

Abstract
4-Nitroquinoline 1-oxide (4-NQO) is a highly carcinogenic chemical that induces mutations in bacteria, fungi, and animals through the formation of bulky purine adducts. 4-NQO has been used as a mutagen for genetic screens and in both the study of DNA damage and DNA repair. In the model eukaryote Aspergillus nidulans, 4-NQO−based genetic screens have been used to study diverse processes, including gene regulation, mitosis, metabolism, organelle transport, and septation. Early work during the 1970s using bacterial and yeast mutation tester strains concluded that 4-NQO was a guanine-specific mutagen. However, these strains were limited in their ability to determine full mutagenic potential, as they could not identify mutations at multiple sites, unlinked suppressor mutations, or G:C to C:G transversions. We have now used a whole genome resequencing approach with mutant strains generated from two independent genetic screens to determine the full mutagenic spectrum of 4-NQO in A. nidulans. Analysis of 3994 mutations from 38 mutant strains reveals that 4-NQO induces substitutions in both guanine and adenine residues, although with a 19-fold preference for guanine. We found no association between mutation load and mutagen dose and observed no sequence bias in the residues flanking the mutated purine base. The mutations were distributed randomly throughout most of the genome. Our data provide new evidence that 4-NQO can potentially target all base pairs. Furthermore, we predict that current practices for 4-NQO−induced mutagenesis are sufficient to reach gene saturation for genetic screens with feasible identification of causative mutations via whole genome resequencing.
Keywords: filamentous fungi, genetic screen, chemical mutagenesis, 4-nitroquinoline 1-oxide, whole genome sequencing
4-Nitroquinoline 1-oxide (4-NQO) is a highly carcinogenic chemical that causes mutations in bacteria, fungi, and animals. 4-NQO has been used widely in the study of DNA damage and DNA repair and to generate mutants for genetic screens. 4-NQO induces mutagenesis after metabolic conversion to 4-hydroxyaminoquinolone 1-oxide (4-HAQO) (Miller 1970), which forms stable bulky adducts on purines (Tada and Tada 1976). Based on in vitro studies as well as in Escherichia coli and animal cells, 4-HAQO forms the majority of adducts (~50%) on the second nitrogen (N2) of guanine (Tada and Tada 1971; Galiegue-Zouitina et al. 1986; Bailleul et al. 1989). However, carbon eight (C8) guanine adducts (Bailleul et al. 1981; Galiègue-Zouitina et al. 1984; Tada et al. 1984) and nitrogen six (N6) adenine adducts (Galiegue-Zouitina et al. 1985, 1986) also occur at a lower frequency, ~30% and ~10%, respectively (Bailleul et al. 1989). Additional lesions were thought to be caused by production of reactive oxygen species (Kohda et al. 1986). In E. coli and mammalian cells, 4-HAQO adducts are repaired by the nucleotide excision repair pathway (Ikenaga et al. 1975a,b, 1977; Ikenaga and Kakunaga 1977), and in E. coli the error prone DNA polymerase IV (Pol IV) is the likely cause of sequence changes (Williams et al. 2010). Early work to characterize the mutagenic effects of 4-NQO in the yeasts Saccharomyces cerevisiae and Schizosaccharomyces pombe, as well as in the bacteria Salmonella typhimurium and E. coli, relied upon reversion of characterized auxotrophic tester strains, as DNA sequencing technology was not yet readily available (Prakash et al. 1974; Janner et al. 1979; Rosenkranz and Poirier 1979). These experiments identified the changes induced by 4-NQO as G:C to A:T transitions, G:C to T:A transversions, and frameshifts (Prakash et al. 1974; Janner et al. 1979; Rosenkranz and Poirier 1979). However, differences in frequency and mutation type varied between species and with 4-NQO concentration (Rosenkranz and Poirier 1979). Studies relying on reversion tester strains are limited by their inability to detect or determine multiple mutations in the same target gene as well as unlinked suppressor mutations, and the lack of strains to specifically detect G:C to C:G transversions (Prakash and Sherman 1973). In addition, these strains were not informative as to how flanking sequence affects mutagenic potential. Furthermore, auxotrophic reversion tester strains may show mutational bias due to functional constraints. Therefore, the full 4-NQO mutagenic spectrum, including type and relative frequency of induced mutations as well as the effect of flanking sequence, remains to be determined.
The genetic model filamentous fungus Aspergillus nidulans has been invaluable for advances in understanding a variety of eukaryotic cellular processes, including cell-cycle progression, development, response to DNA damage and pH changes, gene regulation, and metabolism (Clutterbuck 1969; Arst and Cove 1973; Morris 1975; Oakley and Oakley 1989; Harris et al. 1994; Goldman and Kafer 2004; Penalva et al. 2008; Wong et al. 2008). Many of these advances have been made using genetic screens. The versatility of A. nidulans for genetic analysis is due to several amenable characteristics, including stable haploid and diploid life stages as well as asexual and sexual reproduction (Pontecorvo et al. 1953). Heterozygous diploid strains, constructed via the parasexual cycle, can be used for analysis of dominance or complementation and to map novel mutations to a chromosome by haploidization (Todd et al. 2007a). Mutations can then be mapped more finely by classical genetic mapping via the sexual cycle (Todd et al. 2007b). Furthermore, the well-developed DNA-mediated transformation system, with homologous gene targeting and multiple selectable markers, enables construction of strains for mutational analysis and selection of mutants in genetic screens, and reconstruction of identified candidate mutations to identify the causative mutation associated with the mutant phenotype (Nayak et al. 2006). A. nidulans has been used extensively in genetic screens for mutants generated by a variety of chemical and physical mutagens, including N-methyl-N′-nitro-N-nitrosoguanidine (MNNG) (Clutterbuck 1969; Hynes and Pateman 1970a,b; Arst and Cove 1973; Osman et al. 1993), nitrous acid (Apirion 1965; Clutterbuck 1969), diethyl sulfate (Clutterbuck 1969), ultraviolet (UV) light (Pontecorvo et al. 1953; Clutterbuck 1969; Axelrod et al. 1973; Morris 1975; Osman et al. 1993), and X-rays (Pontecorvo et al. 1953). However, many genetic screens in A. nidulans use 4-NQO (Harris et al. 1994; Wu et al. 1998; Pokorska et al. 2000; Conlon et al. 2001; Heck et al. 2002; Kinghorn et al. 2005; Cecchetto et al. 2012; Larson et al. 2014; Tan et al. 2014) because it is safer and more stable than MNNG and it is thought to produce primarily single base-pair substitutions, which can generate both loss-of-function and altered function mutants. These altered function mutants are important for identifying essential genes in which larger mutations would be lethal. The utility and application of 4-NQO as a mutagen in genetic screens highlight the importance of understanding the full consequences of 4-NQO mutagenesis.
To fully characterize the mutagenic potential of any chemical, analysis of mutations that are unbiased by the selection method or gene function is required. A genomics approach, rather than sampling a single gene target by reversion of auxotrophies, overcomes limitations imposed by functional constraints, as mutations in noncoding regions and mutations unrelated to the selection and independent of function also can be detected. Whole genome sequencing has been used to identify the effects of ethyl methanesulfonate, ethylnitrosourea, and UV light in several eukaryotes, including Arabidopsis thaliana (Uchida et al. 2011), Danio rerio (Voz et al. 2012), Caenorhabditis elegans (Flibotte et al. 2010), and the apicomplexan parasite Toxoplasma gondii (Farrell et al. 2014). Recent advances in sequencing technology have permitted rapid and affordable resequencing of fungal genomes, and this has enabled identification of causative mutations in mutants generated in genetic screens (McCluskey et al. 2011; Pomraning et al. 2011; Nowrousian et al. 2012; Bielska et al. 2014; Tan et al. 2014; Yao et al. 2014; Zhang et al. 2014). In this work, we have used a genome resequencing approach to fully characterize the 4-NQO mutagenic spectrum at a whole genome level using almost 4000 4-NQO−induced mutations arising from independent genetic screens (Tan et al. 2014; this study). 4-NQO causes all possible base-pair substitutions with a 19-fold preference for guanine over adenine residues.
Materials and Methods
A. nidulans strains, media, growth conditions
A. nidulans strains RT244 (biA1 pyrG89 gpdA(p)areAHA fmdS-lacZ pyroA4 nkuA∆::Bar [prnA::areANES::gfp::AfpyroA] crmAT525C::pyrG) and RPA520 (yA::[gpdA(p)mCherry::FLAG::PTS1::Afpyro] pabaA1 pyrG89 [TagGFP2::rabA::AfpyrG] pyroA4 nkuA∆::argB [HH1::TagBFP::Afribo]) were used for mutagenesis. Mutant strains generated from RPA520 were outcrossed to RPA478 (pyrG89 [TagGFP2::rabA::AfpyrG] pyroA4 nkuA∆::argB [HH1::TagBFP::Afribo] riboB2) or RPA496 (pyrG89 [TagGFP2::rabA::AfpyrG] pyroA4 nkuA∆::argB [HH1::TagBFP::Afribo]). A. nidulans growth conditions and media adjusted to pH 6.5 were as described (Cove 1966). Aspergillus nitrogen-free minimal media containing 1% w/v glucose and nitrogen sources (ammonium tartrate, sodium nitrate, or L-proline) added to a final concentration of 10 mM (Cove 1966), or rich yeast and glucose media (Szewczyk et al. 2006), supplemented for auxotrophies, were used for growth.
Mutagenesis and sequencing
Mutagenesis using 4-NQO (Sigma-Aldrich) was carried out primarily as described (Holt and May 1996; Tan et al. 2014). In summary, ~107 or ~108 conidia, suspended in phosphate buffer (0.1 M potassium phosphate pH 7.0, 0.01% Tween 80) and quantified using a hemocytometer, were exposed to 0.24−4.0 µg mL−1 4-NQO at 37° for 30 min. 4-NQO was quenched with an equal volume of 0.5 M sodium thiosulfate and washed twice in phosphate buffer. Strains were recovered from 50%, 10%, and 3% survival treatments (0.24 µg mL−1 4-NQO per 107 spores, 0.45 µg mL−1 4-NQO per 107 spores, and 4.0 µg mL−1 4-NQO per 108 spores, respectively) after 2−4 days’ growth on either yeast and glucose media or supplemented Aspergillus nitrogen-free minimal media containing 10 mM L-proline and tested for mutant phenotypes. Proline-using mutant phenotypes in strains derived from RT244 were mapped by meiotic crossing to RT250 (yA1 pabaA1 pyrG89 gpdA(p)areAHA fmdS-lacZ prn-309). Genomic DNA was isolated as described (Lee and Taylor 1990). The genomes of RT244 and a derivative mutant strain were sequenced by the Genome Sequencing Facility (Kansas University Medical Center, Kansas City, Kansas) on an Illumina HiSEQ 2500 platform using single-end 50-bp reads. The genomes of RPA478, RPA496, RPA520, and bulked segregrant progeny of derivative mutant strains were sequenced by single-end, whole genome sequencing on the Illumina Genome Analyzer HiSeq 2000 platform, generating sequence reads ~50 base pairs in length (Tan et al. 2014). For mutant strains from RT244 showing tight linkage of the causative mutation and prnA, the mutations were identified by amplification of the prnA::areANES::gfp regions with prn3′-F (5′-TCACGGCTATTCCGTGCTTTGA-3′) and gfp5′-R (5′-ACGCTGAACTTGTGGCCGTTA-3′) using Ex Taq (TaKaRa) and sequencing at Kansas State University DNA Sequencing and Genotyping Facility.
In silico analysis
In silico analysis used the Galaxy platform (galaxyproject.org) (Blankenberg et al. 2010b) and Broad Genome Analysis Toolkit (GATK; broadinstitute.org/gatk) (McKenna et al. 2010). FASTA files were converted to FASTQ format using FASTQ Groomer (Blankenberg et al. 2010a). Sequence quality was determined using FastQC (Li and Durbin 2009) (bioinformatics.babraham.ac.uk/projects/fastqc/). Nucleotide sequence reads were aligned using Burrows-Wheeler Alignment for Illumina with default settings to the A. nidulans FGSC_A4 genome (Version S10) downloaded from AspGD (Cerqueira et al. 2014). Genome coverage was determined using BEDTools (Quinlan and Hall 2010). Sequence coverage was lacking or not aligned for the centromeres, the ribosomal rRNA repeats, and mitochondrial sequences. Variants were identified using FreeBayes (Garrison and Marth 2012) with default settings except for report polymorphism probability (−P: 0.01), ploidy (−p: 1), minimum observations (−F: 0.5), and minimum coverage (−!: 4) or using GATK (Depristo et al. 2011) with default settings except for quality score >50 (−stand_call_conf: 50.0, −stand_emit_conf: 10.0) and down sampling to 50 fold coverage (−dcov: 50.0). Variants unique to mutant strains were identified using Select Variants (Depristo et al. 2011). Aligned sequence reads from wild-type strains were manually inspected to confirm the absence of all identified unique variants. Box plots were generated using JMP 11 (SAS), outliers in boxplots are points lying 1.5 × interquartile range (third quartile to first quartile) above the third quartile or below the first quartile. The Student’s t-test and simple χ2 test were computed in Excel (Microsoft Office). SAS 9.4 (SAS) was used for exponential quantile-quantile plots (CAPABILITY procedure: QQplot / exponential, σ = est, θ = est), Kolmogorov-Smirnov tests (UNIVARIATE procedure with histogram & exponential settings), and categorical χ2 tests (FREQ and GENMOD procedures). Consensus motifs of mutated sites were generated using WebLogo (Crooks et al. 2004) (weblogo.berkeley.edu). A. nidulans sequence annotation of transcribed and intergenic regions, and gene function descriptions were obtained from AspGD (Cerqueira et al. 2014), and descriptions of yeast orthologs were obtained from SGD (Cherry et al. 2012).
Prediction of saturation
We derived the following random sampling with replacement equation that can be adjusted to calculate the probability of a specific mutation of every nucleotide (nucleotide saturation) or every possible substitution at every nucleotide (substitution saturation):
The standard equation for probability of a specific event (X) given multiple random samples with replacement is P(X) = 1 − (1 − N-1)n, where N−1 is the probability of the specific event given a single sample was taken, and n is the number of samples taken. For our equation, N−1 is replaced with the relative frequency with which a specific mutation arises (f) divided by the total number of base pairs at which it could have arisen (b). The number of samples is the mean number of mutations arising per spore (m), multiplied by the number of treated spores (s), multiplied by the number of surviving spores (1 − k), where k is the proportion kill, i.e., for a mutation at a single base-pair PS(X) = 1-(1-f.b-1)m.s.(1-k). To determine the probability of a mutation at every possible base pair, where the likelihood of mutating any base pair is equivalent due to random mutagenesis, the probability of a single event is raised to the power of the number of base pairs (b), giving the final equation PS(X) for the probability of saturation of a specific mutation (X). The following values were used: PS(G→H) f = 0.95, PS(G→A) f = 0.53, PS(G→T) f = 0.276, PS(G→C) f = 0.14, PS(A→B) f = 0.05, PS(A→C) f = 0.01, PS(A→G) f = 0.03, PS(A→T) f = 0 0.01), b = 15241995.5 using a 50% GC content in A. nidulans (Galagan et al. 2005), m = 105, s is variable and k = 0.5 (50% kill) or 0.9 (90% kill).
The probability of nucleotide saturation of both guanine and adenine is therefore:
And the probability of substitution saturation of both guanine and adenine is as follows:
Results and Discussion
4-NQO mutations are distributed across the genome
To determine the effects of 4-NQO mutagenesis on A. nidulans DNA, we used whole genome sequence data from two independent genetic screens. The first mutagenesis involved direct selection for reversion of a proline nonutilization phenotype conferred by fusion of a nuclear export signal to the transcription factor PrnA (D. J. Downes and R. B. Todd, unpublished data). Mutant strains were generated with a dose of 4-NQO resulting in 97% kill. We isolated nine mutant strains from this screen by direct selection for proline utilization. For eight mutant strains, the causative mutations mapped to the prnA locus, whereas for the ninth mutant strain the proline utilization phenotype was unlinked to prnA. Mutations in prnA were identified by sequencing polymerase chain reaction products (Table 1). The strain containing the unlinked mutation and the mutagenesis parent were used for whole genome sequencing. The second mutagenesis was for a microscopy-based screen for defective organelle transport on rich media (Tan et al. 2014). Conidia were treated with doses of 4-NQO conferring 50% or 90% kill. Mutant strains of interest were identified by visual screening for mislocalization of fluorescently labeled nuclei, endosomes and peroxisomes (Tan et al. 2014). To identify all lesions induced in this screen bulked segregant progeny of 40 mutant strains, 17 from 50% kill, and 23 from 90% kill, the mutagenesis parent and the outcross parents were sequenced. Reads from both screens were mapped to the A. nidulans FGSC_A4 reference genome (Galagan et al. 2005), providing sufficient coverage high quality variant calling in all regions excluding centromeres and the nucleolar organizing region ribosomal DNA repeats on Chromosome V (Brody et al. 1991; Clutterbuck and Farman 2008). Although our mutant strains were selected or chosen for specific phenotypes and therefore bias may occur for the causative mutation, most of the mutations arising throughout the genome will be random mutations unrelated to the observed phenotypes. Therefore, these mutations represent a data set of 4-NQO−derived sequence changes that are neither biased by selection nor constrained by function. In total we identified almost 7000 mutations in the 41 mutant strains that were absent in the parents. However, ~42% of these mutations were in just three strains. These three mutant strains each carried a substitution or nonsense mutation in at least one DNA repair gene (Supporting Information, File S1). These genes either lacked mutations in the 38 mutant strains with a lower mutation load, or in three cases carried only silent mutations or conservative substitutions. As the mutations arising in the three high mutation load strains may be due to defective DNA repair, rather than resulting directly from 4-NQO−induced mutagenesis, they were excluded from further analysis. Of the remaining 3994 4-NQO−induced mutations distributed across the genomes of 38 mutant strains, 3993 were single-nucleotide substitutions and one was a ∆G:C single base-pair deletion (File S2). The total number of mutations per strain ranged from 23 to 240; however, there was no significant difference in the mutation load arising from different 4-NQO doses and kill percentages (Figure 1). Therefore, we pooled the data for mutants isolated following different mutagen doses for subsequent analyses. The lack of a dose effect on the number of observed mutations per strain in our dataset seems somewhat counterintuitive. It is possible that this could result from the sample size of our data, or our inability to determine the number of mutations in the unrecovered strains killed or selected against.
Table 1. 4-NQO mutations selected by phenotype at specific loci in A. nidulans.
4-NQO, 4-nitroquinoline 1-oxide.
−, not reported.
+N, insertion.
∆N, deletion.
Figure 1.

Number of point mutations per strain is not dose-dependent. Distribution of the number of mutations per strain resulting from 50% kill (N = 17; 0.24 µg mL−1 4-NQO per 107 spores) and 90% kill (N = 20; 0.45 µg mL−1 4-NQO per 107 spores) as well as combined data (N = 38). There was no significant difference between the number of mutations induced by 50% kill compared with 90% kill using unpaired unequal distribution Student’s t-test. The combined data includes the single mutant from 97% kill (4.0 µg mL−1 4-NQO per 108 spores) with 70 mutations. Boxplots show minimum and maximum (whiskers), median (dividing line), mean (circle), and 95% confidence interval of mean (diamond).
To determine whether the effects of 4-NQO are biased toward particular regions of the genome or occur randomly, we classified each of the 3994 mutations as affecting either predicted transcribed regions (5′ untranslated region, coding, intron and 3′ untranslated region sequences) or intergenic regions (all other sequences). We found 2724 mutations within predicted transcribed regions and 1270 mutations in intergenic regions, consistent with relative genome content for each class. The mutations mapped to all regions of the genome, excluding mitochondrial DNA, the centromeres, and ribosomal repeats, where low coverage limited single-nucleotide polymorphism (SNP) calling (Figure 2A). The observed number of mutations per chromosome was not significantly different from that expected, calculated based on DNA content under random distribution (χ2 = 4.7, d.f. = 7, P = 0.695) (Figure 2A). The distances between randomly occurring mutations are expected to follow an exponential distribution with a rate of λ, where λ-1 is the mean distance between mutations (Sun et al. 2006; Farrell et al. 2014). The majority of the mutations were 3−11 kbp apart with a mean spacing of 7461 bp (Figure 2B). An exponential quantile-quantile plot comparing the observed distances between mutations in the whole genome against the expected exponential distribution shows a close match with the theoretical distribution (Figure 2C). However, a one-sample Kolmogorov-Smirnov goodness-of-fit test has a P-value < 0.01 (N = 3977, mean = 7,461.44, D = 0.0247) suggesting the observed data differ significantly from the expected trend. To determine whether this was consistent across the genome, we constructed quantile-quantile plots for each of the eight chromosomes (Figure 2D). Like the whole genome data, the observed distribution for each chromosome follows the exponential line closely. For all chromosomes except Chromosome II, the Kolmogorov-Smirnov test statistically supports an exponential distribution. Therefore, the majority of 4-NQO−generated mutations conform to the expected exponential distribution and are randomly distributed. We observed 71 mutations in very close proximity (<10 bp) to another mutation in the same mutant (File S3). These mutations may have arisen either independently from multiple bulky adducts or from a single adduct and an additional repair-based error. Because these two events cannot be distinguished and these mutations comprise <2% of the total data pool, they are considered individual events for all further analyses.
Figure 2.

4-NQO mutations are randomly distributed across the genome. (A) A. nidulans chromosome map showing locations of 3994 mutations arising from 4-NQO mutagenesis and %GC content (Galagan et al. 2005). Mutations within genes (transcribed regions) are red and those outside genes are blue. Centromeres are marked as circles. Expected (Exp.) number of mutations per chromosome was calculated by dividing 3994 by the proportion of genome content in each chromosome. Obs., observed. (B) Boxplot of distance between mutations showing minimum and maximum values within 1.5 × interquartile range of the box (whiskers), median (dividing line), mean (circle), 95% confidence interval of mean (diamond), and outliers (squares). (C−D) Exponential quantile-quantile plot of distances between mutations compared with theoretical exponential distribution (red line) where λ−1 = mean. P-value shown for Kolmogorov-Smirnov test D statistic. N is the number of distances between mutations. Distances between mutations flanking centromeres and the ribosomal repeats were excluded. Genome (N = 3977, mean = 7461.44, D = 0.0247), I (N = 505, mean = 7332.67, D = 0.0388), II (N = 524, mean = 7610.55, D = 0.0584), III (N = 472, mean = 7226.80, D = 0.0297), IV (N = 367, mean = 7717.14, D = 0.0375), V (N = 419, mean = 7411.85, D = 0.0529), VI (N = 465, mean = 7167.69, D = 0.0251), VII (N = 603, mean = 7397.4, D = 0.0389), VIII (N = 622, mean = 7782.67, D = 0.0201).
4-NQO confers all six possible transitions and transversions
4-NQO was previously reported to induce transitions or transversions of guanine residues and frameshifts in bacteria and yeasts (Prakash et al. 1974; Janner et al. 1979; Rosenkranz and Poirier 1979). However, adducts of adenine are also formed and therefore adenine is a possible target (Galiègue-Zouitina et al. 1984, 1985; Bailleul et al. 1989; Menichini et al. 1989). Of the 3994 mutations identified from our screens, 3799 (95.12%) resulted from mutation of a guanine and only 195 (4.88%) from mutation of an adenine, consistent with the preference for guanine adduct formation (Figure 3, A and B). For SNPs of both guanine and adenine transition mutations were more frequent than transversions, with 56.27% (2137/3798) transitions for guanine (χ2 = 59.65, d.f. = 1, P < 0.0001) and 55.90% (109/195) transitions for adenine (χ2 = 2.71, d.f. = 1, P = 0.099). The most common mutation was G:C to A:T. Conversion of G:C to T:A, or conversion of G:C to C:G occurred at intermediate frequencies (Figure 3A). Mutation of A:T was rare (<5%) and in some individual mutant strains was not detected, but all three possible substitutions were observed in the complete data set (Figure 3B). To ensure the low frequency of adenine mutations was consistent with chemical mutagenesis rather than spontaneous mutation, we estimated the predicted level of spontaneous changes. Although studies of spontaneous mutation rate have been carried out in A. nidulans, they provide rates only for specific loci and not the whole genome (Lilly 1965; Alderson and Hartley 1969; Babudri and Morpurgo 1990; Baracho and Baracho 2003). Spontaneous mutation rates are very similar in Aspergillus spp., Neurospora crassa, and S. cerevisiae (Drake et al. 1998). Using an estimate of 0.0034 mutations per replication (Drake et al. 1998) with 30 days active growth between mutagenesis and sequencing and 1 hr per nuclear division (Bainbridge 1976), we predict an average of 2.5 spontaneous mutations may have arisen per strain. Similarly, calculations using sequence length and number of generations based on two whole genome studies in S. cerevisiae (Lynch et al. 2008; Zhu et al. 2014) predict just 3.5 spontaneous mutations per strain. By distributing the number of predicted spontaneous mutations across the six possible changes at the ratio described in the whole genome studies (Lynch et al. 2008; Zhu et al. 2014), we found all three types of A:T substitutions were more frequent than the expected spontaneous mutation level (Figure 3B). Therefore 4-NQO mutagenesis can cause all possible single-nucleotide substitutions. In previous 4-NQO mutagenesis studies using tester strains, mutations of adenine were reported as either absent (Prakash et al. 1974) or low-frequency events (~7%) and were only significantly different to nonmutagenized control strains in three of six experiments (Janner et al. 1979). We found only one occurrence of a deletion and no insertions. This low indel frequency suggests that this mutation may have arisen spontaneously. Therefore, we found no evidence for 4-NQO−induced frameshift mutations.
Figure 3.

4-NQO induces all six possible base pair substitutions. Distribution of the number of substitutions affecting guanine-cytosine (A) and adenine-thymine (B) base pairs per mutant. Note the different scales on the x-axis for A and B. The dashed line in B shows the predicted number of spontaneous mutations per individual. Boxplots show minimum and maximum values within 1.5 × interquartile range of the box (whiskers), median (dividing line), mean (circle), 95% confidence interval of mean (diamond), and outliers (squares). Using unpaired unequal distribution Student’s t-test: NS, not significantly different, *P < 0.05 and ***P < 0.001.
4-NQO−induced mutations are not influenced by nucleotide flanking sequence
For some mutagens, such as UV light and methyl-nitroso urea, the sequence context can influence the outcome of mutagenesis (Kurowska et al. 2012; Setlow et al. 1963). We analyzed the adjacent sequence for each of the six mutation types using the 10 upstream and 10 downstream nucleotides of all 3993 SNPs (Figure S1). For all six substitutions, there was no consensus outside of the affected residue, suggesting that only the adenine or guanine is required for efficient adduct formation. Therefore, 4-NQO can potentially target any nucleotide pair within the A. nidulans genome.
Phenotype-associated 4-NQO mutation spectrum frequencies differ from nonbiased whole genome data
Although mutant strains arising from the screens in this work were selected for specific restoration of proline utilization or defective organelle transport phenotypes, we expect only one or a few of the mutations identified by whole genome sequencing of each mutant strain to contribute to the selected phenotype as causative mutations (Nowrousian et al. 2012; Tan et al. 2014). Although mutations at some loci will be constrained by function due to their requirement for growth or viability under the selection conditions, normal morphology, or ability to cross for genetic analysis, for example, the majority of mutations are expected to be unrelated to the selection. 4-NQO has been used in many mutagenic screens since being reported as a good mutagen for producing both loss-of-function and altered function mutants in A. nidulans (Bal et al. 1977). We collated data from the literature and from this study for genetic screens in which mutants were selected for a diverse range of phenotypes and where sequence data were reported or the exact mutation associated with the selected phenotype could be inferred (Table 1). To compare our whole genome mutation frequencies with phenotype-selected mutation frequencies, we used a one-way frequency table with χ2 analysis. The distribution of mutation types for the two data sets was significantly different (χ2 = 22.50, d.f. = 5, P = 0.0004). Interestingly, G:C to C:G and A:T to T:A transversions were significantly more common, whereas G:C to A:T and A:T to G:C transitions were less common in the phenotype-selected data compared with the whole genome data set (Figure 4). These differences may be accounted for by the functional constraints of the selection of these mutations. For 24 amino acid codons (those encoding Phe, Leu Tyr, His, Gln, Asn, Lys, Asp, Glu, Cys, Ser, Arg) a transition in the third base position results in a synonymous change unlikely to alter the phenotype, whereas a transversion causes a nonsynonymous change. To test this hypothesis, we performed one-way frequency analysis on the number of transitions and transversions in the two data sets (χ2 = 3.60, d.f. = 1, P = 0.057). Although not significantly different by the conventional 95% confidence level, this test raises the possibility that functional constraints in the selection of mutants could be an important parameter. Therefore, the rates and types of mutations identified by whole genome sequencing of mutants likely approximate the true mutagenic spectrum for survivors of 4-NQO mutagenesis in A. nidulans, whereas the historical data are impacted by the constraints of phenotypic selection at the specific loci studied.
Figure 4.
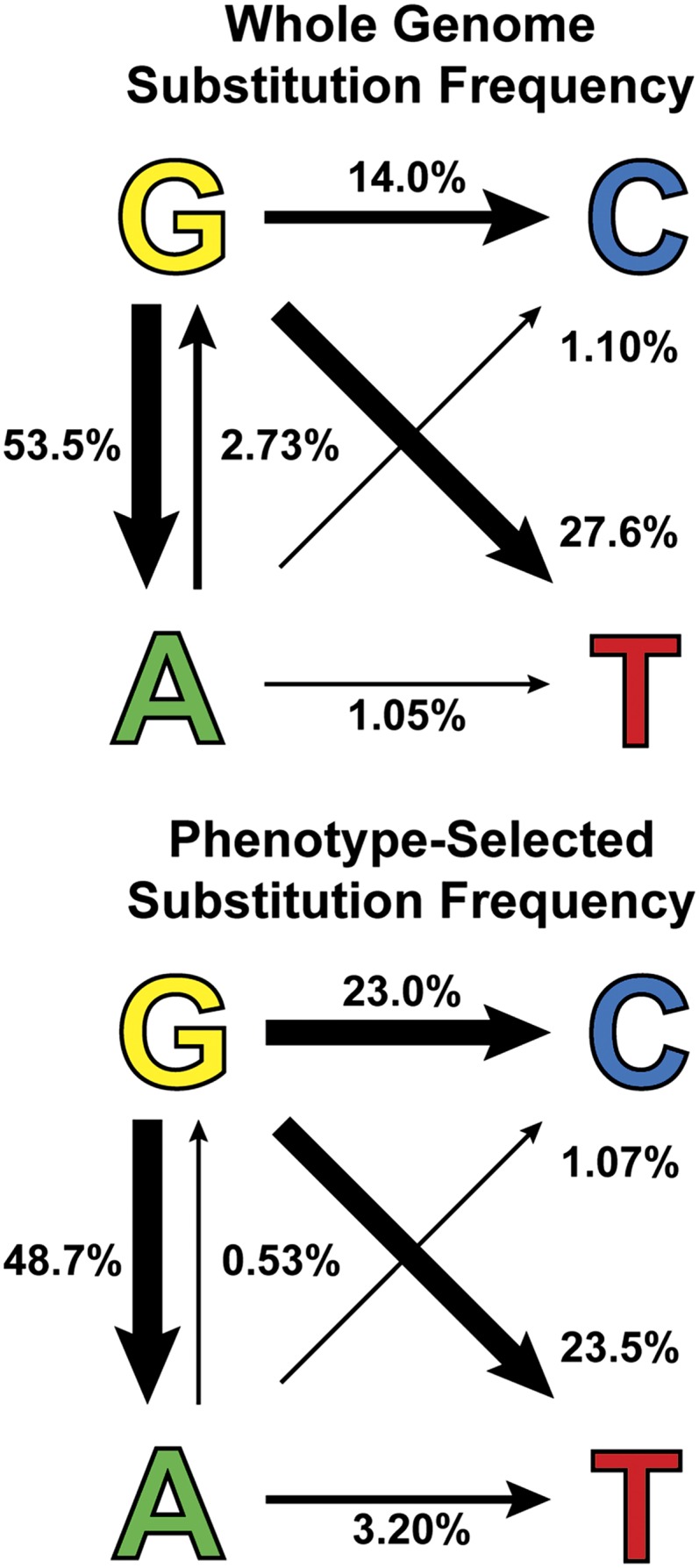
4-NQO affects primarily guanine nucleotides. Relative frequency (percent) of nucleotide substitutions identified by whole genome sequencing of random mutations and in phenotype-selected changes from published screens and this study (Table 1). Weighted arrows indicate change from wild type to mutant nucleotide.
Prediction of 4-NQO screen saturation
The purpose of a genetic screen is to identify genes contributing to a particular phenotype. Generally, a screen that has identified every gene associated with a pathway or phenotype is considered a saturation screen, as was most elegantly demonstrated in the seminal Drosophila melanogaster developmental screen carried out by Nüsslein-Volhard and Wieschaus (1980). Even though estimating the number of possible genes involved in the pathway or phenotype is difficult, several methods, which use gamma or Poisson distributions, have been used to predict gene saturation (Pollock and Larkin 2004). Our whole genome characterization of 4-NQO mutagenesis identified both the mean number and relative frequencies of nucleotide substitutions and therefore allows prediction of the probability of saturation by using a random sampling with replacement equation (see the section Materials and Methods). Our approach calculates the number of spores required to mutate every nucleotide (nucleotide saturation), which is an overestimate of the number of spores required to reach gene saturation. Using our equation, we calculate 2 × 107 or 1 × 108 spores with a kill of 50% and 90%, respectively, are sufficient to isolate a mutation in every A:T and G:C pair and in effect reach nucleotide saturation (Figure 5A).
Figure 5.

Number of spores required for screen saturation. (A) Probability of mutating every G:C (solid), A:T (dashed), and every nucleotide (dotted) in the A. nidulans genome at least once (nucleotide saturation) with 4-NQO doses causing 50% (red), and 90% kill (blue) were calculated using a random sampling with replacement equation. Note, PS(G→H and A→B) = PS(A→B) as for this number of treated spores PS(G→H) = 1. (B) The same equation was used to calculate the number of spores required to generate every possible substitution at every nucleotide (substitution saturation) under the same conditions. Note, PS(G→A,C,T and A→C,G,T) = PS(A→C,G,T) as for this number of treated spores PS(G→A,C,T) = 1.
How many spores would need to be used to isolate every possible mutation at every possible site? Using the same equation, we determined the number of spores required to generate every possible substitution at every nucleotide (substitution saturation). Interestingly, only 4 × 107 spores are required with a 50% kill to reach substitution saturation for guanine, and only 15 times as many spores (6 × 108) are required to reach substitution saturation of both guanine and adenine (Figure 5B). Using a 90% kill, substitution saturation of guanine can be achieved with 2 × 108 spores; however, 4 × 109 spores are required to saturate adenines. Current 4-NQO mutagenesis protocols in A. nidulans use between 107 and 108 spores, and therefore easily reach nucleotide saturation or even substitution saturation. Many laboratories use alternative physical or chemical mutagenesis methods for A. nidulans, including UV light and MNNG. It will be interesting to use the approach we used here to do a comparative study of the outcomes and efficacy of these mutagens.
Mutant screens in A. nidulans to characterize diverse cellular processes, including metabolism, mitosis, and organelle transport have used the highly carcinogenic chemical mutagen 4-NQO to induce sequence changes. Using a whole genome approach, we have characterized the mutagenic spectrum of 4-NQO and determined that its effects are distributed across the genome in a manner unbiased by sequence other than a preference for guanine over adenine at a ratio of 19:1. Interestingly, 4-NQO dose did not impact the number of mutations caused within a single surviving strain for 50% and 90% kill percentages. Therefore, future screens and kill percentages can be designed to suit whether selection or manual screening is required to identify a trait of interest. The number of mutations ranged between 23 and 240 per mutant. Importantly for A. nidulans mutant screens, this is a manageable number of candidate mutations to test for causation of the selected phenotype when combined with the power of haploidization and/or meiotic mapping, or with bulk segregant analysis. Additionally, we have shown that all six possible sequence transitions and transversions are induced by 4-NQO adduct repair, making it possible to conduct saturation screens with this chemical. We conclude that current practices using 4-NQO mutagenesis are sufficient to reach gene saturation in genetic screens. Therefore, our findings provide genome-wide evidence for the assertion of Bal et al., (Bal et al. 1977) that “4-NQO is a good mutagen for A. nidulans.”
Supplementary Material
Acknowledgments
We greatly appreciate the expert assistance of Andrew Princep, Bethany Grabow, and Tim Todd with statistical and probability analyses; technical assistance of Ian Hollyer; and technical advice of Koon Ho Wong. This work was supported by a Kansas NSF EPSCoR First Award EPS-0903806 (R.B.T.), a K-INBRE Scholarship funded by National Institute for General Medical Sciences award P20GM103418 (B.T.P.), a Kansas State University (KSU) Integrated Genomics Facility Competitive Seed Grant (R.B.T.), the KSU Plant Biotechnology Center (R.B.T.), the KSU Johnson Center for Basic Cancer Research (R.B.T., B.T.P.), and a National Institutes of Health New Innovator award OD004268 (S.L.R.-P.). Publication of this article was funded in part by the KSU Open Access Publishing Fund. This is contribution number 15-090-J from the Kansas Agricultural Experiment Station.
Footnotes
Supporting information is available online at http://www.g3journal.org/lookup/suppl/doi:10.1534/g3.114.014712/-/DC1
Sequence data from this article have been deposited with the NCBI Sequence Read Archive under the accession number SRP047490.
Communicating editor: A. Rokas
Literature Cited
- Al Taho N. M., Sealy-Lewis H. M., Scazzocchio C., 1984. Suppressible alleles in a wide domain regulatory gene in Aspergillus nidulans. Curr. Genet. 8: 245–251. [DOI] [PubMed] [Google Scholar]
- Alderson T., Hartley M. J., 1969. Specificity for spontaneous and induced forward mutation at several gene loci in Aspergillus nidulans. Mutat. Res. 8: 255–264. [DOI] [PubMed] [Google Scholar]
- Apirion D., 1965. The two-way selection of mutants and revertants in respect of acetate utilization and resistance to fluoro-acetate in Aspergillus nidulans. Genet. Res. 6: 317–329. [DOI] [PubMed] [Google Scholar]
- Arst H. N., Jr, Cove D. J., 1973. Nitrogen metabolite repression in Aspergillus nidulans. Mol. Gen. Genet. 126: 111–141. [DOI] [PubMed] [Google Scholar]
- Axelrod D. E., Gealt M., Pastushok M., 1973. Gene control of developmental competence in Aspergillus nidulans. Dev. Biol. 34: 9–15. [DOI] [PubMed] [Google Scholar]
- Babudri N., Morpurgo G., 1990. Frequency of spontaneous and induced recessive mutations in a diploid strain of Aspergillus nidulans. Mutat. Res. 230: 187–195. [DOI] [PubMed] [Google Scholar]
- Bailleul B., Galiegue S., Loucheux-Lefebvre M. H., 1981. Adducts from the reaction of O,O’-diacetyl or O-acetyl derivatives of the carcinogen 4-hydroxyaminoquinoline 1-oxide with purine nucleosides. Cancer Res. 41: 4559–4565. [PubMed] [Google Scholar]
- Bailleul B., Daubersies P., Galiègue-Zouitina S., Loucheux-Lefebvre M. H., 1989. Molecular basis of 4-nitroquinoline 1-oxide carcinogenesis. Jpn. J. Cancer Res. 80: 691–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bainbridge B. W., 1976. Estimation of the generation time and peripheral growth zone of Aspergillus nidulans and Alternaria solani hyphae from radial growth rates and ranges in apical cell length. J. Gen. Microbiol. 97: 125–127. [DOI] [PubMed] [Google Scholar]
- Bal J., Kajtaniak E. M., Pieniążek N. J., 1977. 4-Nitroquinoline-1-oxide: a good mutagen for Aspergillus nidulans. Mutat. Res. 56: 153–156. [Google Scholar]
- Baracho M. S., Baracho I. R., 2003. An analysis of the spontaneous mutation rate measurement in filamentous fungi. Genet. Mol. Biol. 26: 83–87. [Google Scholar]
- Bielska E., Schuster M., Roger Y., Berepiki A., Soanes D. M., et al. , 2014. Hook is an adapter that coordinates kinesin-3 and dynein cargo attachment on early endosomes. J. Cell Biol. 204: 989–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blankenberg D., Gordon A., Von Kuster G., Coraor N., Taylor J., et al. , 2010a Manipulation of FASTQ data with Galaxy. Bioinformatics 26: 1783–1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blankenberg, D., G. Von Kuster, N. Coraor, G. Ananda, R. Lazarus et al., 2010b Galaxy: a web-based genome analysis tool for experimentalists. Curr. Protoc. Mol. Biol. Chapter 19: Unit 19.10.1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brody H., Griffith J., Cuticchia A. J., Arnold J., Timberlake W. E., 1991. Chromosome-specific recombinant DNA libraries from the fungus Aspergillus nidulans. Nucleic Acids Res. 19: 3105–3109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruno K. S., Morrell J. L., Hamer J. E., Staiger C. J., 2001. SepH, a Cdc7p orthologue from Aspergillus nidulans, functions upstream of actin ring formation during cytokinesis. Mol. Microbiol. 42: 3–12. [DOI] [PubMed] [Google Scholar]
- Cecchetto G., Richero M., Oestreicher N., Muro-Pastor M. I., Pantano S., et al. , 2012. Mutations in the basic loop of the Zn binuclear cluster of the UaY transcriptional activator suppress mutations in the dimerisation domain. Fungal Genet. Biol. 49: 731–743. [DOI] [PubMed] [Google Scholar]
- Cerqueira G. C., Arnaud M. B., Inglis D. O., Skrzypek M. S., Binkley G., et al. , 2014. The Aspergillus Genome Database: multispecies curation and incorporation of RNA-Seq data to improve structural gene annotations. Nucleic Acids Res. 42(Database issue): D705–D710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherry J. M., Hong E. L., Amundsen C., Balakrishnan R., Binkley G., et al. , 2012. Saccharomyces Genome Database: the genomics resource of budding yeast. Nucleic Acids Res. 40(Database issue): D700–D705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clutterbuck A. J., 1969. A mutational analysis of conidial development in Aspergillus nidulans. Genetics 63(2): 317–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clutterbuck A. J., Farman M. L., 2008. Aspergillus nidulans: linkage map and genome sequence: closing gaps and adding telomeres, pp. 57–73 in The Aspergilli: Genomics, Medical Aspects, Biotechnology, and Research Methods, edited by Goldman G. H., Osmani S. A. CRC Press, Boca Raton, FL. [Google Scholar]
- Conlon H., Zadra I., Haas H., H. N. Arst, Jr, M. G. Jones et al., 2001. The Aspergillus nidulans GATA transcription factor gene areB encodes at least three proteins and features three classes of mutation. Mol. Microbiol. 40: 361–375. [DOI] [PubMed] [Google Scholar]
- Cove D. J., 1966. The induction and repression of nitrate reductase in the fungus Aspergillus nidulans. Biochim. Biophys. Acta 113: 51–56. [DOI] [PubMed] [Google Scholar]
- Crooks G. E., Hon G., Chandonia J. M., Brenner S. E., 2004. WebLogo: a sequence logo generator. Genome Res. 14: 1188–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePristo M. A., Banks E., Poplin R., Garimella K. V., Maguire J. R., et al. , 2011. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 43: 491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake J. W., Charlesworth B., Charlesworth D., Crow J. F., 1998. Rates of spontaneous mutation. Genetics 148: 1667–1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrell A., Coleman B. I., Benenati B., Brown K. M., Blader I. J., et al. , 2014. Whole genome profiling of spontaneous and chemically induced mutations in Toxoplasma gondii. BMC Genomics 15: 354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flibotte S., Edgley M. L., Chaudhry I., Taylor J., Neil S. E., et al. , 2010. Whole-genome profiling of mutagenesis in Caenorhabditis elegans. Genetics 185: 431–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galagan J. E., Calvo S. E., Cuomo C., Ma L. J., Wortman J. R., et al. , 2005. Sequencing of Aspergillus nidulans and comparative analysis with A. fumigatus and A. oryzae. Nature 438: 1105–1115. [DOI] [PubMed] [Google Scholar]
- Galiègue-Zouitina S., Bailleul B., Loucheux-Lefebvre M. H., 1984. Guanyl-C8-arylamination of DNA by the ultimate carcinogen of 4-nitroquinoline-1-oxide: a spectrophotometric titration. Anal. Biochem. 138: 454–457. [DOI] [PubMed] [Google Scholar]
- Galiègue-Zouitina S., Bailleul B., Loucheux-Lefebvre M. H., 1985. Adducts from in vivo action of the carcinogen 4-hydroxyaminoquinoline 1-oxide in rats and from in vitro reaction of 4-acetoxyaminoquinoline 1-oxide with DNA and polynucleotides. Cancer Res. 45: 520–525. [PubMed] [Google Scholar]
- Galiègue-Zouitina S., Bailleul B., Ginot Y. M., Perly B., Vigny P., et al. , 1986. N2-guanyl and N6-adenyl arylation of chicken erythrocyte DNA by the ultimate carcinogen of 4-nitroquinoline 1-oxide. Cancer Res. 46: 1858–1863. [PubMed] [Google Scholar]
- Garrison, E., and G. Marth, 2012 Haplotype-based variant detection from short-read sequencing. Available at: http://arxiv.org/abs/1207.3907. Accessed November 4, 2014.
- Goldman G. H., Kafer E., 2004. Aspergillus nidulans as a model system to characterize the DNA damage response in eukaryotes. Fungal Genet. Biol. 41: 428–442. [DOI] [PubMed] [Google Scholar]
- Harris S. D., Morrell J. L., Hamer J. E., 1994. Identification and characterization of Aspergillus nidulans mutants defective in cytokinesis. Genetics 136: 517–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heck I. S., Schrag J. D., Sloan J., Millar L. J., Kanan G., et al. , 2002. Mutational analysis of the gephyrin-related molybdenum cofactor biosynthetic gene cnxE from the lower eukaryote Aspergillus nidulans. Genetics 161: 623–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt C. L., May G. S., 1996. An extragenic suppressor of the mitosis-defective bimD6 mutation of Aspergillus nidulans codes for a chromosome scaffold protein. Genetics 142: 777–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hynes M. J., Pateman J. A., 1970a The genetic analysis of regulation of amidase synthesis in Aspergillus nidulans. I. Mutants able to utilize acrylamide. Mol. Gen. Genet. 108: 97–106. [DOI] [PubMed] [Google Scholar]
- Hynes M. J., Pateman J. A., 1970b The genetic analysis of regulation of amidase synthesis in Aspergillus nidulans. II. Mutants resistant to fluoroacetamide. Mol. Gen. Genet. 108: 107–116. [DOI] [PubMed] [Google Scholar]
- Ikenaga M., Ichikawa-Ryo H., Kondo S., 1975a The major cause of inactivation and mutation by 4-nitroquinoline 1-oixde in Escherichia coli: excisable 4NQO-purine adducts. J. Mol. Biol. 92: 341–356. [DOI] [PubMed] [Google Scholar]
- Ikenaga M., Ishii Y., Tada M., Kakunaga T., Takebe H., 1975b Excision-repair of 4-nitroquinolin-1-oxide damage responsible for killing, mutation, and cancer. Basic Life Sci. 5B: 763–771. [DOI] [PubMed] [Google Scholar]
- Ikenaga M., Kakunaga T., 1977. Excision of 4-nitroquinoline 1-oxide damage and transformed in mouse cells. Cancer Res. 37: 3672–3678. [PubMed] [Google Scholar]
- Ikenaga M., Takebe H., Ishii Y., 1977. Excision repair of DNA base damage in human cells treated with the chemical carcinogen 4-nitroquinoline 1-oxide. Mutat. Res. 43: 415–427. [DOI] [PubMed] [Google Scholar]
- Janner F., Flury F., Leupold U., 1979. Reversion of nonsense mutants induced by 4-nitroquinoline-1-oxide in Schizosaccharomyces pombe. Mutat. Res. 63: 11–19. [DOI] [PubMed] [Google Scholar]
- Kaminskyj S. G., Hamer J. E., 1998. hyp loci control cell pattern formation in the vegetative mycelium of Aspergillus nidulans. Genetics 148: 669–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinghorn J. R., Sloan J., Kana’n G. J., Dasilva E. R., Rouch D. A., et al. , 2005. Missense mutations that inactivate the Aspergillus nidulans nrtA gene encoding a high-affinity nitrate transporter. Genetics 169: 1369–1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohda K., Tada M., Kasai H., Nishimura S., Kawazoe Y., 1986. Formation of 8-hydroxyguanine residues in cellular DNA exposed to the carcinogen 4-nitroquinoline 1-oxide. Biochem. Biophys. Res. Commun. 139: 626–632. [DOI] [PubMed] [Google Scholar]
- Kudla B., Caddick M. X., Langdon T., Martinez-Rossi N. M., Bennett C. F., et al. , 1990. The regulatory gene areA mediating nitrogen metabolite repression in Aspergillus nidulans. Mutations affecting specificity of gene activation alter a loop residue of a putative zinc finger. EMBO J. 9: 1355–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurowska M., Labocha-Pawlowska A., Gnizda D., Maluszynski M., Szarejko I., 2012. Molecular analysis of point mutations in a barley genome exposed to MNU and gamma rays. Mutat. Res. 738–739: 52–70. [DOI] [PubMed] [Google Scholar]
- Larson J. R., Facemyer E. M., Shen K. F., Ukil L., Osmani S. A., 2014. Insights into dynamic mitotic chromatin organization through the NimA kinase suppressor SonC, a chromatin-associated protein involved in the DNA damage response. Genetics 196: 177–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S. B., Taylor J. W., 1990. Isolation of DNA from fungal mycelia and single spores, pp. 282–287 in PCR Protocols: A guide to Methods and Applications, edited by Innis M. A., Gelfand D. H., Sninsky J. S., White T. J. Academic Press, San Diego, CA. [Google Scholar]
- Li H., Durbin R., 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25: 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lilly J. L., 1965. An investigation of the suitability of the suppressors of meth1 in Aspergillus nidulans for the study of induced and spontaneous mutation. Mutat. Res. 2: 192–195. [DOI] [PubMed] [Google Scholar]
- Lin X., Momany M., 2003. The Aspergillus nidulans swoC1 mutant shows defects in growth and development. Genetics 165: 543–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin X., Momany C., Momany M., 2003. SwoHp, a nucleoside diphosphate kinase, is essential in Aspergillus nidulans. Eukaryot. Cell 2: 1169–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M., Sung W., Morris K., Coffey N., Landry C. R., et al. , 2008. A genome-wide view of the spectrum of spontaneous mutations in yeast. Proc. Natl Acad. Sci. USA 105: 9272–9277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCluskey K., Wiest A. E., Grigoriev I. V., Lipzen A., Martin J., et al. , 2011. Rediscovery by whole genome sequencing: classical mutations and genome polymorphisms in Neurospora crassa. G3 (Bethesda) 1: 303–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., et al. , 2010. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20: 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menichini P., Fronza G., Tornaletti S., Galiègue-Zouitina S., Bailleul B., et al. , 1989. In vitro DNA modification by the ultimate carcinogen of 4-nitroquinoline-1-oxide: influence of superhelicity. Carcinogenesis 10: 1589–1593. [DOI] [PubMed] [Google Scholar]
- Miller J. A., 1970. Carcinogenesis by chemicals: an overview—G. H. A. Clowes memorial lecture. Cancer Res. 30: 559–576. [PubMed] [Google Scholar]
- Momany M., Westfall P. J., Abramowsky G., 1999. Aspergillus nidulans swo mutants show defects in polarity establishment, polarity maintenance and hyphal morphogenesis. Genetics 151: 557–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monahan B. J., Unkles S. E., Tsing I. T., Kinghorn J. R., Hynes M. J., et al. , 2002. Mutation and functional analysis of the Aspergillus nidulans ammonium permease MeaA and evidence for interaction with itself and MepA. Fungal Genet. Biol. 36: 35–46. [DOI] [PubMed] [Google Scholar]
- Morris N. R., 1975. Mitotic mutants of Aspergillus nidulans. Genet. Res. 26: 237–254. [DOI] [PubMed] [Google Scholar]
- Nayak T., Szewczyk E., Oakley C. E., Osmani A., Ukil L., et al. , 2006. A versatile and efficient gene-targeting system for Aspergillus nidulans. Genetics 172: 1557–1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowrousian M., Teichert I., Masloff S., Kuck U., 2012. Whole-genome sequencing of Sordaria macrospora mutants identifies developmental genes. G3 (Bethesda) 2: 261–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nüsslein-Volhard C., Wieschaus E., 1980. Mutations affecting segment number and polarity in Drosophila. Nature 287: 795–801. [DOI] [PubMed] [Google Scholar]
- Oakley C. E., Oakley B. R., 1989. Identification of gamma-tubulin, a new member of the tubulin superfamily encoded by mipA gene of Aspergillus nidulans. Nature 338: 662–664. [DOI] [PubMed] [Google Scholar]
- Oestreicher N., Scazzocchio C., 2009. Phenotypes of mutations in the 5′-UTR of a limiting transcription factor in Aspergillus nidulans can be accounted for by translational inhibition and leaky scanning. Genetics 181: 1261–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osman F., Tomsett B., Strike P., 1993. The isolation of mutagen-sensitive nuv mutants of Aspergillus nidulans and their effects on mitotic recombination. Genetics 134: 445–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penalva M. A., Tilburn J., Bignell E., and H. N. Arst, Jr., 2008. Ambient pH gene regulation in fungi: making connections. Trends Microbiol. 16: 291–300. [DOI] [PubMed] [Google Scholar]
- Pokorska A., Drevet C., Scazzocchio C., 2000. The analysis of the transcriptional activator PrnA reveals a tripartite nuclear localisation sequence. J. Mol. Biol. 298: 585–596. [DOI] [PubMed] [Google Scholar]
- Pollock D. D., Larkin J. C., 2004. Estimating the degree of saturation in mutant screens. Genetics 168: 489–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomraning K. R., Smith K. M., Freitag M., 2011. Bulk segregant analysis followed by high-throughput sequencing reveals the Neurospora cell cycle gene, ndc-1, to be allelic with the gene for ornithine decarboxylase, spe-1. Eukaryot. Cell 10: 724–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pontecorvo G., Roper J. A., Hemmons L. M., Macdonald K. D., Bufton A. W., 1953. The genetics of Aspergillus nidulans. Adv. Genet. 5: 141–238. [DOI] [PubMed] [Google Scholar]
- Prakash L., Sherman F., 1973. Mutagenic specificity: reversion of iso-1-cytochrome c mutants of yeast. J. Mol. Biol. 79: 65–82. [DOI] [PubMed] [Google Scholar]
- Prakash L., Stewart J. W., Sherman F., 1974. Specific induction of transitions and transversions of GC base pairs by 4-nitroquinoline-1-oxide in iso-1-cytochrome c mutants of yeast. J. Mol. Biol. 85: 51–56. [DOI] [PubMed] [Google Scholar]
- Quinlan A. R., Hall I. M., 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenkranz H. S., Poirier L. A., 1979. Evaluation of the mutagenicity and DNA-modifying activity of carcinogens and noncarcinogens in microbial systems. J. Natl Cancer Inst. 62: 873–891. [PubMed] [Google Scholar]
- Setlow R. B., Swenson P. A., Carrier W. L., 1963. Thymine dimers and inhibition of DNA synthesis by ultraviolet irradiation of cells. Science 142: 1464–1466. [DOI] [PubMed] [Google Scholar]
- Shaw B. D., Momany M., 2002. Aspergillus nidulans polarity mutant swoA is complemented by protein O-mannosyltransferase pmtA. Fungal Genet. Biol. 37: 263–270. [DOI] [PubMed] [Google Scholar]
- Shaw B. D., Momany C., Momany M., 2002. Aspergillus nidulans swoF encodes an N-myristoyl transferase. Eukaryot. Cell 1: 241–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi X., Sha Y., Kaminskyj S., 2004. Aspergillus nidulans hypA regulates morphogenesis through the secretion pathway. Fungal Genet. Biol. 41: 75–88. [DOI] [PubMed] [Google Scholar]
- Sun Y. V., Levin A. M., Boerwinkle E., Robertson H., Kardia S. L., 2006. A scan statistic for identifying chromosomal patterns of SNP association. Genet. Epidemiol. 30: 627–635. [DOI] [PubMed] [Google Scholar]
- Szewczyk E., Nayak T., Oakley C. E., Edgerton H., Xiong Y., et al. , 2006. Fusion PCR and gene targeting in Aspergillus nidulans. Nat. Protoc. 1: 3111–3120. [DOI] [PubMed] [Google Scholar]
- Tada M., Tada M., 1971. Interaction of a carcinogen, 4-nitroquinoline-1-oxide, with nucleic acids: chemical degradation of the adducts. Chem. Biol. Interact. 3: 225–229. [DOI] [PubMed] [Google Scholar]
- Tada M., Tada M., 1976. Main binding sites of the carcinogen, 4-nitroquinoline 1-oxide in nucleic acids. Biochim. Biophys. Acta 454: 558–566. [DOI] [PubMed] [Google Scholar]
- Tada M., Kohda K. H., Kawazoe Y., 1984. Biomimetic preparation and structure determination of QGI, one of the quinoline-DNA base adducts formed in cells treated with 4-nitroquinoline 1-oxide. Gann 75: 976–985. [PubMed] [Google Scholar]
- Tan K., Roberts A. J., Chonofsky M., Egan M. J., Reck-Peterson S. L., 2014. A microscopy-based screen employing multiplex genome sequencing identifies cargo-specific requirements for dynein velocity. Mol. Biol. Cell 25: 669–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd R. B., Davis M. A., Hynes M. J., 2007a Genetic manipulation of Aspergillus nidulans: heterokaryons and diploids for dominance, complementation and haploidization analyses. Nat. Protoc. 2: 822–830. [DOI] [PubMed] [Google Scholar]
- Todd R. B., Davis M. A., Hynes M. J., 2007b Genetic manipulation of Aspergillus nidulans: meiotic progeny for genetic analysis and strain construction. Nat. Protoc. 2: 811–821. [DOI] [PubMed] [Google Scholar]
- Uchida N., Sakamoto T., Kurata T., Tasaka M., 2011. Identification of EMS-induced causal mutations in a non-reference Arabidopsis thaliana accession by whole genome sequencing. Plant Cell Physiol. 52: 716–722. [DOI] [PubMed] [Google Scholar]
- Voz M. L., Coppieters W., Manfroid I., Baudhuin A., Von Berg V., et al. , 2012. Fast homozygosity mapping and identification of a zebrafish ENU-induced mutation by whole-genome sequencing. PLoS ONE 7: e34671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams A. B., Hetrick K. M., Foster P. L., 2010. Interplay of DNA repair, homologous recombination, and DNA polymerases in resistance to the DNA damaging agent 4-nitroquinoline-1-oxide in Escherichia coli. DNA Repair (Amst.) 9: 1090–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong K. H., Hynes M. J., Davis M. A., 2008. Recent advances in nitrogen regulation: a comparison between Saccharomyces cerevisiae and filamentous fungi. Eukaryot. Cell 7: 917–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L., Osmani S. A., Mirabito P. M., 1998. A role for NIMA in the nuclear localization of cyclin B in Aspergillus nidulans. J. Cell Biol. 141: 1575–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y., El-Ganiny A. M., Bray G. E., Sanders D. A., Kaminskyj S. G., 2008. Aspergillus nidulans hypB encodes a Sec7-domain protein important for hyphal morphogenesis. Fungal Genet. Biol. 45: 749–759. [DOI] [PubMed] [Google Scholar]
- Yao X., Wang X., Xiang X., 2014. FHIP and FTS proteins are critical for dynein-mediated transport of early endosomes in Aspergillus. Mol. Biol. Cell 25: 2181–2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J., Qiu R., Arst H. N., Jr, Penalva M. A., Xiang X., 2014. HookA is a novel dynein-early endosome linker critical for cargo movement in vivo. J. Cell Biol. 204: 1009–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y. O., Siegal M. L., Hall D. W., Petrov D. A., 2014. Precise estimates of mutation rate and spectrum in yeast. Proc. Natl Acad. Sci. USA 111: E2310–E2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.