

Abstract
The MAPK phosphatase MKP1 (DUSP1) is overexpressed in many human cancers, including chemoresistant and radioresistant breast cancer cells, but its functional contributions in these settings are unclear. Here we report that after cell irradiation MKP1 translocates into mitochondria, where it prevents apoptotic induction by limiting accumulation of phosphorylated active forms of the stress kinase JNK. Increased levels of mitochondrial MKP1 after irradiation occurred in the mitochondrial inner membrane space. Notably, cell survival regulated by mitochondrial MKP1 was responsible for conferring radioresistance in HER2-overexpressing breast cancer cells, due to the fact that MKP1 serves as a major downstream effector in the HER2-activated RAF-MEK-ERK pathway. Clinically, we documented MKP1 expression exclusively in HER2-positive breast tumors, relative to normal adjacent tissue from the same patients. MKP1 overexpression was detected also in irradiated HER2-positive breast cancer stem-like cells (HER2+/CD44+/CD24−/low) isolated from a radioresistant breast cancer cell population after long-term radiation treatment. MKP1 silencing reduced clonogenic survival and enhanced radiosensitivity in these stem-like cells. Combined inhibition of MKP1 and HER2 enhanced cell killing in breast cancer. Together, our findings identify a new mechanism of resistance in breast tumors and reveal MKP1 as a novel therapeutic target for radiosensitization.
Keywords: MKP1, mitochondrial apoptosis, breast cancer, MAPK, radioresistance, HER2
Introduction
Combined with surgery and chemotherapy, radiotherapy continues to be a powerful tool in the treatment of breast cancer (1); however, the therapy-resistant phenotype in recurrent and metastatic lesions accounts for major failure in many patients and is the barrier for further improvement of the efficacy of anti-cancer therapy (2, 3). A pro-survival network regulated by transcription factor NF-kB is responsible for a fraction of breast cancer cells surviving long-term following irradiation (4, 5). Radiation-induced NF-kB mediates the overexpression of a novel pro-survival gene MAPK phosphatase 1 (MKP1), which is capable of inhibiting radiation-induced apoptosis by attenuating JNK activity (5). Induction of MKP1 is controlled by the growth factor-induced activation of mitogen-activated protein kinases (MAPKs), ERK2/ERK1, suggesting an auto-regulatory mechanism (6). MKP1 is a direct substrate of ERK, which can phosphorylate MKP1 on S359 and S364 (7), and increase its half-life (6). MKP1 is also catalytically activated via ERK binding to its N-terminal non-catalytic moiety (8), indicating various means of feedback exerted by active ERK on MKP1. The role of MKP1 in HER2-overexpressing breast tumors is critical as HER2 overexpression results in the hyper-activation of MAPKs (9, 10), which in turn suggests potential increases in the expression, stability and activation of MKP1. Not surprisingly, MKP1 has been shown to be overexpressed in human breast cancer (11) and is implicated as a significant mediator of breast cancer chemoresistance (12).
Mitochondria play a crucial role in the regulation of cell death, namely apoptosis (13). Cancer-specific mitochondrial alterations are responsible for the resistance to mitochondria-mediated apoptosis (5, 14). Many nuclear-encoded proteins have been identified in the mitochondria of mammals to regulate mitochondria-mediated apoptosis (14). Among the MAPKs induced in response to irradiation, JNK was shown to localize into mitochondria upon γ-irradiation and initiate mitochondria-mediated apoptosis via the phosphorylation of Bcl-xl (15). JNK is targeted by MKP1 in irradiated cells to block radiation-induced apoptosis, suggesting a potential pro-survival role for MKP1 in mitochondria.
Recent evidence suggest that cancer stem cells (CSCs) are present in breast tumors (16) and are linked with tumor resistance due to their increased survival (16-18). Al-Hajj et al. and others showed that breast CSCs with the feature of CD44+/CD24− are more tumorigenic (16) under therapeutic irradiation (19-26). Recent findings from our lab demonstrated that HER2+/CD44+/CD24− breast CSCs are more aggressive, invasive, tumorigenic, and radioresistant compared to HER2−/CD44+/CD24− cells (22). Interestingly, we found that HER2-positive breast CSCs overexpress MKP1 and depend on MKP1 for survival.
Approximately 25% of human breast cancers overexpress HER2, which is associated with poor prognosis and a more aggressive phenotype in patients (27, 28). Current clinical therapies targeting HER2 consists of the monoclonal antibody Trastuzumab and the tyrosine kinase inhibitor Lapatinib (29-31); however, due to adaptive resistance that tumors acquire against anti-HER2 therapy, breast cancer recurrence and metastasis eventually develop in a fraction of HER2-positive breast cancer patients (27, 29, 32). It is an urgent need to define alternative approaches to treat HER2-positive breast tumors with therapy-resistance and history of anti-HER2 therapy. Combination therapies targeting HER2 and therapy resistance pathways could efficiently overcome the resistance and potentially prevent the recurrence.
Herein, MKP1 was identified in the mitochondria of MEFs and a variety of human cancer cells. The mitochondrial MKP1 was enhanced under the genotoxic stress following γ-irradiation, and was able to dephosphorylate and inactivate mitochondrial JNK resulting in decreased apoptosis and radioresistance. Furthermore, MKP1 expression in clinical breast tumor samples showed a strong correlation with HER2 expression. MKP1-mediated survival of breast cancer cells varied according to their HER2 status. These data suggest MKP1 mitochondrial localization as a mechanism of therapy resistance in breast cancer and offers MKP1 as a potentially effective target for re-sensitizing tumor cells for anti-cancer therapy.
Materials and Methods
Cell lines and clinical tumor samples
Wild type (wt) and MKP1−/− (MKP1 knock-out) MEFs were kindly provided by Robert Z. Orlowski at the University of North Carolina. These cells were maintained in DMEM supplemented with 10% FBS. MDA-MB-231, MCF7 wt, MCF7/HER2 and MCF7/C6 cells were kept in 1% non-essential amino acid containing MEM supplemented with 10% FBS. SKBR3 cells were maintained in RPMI 1640 containing 10% FBS. HCT116 cells were obtained from Dr. Bert Vogelstein at Johns Hopkins University and kept in McCoy’s 5A supplemented with 10% FBS. HER2+/CD44+/CD24−/low and HER2−/CD44+/CD24−/low cells were sorted from MCF7/C6 cell line and maintained in high serum (20%) containing MEM media supplemented with 1% non-essential amino acids. Clinical specimens were provided by the UC Davis Comprehensive Cancer Center Biorepository, which is funded by the National Cancer Institute.
Reagents and antibodies
U0126 was purchased from VWR International (West Chester, PA), JC-1 from Invitrogen (Grand Island, NY), Sanguinarine from Tocris Biosciences (Minneapolis, MN), and Lapatinib from Selleckchem (Houston, TX). Antibodies for MKP1, ERK, pERK, JNK, pJNK, HER2, and TOM40 were purchased from Sigma (St. Louis, MO); COXIV and pMKP1 were purchased from Cell Signaling (Beverly, MI).
Plasmids and siRNA
The details of recombinant plasmid construction and siRNA synthesis were included in the supplemental information.
Mitochondria Isolation
Cells were harvested and resuspended in Buffer A (134 mM NaCl, 5 mM KCl, 0.7 mM Na2HPO4, 2.5 mM Tris HCl pH 7.5). After centrifugation at 600 × g, the pellet was resuspended in Buffer B (10 mM NaCl, 1.5 mM MgCl2, 10 mM Tris HCl pH 7.5) and the cells were lysed using a glass homogenizer followed by the addition of Buffer C (2 M Sucrose, 35 mM EDTA, 50 mM Tris HCl pH 7.5). Centrifugation at 600 × g removed cell debris and the supernatant was centrifuged at 10,000 rpm to pellet the mitochondria. The pellet was washed with Buffer D (0.33 M Sucrose, 1 mM EDTA, 8.3 mM Tris HCl pH 7.5), lysed and stored.
Alkaline extraction and mitoplasting
Alkaline extraction was performed as previously reported (33). Briefly, the mitochondria was incubated in 0.1 M Na2CO3 (pH 11) for 20 min at 4 °C. The membrane was then centrifuged at 100,000 × g. Proteins in the supernatant were precipitated using a final volume of 10% trichloroacetic acid (TCA). The pellets were re-suspended in dissolving buffer containing 7 M urea, 3 M thiourea, 2% CHAPS, 30 mM Tris, pH 8.5. Mitoplasting was performed by diluting mitochondria in hypotonic sucrose buffers (1 mM EDTA, 10 mM MOPS-KOH, pH 7.2; sucrose concentration ranging from 25 mM to 200 mM) with or without 50 μg/ml soybean trypsin. Proteolysis was stopped by adding 1 mM PMSF. The mitoplasts were separated by centrifugation at 16,000 × g. Pellets were lysed and all supernatant fractions were precipitated with 10% TCA and resuspended in dissolving buffer.
Mitochondrial Membrane Potential Assay (Δψm)
Cells (104 cells/well) were seeded on 96-well plates and treated with 10 Gy or 0 Gy irradiation. At indicated time points, 2μg/ml of JC-1 dye was added for 30 min at 37 °C. Cationic dye taken up by mitochondria was detected by the formation of red precipitate in the cells. The dye was excluded and detected as a green monomer in the cytoplasm of the cells with disrupted mitochondria. After washing with phosphate-buffered saline (pH 7.4), the fluorescence intensity of the red precipitate (JC-1 red) and green monomer (JC-1 green) were detected using Spectra Max M2e plate reader (Molecular Devices Co., Sunnyvale, CA) at excitation 485/emission 595 or excitation 485/emission 525, respectively. The ratio of JC-1 red/JC-1 green was calculated as Δψm.
Immunoprecipitation
Approximately 107 cells were incubated with 10 μl of Protein A/G agarose beads (Roche, Germany) and normal IgG at 4 °C for 1 h for pre-clearance. After centrifuging at 1000 rpm, the supernatant was incubated with 10 μg of antibody overnight at 4 °C. Subsequently, 50 μl of Protein A beads was added for 1 h at 4 °C to capture the antibody-protein complexes. The mixture was then spun to collect the beads. After the beads were washed four times with lysis buffer, 1X protein sample buffer was added and the beads were boiled at 95 °C for 10 min to separate beads and the pulled down proteins.
BCSC sorting
Following described procedures (16), the cell suspensions were rinsed with PBS with 2% FBS, re-suspended in PBS containing 0.5% FBS and PI (0.5 mg/ml) and sorted using the Cytopeia influx Cell Sorter (BD Biosciences, San Jose, CA).
Trypan Blue Assay
Cells were collected in PBS, and 4% Trypan Blue solution was added to the cells. The percentage of viable cells was determined by counting the cells that are white (live cells that has excluded the dye) and blue (dead cells that cannot exclude the dye) under the microscope using a hematocytometer.
Clonogenic Survival Assay
The standard clonogenic survival assay was performed as previously described (4). Survival fraction was assessed by colony formation following exposure to sham or 10Gy of radiation. The colonies were stained with Coomassie blue, and colonies with more than 50 cells were scored and normalized against the plating efficiency of cells.
Statistical Analysis
The data are presented as means +/− S.E; findings were considered significant at p < 0.05.
Results
MKP1 translocates into mitochondria and inhibits pro-apoptotic signals from JNK
Mitochondrial fractions isolated from irradiated MEFs showed that mitochondrial MKP1 levels increased after 10 Gy irradiation until 4 h post-irradiation, and then gradually returned to basal levels (Fig. 1A, top panel), while the overall expression of MKP1 remained constant until after 8 h post-irradiation (Fig. 1A, bottom panel). The purity of mitochondrial preparations was confirmed by the absence of markers of other cellular compartments such as cytoplasm, nucleus and Golgi, in the mitochondrial preparations (Fig. 1B). The alkaline extraction method (33) was used to prepare sub-mitochondrial fractions to determine the mitochondrial sub-localization of MKP1. After alkaline extraction, membrane proteins are retained in the pellet while soluble proteins are recovered in the supernatant. TIMM13 subunit of the translocase of inner membrane (IM) complex, TOM40 subunit of the translocase of outer membrane (OM) complex, and HSP60 chaperon were utilized as markers of the sub-mitochondrial compartments; inner membrane space (IMS), OM, and matrix, respectively. The results indicated that MKP1 is either a soluble protein that localizes into the matrix like HSP60; or an integral membrane protein that localizes into the inner membrane like TIMM13 (Fig. 1C). To further define the sub-mitochondrial location of MKP1, mitoplasting was performed by diluting mitochondria in hypotonic buffers with decreasing concentrations of the osmoticum sucrose from 200 mM to 25 mM. The outer membrane began to rupture at 200 mM of sucrose, while the inner membrane remained intact until the final concentration at 25 mM of sucrose (Fig. 1D). In combination with mitoplasting, a protease protection assay was performed using trypsin to digest exposed proteins after outer membrane rupture. While HSP60 was protected from trypsin digestion; MKP1, similar to TIMM13, was digested in the supernatant of the trypsin-treated samples, indicating its mitochondrial IMS localization (Fig. 1D).
Figure 1.
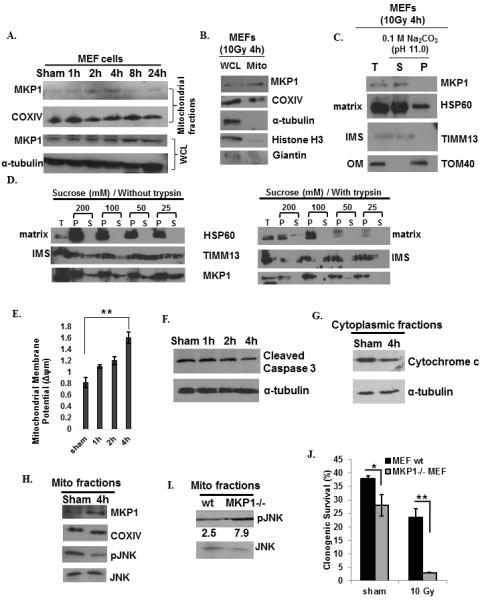
Mitochondrial MKP1 is enhanced by genotoxic stress in MEFs to inhibit mitochondria-initiated apoptosis via reduction of pJNK. A, Mitochondrial translocation of MKP1 in sham or 10 Gy irradiated MEFs. COXIV was used as the marker for mitochondria and IκB was used as the cytoplasmic marker. B, The purity of mitochondrial preparations in these experiments were further analyzed by immunoblots of MKP1, COXIV, α-tubulin (cytoplasmic marker), Histone H3 (nuclear marker), and Giantin (Golgi marker). C, Sub-mitochondrial localization of MKP1 detected by alkaline extraction (33). Total input (T), soluble matrix proteins (S), and membrane pellets (P) were immunoblotted for MKP1, TOM40 (an outer membrane protein), TIMM13 (an inter-space protein), and HSP60 (a matrix protein). D, Sub-mitochondrial localization of MKP1 detected via mitoplasting and protease protection assay (49). The total (T), pellet (P), and supernatant (S) fractions were subjected to western blotting with indicated antibodies. m (E, measured by fluorescent probe JC-1; n=3, **p<0.01), Caspase 3 cleavage (F), and cytochrome c release (G) in sham or 10 Gy irradiated MEFs. H, Increased MKP1 and decreased pJNK levels in mitochondrial fractions 4h post 10 Gy IR. I, JNK phosphorylation in the mitochondria of MKP1 knock-out and wt MEFs 4h after 10 Gy of radiation. pJNK levels were normalized to that of JNK levels and represented under the blots. J, Clonogenic survival analysis of wt versus MKP1−/− MEFs (n=3, *p<0.05, **p<0.01).
Elevation of mitochondrial MKP1 levels correlated with an increase in the mitochondrial membrane potential when compared to basal levels in untreated sham control cells. At peak accumulation of MKP1 in the mitochondria (4 h post IR), a higher mitochondrial membrane potential, indicative of lower apoptosis, was observed (Fig. 1E). Cleaved caspase 3 levels (Fig. 1F), and cytochrome c release (Fig. 1G) were both reduced at 4 h post-IR compared to their levels in untreated sham control cells. The reduced levels of apoptosis of MEFs at 4 h post-IR, as shown by reduced caspase cleavage and cytochrome c release to cytoplasm, may have resulted from increased levels of MKP1 and consequently, reduced phosphorylated JNK (pJNK) levels in the mitochondria (Fig. 1H). To further address whether mitochondrial MKP1 targets pJNK in irradiated cells, we compared the pJNK levels in the mitochondria of wild type (wt) and MKP1−/− MEF cells upon irradiation. As expected, pJNK levels were higher in the mitochondria of MKP1 knock-out cells (Fig. 1I). These cells exhibited dramatically low clonogenic survival ability when compared to wt MEF cells following irradiation (92% lower survival). While the absence of MKP1 alone resulted in 29% lower survival in non-irradiated cells, irradiation alone caused 39% lower survival in wt MEFs and 88% lower survival in MKP1 knock-out cells, indicating the importance of MKP1 for survival following irradiation (Fig. 1J). Taken together, these data show that MKP1 translocates into the IMS of mitochondria upon irradiation, and functions to attenuate pro-apoptotic signals from pJNK leading to increased survival.
Mitochondrial localization of MKP1 in breast cancer cells
To evaluate whether MKP1 translocation to mitochondria is a survival strategy also adopted by other cells, we tested the radiation-induced mitochondrial localization of MKP1 in a variety of normal and cancer cells. In MCF7 breast cancer cells, mitochondrial MKP1 levels were increased shortly after irradiation, 1-2 h, and then returned to basal levels; while the total MKP1 levels remained unchanged (Fig. 2A). Mitochondrial MKP1 localization was also observed in MDA-MB-231 and SKBR3 breast cancer cell lines (Fig. 2B) and in HK18 human skin keratinocytes (data not shown). Although with different dynamics, MKP1 was also localized to mitochondria in HCT116 colon cancer cells at 8 h post IR (Fig. 2C). Co-immunoprecipitation experiments showed that MKP1 physically interacts with JNK in the mitochondria of MCF7 cells (Fig. 2D). These data indicate mitochondrial translocation of MKP1 as a common survival strategy for diverse mammalian cell types, including normal mouse (MEFs) and human tumor cells.
Figure 2.
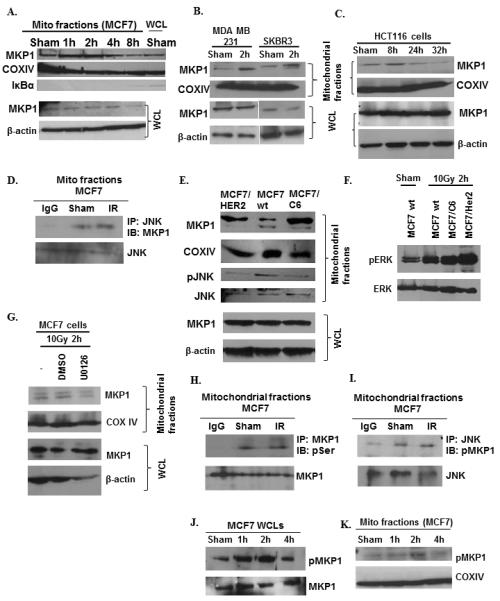
ERK1/2-dependent mitochondrial localization and phosphorylation of MKP1 under radiation insult. Immunoblots of mitochondrial MKP1 in IR-treated breast cancer MCF7 (A), MDA-MB-231 and SKBR3 (B) cells and colon cancer HCT116 cell (C). D, Co-IP of mitochondrial MKP1 and JNK in sham or IR-treated MCF7 cells. E, Enhanced mitochondrial MKP1 and decreased pJNK levels in radioresistant MCF7/C6 and MCF7/HER2 cells compared to wt MCF7s. F, pERK and ERK levels in sham or 10 Gy irradiated wt and radioresistant MCF7s. G, MKP1 mitochondrial translocation was inhibited by the MEK inhibitor, U0126. MCF7 cells were pre-treated with 10μM of U0126 for 1 h before IR and harvested 2 h post-IR. H, ERK-mediated phosphorylation of MKP1 determined via co-IP using anti-MKP1 for IP and anti-pSer antibody for IB; and I, anti-JNK for IP and anti-pMKP for IB. MKP1 phosphorylation in irradiated MCF7 cells (J) and their mitochondria (K).
Mitochondrial MKP1 levels are elevated in radioresistant breast cancer cells
The pro-survival role of mitochondrial MKP1 points to its potential role in radioresistance. We compared the mitochondrial MKP1 levels in MCF7/HER2 (HER2 overexpressing radioresistant cells), MCF7/C6 (FIR-derived radioresistant cells (4)) and wt MCF7. As expected, MKP1 translocation to mitochondria was enhanced in the radioresistant breast cancer cells (Fig. 2E). In accordance with the previous results, these radioresistant cells showed low levels of mitochondrial phosphoJNK, indicating that MKP1 may contribute to the radioresistance of breast cancer cells by blocking the pro-apoptotic activity of pJNK (Fig. 2E). The mitochondrial membrane potential of radioresistant cell lines was significantly higher, consistent with the lower levels of apoptosis expected in radioresistant cells with decreased mitochondrial pJNK (Fig. S1).
ERK1/2 is an upstream regulator of MKP1 in breast cancer cells
Similar to many cell signaling molecules, MKP1 is also regulated by phosphorylation/dephosphorylation events (6, 7). One of the upstream regulators of MKP1 was identified as ERK1/2, which binds to and phosphorylates MKP1 at Ser359 (7, 8). Thus, we addressed whether ERK is responsible for activation of MKP1 in MCF7 cells. The phosphorylated ERK levels were higher in radioresistant breast cancer cells compared to wt MCF7 following irradiation (Fig. 2F), indicating a potential role for active ERK1/2 in radioresistance. The analysis of mitochondrial fractions isolated from MEK inhibitor (U0126)-treated cells revealed that mitochondrial localization of MKP1 was reduced by ERK inactivation, while the expression of MKP1 remained constant (Figs. S2 and 2G). Mitochondrial MKP1 was found to be serine-phosphorylated (Fig. 2H), and mitochondrial JNK was shown to interact with Ser359-phosphorylated MKP1 (Fig. 2I). While Ser359 phosphorylation of MKP1 was enhanced in cells as early as 1 h post-IR (Fig. 2J), the mitochondrial MKP1 phosphorylation was increased at 2 h post-IR (Fig. 2K). Together, the results suggest that ERK-dependent phosphorylation/activation of MKP1 may be required for translocation of MKP1 into the mitochondria.
MKP1 expression in HER2-expressing breast cancer stem cells and clinical breast tumor samples
Next, we analyzed 6 pairs of normal and tumor tissue samples from 6 breast cancer patients for MKP1 expression (Fig. 3A). MKP1 expression was induced in tumor tissues in 4 of the patients tested, while their normal tissues lacked MKP1 (Patients 1, 2, 4, and 5). One patient showed reduced MKP1 levels in tumor tissue (Patient 6), while MKP1 was overexpressed in the normal tissue. Although there is no consensus for the expression of MKP1 being specific to normal or tumor tissues within the limits of small sample size, MKP1 expression strictly correlated with HER2 expression in these patients (Fig. 3A). HER2-overexpressing tumors also exhibited MKP1 overexpression (Patients 1, 2, 4, and 5), and patients that lost HER2 expression in their tumors also lost MKP1 expression (Patient 6). We then tested the expression of MKP1 in a panel of clinical tumor samples with differential HER2 status. The results showed that MKP1 expression is limited exclusively to HER2-positive tumors (Fig. 3B). MKP1 phosphorylation was also observed in these HER2-positive samples.
Figure 3.
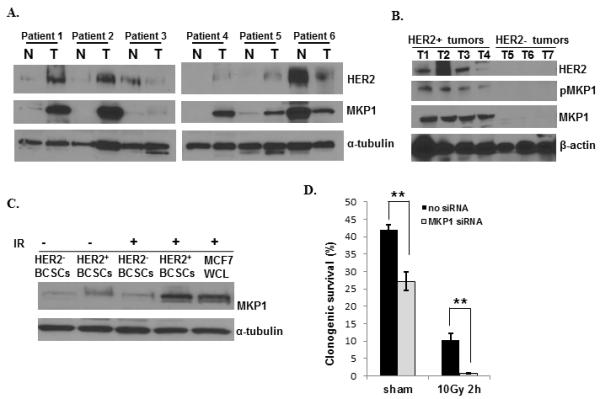
Co-activation of HER2 and MKP1 in HER2+ and HER2− breast cancer tissues and radioresistant HER+ breast cancer stem cells (HER2+ BCSCs). A, The co-expression of HER2 and MKP1 in a panel of paired human normal and tumor breast tissues (N = tumor surrounding normal breast tissue). B, The expression of MKP1, pMKP1 (S359) and HER2 in clinically diagnosed breast cancer patients with differential HER2 status. C, MKP1 expression levels in HER2+/CD44+/CD24− and HER2−/CD44+/CD24− breast cancer stem cells (HER2+ BCSCs and HER2− BCSCs) that were live-sorted from MCF7/C6 cells according to Duru et al. (22). D, Clonogenic survival of HER2-positive breast CSCs with or without MKP1 siRNA transfection (n=3, **p<0.01).
A recent study showed that HER2-positive breast cancer stem cells (BCSC), HER2+/CD44+/CD24−/low, are radiotherapy resistant (22). We analyzed the expression of MKP1 in HER2-positive and HER2-negative BCSCs, originally isolated from MCF7/C6 cells, with or without irradiation. The results showed that although the basal MKP1 expression was low in both HER2-positive and HER2-negative BCSCs, MKP1 expression was greatly induced in irradiated HER2+/CD44+/CD24−/low cells (Fig. 3C). Knocking down MKP1 in HER2+/CD44+/CD24−/low cells resulted in reduced survival (35% decrease), while irradiation alone caused a 70% decrease (Fig. 3D). Irradiation of MKP1-depleted HER2-positive BCSCs resulted in a dramatic reduction in their clonogenic survival (95% decrease), indicating a role for radiation-induced MKP1 in the radioresistant phenotype of HER2-positive BCSCs. These results suggest that MKP1 expression may be regulated by HER2 to manage the therapy resistance of HER2-expressing breast cancer cells.
MKP1 is required for breast cancer cell survival
The role of MKP1 in radioresistance of breast cancer cells was further studied via MKP1 siRNA treatment of wt or radioresistant MCF7 cells (Fig. 4A). Knockdown of MKP1 in MCF7 cells resulted in a 51% decrease in their survival, while radiation treatment reduced their survival by 64%. Together, irradiation and MKP1 depletion caused 89% reduction in the survival of breast cancer MCF7 cells (Fig. 4B). The knockdown of MKP1 in radioresistant MCF7/C6 cells resulted in a 45% reduction in clonogenic survival; radiation treatment alone caused a 60% decrease, while the combination of MKP1 siRNA treatment plus irradiation resulted in a dramatic 78% decrease (Fig. 4C). These results indicate that MKP1 is required for the survival of breast cancer cells and knockdown of MKP1 can re-sensitize radioresistant breast cancer cells.
Figure 4.
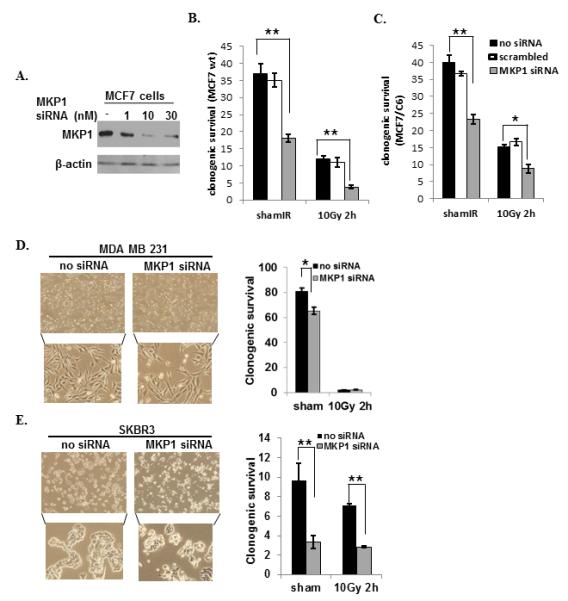
MKP1 is a potential therapeutic target for HER2-positive breast cancer cells. A, Immunoblots of MKP1 in MCF7 cells incubated with indicated concentrations of siRNA for 48h. Clonogenic survival of MCF7 (B), MCF7/C6 (C), HER2-negative breast cancer MDA-MB-231 cells (D); and HER2-positive breast cancer SKBR3 cells (E) treated with MKP1 siRNA for 48h followed by sham or 10 Gy IR. Cells were seeded 2 h post-IR and clonogenic survival was determined on 14th day (n=3, *p<0.05, **p<0.01). The bright field images of cells 48h post siRNA treatment were shown in left panels of D and E.
The strong correlation of HER2 and MKP1 expression in breast cancer encouraged us to test the MKP1-mediated survival mechanism in a panel of breast cancer cells with differential HER2 status. We utilized MCF7 wt (HER2-low), MDA-MB-231 (HER2-negative), SKBR3 (HER2-positive), MCF7/C6 (HER2-overexpressing) and HER2+/CD44+/CD24−/low (HER2-positive BCSCs). The results showed that HER2-positive breast cancer cells were more sensitive to MKP1 siRNA treatment compared to HER2 low/negative cell lines, as they exhibited massive cell death after 48 h of siRNA treatment (Fig. S3A-B). Comparison of the clonogenic survival of the HER2-negative MDA-MB-231 and HER2-overexpressing SKBR3 cells revealed that knocking down MKP1 in both MDA-MB-231 and SKBR3 cells reduced their clonogenic survival; however, the effect of MKP1 inhibition in SKBR3 cells (~65%) was significantly greater than that of MDA-MB-231 cells (~20%) (Fig. 4D-E). MDA-MB-231 cells were highly radiosensitive and knock-down of MKP1 in these cells did not further change their clonogenic survival upon irradiation. In contrast, SKBR3 cell survival was greatly reduced by MKP1 siRNA treatment after irradiation, indicating that HER2-positive breast cancer cells may rely more on MKP1 for survival than their HER2-negative counterparts. Even though MKP1 depletion dramatically reduced the survival of the cells, both wt and MKP1 siRNA-treated cells showed similar reduction in their survival after irradiation. This may have resulted from the compensation by the residual MKP1 in these knock-down cells as the siRNA-mediated knock-down did not completely wipe out MKP1 from the cells (Fig. 4A).
Furthermore, we utilized a selective inhibitor of MKP1, Sanguinarine (34), and tested cell survival in a variety of breast cancer cell lines after inhibitor treatment. The optimum time and concentration of Sanguinarine to be used was determined; 5 μM and 10 μM Sanguinarine killed 50% and 75% of SKBR3 cells as early as 2 h. (Fig. S4). A similar pattern was observed with MCF7/C6 cells (Fig. S5). The comparison of cell killing ability of MKP1 inhibitor revealed that all breast cancer cell lines are sensitive to Sanguinarine treatment (Fig. 5A-D); however, MDA-MB-231 cells were slightly less sensitive to MKP1 inhibition, suggesting HER2-expressing cell lines may rely more on MKP1 for survival than HER2-negative cells. The clonogenic survival of these cells confirmed that the cells treated with MKP1 inhibitor showed reduced clonogenic ability compared to no treatment and solvent control cells (Fig. 5E). Sanguinarine treatment alone resulted in 70%, 80%, and 74% decrease in the survival of MCF7/C6, MCF7 wt and SKBR3 cells, respectively; while radiation treatment alone reduced their survival by 77%, 78%, and 82%. Combination of inhibitor and radiation treatment resulted in 90% reduction in their survival rate.
Figure 5.
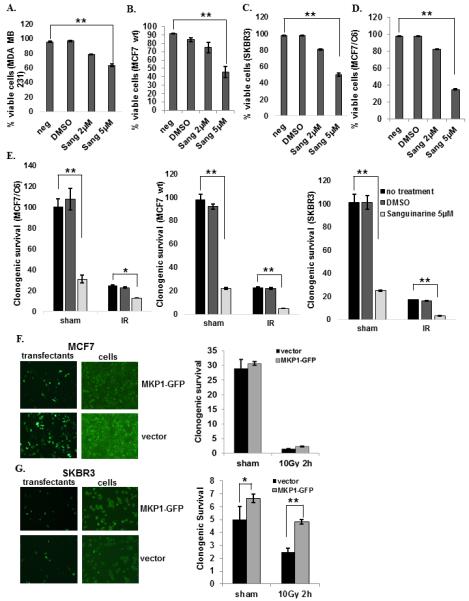
MKP1 inhibition via Sanguinarine treatment enhances cell death and radiosensitivity in breast cancer cells. A, MDA-MB-231 (HER2 negative), B, MCF7 (HER2 low), C, SKBR3 (HER2 overexpressing), and D, MCF7/C6 (HER2 overexpressing) cells were treated with 2μM and 5μM of Sanguinarine and the cell viability was determined by trypan blue assay (n=3, **p<0.01). E, Clonogenic survival of the MCF7, SKBR3, and MCF7/C6 cells after 24h of 2μM Sanguinarine treatment with or without 10 Gy IR (n=3, *p<0.05, **p<0.01). F, MCF7 (HER2 low) and G, SKBR3 (HER2 overexpressing) cells were transfected with GFP-tagged MKP1 construct and the clonogenic survival was determined with or without 10 Gy of IR (n=3, *p<0.05, **p<0.01). The transfection efficiency was shown in left panels.
Finally, to determine whether MKP1 overexpression provides a survival advantage to breast cancer cells, we constructed a GFP-tagged MKP1 vector and transfected MCF7 and SKBR3 cells (Fig. 5F-G, left panels). Overexpression of MKP1 did not provide a significant survival advantage to MCF7 cells with or without irradiation (Fig. 5F, right panel); however, SKBR3 cells showed significant enhancement in their clonogenic survival ability after radiation treatment when MKP1 is overexpressed (85% recovery in survival; Fig. 5G, right panel). These results suggest that the MKP1-mediated survival mechanism may require HER2 expression to be activated. It is likely that MCF7 cells with low HER2 expression are not able to activate overexpressed GFP-MKP1 to exhibit the expected levels of enhanced survival. Conversely, SKBR3 cells with HER2 overexpression were able to take advantage of enhanced MKP1 levels to survive the subsequent radiation insult. All in all, the data suggests that HER2 expressing cells depend on MKP1 for survival, demonstrating MKP1 as a potential target in HER2-positive breast cancer for tumor control.
Targeting MKP1 and HER2 for efficent killing of breast cancer cells
The role of MKP1 in the survival of HER2-positive breast cancer cells suggested that combinatory targetting of MKP1 and HER2 may provide more efficient cell killing than HER2 inhibition alone. We treated the MCF7/C6 (Fig. 6A), SKBR3 (Fig. 6B) and MCF7 wt (Fig. 6C) cells with increasing concentrations of Lapatinib (a clinically used HER2 inhibitor) alone or in combination with MKP1 siRNA to test if MKP1 depletion would provide any benefit over Lapatinib alone. Inhibition of MKP1 along with Lapatinib treatment further reduced the viability in HER2 expressing breast cancer cells (Fig. 6A-C, Fig. S6). A similar combinatorial treatment targeting study with HER2 siRNA and MKP1 inhibitor, Sanguinarine, resulted in increased cell killing in breast cancer cells compared to HER2 siRNA treatment alone (Fig. 6D-F). Last, we explored the efficacy of combination of chemo- and radiotherapy in HER2-positive breast cancer cells and breast cancer stem cells using a combination of Sanguinarine and Lapatinib inhibitors and IR (Fig. 6G-I). The results revealed that the combination of Sanguinarine and Lapatinib killed more cells when, which was further exacerbated by radiation treatment. Lapatinib inhibits the receptor tyrosine kinase activity of HER2 (35), which is also shown here by the inhibition of phosphorylation of downstream effector ERK (Fig. S7A). Expectedly, Lapatinib treatment resulted in decreased mitochondrial localization of MKP1 (Fig. S7B), which accounts for the reduced survival of Lapatinib-treated cells. These combinatorial treatment studies performed in a panel of breast cancer cell lines demonstrated that combination therapy is a more efficacious anti-cancer strategy than the individual inhibition of MKP1 or HER2 in the control of breast cancer.
Figure 6.
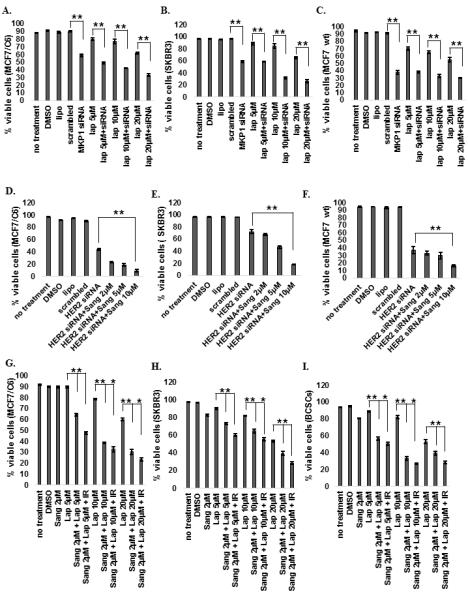
Simultaneous inhibition of HER2 and MKP1 in HER2-positive breast cancer cells. A, MCF7/C6, B, SKBR3 and C, MCF7 wt cells were treated with MKP1 siRNA (10nM for 48h) alone or in combination with Lapatinib (RTK inhibitor, indicated concentrations for 72 h) and cell viability was determined by trypan blue assay. D, MCF7/C6, E, SKBR3 and F, MCF7 wt cells were treated with HER2 siRNA alone or in combination with Sanguinarine (MKP1 inhibitor) and cell viability was determined by trypan blue assay. G, MCF7/C6, H, SKBR3 and I, HER2+/CD44+/CD24− BCSCs were treated with Sanguinarine, Lapatinib, or their combination and cell viability was determined by trypan blue assay before and 24h after 10 Gy of IR (n=3, *p<0.05, **p<0.01).
Discussion
This study reveals a mechanism by which MAPK phosphatase, MKP1, mediates a pro-survival response in breast cancer cells overexpressing HER2 (Fig. 7), suggesting an alternative therapeutic approach for HER2-positive tumors, especially the recurrent and metastatic tumors with an acquired resistance to previously administered anti-HER2 therapy. MKP1 expression was found to correlate with HER2 expression in breast cancer stem cells and clinical breast tumor specimens. Comparison of MKP1 inhibition in a panel of breast cancer cells with differential HER2 status revealed that HER2-positive breast cancer cells rely on MKP1 for survival. Therefore, targeting of MKP1 along with HER2 in HER2-expressing breast cancer cells resulted in increased cell killing and suggest MKP1 as a therapeutic target for controlling breast cancer cell survival. The data showing that MKP1 is overexpressed in HER2-expressing breast tumors, but not in the adjacent normal tissue of the same patients, suggest that MKP1 may be a potential target to treat resistant breast tumors with minimal side effects to the normal tissue. Our current studies reveal that MKP1 is not only expressed in clinical tumor tissues compared to their counterpart normal tissues from the same patients, but also that MKP1 is significantly enhanced in tumor cells under the stress of therapeutic radiation (5). Although a larger cohort of patient studies will be essential to further investigate the expression of MKP1 in tumor and normal tissues, our data clearly indicate that MKP1 is an important survival protein in breast cancer cells and a critical down-stream element of HER2 signaling.
Figure 7.
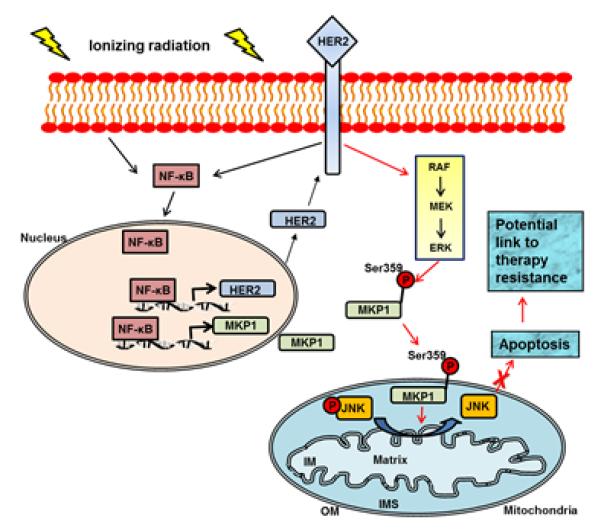
Activation of HER2/ERK/MKP1 pathway in therapy-resistant breast cancer cells. We have previously reported that radiation therapy induces the expression of HER2 and MKP1 via NF-κB-mediated gene promoter activation in breast cancer cells (4, 5, 50). Here we identified that MKP1 mitochondrial relocation is enhanced by radiation to target mitochondrial pJNK. The dephosphorylation and inactivation of JNK leads to the attenuation of the pro-apoptotic signals from JNK, resulting in the inhibition of mitochondria-mediated apoptosis and radioresistant phenotype of breast cancer cells with HER2 status, including the HER2-positive breast cancer stem cells. The mitochondrial MKP1 is thus a potential target for therapy-resistant breast cancer cells, especially for recurrent/metastatic lesions with adaptive resistance to anti-HER2 therapy.
Elucidation of the mechanism in which MKP1 functions in breast tumors may prove to be beneficial in the clinic. It will be of great importance to understand why radiation-induced MKP1 particularly targets JNK of the three MAPKs (5). MKP1 dephosphorylates proteins of the MAPK family in the following order of affinity: p38MAPK ≥ JNK >> ERK1/2 (36-38). Although MKP1 binding to ERK1/2 and p38αMAPK relies on the same arginine residues (arginine 53-55, (39)), its binding to JNK depends on presently unidentified residues within the first N-terminal 188 residues of MKP1 and not arginine 53-55 (8). This may have implications for why radiation-induced MKP1 specifically targets JNK (5). Nevertheless, the identification of the MKP1 residues that are responsible for its interaction with JNK may be beneficial for eventual therapeutics. More importantly, the ability to interfere with binding of each MAPK to MKP1 independently will contribute greatly to the clinic. The role of ERK MAPK in the induction of MKP1 mitochondrial translocation suggests that hindering the interaction between ERK and MKP1 may be a valuable strategy to block the survival pathway initiated by MKP1 in order to increase cell death.
MKP1 is currently believed to be a nuclear phosphatase with its dephosphorylating activity restricted to within the nucleus (36, 40). MKP1 is targeted to the nucleus via its LXXLL motif (40), which is different than any known consensus nuclear targeting sequences (41, 42). How MKP1 is targeted to the nucleus, whether on its own or via binding to a facilitator molecule, remains elusive. In addition to literature on nuclear MKP1, our study revealed MKP1 localization into the mitochondria. MKP1 lacks an N-terminal mitochondria targeting sequence, which suggests the presence of an internal targeting sequence. Proteins without N-terminal mitochondria targeting sequences are translocated into the mitochondria via binding to heat shock proteins (Hsps) and/or 14-3-3 chaperons (43). The elucidation of the detailed mechanisms underlying nuclear or mitochondrial targeting of MKP1 will offer additional control over this phosphatase via the regulation of its intracellular localization. The possibility of sequestering MKP1 in the cytosol as a means to limit its phosphatase activity in the mitochondria, via modification of the molecules involved in its mitochondrial localization, carries a great potential for anti-cancer therapeutics.
Lapatinib is a reversible inhibitor of the tyrosine kinase activity of HER2 and EGFR (27, 30); however, its anti-tumor activity in breast cancer is more dependent on HER2 overexpression than EGFR (44, 45). Although Lapatinib and other HER2-targeting agents produce a positive outcome in breast cancer patients, the development of acquired resistance inevitably causes a fraction of these patients to recur. Understanding the survival networks underlying the acquired resistance may enable the use of agents targeted against these pro-survival pathways and yield clinically relevant neo-adjuvant regimens to improve the efficacy of anti-cancer treatment. Identification of a MKP1-mediated pro-survival pathway downstream of HER2 in breast cancer cells reveals MKP1 as a rational candidate for targeted agents. The availability of a selective MKP1 inhibitor, Sanguinarine (34), is a valuable tool for the assessment of combination strategies to overcome tumor resistance.
This study showed that HER2-positive breast cancer cells are exclusively dependent on MKP1 for their survival. This could be a significant finding as ~50% of patients that receive chemotherapy are also given radiotherapy (46), suggesting that combinatorial strategies using specific molecules to inhibit the cell-protecting function of MKP1 along with conventional chemo- or radiotherapy may offer a better anti-cancer approach. In human breast cancers, there is a strong correlation between the expression of the HER2 oncogene and ERK1/2 and MKP1 protein expression (47). It is suggested that HER2, by stimulating the Raf-MEK-ERK pathway (48), protects ERK1/2 from inactivation by MKP1. This scenario can be deleterious when considering our findings that ERK is upstream of MKP1 mitochondrial localization. Constitutive ERK activation in HER2-overexpressing breast cancers would result in enhanced MKP1 mitochondrial localization and increased survival. In accordance with this scenario, we found that MKP1 is overexpressed in HER2 overexpressing breast cancer stem cells. Targeting MKP1 by siRNAs or chemical inhibitors in a variety of breast cancer cells proved to be an efficient mechanism to sensitize breast cancer cells to radiation and chemotherapy. In the clinic, it has been well-observed that many breast cancer patients develop tumor resistance to anti-HER2 therapy (27, 29-31).Our combinatorial inhibition studies showed that targeting of MKP1 and HER2 together is a more efficient anti-cancer strategy and that the addition of radiation therapy to the chemical inhibition of MKP1 and HER2 further reduced tumor cell survival. Further elucidation of the mechanisms of HER2-MKP1 pro-survival network may generate alternative approaches to treat resistant breast cancer.
Supplementary Material
Acknowledgements
The authors acknowledge the technical assistance of Dr. Zhien Wang at the University of California San Francisco in sorting the BCSCs. We appreciate the help of Irmgard Feldman at the UC Davis Comprehensive Cancer Center Biorepository in obtaining left-over tissue specimens. We thank Dr. Dominik J. Green for his critical insights into the manuscript preparation and proofreading.
Grant Support
This work was supported by National Institutes of Health Grants RO1 CA133402 and RO1 CA152313.
Footnotes
The authors disclose no potential conflicts of interest.
Authors’ Contributions
Concept and design: D.C., C. S., A.D.B., and J.J.L
Development of methodology: D.C., C.L., M.F., and F.Y.S.C.
Acquisition of data: D.C., C.L., M.F., C. S., and A.D.B.
Writing of the manuscript: D.C., C.S. and J.J.L
Study supervision: J.J.L
References
- 1.Recht A, Come SE, Henderson IC, Gelman RS, Silver B, Hayes DF, et al. The sequencing of chemotherapy and radiation therapy after conservative surgery for early-stage breast cancer. N Engl J Med. 1996;334:1356–61. doi: 10.1056/NEJM199605233342102. [DOI] [PubMed] [Google Scholar]
- 2.Liang K, Lu Y, Jin W, Ang KK, Milas L, Fan Z. Sensitization of breast cancer cells to radiation by trastuzumab. Mol Cancer Ther. 2003;2:1113–20. [PubMed] [Google Scholar]
- 3.Debeb BG, Xu W, Woodward WA. Radiation resistance of breast cancer stem cells: understanding the clinical framework. J Mammary Gland Biol Neoplasia. 2009;14:11–7. doi: 10.1007/s10911-009-9114-z. [DOI] [PubMed] [Google Scholar]
- 4.Cao N, Li S, Wang Z, Ahmed KM, Degnan ME, Fan M, et al. NF-kappaB-mediated HER2 overexpression in radiation-adaptive resistance. Radiat Res. 2009;171:9–21. doi: 10.1667/RR1472.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang Z, Cao N, Nantajit D, Fan M, Liu Y, Li JJ. Mitogen-activated protein kinase phosphatase-1 represses c-Jun NH2-terminal kinase-mediated apoptosis via NF-kappaB regulation. J Biol Chem. 2008;283:21011–23. doi: 10.1074/jbc.M802229200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pouyssegur J, Lenormand P. Fidelity and spatio-temporal control in MAP kinase (ERKs) signalling. Eur J Biochem. 2003;270:3291–9. doi: 10.1046/j.1432-1033.2003.03707.x. [DOI] [PubMed] [Google Scholar]
- 7.Brondello JM, Pouyssegur J, McKenzie FR. Reduced MAP kinase phosphatase-1 degradation after p42/p44MAPK-dependent phosphorylation. Science. 1999;286:2514–7. doi: 10.1126/science.286.5449.2514. [DOI] [PubMed] [Google Scholar]
- 8.Slack DN, Seternes OM, Gabrielsen M, Keyse SM. Distinct binding determinants for ERK2/p38alpha and JNK map kinases mediate catalytic activation and substrate selectivity of map kinase phosphatase-1. J Biol Chem. 2001;276:16491–500. doi: 10.1074/jbc.M010966200. [DOI] [PubMed] [Google Scholar]
- 9.Kurokawa H, Arteaga CL. Inhibition of erbB receptor (HER) tyrosine kinases as a strategy to abrogate antiestrogen resistance in human breast cancer. Clin Cancer Res. 2001;7:4436s–42s. discussion 11s-12s. [PubMed] [Google Scholar]
- 10.Kurokawa H, Lenferink AE, Simpson JF, Pisacane PI, Sliwkowski MX, Forbes JT, et al. Inhibition of HER2/neu (erbB-2) and mitogen-activated protein kinases enhances tamoxifen action against HER2-overexpressing, tamoxifen-resistant breast cancer cells. Cancer Res. 2000;60:5887–94. [PubMed] [Google Scholar]
- 11.Wang HY, Cheng Z, Malbon CC. Overexpression of mitogen-activated protein kinase phosphatases MKP1, MKP2 in human breast cancer. Cancer Lett. 2003;191:229–37. doi: 10.1016/s0304-3835(02)00612-2. [DOI] [PubMed] [Google Scholar]
- 12.Small GW, Shi YY, Higgins LS, Orlowski RZ. Mitogen-activated protein kinase phosphatase-1 is a mediator of breast cancer chemoresistance. Cancer Res. 2007;67:4459–66. doi: 10.1158/0008-5472.CAN-06-2644. [DOI] [PubMed] [Google Scholar]
- 13.Wang C, Youle RJ. The role of mitochondria in apoptosis*. Annu Rev Genet. 2009;43:95–118. doi: 10.1146/annurev-genet-102108-134850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fulda S, Galluzzi L, Kroemer G. Targeting mitochondria for cancer therapy. Nat Rev Drug Discov. 2010;9:447–64. doi: 10.1038/nrd3137. [DOI] [PubMed] [Google Scholar]
- 15.Kharbanda S, Saxena S, Yoshida K, Pandey P, Kaneki M, Wang Q, et al. Translocation of SAPK/JNK to mitochondria and interaction with Bcl-x(L) in response to DNA damage. J Biol Chem. 2000;275:322–7. doi: 10.1074/jbc.275.1.322. [DOI] [PubMed] [Google Scholar]
- 16.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100:3983–8. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3:730–7. doi: 10.1038/nm0797-730. [DOI] [PubMed] [Google Scholar]
- 18.Weisenthal LM, Lippman ME. Clonogenic and nonclonogenic in vitro chemosensitivity assays. Cancer Treat Rep. 1985;69:615–32. [PubMed] [Google Scholar]
- 19.Al-Hajj M. Cancer stem cells and oncology therapeutics. Curr Opin Oncol. 2007;19:61–4. doi: 10.1097/CCO.0b013e328011a8d6. [DOI] [PubMed] [Google Scholar]
- 20.Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444:756–60. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- 21.Diehn M, Cho RW, Lobo NA, Kalisky T, Dorie MJ, Kulp AN, et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature. 2009;458:780–3. doi: 10.1038/nature07733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Duru N, Fan M, Candas D, Menaa C, Liu HC, Nantajit D, et al. HER2-associated radioresistance of breast cancer stem cells isolated from HER2-negative breast cancer cells. Clin Cancer Res. 2012;18:6634–47. doi: 10.1158/1078-0432.CCR-12-1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eyler CE, Rich JN. Survival of the fittest: cancer stem cells in therapeutic resistance and angiogenesis. J Clin Oncol. 2008;26:2839–45. doi: 10.1200/JCO.2007.15.1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Phillips TM, McBride WH, Pajonk F. The response of CD24(−/low)/CD44+ breast cancer-initiating cells to radiation. J Natl Cancer Inst. 2006;98:1777–85. doi: 10.1093/jnci/djj495. [DOI] [PubMed] [Google Scholar]
- 25.Rich JN. Cancer stem cells in radiation resistance. Cancer Res. 2007;67:8980–4. doi: 10.1158/0008-5472.CAN-07-0895. [DOI] [PubMed] [Google Scholar]
- 26.Visvader JE, Lindeman GJ. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer. 2008;8:755–68. doi: 10.1038/nrc2499. [DOI] [PubMed] [Google Scholar]
- 27.Rexer BN, Arteaga CL. Intrinsic and acquired resistance to HER2-targeted therapies in HER2 gene-amplified breast cancer: mechanisms and clinical implications. Crit Rev Oncog. 2012;17:1–16. doi: 10.1615/critrevoncog.v17.i1.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 1987;235:177–82. doi: 10.1126/science.3798106. [DOI] [PubMed] [Google Scholar]
- 29.Chen FL, Xia W, Spector NL. Acquired resistance to small molecule ErbB2 tyrosine kinase inhibitors. Clin Cancer Res. 2008;14:6730–4. doi: 10.1158/1078-0432.CCR-08-0581. [DOI] [PubMed] [Google Scholar]
- 30.Geyer CE, Forster J, Lindquist D, Chan S, Romieu CG, Pienkowski T, et al. Lapatinib plus capecitabine for HER2-positive advanced breast cancer. N Engl J Med. 2006;355:2733–43. doi: 10.1056/NEJMoa064320. [DOI] [PubMed] [Google Scholar]
- 31.Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med. 2001;344:783–92. doi: 10.1056/NEJM200103153441101. [DOI] [PubMed] [Google Scholar]
- 32.Xia W, Bacus S, Hegde P, Husain I, Strum J, Liu L, et al. A model of acquired autoresistance to a potent ErbB2 tyrosine kinase inhibitor and a therapeutic strategy to prevent its onset in breast cancer. Proc Natl Acad Sci U S A. 2006;103:7795–800. doi: 10.1073/pnas.0602468103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lu G, Ren S, Korge P, Choi J, Dong Y, Weiss J, et al. A novel mitochondrial matrix serine/threonine protein phosphatase regulates the mitochondria permeability transition pore and is essential for cellular survival and development. Genes Dev. 2007;21:784–96. doi: 10.1101/gad.1499107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vogt A, Tamewitz A, Skoko J, Sikorski RP, Giuliano KA, Lazo JS. The benzo[c]phenanthridine alkaloid, sanguinarine, is a selective, cell-active inhibitor of mitogen-activated protein kinase phosphatase-1. J Biol Chem. 2005;280:19078–86. doi: 10.1074/jbc.M501467200. [DOI] [PubMed] [Google Scholar]
- 35.Higa GM, Abraham J. Lapatinib in the treatment of breast cancer. Expert Rev Anticancer Ther. 2007;7:1183–92. doi: 10.1586/14737140.7.9.1183. [DOI] [PubMed] [Google Scholar]
- 36.Camps M, Nichols A, Arkinstall S. Dual specificity phosphatases: a gene family for control of MAP kinase function. FASEB J. 2000;14:6–16. [PubMed] [Google Scholar]
- 37.Farooq A, Zhou MM. Structure and regulation of MAPK phosphatases. Cell Signal. 2004;16:769–79. doi: 10.1016/j.cellsig.2003.12.008. [DOI] [PubMed] [Google Scholar]
- 38.Franklin CC, Kraft AS. Conditional expression of the mitogen-activated protein kinase (MAPK) phosphatase MKP-1 preferentially inhibits p38 MAPK and stress-activated protein kinase in U937 cells. J Biol Chem. 1997;272:16917–23. doi: 10.1074/jbc.272.27.16917. [DOI] [PubMed] [Google Scholar]
- 39.Boutros T, Chevet E, Metrakos P. Mitogen-activated protein (MAP) kinase/MAP kinase phosphatase regulation: roles in cell growth, death, and cancer. Pharmacol Rev. 2008;60:261–310. doi: 10.1124/pr.107.00106. [DOI] [PubMed] [Google Scholar]
- 40.Wu JJ, Zhang L, Bennett AM. The noncatalytic amino terminus of mitogen-activated protein kinase phosphatase 1 directs nuclear targeting and serum response element transcriptional regulation. Mol Cell Biol. 2005;25:4792–803. doi: 10.1128/MCB.25.11.4792-4803.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cartier R, Reszka R. Utilization of synthetic peptides containing nuclear localization signals for nonviral gene transfer systems. Gene Ther. 2002;9:157–67. doi: 10.1038/sj.gt.3301635. [DOI] [PubMed] [Google Scholar]
- 42.Christophe D, Christophe-Hobertus C, Pichon B. Nuclear targeting of proteins: how many different signals? Cell Signal. 2000;12:337–41. doi: 10.1016/s0898-6568(00)00077-2. [DOI] [PubMed] [Google Scholar]
- 43.Young JC, Hoogenraad NJ, Hartl FU. Molecular chaperones Hsp90 and Hsp70 deliver preproteins to the mitochondrial import receptor Tom70. Cell. 2003;112:41–50. doi: 10.1016/s0092-8674(02)01250-3. [DOI] [PubMed] [Google Scholar]
- 44.Johnston S, Trudeau M, Kaufman B, Boussen H, Blackwell K, LoRusso P, et al. Phase II study of predictive biomarker profiles for response targeting human epidermal growth factor receptor 2 (HER-2) in advanced inflammatory breast cancer with lapatinib monotherapy. J Clin Oncol. 2008;26:1066–72. doi: 10.1200/JCO.2007.13.9949. [DOI] [PubMed] [Google Scholar]
- 45.Konecny GE, Pegram MD, Venkatesan N, Finn R, Yang G, Rahmeh M, et al. Activity of the dual kinase inhibitor lapatinib (GW572016) against HER-2-overexpressing and trastuzumab-treated breast cancer cells. Cancer Res. 2006;66:1630–9. doi: 10.1158/0008-5472.CAN-05-1182. [DOI] [PubMed] [Google Scholar]
- 46.Bentzen SM. Preventing or reducing late side effects of radiation therapy: radiobiology meets molecular pathology. Nat Rev Cancer. 2006;6:702–13. doi: 10.1038/nrc1950. [DOI] [PubMed] [Google Scholar]
- 47.Loda M, Capodieci P, Mishra R, Yao H, Corless C, Grigioni W, et al. Expression of mitogen-activated protein kinase phosphatase-1 in the early phases of human epithelial carcinogenesis. Am J Pathol. 1996;149:1553–64. [PMC free article] [PubMed] [Google Scholar]
- 48.Grant S, Qiao L, Dent P. Roles of ERBB family receptor tyrosine kinases, and downstream signaling pathways, in the control of cell growth and survival. Front Biosci. 2002;7:d376–89. doi: 10.2741/grant. [DOI] [PubMed] [Google Scholar]
- 49.Antonyuk SV, Han C, Eady RR, Hasnain SS. Structures of protein-protein complexes involved in electron transfer. Nature. 2013;496:123–6. doi: 10.1038/nature11996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Guo G, Wang T, Gao Q, Tamae D, Wong P, Chen T, et al. Expression of ErbB2 enhances radiation-induced NF-kappaB activation. Oncogene. 2004;23:535–45. doi: 10.1038/sj.onc.1207149. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.