/− growth plate phenotype is the manifestation of a hypocellular core in the growth plate at mid-gestation. This was accompanied by increased intracellular retention of type II collagen, as well as increased chondrocyte death. These phenotypes were localized to the most hypoxic region of the growth plate (Schipani et al., 2001). Impaired adaptation of cells to a hypoxic environment is detrimental to chondrocyte survival; cartilage-specific deletion of HIF1α results in cell death in the center of the growth plate (Schipani et al., 2001). Thus, we examined whether loss of Smad7 leads to an impaired hypoxic response by analyzing HIF1α expression at the stage when the hypocellular core became apparent. Contrary to expectations, we found increased rather than decreased levels of HIF1α in Smad7−/− growth plates. We then evaluated candidate signaling pathways that could contribute to increased HIF1α levels in chondrocytes. Given that TGFβ signaling promotes HIF1α stabilization in tumor cells (McMahon et al., 2006), we assessed whether similar mechanisms were intact in chondrocytic cells. Indeed, we found that ALK5-mediated TGFβ signaling can promote HIF1α stabilization under both normoxic and hypoxic conditions. Thus, elevated HIF1α levels in Smad7 mutant growth plates may be due to increased TGFβ signaling in Smad7-deficient chondrocytes.
Chondrocytes are highly synthetic cells that constantly synthesize ECM. HIF1α is an essential regulator of ECM synthesis in chondrocytes (Pfander et al., 2003). Moreover, elevation of HIF1α activity in mice due to loss of von Hippel Lindau (pVHL) protein led to hypocellularity and increased expression of ECM proteins (Pfander et al., 2004). Whether or not there were defects in ECM secretion was not examined. Nonetheless, too much HIF1α can be as detrimental as too little, and the enhanced matrix synthesis would be expected to render chondrocytes more susceptible to ER stress. In fact, hypocellularity owing to increased activation of HIF1α was accompanied by ER stress in mice lacking the tumor suppressor PTEN in cartilage, and the evidence suggested that overactivation of HIF1α signaling was responsible for the emergence of ER stress (Yang et al., 2008). Thus, given that we found increased intracellular retention of types II and X collagen, along with elevated BiP expression in Smad7−/− medial growth plates, the observed Smad7−/− hypocellular phenotype may result from ER stress, in part, due to elevated HIF1α levels. However, we cannot exclude the possibility that loss of Smad7 leads to increased ER stress as a result of increased ECM synthesis that is restricted by an unknown mechanism to core chondrocytes, and that the hypoxic stress is thus secondary to ER stress.
In summary, Smad7 plays an important role in axial and appendicular skeletal development. Smad7 deficiency leads to defects in A/P patterning, and in terminal maturation of chondrocytes. We also show that Smad7 regulates these processes by inhibiting both BMP and TGFβ signaling. Finally, we show that Smad7 regulates stress pathways in the growth plate, in part via ALK5-mediated TGFβ signaling. Additional studies are warranted to determine the mechanisms by which TGFβ signaling promotes HIF1α stabilization and regulates cellular stress responses in chondrocytes.
Supplementary Material
Acknowledgments
We thank Alana Chin, Christina Hong, and Mathew Hilton for technical assistance and are grateful to members of the laboratory for helpful discussions and critical comments on the manuscript. This work was supported by the National Institutes of Health Grants R01 AR044528 (to K.M.L) and F31-AR060147 (to K.D.E).
Appendix A. Supporting information
Supplementary data associated with this article can be found in the online version at http://dx.doi.org/10.1016/j.ydbio.2013.08.021.
References
- Andersson O, Reissmann E, Ibanez CF. Growth differentiation factor 11 signals through the transforming growth factor-beta receptor ALK5 to regionalize the anterior-posterior axis. EMBO Rep. 2006;7:831–837. doi: 10.1038/sj.embor.7400752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arikawa-Hirasawa E, Watanabe H, Takami H, Hassell JR, Yamada Y. Perlecan is essential for cartilage and cephalic development. Nat Genet. 1999;23:354–358. doi: 10.1038/15537. [DOI] [PubMed] [Google Scholar]
- Baffi MO, Slattery E, Sohn P, Moses HL, Chytil A, Serra R. Conditional deletion of the TGF-beta type II receptor in Col2a expressing cells results in defects in the axial skeleton without alterations in chondrocyte differentiation or embryonic development of long bones. Dev Biol. 2004;276:124–142. doi: 10.1016/j.ydbio.2004.08.027. [DOI] [PubMed] [Google Scholar]
- Clark CA, Li TF, Kim KO, Drissi H, Zuscik MJ, Zhang X, O’Keefe RJ. Prostaglandin E2 inhibits BMP signaling and delays chondrocyte maturation. J Orthop Res. 2009;27:785–792. doi: 10.1002/jor.20805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. 2003;425:577–584. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- Estrada KD, Retting KN, Chin AM, Lyons KM. Smad6 is essential to limit BMP signaling during cartilage development. J Bone Miner Res. 2011;26:2498–2510. doi: 10.1002/jbmr.443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimsrud CD, Romano PR, D’Souza M, Puzas JE, Reynolds PR, Rosier RN, O’ Keefe RJ. BMP-6 is an autocrine stimulator of chondrocyte differentiation. J Bone Miner Res. 1999;14:475–482. doi: 10.1359/jbmr.1999.14.4.475. [DOI] [PubMed] [Google Scholar]
- Grimsrud CD, Romano PR, D’Souza M, Puzas JE, Schwarz EM, Reynolds PR, Roiser RN, O’Keefe RJ. BMP signaling stimulates chondrocyte maturation and the expression of Indian hedgehog. J Orthop Res. 2001;19:18–25. doi: 10.1016/S0736-0266(00)00017-6. [DOI] [PubMed] [Google Scholar]
- Hall-Glenn F, De Young RA, Huang BL, van Handel B, Hofmann JJ, Chen TT, Choi A, Ong JR, Benya PD, Mikkola H, Iruela-Arispe ML, Lyons KM. CCN2/connective tissue growth factor is essential for pericyte adhesion and endothelial basement membrane formation during angiogenesis. PLoS One. 2012;7:e30562. doi: 10.1371/journal.pone.0030562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanyu A, Ishidou Y, Ebisawa T, Shimanuki T, Imamura T, Miyazono K. The N domain of Smad7 is essential for specific inhibition of transforming growth factor-beta signaling. J Cell Biol. 2001;155:1017–1027. doi: 10.1083/jcb.200106023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi H, Abdollah S, Qiu Y, Cai J, Xu YY, Grinnell BW, Richardson MA, Topper JN, Gimbrone MA, Jr, Wrana JL, Falb D. The MAD-related protein Smad7 associates with the TGFbeta receptor and functions as an antagonist of TGFbeta signaling. Cell. 1997;89:1165–1173. doi: 10.1016/s0092-8674(00)80303-7. [DOI] [PubMed] [Google Scholar]
- Horiki M, Imamura T, Okamoto M, Hayashi M, Murai J, Myoui A, Ochi T, Miyazono K, Yoshikawa H, Tsumaki N. Smad6/Smurf1 overexpression in cartilage delays chondrocyte hypertrophy and causes dwarfism with osteopenia. J Cell Biol. 2004;165:433–445. doi: 10.1083/jcb.200311015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imamura T, Takase M, Nishihara A, Oeda E, Hanai J, Kawabata M, Miyazono K. Smad6 inhibits signalling by the TGF-beta superfamily. Nature. 1997;389:622–626. doi: 10.1038/39355. [DOI] [PubMed] [Google Scholar]
- Inoue Y, Imamura T. Regulation of TGF-beta family signaling by E3 ubiquitin ligases. Cancer Sci. 2008;99:2107–2112. doi: 10.1111/j.1349-7006.2008.00925.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishisaki A, Yamato K, Hashimoto S, Nakao A, Tamaki K, Nonaka K, ten Dijke P, Sugino H, Nishihara T. Differential inhibition of Smad6 and Smad7 on bone morphogenetic protein- and activin-mediated growth arrest and apoptosis in B cells. J Biol Chem. 1999;274:13637–13642. doi: 10.1074/jbc.274.19.13637. [DOI] [PubMed] [Google Scholar]
- Iwai T, Murai J, Yoshikawa H, Tsumaki N. Smad7 Inhibits chondrocyte differentiation at multiple steps during endochondral bone formation and down-regulates p38 MAPK pathways. J Biol Chem. 2008;283:27154–27164. doi: 10.1074/jbc.M801175200. [DOI] [PubMed] [Google Scholar]
- Jacenko O, LuValle P, Solum K, Olsen BR. A dominant negative mutation in the alpha 1 (X) collagen gene produces spondylometaphyseal defects in mice. Prog Clin Biol Res. 1993;383B:427–436. [PubMed] [Google Scholar]
- Kronenberg HM. Developmental regulation of the growth plate. Nature. 2003;423:332–336. doi: 10.1038/nature01657. [DOI] [PubMed] [Google Scholar]
- Li R, Rosendahl A, Brodin G, Cheng AM, Ahgren A, Sundquist C, Kulkarni S, Pawson T, Heldin CH, Heuchel RL. Deletion of exon I of SMAD7 in mice results in altered B cell responses. J Immunol. 2006a;176:6777–6784. doi: 10.4049/jimmunol.176.11.6777. [DOI] [PubMed] [Google Scholar]
- Li TF, Darowish M, Zuscik MJ, Chen D, Schwarz EM, Rosier RN, Drissi H, O’ Keefe RJ. Smad3-deficient chondrocytes have enhanced BMP signaling and accelerated differentiation. J Bone Miner Res. 2006b;21:4–16. doi: 10.1359/JBMR.050911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Ionescu AM, Schwarz EM, Zhang X, Drissi H, Puzas JE, Rosier RN, Zuscik MJ, O’Keefe RJ. Smad6 is induced by BMP-2 and modulates chondrocyte differentiation. J Orthop Res. 2003;21:908–913. doi: 10.1016/S0736-0266(03)00008-1. [DOI] [PubMed] [Google Scholar]
- Liu GX, Li YQ, Huang XR, Wei L, Chen HY, Shi YJ, Heuchel RL, Lan HY. Disruption of Smad7 promotes ANG II-mediated renal inflammation and fibrosis via Sp1-TGF-beta/Smad3-NF. kappaB-dependent mechanisms in mice. PLoS One. 2013;8:e53573. doi: 10.1371/journal.pone.0053573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massague J. TGF-beta signal transduction. Annu Rev Biochem. 1998;67:753–791. doi: 10.1146/annurev.biochem.67.1.753. [DOI] [PubMed] [Google Scholar]
- Massague J, Seoane J, Wotton D. Smad transcription factors. Genes Dev. 2005;19:2783–2810. doi: 10.1101/gad.1350705. [DOI] [PubMed] [Google Scholar]
- Matsunobu T, Torigoe K, Ishikawa M, de Vega S, Kulkarni AB, Iwamoto Y, Yamada Y. Critical roles of the TGF-beta type I receptor ALK5 in perichondrial formation and function, cartilage integrity, and osteoblast differentiation during growth plate development. Dev Biol. 2009;332:325–338. doi: 10.1016/j.ydbio.2009.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon S, Charbonneau M, Grandmont S, Richard DE, Dubois CM. Transforming growth factor beta1 induces hypoxia-inducible factor-1 stabilization through selective inhibition of PHD2 expression. J Biol Chem. 2006;281:24171–24181. doi: 10.1074/jbc.M604507200. [DOI] [PubMed] [Google Scholar]
- Milenkovic L, Goodrich LV, Higgins KM, Scott MP. Mouse patched1 controls body size determination and limb patterning. Development. 1999;126:4431–4440. doi: 10.1242/dev.126.20.4431. [DOI] [PubMed] [Google Scholar]
- Minina E, Wenzel HM, Kreschel C, Karp S, Gaffield W, McMahon AP, Vortkamp A. BMP and Ihh/PTHrP signaling interact to coordinate chondrocyte proliferation and differentiation. Development. 2001;128:4523–4534. doi: 10.1242/dev.128.22.4523. [DOI] [PubMed] [Google Scholar]
- Mochizuki T, Miyazaki H, Hara T, Furuya T, Imamura T, Watabe T, Miyazono K. Roles for the MH2 domain of Smad7 in the specific inhibition of transforming growth factor-beta superfamily signaling. J Biol Chem. 2004;279:31568–31574. doi: 10.1074/jbc.M313977200. [DOI] [PubMed] [Google Scholar]
- Moriguchi T, Kuroyanagi N, Yamaguchi K, Gotoh Y, Irie K, Kano T, Shirakabe K, Muro Y, Shibuya H, Matsumoto K, Nishida E, Hagiwara M. A novel kinase cascade mediated by mitogen-activated protein kinase kinase 6 and MKK3. J Biol Chem. 1996;271:13675–13679. doi: 10.1074/jbc.271.23.13675. [DOI] [PubMed] [Google Scholar]
- Murakami G, Watabe T, Takaoka K, Miyazono K, Imamura T. Cooperative inhibition of bone morphogenetic protein signaling by Smurf1 and inhibitory Smads. Mol Biol Cell. 2003;14:2809–2817. doi: 10.1091/mbc.E02-07-0441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakao A, Afrakhte M, Moren A, Nakayama T, Christian JL, Heuchel R, Itoh S, Kawabata M, Heldin NE, Heldin CH, ten Dijke P. Identification of Smad7, a TGFbeta-inducible antagonist of TGF-beta signalling. Nature. 1997;389:631–635. doi: 10.1038/39369. [DOI] [PubMed] [Google Scholar]
- Pfander D, Cramer T, Schipani E, Johnson RS. HIF-1alpha controls extracellular matrix synthesis by epiphyseal chondrocytes. J Cell Sci. 2003;116:1819–1826. doi: 10.1242/jcs.00385. [DOI] [PubMed] [Google Scholar]
- Pfander D, Kobayashi T, Knight MC, Zelzer E, Chan DA, Olsen BR, Giaccia AJ, Johnson RS, Haase VH, Schipani E. Deletion of Vhlh in chondrocytes reduces cell proliferation and increases matrix deposition during growth plate development. Development. 2004;131:2497–2508. doi: 10.1242/dev.01138. [DOI] [PubMed] [Google Scholar]
- Retting KN, Song B, Yoon BS, Lyons KM. BMP canonical Smad signaling through Smad1 and Smad5 is required for endochondral bone formation. Development. 2009;136:1093–1104. doi: 10.1242/dev.029926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg L. Chemical basis for the histological use of safranin O in the study of articular cartilage. J Bone Joint Surg Am. 1971;53:69–82. [PubMed] [Google Scholar]
- Scharstuhl A, Diepens R, Lensen J, Vitters E, van Beuningen H, van der Kraan P, van den Berg W. Adenoviral overexpression of Smad-7 and Smad-6 differentially regulates TGF-beta-mediated chondrocyte proliferation and pro-teoglycan synthesis. Osteoarthritis Cartilage. 2003;11:773–782. doi: 10.1016/s1063-4584(03)00165-1. [DOI] [PubMed] [Google Scholar]
- Schipani E, Ryan HE, Didrickson S, Kobayashi T, Knight M, Johnson RS. Hypoxia in cartilage: HIF-1alpha is essential for chondrocyte growth arrest and survival. Genes Dev. 2001;15:2865–2876. doi: 10.1101/gad.934301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serra R, Johnson M, Filvaroff EH, LaBorde J, Sheehan DM, Derynck R, Moses HL. Expression of a truncated, kinase-defective TGF-beta type II receptor in mouse skeletal tissue promotes terminal chondrocyte differentiation and osteoarthritis. J Cell Biol. 1997;139:541–552. doi: 10.1083/jcb.139.2.541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi X, Yang X, Chen D, Chang Z, Cao X. Smad1 interacts with homeobox DNA-binding proteins in bone morphogenetic protein signaling. J Biol Chem. 1999;274:13711–13717. doi: 10.1074/jbc.274.19.13711. [DOI] [PubMed] [Google Scholar]
- Shim JH, Greenblatt MB, Xie M, Schneider MD, Zou W, Zhai B, Gygi S, Glimcher LH. TAK1 is an essential regulator of BMP signalling in cartilage. EMBO J. 2009;28:2028–2041. doi: 10.1038/emboj.2009.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St-Jacques B, Hammerschmidt M, McMahon A. Indian hedgehog signaling regulates proliferation and differentiation of chondrocytes and is essential for bone formation. Genes Dev. 1999;13:2072–2086. doi: 10.1101/gad.13.16.2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valcourt U, Gouttenoire J, Moustakas A, Herbage D, Mallein-Gerin F. Functions of transforming growth factor-beta family type I receptors and Smad proteins in the hypertrophic maturation and osteoblastic differentiation of chondrocytes. J Biol Chem. 2002;277:33545–33558. doi: 10.1074/jbc.M202086200. [DOI] [PubMed] [Google Scholar]
- Wang J, Zhao J, Chu ES, Mok MT, Go MY, Man K, Heuchel R, Lan HY, Chang Z, Sung JJ, Yu J. Inhibitory role of Smad7 in hepatocarcinogenesis in mice and in vitro. J Pathol. 2013 doi: 10.1002/path.4206. [DOI] [PubMed] [Google Scholar]
- Wang W, Lian N, Li L, Moss HE, Wang W, Perrien DS, Elefteriou F, Yang X. Atf4 regulates chondrocyte proliferation and differentiation during endochondral ossification by activating Ihh transcription. Development. 2009;136:4143–4153. doi: 10.1242/dev.043281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wouters BG, Koritzinsky M. Hypoxia signalling through mTOR and the unfolded protein response in cancer. Nat Rev Cancer. 2008;8:851–864. doi: 10.1038/nrc2501. [DOI] [PubMed] [Google Scholar]
- Yamaguchi K, Shirakabe K, Shibuya H, Irie K, Oishi I, Ueno N, Taniguchi T, Nishida E, Matsumoto K. Identification of a member of the MAPKKK family as a potential mediator of TGF-beta signal transduction. Science. 1995;270:2008–2011. doi: 10.1126/science.270.5244.2008. [DOI] [PubMed] [Google Scholar]
- Yang G, Sun Q, Teng Y, Li F, Weng T, Yang X. PTEN deficiency causes dyschondroplasia in mice by enhanced hypoxia-inducible factor 1alpha signaling and endoplasmic reticulum stress. Development. 2008;135:3587–3597. doi: 10.1242/dev.028118. [DOI] [PubMed] [Google Scholar]
- Yang X, Chen L, Xu X, Li C, Huang C, Deng CX. TGF-beta/Smad3 signals repress chondrocyte hypertrophic differentiation and are required for maintaining articular cartilage. J Cell Biol. 2001;153:35–46. doi: 10.1083/jcb.153.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.