

Abstract
The membrane landscape of a cell often changes drastically upon infection by a virus. In the case of the well-studied positive strand RNA virus poliovirus, the short infection cycle induces vesicles and tubular structures early in infection, and double-membraned vesicles late in infection. In this review, the current understanding of membrane changes in a PV-infected cell, the host and viral factors that facilitate these changes, and how these changes may promote virus replication will be discussed. Host factors involved in membrane rearrangement during infection include components of the COPI and COPII secretory pathways, lipid kinases, and the autophagy pathway. The roles of cellular membranes include acting as a scaffold for the RNA replication complex and roles in exit of mature virus. Finally, recent studies suggesting that not all picornaviruses are truly “non-enveloped” are discussed in the context of the field, raising the possibility that cell-derived membranes play a role in delivering poliovirus particles to the extracellular space.
Keywords: Poliovirus, membranes, secretory pathway, COPI, COPII, Arf1, GBF1, Autophagy
Introduction
Poliovirus (PV), the causative agent of poliomyelitis, is a picornavirus, traditionally described as a non-enveloped particle containing a short positive-sense RNA genome. Like all positive-strand RNA viruses, PV physically alters the organelle composition of host cells to support its replication. Models of nucleic acid replication tend to describe virtually all steps taking place “in solution.” For the RNA-dependent RNA replication of viruses such as poliovirus, however, this is not the case. PV RNA replication complexes are membrane associated, although we do not understand the benefit this affords to the virus. [1,2] The membrane association is achieved through multiple interactions. The small poliovirus 3A protein has a transmembrane domain, and the vast majority of the protein is oriented toward the cytosol. [3] Many other poliovirus proteins, including 2BC and the 3D polymerase, are associated with membranes, presumably through interactions with transmembrane proteins or, perhaps, in the case of 3D, specific classes of lipid. [3–11]
The scope and focus of this review will be to describe what is known about how poliovirus effects changes in cellular membrane composition and organization. The limited space makes it difficult to incorporate all data from other picornaviruses; however, the review would not be complete without comparing and contrasting some of what has been learned about other picornaviruses, especially, in recent years, coxsackievirus B3 (CVB3). Several key questions have been raised by the literature, and answers are beginning to be found. First, how are the proliferating membranes in PV-infected cells generated? Are they primarily new synthesis, or are they derived from existing membranes? More importantly, why does the cell need to be so dramatically rearranged to support RNA virus replication?
Ultrastructural studies
Electron microscopy remains the most reliable way to study cellular ultrastructure. One early electron microscopy study of fragments from PV-infected cells was carried out in 1959. Although the study is largely focused on the virions themselves, bounding membranes around the virions are frequently observed and commented upon by the authors. [12] In 1965 Dales and Palade performed a seminal study focused on the effect of poliovirus infection on intact HeLa cells, revealing a dramatically altered cytoplasm. [13] At three hours post-infection, large numbers of polysomes are observed near the ER and nucleus, and some vesicles, with an interior density consistent with cytoplasm, are observed. This time point roughly correlates with the peak of viral RNA replication. [14] Viral particles, some of which are identified as empty capsids, are observed near and within these vesicles by five hours post-infection. Images taken at seven hours post-infection reveal vesicles delineated by two lipid bilayers, many of which have virus within the interior membrane. In or near these double-membraned vesicles are large groups of viral particles. The dramatic and synchronous rearrangements observed at distinct time points post-infection have been a consistent feature of PV membrane studies, and have sometimes led to confusion as different groups compare studies at different stages of the virus infection cycle.
Almost fifty years after the Dales and Palade paper, the Ehrenfeld group performed similar studies using modern EM tomography, examining cells at three, four, and seven hours post-infection. [15] The 3D-reconstructions reveal that at least some of the early “vesicles” observed in two dimensions are in fact convoluted tubular structures at both three and four hpi, with the structures appearing more invaginated and convoluted at 4hpi. At seven hpi, double-membraned structures, similar to those seen by Dales and Palade, are observed. Evidence of active RNA replication can be observed on both early single-membraned and late double-membraned structures. These data, and images of membrane loops that appear to be near-fusion (as illustrated in Fig. 1, “PAS”) lead the authors to suggest that the early structures may develop into the later multi-lamellar structures.
Figure 1. Model timeline of PV membrane-alteration events.
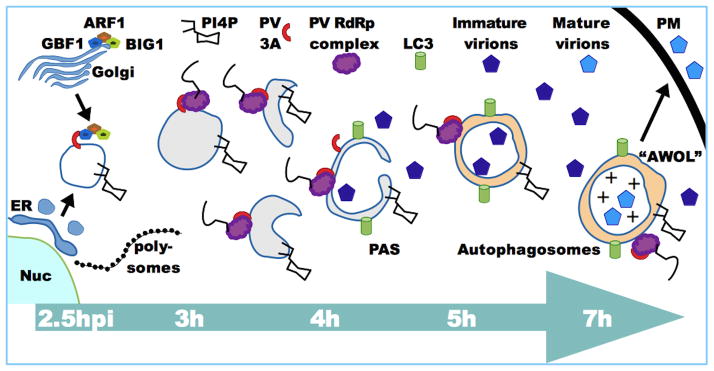
Infection proceeds from left to right. At 2.5hpi, novel organelles form with contributions from COPII vesicles leaving ER exit sites and the Golgi COPI complex Arf1/BIG1/GBF1, recruited by PV 3A and 3CD proteins. The presence of PI4KIIIβ at the newly forming membranes enriches them for PI4P. Large numbers of polysomes can be observed near these vesicles, as viral translation proceeds, using the vast majority of the available ribosomal machinery. By 3hpi Arf1/GBF1/BIG1 are no longer directly associated with the membranes, which are often tubular and convoluted. RNA replication is associated with these membranes at this time, which is the peak of RNA replication. By 4hpi more invaginations can be seen in these vesicles as they begin to resemble pre-autophagosomal structures (PAS) and attract the lipidated form of the autophagy marker LC3. By 5h double-membraned autophagosome-like vesicles predominate, often containing virions. These vesicles remain associated with active RNA replication. Maturation of autophagosomes through acquisition of endosomal VTPase causes vesicle acidification (+) and promotes maturation of encapsulated virions. The autophagy pathway promotes exit of these virions though autophagosome-mediated exit with out lysis (AWOL) at the plasma membrane (PM).
Individual expression of PV proteins has been investigated as a way to understand the roles of the proteins in cellular changes during infection. Expression of PV 2B, for example, leads to Golgi complex disassembly in Vero, NRK, and COS-7 cells, a phenomenon also observed during infection of a variety of cells. [7,16] Expression of 2C or its precursor, 2BC, induces a variety of dramatic changes in the cytosol of HeLa and COS-1 cells, including formation of smooth single-membraned vesicles with electron-light contents. [10,17] 2C expression also induces changes to the ER, resulting in densely packed, anastomotic regions. [6] Double-membraned vesicles resembling those formed during infection can be induced by co-expression of PV 3A and 2BC, indicating that these vesicles are unlikely to be a cellular response to infection but specifically induced by the presence of virus proteins. [17] The wide variety of observations, dependent upon the time point post-infection, technique used, and viral proteins being expressed, indicate that the host-virus interactions leading to membrane rearrangements are complex and dynamic.
PV and COPII-like membranes
Poliovirus efficiently inhibits ER-to-Golgi trafficking. This led to a hypothesis that PV might divert anterograde COPII-dependent trafficking vesicles for use as sites of RNA replication. A light microscopy approach was taken to observe vesicle formation at ER exit sites in poliovirus-infected cells. [18] This study generated maximal-intensity projections of images first captured by confocal microscopy, then subjected to deconvolution. The resulting images showed proteins of the COPII complex co-localizing with antibody against the poliovirus protein 2B on vesicles budding from the ER. Sec13 and Sec31, two outer coat component proteins involved in ER exit site budding, co-localized with 2B. 2B, although not a transmembrane protein, has been shown to be almost entirely membrane-associated. [10] [19] [1] [11] More recently it was shown that COPII budding increases during the early part of the poliovirus replication cycle. [20] Furthermore, expression of a dominant negative mutant form (T39N) of the Sar1 ER-resident GTPase confirmed that PV replicons require functional ER exit sites for normal levels of RNA production and expression. [21] These data, following up on the earlier studies, have been interpreted to indicate that PV replication vesicles are derived from a precursor pool including, among other things, COPII vesicles.
However, studies with any virus protein, be it 3A, 2B, or another, do not necessarily identify active RNA replication. Recently a pan-double stranded RNA antibody has been developed (English and Scientific Consulting). dsRNA is a necessary intermediate in virus RNA replication as positive strands are copied to negative strands and vice versa, but no significant dsRNA signal is present in uninfected cells. Neither Sec31 nor β-COP, another component of the COPII coat complex, co-localize with the dsRNA signal associated with active RNA-RNA replication during PV infection. [22] On a western blot, Sec31 shifts to a higher mobility form late in poliovirus infection, and this change can be inhibited with the proteasome inhibitor MG132. This suggests that a proteasome-dependent Sec31 cleavage event takes place. Although increased ER exit site activity may contribute to a membrane pool that generate RNA replication organelles, the recent evidence argues against the model that “standard” COPII vesicles are the sites of RNA replication. Several questions about COPII components and PV membranes remain. For one, why is 2B associated with COPII membranes as they bud? Does 2B association alter the fate of COPII vesicles, or otherwise redirect them in the cell? Is the Sec31 imaged in the Rust et. al paper full-length, or the faster-migrating form seen in Richards et. al? Do poliovirus RNA replication complexes partially form on precursor membranes before becoming active?
PV and COPI components
Even more extensive studies have gone into the Golgi resident proteins involved in initiation of retrograde traffic from the Golgi to the ER. One reason for this is that the lactone antibiotic Brefeldin A has been shown to be a potent inhibitor of poliovirus RNA replication. [23] Brefeldin A acts by restricting retrograde (COPI) protein transport, largely by inhibiting the activity of GBF1, a Guanine nucleotide exchange factor (GEF) for the Golgi-resident) small G protein Arf1 (ADP-ribosylation factor 1.) [24] Membrane association of Arf1 can be induced either by PV infection, or by individual expression of PV 3A or 3CD proteins. [25] This membrane association is thought to be mediated by recruitment of three different Arf1 GEFs: GBF1, BIG1, and BIG2. The PV 3A protein is responsible for membrane association of GBF1, and PV 3CD is responsible for BIG1 and BIG2 recruitment. [4,26–28] This led to a hypothesis that Arf1 and all three associated GEFs are critical for generating PV replication membranes, possibly through its role in generating COPI vesicles. GBF1 briefly and transiently co-localizes with dsRNA signal at 2.5hpi, while the signals are close but separate at later time points. [22]
However, PV does not appear to require the catalytic activity of GBF1 for replication. [29] In addition, BFA-resistant viruses, which do not require GBF1 for replication, are readily obtained using in vitro selection of poliovirus, indicating that the viral requirement for GBF1 is easily bypassed. [30] It is, of course, possible that in vitro-selected BFA-resistant viruses would be unsuccessful in vivo, and the requirement for GBF1 fills a role that has not yet been explored. One possibility is that 3A may interact with GBF1 as part of a virus strategy to inhibit cellular secretion. Although GBF1 is involved in retrograde trafficking, shutting down retrograde flow results in a block of anterograde flow as well due to inhibited recycling of trafficking components. [31] The 3A-GBF1 interaction would therefore blunt immune responses by reducing secretion of cytokines and presentation of viral antigens at the infected cell surface. [32,33] If this is significant in vivo, then the virus may have multiple reasons for interacting with and recruiting members of the Arf1 complex, and tissue culture studies are only revealing a small part of the story.
Lipids and PI4P
Until recently, studies of PV-induced membrane changes focused on the protein components of the membranes. However, wholesale changes in cellular lipid metabolism and content have long been known to occur during PV infection. [34–37] Recently, however, investigations have begun to identify specific lipid components of PV replication membranes. One major Arf1 effector, phosphoinositol-4-kinase IIIβ (PI4KIIIβ), is responsible for the production of PI4P lipids from PI. [38] Due to the relationship between the Arf1 complex and PV, this pathway has been extensively studied during virus infections. [39]
Inhibition of PI4KIIIβ by the small molecule inhibitor PIK90 blocks PV replicon production efficiently. [21] PI4P co-localizes with dsRNA signal throughout infection, and is the only major cellular marker studied so far to do so. [22] PI4P binds to the PV RNA-dependent RNA polymerase, 3D, in in vitro binding assays, providing a hypothesis for the function of PI4P in PV replication membranes: to induce and maintain association of polymerase with particular membranes. [21] More extensive PI4KIIIβ experiments have been carried out with coxsackievirus, and it remains unclear if some, most, or all of the findings will prove consistent between PV and CVB3. As with Brefeldin A, CVB3 mutant viruses resistant to PI4KIIIβ-inhibiting drugs can be obtained in vitro, calling into question the essential nature of PI4P in virus replication membranes. However, it is unclear if the resistant strains would be attenuated in vivo. {[40]
It was originally suggested that, at least for CVB3, the 3A-GBF1-Arf1 complex might be responsible for recruiting PI4KIIIβ to virus replication membranes. [21] Studies of another picornavirus, Aichi virus, suggest that Aichi proteins interact with the Golgi resident protein acyl-coenzyme A binding domain containing 3 (ACBD3), and that this interaction mediates PI4KIIIβ recruitment to membranes. [41,42] However, PI4KIIIβ is efficiently recruited to CVB3 replication organelles in the absence of GBF1, Arf1, or ACBD3. [43] It is unclear if these data will reflect PV or Aichi virus recruitment of PI4KIIIβ, but at a minimum, the mechanism and requirement for PI4P-enrichment at CVB3 membranes remains unclear.
It has recently been shown that the host endocytic pathway plays a role in altering the balance of cholesterol in the cell during infection with enteroviruses. [44] The PV 3AB protein co-localizes with cholesterol and PI4P during infection. Changes in the lipid and cholesterol landscape during positive strand RNA virus infection will be covered in more detail in other reviews in this issue; the reader is referred to those articles for more details on these events.
The Autophagy pathway and AWOL
Dales and Palade presciently suggested that the double-membraned vesicles they observed late in PV infection might be degradative compartments. [13] We now know that double-membraned compartments strongly resemble autophagosomes, the hallmark vesicles of the constitutive degradative process known as autophagy, or “self-eating”. [45] Autophagy is an essential and constitutive cellular process that regulates turnover of organelles, lipid, and proteins, and plays a role in cell stress responses, human disease, and innate immunity. [46] It has been shown that a subset of viruses and bacteria, including many enteroviruses, do not succumb to the innate immune response of autophagy but subvert the pathway to promote their own replication. [47,48] Observations of autophagosomes were not limited to PV; by 1972, in mice infected with CVB3, “autophagic vacuoles” are described in pancreatic acinar cells, and images of such vesicles appear throughout the literature. [49,50] Using high-pressure freezing techniques to maximize resolution, the Kirkegaard group was able to identify double-membraned vesicles as early as 4hpi in COS-1 cells. [17,51] These vesicles, which are decorated with the PV 2C and 3A proteins, have apparent cytosolic contents and are in the range of 500nM in diameter. In images taken for both PV and CVB3 by multiple groups, the virions appear to be within the double-membraned vesicles. The autophagic marker LC3 and PV capsid are both associated with vesicles that appear to be in the act of budding from the cell surface during infection. [52]
The presence of double-membraned vesicles in PV-infected cells led to two major questions. First, are these vesicles bona fide autophagosomes? The PV-induced double membraned vesicles, which begin to appear at the tail end of the peak of maximum viral RNA synthesis, contain protein markers, such as LC3, associated with autophagosomes. [52] RNA replication occurs on these double membraned vesicles. [15,22] In addition, active autophagic degradation occurs during PV infection, indicating that they are functional organelles. [53] It is interesting to note how different this is from coxsackievirus B3, which induces massive, immature autophagosomes but prevents them from maturing into functional degradative vesicles. [54,55] This difference highlights the danger in drawing conclusions from one picornavirus based on data from another.
The next question, then, is what role these vesicles play in the PV replication cycle. Increased autophagy promotes replication of PV, while autophagic inhibition reduces PV production. [52] Furthermore, exit of virus prior to cell lysis is dramatically enhanced when autophagy increases, a process we originally termed “AWOL” for autophagosome-mediated exit with out lysis. Autophagosomes are microtubule-associated and immobilized for much of infection, but if microtubules are disrupted, AWOL increases. [56] Vesicle acidification, a hallmark of autophagosome maturation, promotes the final cleavage maturation of PV. [53] This cleavage event, which is poorly understood, occurs on the interior of fully formed viral particles that contain an RNA genome. [57] In the past, models of PV release regularly described virus-containing vesicles as the agents of virus release prior to cell lysis, although this model has fallen out of favor, replaced by a model of virus release exclusively mediated by cell lysis. [58] Autophagy is known to feed into a pathway of non-canonical secretion, and AWOL may represent PV hitching a ride on this pathway. [59,60] The valosin-containing protein (VCP), a cellular ATPase with roles in autophagy and the endocytic pathway, is a crucial host factor for PV replication and may provide a link between autophagy, cellular exit, and viral replication. [61,62]
Conclusion - Enveloped Picornaviruses, the new paradigm?
Historically, studies of PV-induced membranes have focused on their role as RNA replication scaffolds. While this is clearly one important role of membranes, we do not yet understand why this is important. Possibly there are genetic and/or energetic benefits of keeping genomic replication complexes close to one another and on a physical surface. Alternatively, keeping RNA replication in close proximity to membranes, particularly later in infection, could facilitate assembly, maturation, and exit of virions through a vesicle-dependent pathway.
Enveloped viruses often bud from the plasma membrane or use the cell secretory pathway to facilitate viral exit and acquisition of membranes. In traditional models, non-enveloped viruses like PV do not bud or enter the secretory pathway because they do not acquire a membrane. Recently, however, two studies have challenged that notion. It was recently shown that a significant portion of the non-lytic picornavirus Hepatitis A Virus, long thought to be non-enveloped, is actually shed in exosome-like vesicles. [63] Formation of these vesicles is not dependent on the autophagy pathway, but it is dependent on proteins associated with endosomal-sorting complexes required for transport (ESCRT) complexes. Another study showed that CVB3 is also released in vesicles, and that these vesicles are decorated with the autophagy marker LC3, indicating that something like AWOL may be occurring in CVB3. [64] Clearly the idea that picornaviruses are “non-enveloped” is being challenged on multiple fronts. Although there are, as yet, no reports of enveloped PV particles, the role of cellular vesicles in producing and releasing extracellular virus particles is an exciting future direction for the field.
Highlights.
Ultrastructural studies observe distinct membrane rearrangements at early and late time points post-poliovirus infection.
The lipid kinase PI4KIIIβ and its product PI4P are consistent features of virus-induced membranes.
Components of the COPI and COPII secretory pathways generate early single-membraned RNA replication vesicles.
Cell-derived autophagic membranes play a role in delivering poliovirus particles to the extracellular space.
Acknowledgments
The author is supported by NIAID grant AI104928. Thanks to past and present members of the Jackson Lab, especially Alexsia Richards, Claire Quiner, Jamária A P Soares-Martins, and Geoffery Riddell, for many spirited discussions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bienz K, Egger D, Pfister T. Characteristics of the poliovirus replication complex. Arch Virol Suppl. 1994;9:147–157. doi: 10.1007/978-3-7091-9326-6_15. [DOI] [PubMed] [Google Scholar]
- 2.Caliguiri LA. Analysis of RNA associated with the poliovirus RNA replication complexes. Virology. 1974;58:526–535. doi: 10.1016/0042-6822(74)90086-5. [DOI] [PubMed] [Google Scholar]
- 3.Choe SS, Kirkegaard K. Intracellular topology and epitope shielding of poliovirus 3A protein. J Virol. 2004;78:5973–5982. doi: 10.1128/JVI.78.11.5973-5982.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Belov GA, Habbersett C, Franco D, Ehrenfeld E. Activation of cellular Arf GTPases by poliovirus protein 3CD correlates with virus replication. J Virol. 2007;81:9259–9267. doi: 10.1128/JVI.00840-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Echeverri A, Banerjee R, Dasgupta A. Amino-terminal region of poliovirus 2C protein is sufficient for membrane binding. Virus Res. 1998;54:217–223. doi: 10.1016/s0168-1702(98)00016-1. [DOI] [PubMed] [Google Scholar]
- 6.Teterina NL, Gorbalenya AE, Egger D, Bienz K, Ehrenfeld E. Poliovirus 2C protein determinants of membrane binding and rearrangements in mammalian cells. J Virol. 1997;71:8962–8972. doi: 10.1128/jvi.71.12.8962-8972.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sandoval IV, Carrasco L. Poliovirus infection and expression of the poliovirus protein 2B provoke the disassembly of the Golgi complex, the organelle target for the antipoliovirus drug Ro-090179. J Virol. 1997;71:4679–4693. doi: 10.1128/jvi.71.6.4679-4693.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barco A, Carrasco L. A human virus protein, poliovirus protein 2BC, induces membrane proliferation and blocks the exocytic pathway in the yeast Saccharomyces cerevisiae. EMBO J. 1995;14:3349–3364. doi: 10.1002/j.1460-2075.1995.tb07341.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Echeverri AC, Dasgupta A. Amino terminal regions of poliovirus 2C protein mediate membrane binding. Virology. 1995;208:540–553. doi: 10.1006/viro.1995.1185. [DOI] [PubMed] [Google Scholar]
- 10.Aldabe R, Carrasco L. Induction of membrane proliferation by poliovirus proteins 2C and 2BC. Biochem Biophys Res Commun. 1995;206:64–76. doi: 10.1006/bbrc.1995.1010. [DOI] [PubMed] [Google Scholar]
- 11.Cho MW, Teterina N, Egger D, Bienz K, Ehrenfeld E. Membrane rearrangement and vesicle induction by recombinant poliovirus 2C and 2BC in human cells. Virology. 1994;202:129–145. doi: 10.1006/viro.1994.1329. [DOI] [PubMed] [Google Scholar]
- 12.Horne RW, Nagington J. Electron Microscope Studies of the Development and Structure of Poliomyelitis Virus. J Mol Biol. 1959;1:333–338. [Google Scholar]
- ••13.Dales S, Eggers HJ, Tamm I, Palade GE. Electron Mcroscopic Study of the Formation of Poliovirus. Virology. 1965;26:379–389. doi: 10.1016/0042-6822(65)90001-2. The seminal study of poliovirus-infected cells using electron microscopy. A must-read for anyone interested in the field. [DOI] [PubMed] [Google Scholar]
- 14.Bolten R, Egger D, Gosert R, Schaub G, Landmann L, Bienz K. Intracellular localization of poliovirus plus- and minus-strand RNA visualized by strand-specific fluorescent In situ hybridization. J Virol. 1998;72:8578–8585. doi: 10.1128/jvi.72.11.8578-8585.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ••15.Belov GA, Nair V, Hansen BT, Hoyt FH, Fischer ER, Ehrenfeld E. Complex dynamic development of poliovirus membranous replication complexes. J Virol. 2012;86:302–312. doi: 10.1128/JVI.05937-11. Beautiful electron tomography or early and late time points post-infection. Makes an argument for early vesicles developing into late double-membraned vesicles. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Beske O, Reichelt M, Taylor MP, Kirkegaard K, Andino R. Poliovirus infection blocks ERGIC-to-Golgi trafficking and induces microtubule-dependent disruption of the Golgi complex. J Cell Sci. 2007;120:3207–3218. doi: 10.1242/jcs.03483. [DOI] [PubMed] [Google Scholar]
- 17.Suhy DA, Giddings TH, Kirkegaard K. Remodeling the endoplasmic reticulum by poliovirus infection and by individual viral proteins: an autophagy-like origin for virus-induced vesicles. J Virol. 2000;74:8953–8965. doi: 10.1128/jvi.74.19.8953-8965.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rust RC, Landmann L, Gosert R, Tang BL, Hong W, Hauri HP, Egger D, Bienz K. Cellular COPII proteins are involved in production of the vesicles that form the poliovirus replication complex. J Virol. 2001;75:9808–9818. doi: 10.1128/JVI.75.20.9808-9818.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bienz K, Egger D, Pasamontes L. Association of polioviral proteins of the P2 genomic region with the viral replication complex and virus-induced membrane synthesis as visualized by electron microscopic immunocytochemistry and autoradiography. Virology. 1987;160:220–226. doi: 10.1016/0042-6822(87)90063-8. [DOI] [PubMed] [Google Scholar]
- 20.Trahey M, Oh HS, Cameron CE, Hay JC. Poliovirus infection transiently increases COPII vesicle budding. J Virol. 2012;86:9675–9682. doi: 10.1128/JVI.01159-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ••21.Hsu N-Y, Ilnytska O, Belov G, Santiana M, Chen Y-H, Takvorian PM, Pau C, van der Schaar H, Kaushik-Basu N, Balla T, et al. Viral reorganization of the secretory pathway generates distinct organelles for RNA replication. Cell. 2010;141:799–811. doi: 10.1016/j.cell.2010.03.050. The first demonstration of the localization of PI4KIIIβ, and subsequently PI4P, to CVB3 and PV membranes. This paper also provided the picornavirus RNA replication membranes as a novel organelle. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Richards AL, Soares-Martins JAP, Riddell GT, Jackson WT. Generation of unique poliovirus RNA replication organelles. MBio. 2014:5. doi: 10.1128/mBio.00833-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Maynell LA, Kirkegaard K, Klymkowsky MW. Inhibition of poliovirus RNA synthesis by brefeldin A. J Virol. 1992;66:1985–1994. doi: 10.1128/jvi.66.4.1985-1994.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wright J, Kahn RA, Sztul E. Regulating the large Sec7 ARF guanine nucleotide exchange factors: the when, where and how of activation. Cell Mol Life Sci. 2014 doi: 10.1007/s00018-014-1602-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Belov GA, Ehrenfeld E. Involvement of cellular membrane traffic proteins in poliovirus replication. Cell Cycle. 2007;6:36–38. doi: 10.4161/cc.6.1.3683. [DOI] [PubMed] [Google Scholar]
- 26.Belov GA, Fogg MH, Ehrenfeld E. Poliovirus proteins induce membrane association of GTPase ADP-ribosylation factor. J Virol. 2005;79:7207–7216. doi: 10.1128/JVI.79.11.7207-7216.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Belov GA, Altan-Bonnet N, Kovtunovych G, Jackson CL, Lippincott-Schwartz J, Ehrenfeld E. Hijacking components of the cellular secretory pathway for replication of poliovirus RNA. J Virol. 2007;81:558–567. doi: 10.1128/JVI.01820-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wessels E, Duijsings D, Lanke KHW, Melchers WJG, Jackson CL, van Kuppeveld FJM. Molecular determinants of the interaction between coxsackievirus protein 3A and guanine nucleotide exchange factor GBF1. J Virol. 2007;81:5238–5245. doi: 10.1128/JVI.02680-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Belov GA, Kovtunovych G, Jackson CL, Ehrenfeld E. Poliovirus replication requires the N-terminus but not the catalytic Sec7 domain of ArfGEF GBF1. Cell Microbiol. 2010;12:1463–1479. doi: 10.1111/j.1462-5822.2010.01482.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Crotty S, Saleh M-C, Gitlin L, Beske O, Andino R. The poliovirus replication machinery can escape inhibition by an antiviral drug that targets a host cell protein. J Virol. 2004;78:3378–3386. doi: 10.1128/JVI.78.7.3378-3386.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Barlowe CK, Miller EA. Secretory protein biogenesis and traffic in the early secretory pathway. Genetics. 2013;193:383–410. doi: 10.1534/genetics.112.142810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dodd DA, Giddings TH, Kirkegaard K. Poliovirus 3A protein limits interleukin-6 (IL-6), IL-8, and beta interferon secretion during viral infection. J Virol. 2001;75:8158–8165. doi: 10.1128/JVI.75.17.8158-8165.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Deitz SB, Dodd DA, Cooper S, Parham P, Kirkegaard K. MHC I-dependent antigen presentation is inhibited by poliovirus protein 3A. Proc Natl Acad Sci USA. 2000;97:13790–13795. doi: 10.1073/pnas.250483097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Irurzun A, Pérez L, Carrasco L. Enhancement of phospholipase activity during poliovirus infection. J Gen Virol. 1993;74 (Pt 6):1063–1071. doi: 10.1099/0022-1317-74-6-1063. [DOI] [PubMed] [Google Scholar]
- 35.Halperen S. Comparisons of picornavirus-induced modifications of the distribution of radioactive choline among cytoplasmic lipid macromolecules. J Gen Virol. 1983;64 (Pt 2):491–497. doi: 10.1099/0022-1317-64-2-491. [DOI] [PubMed] [Google Scholar]
- 36.Schaefer A, Kühne J, Zibirre R, Koch G. Poliovirus-induced alterations in HeLa cell membrane functions. J Virol. 1982;44:445–449. doi: 10.1128/jvi.44.2.445-449.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vance DE, Trip EM, Paddon HB. Poliovirus increases phosphatidylcholine biosynthesis in HeLa cells by stimulation of the rate-limiting reaction catalyzed by CTP: phosphocholine cytidylyltransferase. J Biol Chem. 1980;255:1064–1069. [PubMed] [Google Scholar]
- 38.Delang L, Paeshuyse J, Neyts J. The role of phosphatidylinositol 4-kinases and phosphatidylinositol 4-phosphate during viral replication. Biochem Pharmacol. 2012;84:1400–1408. doi: 10.1016/j.bcp.2012.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.van der Schaar HM, van der Linden L, Lanke KHW, Strating JRPM, Pürstinger G, de Vries E, de Haan CAM, Neyts J, van Kuppeveld FJM. Coxsackievirus mutants that can bypass host factor PI4KIIIβ and the need for high levels of PI4P lipids for replication. Cell Res. 2012;22:1576–1592. doi: 10.1038/cr.2012.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Greninger AL, Knudsen GM, Betegon M, Burlingame AL, DeRisi JL. The 3A protein from multiple picornaviruses utilizes the golgi adaptor protein ACBD3 to recruit PI4KIIIβ. J Virol. 2012;86:3605–3616. doi: 10.1128/JVI.06778-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sasaki J, Ishikawa K, Arita M, Taniguchi K. ACBD3-mediated recruitment of PI4KB to picornavirus RNA replication sites. EMBO J. 2012;31:754–766. doi: 10.1038/emboj.2011.429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ••42.Dorobantu CM, van der Schaar HM, Ford LA, Strating JRPM, Ulferts R, Fang Y, Belov G, van Kuppeveld FJM. Recruitment of PI4KIIIβ to coxsackievirus B3 replication organelles is independent of ACBD3, GBF1, and Arf1. J Virol. 2014;88:2725–2736. doi: 10.1128/JVI.03650-13. This paper provides an important test of several models in the field, and shows that for CVB3 the host proteins most commonly proposed to recruit PI4KIIIβ to membranes are in fact not required. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ilnytska O, Santiana M, Hsu N-Y, Du W-L, Chen Y-H, Viktorova EG, Belov G, Brinker A, Storch J, Moore C, et al. Enteroviruses harness the cellular endocytic machinery to remodel the host cell cholesterol landscape for effective viral replication. Cell Host Microbe. 2013;14:281–293. doi: 10.1016/j.chom.2013.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Herr D, Finley KD. sciencedirect.com. Elsevier; 2013. Molecular Machinery and Genetics of the Autophagy Pathway; pp. 11–30. [Google Scholar]
- 45.Schneider JL, Cuervo AM. Autophagy and human disease: emerging themes. Curr Opin Genet Dev. 2014;26C:16–23. doi: 10.1016/j.gde.2014.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shi J, Luo H. Interplay between the cellular autophagy machinery and positive-stranded RNA viruses. Acta Biochim Biophys Sin (Shanghai) 2012;44:375–384. doi: 10.1093/abbs/gms010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Huang J, Brumell JH. Bacteria-autophagy interplay: a battle for survival. Nat Rev Microbiol. 2014;12:101–114. doi: 10.1038/nrmicro3160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Harrison AK, Bauer SP, Murphy FA. Viral pancreatitis: ultrastructural pathological effects of Coxsackievirus B3 infection in newborn mouse pancreas. Exp Mol Pathol. 1972;17:206–219. doi: 10.1016/0014-4800(72)90070-6. [DOI] [PubMed] [Google Scholar]
- 49.Burch GE, Harb JM. Electron microscopic studies of viral pancreatitis in coxsackie B4 virus infected mice. Exp Mol Pathol. 1979;31:23–35. doi: 10.1016/0014-4800(79)90004-2. [DOI] [PubMed] [Google Scholar]
- 50.Schlegel A, Giddings TH, Ladinsky MS, Kirkegaard K. Cellular origin and ultrastructure of membranes induced during poliovirus infection. J Virol. 1996;70:6576–6588. doi: 10.1128/jvi.70.10.6576-6588.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ••51.Jackson WT, Giddings TH, Taylor MP, Mulinyawe S, Rabinovitch M, Kopito RR, Kirkegaard K. Subversion of cellular autophagosomal machinery by RNA viruses. PLoS Biol. 2005;3:e156. doi: 10.1371/journal.pbio.0030156. The initial description of autophagy benefitting PV replication and autophagosomes promoting viral exit. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ••52.Richards AL, Jackson WT. Intracellular vesicle acidification promotes maturation of infectious poliovirus particles. PLoS Pathog. 2012;8:e1003046. doi: 10.1371/journal.ppat.1003046. In this paper we showed that the acidification of vesicles regulates virion maturation for poliovirus. This is the first description of a step in PV replication requiring pH change of a cellular compartment. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ••53.Kemball CC, Alirezaei M, Flynn CT, Wood MR, Harkins S, Kiosses WB, Whitton JL. Coxsackievirus infection induces autophagy-like vesicles and megaphagosomes in pancreatic acinar cells in vivo. J Virol. 2010;84:12110–12124. doi: 10.1128/JVI.01417-10. An important study of CVB3 in vivo, and an important contrast to PV. CVB3 is shown to induce large multilamellar megasomes but not autophagic degradation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wong J, Zhang J, Si X, Gao G, Mao I, McManus BM, Luo H. Autophagosome supports coxsackievirus B3 replication in host cells. J Virol. 2008;82:9143–9153. doi: 10.1128/JVI.00641-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Taylor MP, Burgon TB, Kirkegaard K, Jackson WT. Role of microtubules in extracellular release of poliovirus. J Virol. 2009;83:6599–6609. doi: 10.1128/JVI.01819-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Basavappa R, Syed R, Flore O, Icenogle JP, Filman DJ, Hogle JM. Role and mechanism of the maturation cleavage of VP0 in poliovirus assembly: structure of the empty capsid assembly intermediate at 2.9 A resolution. Protein Sci. 1994;3:1651–1669. doi: 10.1002/pro.5560031005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Koch F, Koch G. The molecular biology of poliovirus. Springer; 1985. [Google Scholar]
- 58.Duran JM, Anjard C, Stefan C, Loomis WF, Malhotra V. Unconventional secretion of Acb1 is mediated by autophagosomes. J Cell Biol. 2010;188:527–536. doi: 10.1083/jcb.200911154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Manjithaya R, Anjard C, Loomis WF, Subramani S. Unconventional secretion of Pichia pastoris Acb1 is dependent on GRASP protein, peroxisomal functions, and autophagosome formation. J Cell Biol. 2010;188:537–546. doi: 10.1083/jcb.200911149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Arita M, Wakita T, Shimizu H. Valosin-containing protein (VCP/p97) is required for poliovirus replication and is involved in cellular protein secretion pathway in poliovirus infection. J Virol. 2012;86:5541–5553. doi: 10.1128/JVI.00114-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bug M, Meyer H. Expanding into new markets--VCP/p97 in endocytosis and autophagy. J Struct Biol. 2012;179:78–82. doi: 10.1016/j.jsb.2012.03.003. [DOI] [PubMed] [Google Scholar]
- ••62.Feng Z, Hensley L, McKnight KL, Hu F, Madden V, Ping L, Jeong S-H, Walker C, Lanford RE, Lemon SM. A pathogenic picornavirus acquires an envelope by hijacking cellular membranes. Nature. 2013;496:367–371. doi: 10.1038/nature12029. The first evidence that a picornavirus has an enveloped subpopulation, and a major paradigm shift in the field. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ••63.Robinson SM, Tsueng G, Sin J, Mangale V, Rahawi S, McIntyre LL, Williams W, Kha N, Cruz C, Hancock BM, et al. Coxsackievirus B exits the host cell in shed microvesicles displaying autophagosomal markers. PLoS Pathog. 2014;10:e1004045. doi: 10.1371/journal.ppat.1004045. This paper provides the best direct evidence of a picornavirus exiting the cell in vesicles derived from the autophagic pathway. [DOI] [PMC free article] [PubMed] [Google Scholar]