

Abstract
Respiratory viruses can cause a wide spectrum of pulmonary diseases, ranging from mild, upper respiratory tract infections to severe and life‐threatening lower respiratory tract infections, including the development of acute lung injury (ALI) and acute respiratory distress syndrome (ARDS). Viral clearance and subsequent recovery from infection require activation of an effective host immune response; however, many immune effector cells may also cause injury to host tissues. Severe acute respiratory syndrome (SARS) coronavirus and Middle East respiratory syndrome (MERS) coronavirus cause severe infection of the lower respiratory tract, with 10% and 35% overall mortality rates, respectively; however, >50% mortality rates are seen in the aged and immunosuppressed populations. While these viruses are susceptible to interferon treatment in vitro, they both encode numerous genes that allow for successful evasion of the host immune system until after high virus titres have been achieved. In this review, we discuss the importance of the innate immune response and the development of lung pathology following human coronavirus infection. © 2014 The Authors. The Journal of Pathology published by John Wiley & Sons Ltd on behalf of Pathological Society of Great Britain and Ireland.
Keywords: SARS‐CoV, MERS‐CoV, coronavirus, acute respiratory distress syndrome, ARDS, acute lung injury, type II pneumocytes
Introduction
Acute lung injury (ALI) and its more severe form, acute respiratory distress syndrome (ARDS), can arise after many types of injury to the lung, including sepsis, mechanical and chemical injury and bacterial and viral infections 1. In ALI, the mortality rate is in the range 20–30%, with about 55% of the cases progressing to ARDS within a few days. ARDS causes significant morbidity and approximately 40% mortality, resulting in ∼75 000 deaths/year in the USA alone 2. In the past two decades, five emerging viruses have been known to cause significant ARDS‐related mortality, including influenza H1N1 2009 and, in particular, the highly pathogenic avian influenza H5N1 and H7N9 viruses and the SARS and MERS coronaviruses. In this review we focus on mechanisms of coronavirus‐induced lung pathogenesis and ARDS.
Human coronavirus (CoV) infections have traditionally caused a low percentage of annual upper and lower respiratory infections 3, including severe disease outcomes in the elderly, immunocompromised patients and infants. HCoV‐OC43 (OC43) and HCoV‐229E (229E) were the first documented human CoVs but, more recently, HCoV‐NL63 (NL63) 4 and HCoV‐HKU1 (HKU1) 5 were identified as a consequence of increased viral surveillance efforts in the early twenty‐first century (Table 1). These four viruses usually cause acute infection of the upper respiratory tract and less frequently are associated with lower respiratory tract 6, 7 diseases as well. Severe disease is both rare and typically associated with co‐morbidities and/or immunosenescence. The past 15 years have also seen the emergence of two new human coronaviruses that cause significant disease and mortality. SARS‐CoV was identified in 2003 and caused an acute, atypical pneumonia and diffuse alveolar damage (DAD) in roughly 8000 patients 8, 9. Those over 65 years of age often developed ARDS, resulting in mortality rates that exceeded 50%. Overall, SARS‐CoV infection caused nearly 800 fatalities, representing a nearly 10% mortality rate. More recently, in 2012, a new human coronavirus, designated MERS‐CoV, was identified. MERS‐CoV continues to circulate in camels and humans, with over 857 official cases and 334 deaths, representing an approximately 35% case fatality rate to date in humans 10, 11. MERS‐CoV‐induced disease is particularly severe in aged patients and those with pre‐existing co‐morbidities. MERS‐CoV does not appear to be highly pathogenic or virulent in camels.
Table 1.
Human coronaviruses, their receptors, emergence, disease and infection data
| Virus | Receptor | Discovery and estimated date of divergence | Cell types infected | Disease types caused |
|---|---|---|---|---|
| OC43 | Receptor unknown, sialic acid and HLA class 1 involvement 113, 114 | Divergence from BCoV in 1890 115 | Ciliated airway epithelial cells 116, macrophages in culture 117, neuronal cells 118 | Upper respiratory infection, GI infection, pneumonia 119 |
| 229E | Aminopeptidase N 120 | Divergence in 1700–1800 121, divergence from NL63 in the eleventh century 122 | Non‐ciliated airway epithelial cells 116, human monocytes 123, neuronal cells 118 | Upper respiratory infection 124, GI infection, pneumonia 125 |
| NL63 | Ace2 126 | Discovered in 2004 4, divergence between 1200 and 1500 127 | Ciliated airway epithelial cells 116 | Upper and lower respiratory infection 6, 128, associated with croup in children 129 |
| HKU1 | Unknown | Discovered in 2005 5 | Ciliated airway epithelial cells 116 | Upper respiratory infection and pneumonia 130, enteric symptoms 131 |
| SARS | Ace2 130, role for DC‐sign 132 | Emerged in 2002 133, divergence estimates from 1986–2002 134, 135 | Epithelial cells 136, ciliated cells, type II pneumocytes 137 | Lower respiratory infection 99, 138, pneumonia, DAD, ARDS |
| MERS | DPP4 139 | Emerged in 2012 25, common ancestor from 2011–2012 140 | Airway epithelial cells 141, renal epithelial cells 142, dendritic cells 143 | Lower respiratory infection 25, 144, pneumonia, renal failure |
SARS‐CoV and MERS‐CoV have clear zoonotic origins, although their exact paths from animal reservoir to human infection are not yet clear. Viruses with high nucleotide identity to SARS‐CoV were found in key amplifying hosts such as palm civets and raccoon dogs in Guangdong Province China during the 2002–2003 SARS epidemic 12. Later studies identified highly conserved viruses circulating in horseshoe bats, including some strains that are able to bind to, and infect, human cells 13, 14, 15, 16. The existence of novel bat SARS‐like coronaviruses that also use bat, civet and human angiotensin 1 converting enzyme 2 (ACE2) receptors for entry, such as SARS‐CoV, strongly suggests an opportunity for further zoonotic disease outbreaks in human and animal populations.
SARS causes an atypical pneumonia characterized by cough, fever and infiltrates with a ground‐glass appearance on X‐ray 17, 18. Early‐stage disease was characterized by acute DAD, with oedema, fibrin and hyaline membranes in the alveolar spaces, typical of ALI 19. Other patients predominantly showed an acute fibrinous and organizing pneumonia pattern or a mixture of the two patterns 20, 21. Longer‐term disease courses typically progressed to organizing phase DAD and eventual deposition of fibrous tissue. Autopsy of fatal SARS‐CoV cases also revealed denuded airways, haemorrhage and increased macrophage populations in the lung 22, 23. During the SARS epidemic, researchers noted that late‐term disease progression was unrelated to viraemia but was more likely to be associated with immunopathological damage 24.
MERS‐CoV has caused sporadic infections, along with several local outbreaks throughout the Middle East since its discovery in 2012 25, 26. Although much remains unknown, closely related viruses have been isolated from camels 27 and highly homologous MERS‐like bat CoVs have been identified in African Neoromicia capensis bats 28. Local surveillance efforts have detected high levels of antibodies that recognize MERS‐CoV in dromedary camels 29; furthermore, sampling of archived camel serum samples has revealed MERS antibodies from as early as 1992 30. These data suggest that bat to camel to human transmission routes may have seeded the 2012 outbreak in human populations, perhaps associated with the expanding camel trade that has emerged between equatorial Africa and Saudi Arabia over the past 20 years.
Animal models of human disease should recapitulate many of the pathological and immune outcomes seen in human infections. Numerous models have been established to better enable our understanding of the mechanics of SARS‐CoV infection and pathogenesis, although few recapitulate the human disease phenotypes (Table 2). Initial studies utilized late epidemic strains in non‐human primates 31, 32, 33, where mild to severe disease was observed, depending on the study location and animal age. To date, the differences in disease severity noted in primates have not been reconciled, but may reflect differences in virus strains or infection conditions. Although still under development, MERS‐CoV replication and disease have been reported in both rhesus macaques and common marmosets 34, 35. SARS‐CoV replication resulted in limited disease in young models of immunocompetent mice 36, 37, 38; however, mild clinical disease was noted in 1 year‐old mice 39. A mouse‐adapted SARS (MA‐SARS) strain was also developed that provides a model for moderate to lethal disease, depending on infectious dose, animal age and genetic background of the host 40, 41, 42 (Table 2). The MA‐SARS model faithfully replicates the age‐dependent susceptibility observed in human patients, as well as key features of human lung pathology, including virus tropism to airway epithelial cells and type II pneumocytes, pneumonia, hyaline membrane formation, development of DAD and denudation of airway epithelial cells 40, 43. A limitation may be the rapid clearance of virus titres that is seen in younger and, to a much lesser extent, in aged animals. Development of the MA‐SARS model has allowed for in‐depth studies of viral pathogenesis and the host immune response, taking advantage of immunological tools and reagents for the mouse as well as the existence of knockout mouse strains. Use of these tools has greatly added to our understanding of SARS‐CoV pathogenesis, far beyond what could be learned in in vitro experiments or observational studies of human cases. Because of receptor incompatibilities, MERS‐CoV does not replicate in mice unless the animals are first transduced with adenovirus vectors encoding the receptor for entry, human dipeptidyl peptidase‐4 (DPP4) 44.
Table 2.
Non‐human primate and mouse models of SARS‐CoV and MERS‐CoV infection; less common models include hamster 145, ferret 146 and cat
| Virus | Animal model | Virus modifications? | Disease types caused | Drawbacks | Aged model? |
|---|---|---|---|---|---|
| SARS‐CoV | Rhesus macaque | None | Viral replication, mild pneumonia 147 | Expense, ethical considerations, no severe disease | |
| African green monkey | None | Viral replication, pneumonitis 33, hyaline membrane formation 102 | Expense, ethical considerations, no severe disease | ||
| Cynomolgus macaque | None | Viral replication, upper respiratory symptoms, pneumonia 31, 148 | Expense, ethical considerations | Yes 149 | |
| Ace2 transgenic mice | None | Viral replication, weight loss, inflammatory cell infiltrates 150 | Virus causes encephalitis 150, use of knockout mice requires extensive breeding | ||
| Mouse | None | Virus replication, mild pneumonia in aged mice 39 | Minimal pathogenesis, especially in young mice 36 | Yes 39 | |
| SARS‐MA15 | Mouse | Six point mutations from serial mouse passage 40 | Viral replication, weight loss 40, pneumonia, DAD 43, pulmonary fibrosis 56 | Virus has been adapted from human strains | Yes 41 |
| MERS‐CoV | Rhesus macaque | None | Viral replication 35, transient pneumonia | Expense, ethical considerations, no severe disease | |
| hAd5‐DPP4 mouse | None | Viral replication 44, weight loss in immune knockouts | Requires infection with hAd5 to express human DPP4 | Yes 44 |
In this review we focus solely on hCoV interactions within the context of the respiratory system and infection of relevant cell types. More specifically, we review some CoV–host interactions that alter cell‐intrinsic antiviral defence programmes and other host pathways that contribute to pathological findings of ARDS, with its associated exudative and organizing phase diffuse alveolar damage and pulmonary fibrosis.
Innate immune response
NF‐κB signalling is an important component of numerous cellular responses, including stress, cytokine signalling, response to bacterial or viral infection and apoptosis 45, 46. The SARS‐CoV envelope (E) protein stimulates NF‐κB signalling 47, leading to lung cytokine signalling and inflammatory cell recruitment. The SARS‐CoV papain‐like protease (PLP) has also been shown to antagonize NF‐κB signalling 48 in vitro. Chemical inhibitors of NF‐κB signalling reduce lung pathology and inflammation following MA‐SARS infection, demonstrating the importance of this pathway 47 in pathogenesis. While the SARS‐CoV E protein is not required for viral replication, it is important for inhibition of the host cellular stress response, apoptosis and unfolded protein response 49, 50. The E protein, along with the SARS‐CoV ORF3a and ORF8a proteins, has ion channel activity 50 and may contribute to vascular permeability and fluid accumulation in the lung following SARS‐CoV infection. SARS‐CoV lacking E has been shown to be an effective vaccine 51, 52 and a MERS‐CoV clone lacking E has been generated 53, although replication requires expression of E in trans.
SARS‐CoV, and to a greater extent MERS‐CoV, are highly sensitive to interferon treatment in cell culture. Interestingly, SARS‐CoV pathogenesis does not significantly change in various type I interferon (IFN) knockout mouse models, except for a slight increase in overall virus titres 54, 55, 56. Despite this, STAT1‐ and Myd88‐deficient mice are significantly more vulnerable to lethal outcomes following infection 56, 57. Like many viruses, CoVs encode a suite of genes that antagonize cell‐intrinsic innate immune defence programmes in the infected host cell (reviewed in 58). Numerous in vitro studies have demonstrated the IFN antagonist activity of both SARS‐CoV and MERS‐CoV proteins 59, 60, 61, and a detailed review of SARS‐CoV evasion of the innate immune response was recently published by Totura and Baric 62.
Analysis of IFN‐stimulated gene (ISG) expression in Calu‐3 human airway epithelial cells highlighted the ability of SARS‐CoV and MERS‐CoV to avoid detection by the host 63. As compared with influenza A viruses, ISG transcripts and proteins are not induced until late after SARS‐CoV and MERS‐CoV infection, when peak titres have already occurred in culture (∼18–24 h). Late in infection, ISGs showed nearly universally increased expression following SARS‐CoV infection, except for ACE2 and Serping1. However, a much larger subset of ISGs had significantly decreased expression following MERS‐CoV infection. Like MERS‐CoV, H5N1 VN1203 infection also resulted in significant down‐regulation of subsets of ISGs. No consistent pattern in up‐regulation or down‐regulation of gene expression correlated with transcription factor usage, suggesting that a novel mechanism may be responsible for expression of the ISG subsets. Cells infected with MERS‐CoV and H5N1 avian influenza were shown to have specifically altered open and closed chromatin structures, potentially limiting the ability of transcription factors to access and bind certain ISG promoter regions. The mechanism by which MERS‐CoV induces this chromatin structural alteration is as yet unknown. In contrast, the NS1 protein of H5N1 was responsible for the chromatin changes in influenza‐infected cells. Although speculative, it seems likely that many RNA viruses may encode strategies to epigenetically alter host chromatin structure, influencing host gene expression. This newly identified method of ISG control requires additional study.
SARS‐CoV further evades the host immune response by masking its RNA genome. This mechanism may be partially mediated by the production of double‐membrane vesicles, which could sequester RNA replication intermediates away from the host‐sensing machinery 64, 65. MDA5 and IFIT1 are important host antiviral sensor or antiviral defence ISGs that detect viral RNAs. IFIT1 recognizes unmethylated 2′‐O RNA 66 and alters efficient translation/stability of uncapped viral mRNAs 67. SARS‐CoV and other coronavirus RNAs are protected from IFIT recognition because they encode a 2′‐O‐methyltransferase (2‐OMT) activity in the viral replicase protein, nsp16 68, 69. SARS‐CoV is much more sensitive to interferon treatment in the absence of functional nsp16 methyltransferase activity, and mutant viral titres drop rapidly in both infected epithelial cells and mice. Deletion or knockdown of either MDA5 or IFIT1 restored mutant SARS‐CoV viral loads demonstrated the essential role of these host proteins in detecting pathogen‐associated molecular patterns. Ablation of the 2‐OMT activity may provide a universal strategy to rationally design live attenuated mutants of contemporary and newly emerging CoV.
Both in vivo and in vitro studies have addressed the role of specific proteins in the innate immune system, often ISGs, in SARS‐CoV pathogenesis. Transcriptional analysis on autopsy tissue from SARS‐CoV‐infected patients revealed increased expression of STAT1 along with other IFN‐induced cytokines 70. The SARS‐CoV accessory protein ORF6 was identified as an interferon antagonist important for viral replication in low multiplicity of infection (MOI) in vitro infections 71, 72. ORF6 was subsequently found to sequester Karyopherin 2α, a nuclear import factor, and block the nuclear translocation of STAT1 after SARS‐CoV infection. Interestingly, STAT1 translocation to the nucleus is not blocked in MERS‐CoV‐infected cells, so it remains uncertain whether antagonists of nuclear import are encoded in the viral genome 73. Transcriptional profiling of SARS‐CoV‐infected macaques revealed robust IFN signalling, including STAT1 translocation to the nucleus, in the lung but not in the cells that stained positive for viral antigen 74. These data highlight the importance of in vivo studies versus high MOI in vitro studies. Significantly, they also highlight the need to examine, or at least consider, expressing and signalling differences in specific cell types instead of global transcriptomic studies in those in vivo experiments.
STAT1 knockout mice have been studied extensively in the context of viral infection, typically showing a heightened susceptibility to disease, due to the lack of a type I IFN response 75. These knockouts were first tested for SARS‐CoV susceptibility using the Tor2 strain in a sublethal model; animals deficient in STAT1 were unable to clear virus from the lung and developed a more severe and longer‐lasting pneumonia than the control mice 76. Frieman et al showed that STAT1 knockout mice are highly susceptible to infection with MA‐SARS in a novel, IFN‐independent mechanism 56. MA‐SARS‐infection causes massive inflammatory cell influx in the lungs of STAT1 knockout mice, including large numbers of macrophages, neutrophils and eosinophils. STAT1 knockout mice have gross pathological changes in their lungs, including massive haemorrhage as well as increased lung size and stiffness. As seen in some humans, these mice develop severe pulmonary fibrosis and succumb to disease at late time points after infection. Stained lung sections revealed the presence of collagen protein in alveolar exudates in STAT1 knockouts, indicating development of early‐stage pulmonary fibrosis. Subsequent studies demonstrated that STAT1 knockout animals developed a Th2‐skewed immune response and had significant numbers of alternatively activated or M2 macrophages in their lungs 77. These macrophages were characterized by positive CD11c, arginase and mannose receptor staining. STAT6 is required for the development of alternatively activated macrophages, and STAT1/STAT6 double knockout mice do not develop the severe lung disease and pro‐fibrotic lesions observed in the STAT1 single knockout 78, thus demonstrating that these macrophages are essential for development of the pulmonary fibrosis phenotype. Further elegant experiments showed that it is the STAT1 deficiency in monocyte/macrophage cells, not the infected epithelial cells, that drives alternatively activated macrophage production and induction of fibrotic lung disease following SARS‐CoV infection. Alternatively activated macrophages are typically induced by the Th2 cytokines IL‐4 and IL‐13; they have an anti‐inflammatory role and play an important role in wound‐healing processes 151.
ACE2 is expressed on well‐differentiated airway epithelial cells 79 and its expression increases following type I interferon treatment 63. Both ACE2 protein levels and RNA expression are down‐regulated after either in vitro or in vivo SARS‐CoV infection 63, 80 and NL63 also down‐regulates ACE2 expression following in vitro infection 81. It has previously been reported that ACE2 and angiotensin‐2 protect mice from sepsis‐ and acid aspiration‐induced ALI 82. Additionally, histopathological lung disease worsens when spike‐Fc is inoculated into mice with ALI 80. The normal function of ACE2 is to inactivate angiotensin‐2, a negative regulator of the renin–angiotensin system 83, 84. This system controls blood pressure and is involved in the development of pulmonary hypertension and pulmonary fibrosis. The renin–angiotensin system is involved in lipopolysaccharide (LPS)‐induced neutrophil recruitment to the lung 85. Multiple genome‐wide association studies have investigated an association between genetic variation in ACE and susceptibility to ARDS, with mixed results 86. The role of DPP4 in MERS‐CoV infection is discussed in detail by Haagmans et al in a separate review in this issue 152.
High ISG expression has been linked to development of ARDS 87. Furthermore, it has been suggested that unregulated IFN responses contributed to development of immunopathology and severe disease following SARS‐CoV infection 88. The data discussed above support this hypothesis and suggest that early control of ISG signalling may be a means of preventing or controlling the development of severe lung disease. Expression of the ISG Serping1 is also decreased following SARS‐CoV infection of epithelial cells; it functions by inhibiting the complement system as well as several proteases in the coagulation pathway. The role of the coagulation, fibrinolysis and wound healing in ARDS development are discussed below.
Acute respiratory distress syndrome (ARDS), coagulation, fibrinolysis and respiratory function
The alveoli of the lung are where gas exchange occurs, providing oxygen to blood flowing through capillaries in the alveolar membrane. The alveolar walls are composed of type I and type II pneumocytes, along with alveolar macrophages 89. Type I pneumocytes cover 95% of the alveolar surface area and allow for gas exchange with blood in the capillaries of the lung. Type II pneumocytes are the progenitors of type I pneumocytes and are also responsible for generating pulmonary surfactant 90, a mixture of lipids and surfactant proteins that is crucial in reducing surface tension in the lung. SARS‐CoV infection causes desquamation of pneumocytes in humans and mice, contributing to alveolar dysfunction, oedema and haemorrhage. Alveolar macrophages play an essential role in surveillance of the local environment and inhibit an excessive immune response, although this inhibition can also block an effective response to SARS‐CoV infection 91. The functions of these cell types are critical in maintaining balance between inflammation, coagulation and wound repair, especially following lung injuries such as viral infection 92.
In ARDS patients, uncontrolled inflammation, fluid accumulation and developing fibrosis severely compromise gas exchange and lead to respiratory failure. SARS‐CoV and influenza infect type I and type II pneumocytes in the lung 93, 94. ARDS patients exhibit decreased surfactant levels 95 and MA‐SARS infection results in decreased surfactant transcript and protein levels 43. Decreased surfactant, and the consequent increase in surface tension, reduces the ability of the lung to expand and contract during normal respiration; it also heightens the risk of lung collapse during expiration. Respiratory dysfunction occurs when the alveolar membranes are obstructed, or when the ability of the lung to expand and contract, circulating oxygenated air, is compromised. Lethal SARS‐CoV infection in the mouse and human is characterized by a breakdown of alveolar membrane integrity, resulting in accumulation of fluid exudates in the alveolar spaces. Virus infection also results in an overwhelming cytokine response, severe lung tissue damage and respiratory failure 96, 97, 98, 99. The progression from initial disease to diffuse alveolar damage and the exudative and organizing stage of DAD is often independent of high‐titre viral replication 24, indicating that this severe disease outcome is primarily driven by an immunopathological response, including inflammatory cell recruitment and viral damage to type II pneumocytes. This conclusion is further supported by non‐human primate and mouse models of SARS‐CoV infection, where lethal disease is more often associated with severe pulmonary lesions, alveolar exudates and respiratory dysfunction than with high viral load 43, 100. MA‐SARS infection results in peak viral titres at 1–2 days post‐infection, along with airway denudation and resulting debris, which can occlude the small airways. Severe lung disease, including inflammatory cell infiltrates, haemorrhage, alveolar oedema and hyaline membrane formation typical of the exudative stage of DAD (Figure 1), occurs at days 4–7 post‐infection, when virus loads in the lung are dropping rapidly and/or are below the limit of detection. Many ISGs stimulated by SARS‐CoV infection are involved in wound‐healing responses and thus may contribute to SARS‐induced ALI and ARDS.
Figure 1.

MA‐SARS lung immunopathology. (A) Mock‐infected lung stained with haematoxylin and eosin. (B) Large airway of a C57BL/6 J (B6) mouse, 7 days post‐infection, with 105 plaque‐forming units (PFU) MA‐SARS, shows denudation of the epithelial cells. (C, D) Immunohistochemical staining of the SARS‐CoV N protein at 2 days post‐infection shows staining consistent with infection of airway epithelial cells and type II pneumocytes, respectively. (E) MSB staining highlights fibrin in the parenchyma of the lung (red staining) in B6 mice, 7 days post‐infection with 105 PFU MA‐SARS. (F) Perivascular cuffing in a B6 mouse, 4 days post‐infection with 105 PFU MA‐SARS. (G) Hyaline membranes in the parenchyma of the lung of a B6 mouse, 7 days post‐infection with 105 PFU MA‐SARS. (H) Inflammation in the lung of a B6 mouse, 7 days post‐infection with 104 PFU MA‐SARS. (I) Haemorrhage in the lung of a Serpine1 mouse, 7 days post‐infection with 104 PFU MA‐SARS.
Figure 2.
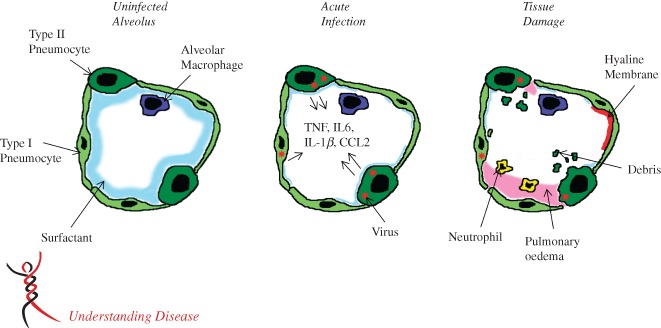
Model of an infected alveolus in the lung. Type I and type II pneumocytes make up the alveolar walls and resident alveolar macrophages and pulmonary surfactant exist in the airspace (A). In the acute phase of SARS‐CoV infection (B), type I and type II pneumocytes are infected and secrete inflammatory cytokines, while surfactant levels decrease. During the late stage/tissue damage portion of viral infection, viral titres decrease, while airway debris, pulmonary oedema and hyaline membrane formation all impede respiration (C).
While SARS‐CoV evades detection by the host immune system and causes minimal changes in transcript and protein levels for the first 24 h of infection 43, it ultimately induces a massive signalling response in infected lungs. Pro‐inflammatory cytokines and chemokines, including IL‐6, TNFα, IL‐1β and CCL2 57, recruit inflammatory cells to the site of infection. Neutrophils and cytotoxic T cells, along with these cytokines, can induce tissue damage, including vascular leakage, and stimulate pulmonary fibrosis 101. Pro‐fibrotic genes, including Tgfβ1, Ctgf and Pdgfa and numerous collagen transcripts, have increased expression following MA‐SARS infection. Fluid exudates, haemorrhage and fibrin are all observed in the alveolar spaces of SARS patients, as well as in animal models of disease 17, 43, 102, increasing in severity as a function of age. In response, the coagulation cascade is activated, including increased factor 10 (FX), F2, F3 (tissue factor), F11, F12 and F7 transcript levels. Activation of the coagulation cascade results in F10 cleavage of prothrombin into thrombin and subsequent thrombin cleavage of fibrinogen into fibrin 103. Fibrin clots in the alveoli are a prominent feature of SARS‐CoV infection in humans and mice. The goal of this coagulation response is likely to protect the host by sealing the alveoli, preventing alveolar flooding and haemorrhage, which limit oxygen exchange and endanger patient survival. Collagen expression is also increased following SARS‐CoV infection 43. Collagen accumulation, fibrin and fibrin clots all contribute to a developing fibrotic lung state, while at the same time stimulating the infected host to up‐regulate fibrinolytic pathways 92.
Profibrinolytic genes include members of the urokinase pathway, such as urokinase (plau), tPA and plasmin (plg) 104. The urokinase signalling pathway leads to cleavage and activation of plasmin into plasminogen; this protease then cleaves fibrin clots. Serpine1 and Serpine2 are negative regulators of urokinase pathway and inhibit urokinase and tissue plasminogen activator (tPA) activity. Urokinase signalling is highly active in the absence of Serpine1, and this imbalance often results in haemorrhage in knockout mice. Serpine1 is highly expressed in SARS patients, non‐human primates and small animal models 43, 74, 105. ARDS studies, independent of coronavirus infection, have attributed this Serpine1 expression to alveolar macrophages and type II pneumocytes 106, 107. MA‐SARS infection in a Serpine1 knockout mouse model results in lethal disease with extreme lung pathology 43. Conversely, MA‐SARS‐infected mice with a genetic deficiency in tPA have increased exudates in the lung. The dysregulation of these coagulation/anti‐coagulation cascades can result in worsening end‐stage lung disease conditions, resulting in death.
Profibrotic and profibrinolytic signalling are part of the wound‐healing response, along with other extracellular matrix (ECM) remodelling pathways 101. SARS‐CoV infection causes massive tissue remodelling through urokinase and coagulation pathways activity, as discussed above. Other important wound‐healing pathways and ECM proteins with altered signalling following SARS‐CoV infection include matrix metalloproteinases, EGFR and collagens 43. Successful recovery from ALI requires a delicate balance of pro‐inflammatory, profibrotic and profibrinolytic responses. By altering ISG expression, including ACE2, STAT1 and Serpine1, SARS‐CoV infection of alveolar epithelial cells sets the stage for the development of severe lung disease, including ARDS.
SARS and MERS patients with severe lung disease exhibited lung consolidation, decreased blood oxygen saturation and often required intubation and ventilation 99, 108, 109. Small animal models of severe lung disease typically lack physiological readouts of respiratory function that can be directly correlated back to human signs of disease. Whole‐body plethysmography captures respiratory data in unrestrained animals, allowing for longitudinal measurement of pulmonary function; these data can also be directly related to some human respiratory metrics 110, 111. SARS‐CoV infection causes increased Penh, a calculated measure of airway resistance and increased EF50 (mid‐breath exhalation force), indicating that respiratory function is compromised and animals must do more work to breathe 69. Unpublished data (Gralinski and Menachery) indicate that it is the exhalation portion of each breath that is impacted by SARS‐CoV infection, likely due to extensive debris clogging the conducting airways. Further experiments have shown that Stat1 knockout mice have increased Penh levels early after infection, and that at late timepoints they have reduced lung capacity, corresponding with the profibrotic histopathological changes observed in the lung (Gralinski, unpublished data).
Concluding thoughts
ARDS is a devastating end‐stage lung disease with no cure. Despite numerous clinical trials, improved clinical outcomes have remained marginal at best 112. Several highly pathogenic emerging virus infections cause ARDS with high frequency, underscoring the critical public health importance of understanding the virological components and molecular mechanisms that drive this devastating end‐stage lung disease. Furthermore, a portion of ARDS cases may progress to pulmonary fibrosis, another clinically devastating end‐stage lung disease with few treatment options. Consequently, understanding the development of ARDS following viral infection remains a high‐priority research topic that is germane to global health and pandemic disease control. The twenty‐first century has demonstrated that zoonotic events will continue to introduce coronaviruses and other viruses into the human population, and that these viruses have the potential to spread rapidly, cause significant disease in communities and disrupt the global economy. An emerging theme is the connectivity between virus infection, complement and coagulation cascade activation, pro‐inflammatory and profibrotic cytokine responses and disease severity. More studies are needed to unravel the complex interactions between these pathways that can interact to promote or dysregulate wound recovery after life‐threatening respiratory virus infection. In particular there is a need for including well‐articulated animal models that faithfully recapitulate disease processes across species. Only through a better understanding of the interplay between a dysregulated host immune response and ALI and ARDS can more effective treatments and therapeutics be developed.
Author contributions
LEG and RSB wrote the paper.
Acknowledgements
This study was supported by funds from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services, contract number HHSN272200800060C, U19 AI109761 and U19 AI100625.
No conflicts of interest were declared.
References
- 1. Johnson ER, Matthay MA. Acute lung injury: epidemiology, pathogenesis, and treatment. J Aerosol Med Pulm Drug Deliv 2010; 23: 243–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Reynolds HN, McCunn M, Borg U, et al. Acute respiratory distress syndrome: estimated incidence and mortality rate in a 5 million‐person population base. Crit Care 1998; 2: 29–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cabeca TK, Granato C, Bellei N. Epidemiological and clinical features of human coronavirus infections among different subsets of patients. Influenza Other Respir Viruses 2013; 7: 1040–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. van der Hoek L, Pyrc K, Jebbink MF, et al. Identification of a new human coronavirus. Nat Med 2004; 10: 368–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Woo PC, Lau SK, Chu CM, et al. Characterization and complete genome sequence of a novel coronavirus, coronavirus HKU1, from patients with pneumonia. J Virol 2005; 79: 884–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Reina J, Lopez‐Causape C, Rojo‐Molinero E, et al. Clinico‐epidemiological characteristics of acute respiratory infections caused by coronavirus OC43, NL63 and 229E. Rev Clin Esp 2014; doi: 10.1016/j.rce.2014.05.020. (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lau SK, Woo PC, Yip CC, et al. Coronavirus HKU1 and other coronavirus infections in Hong Kong. J Clin Microbiol 2006; 44: 2063–2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Drosten C, Gunther S, Preiser W, et al. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N Engl J Med 2003; 348: 1967–1976. [DOI] [PubMed] [Google Scholar]
- 9. http://www.who.int/csr/sars/country/table2004_04_21/en/
- 10. http://www.who.int/csr/don/2014_07_23_mers/en/
- 11.ProMed: http://www.promedmail.org/direct.php?id=20140912.2770252
- 12. Guan Y, Zheng BJ, He YQ, et al. Isolation and characterization of viruses related to the SARS coronavirus from animals in southern China. Science 2003; 302: 276–278. [DOI] [PubMed] [Google Scholar]
- 13. Lau SK, Woo PC, Li KS, et al. Severe acute respiratory syndrome coronavirus‐like virus in Chinese horseshoe bats. Proc Natl Acad Sci USA 2005; 102: 14040–14045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Li W, Shi Z, Yu M, et al. Bats are natural reservoirs of SARS‐like coronaviruses. Science 2005; 310: 676–679. [DOI] [PubMed] [Google Scholar]
- 15. Ren W, Qu X, Li W, et al. Difference in receptor usage between severe acute respiratory syndrome (SARS) coronavirus and SARS‐like coronavirus of bat origin. J Virol 2008; 82: 1899–1907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ge XY, Li JL, Yang XL, et al. Isolation and characterization of a bat SARS‐like coronavirus that uses the ACE2 receptor. Nature 2013; 503: 535–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Franks T, Chong P, Chui P, et al. Lung pathology of severe acute respiratory syndrome (SARS): a study of eight autopsy cases from Singapore. Hum Pathol 2003; 34: 743–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Liu J, Tang X, Jiang S, et al. The chest X‐ray image features of patients with severe SRAS: a preliminary study. Chin Med J (Engl) 2003; 116: 968–971. [PubMed] [Google Scholar]
- 19. Gu J, Korteweg C. Pathology and pathogenesis of severe acute respiratory syndrome. Am J Pathol 2007; 170: 1136–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hwang DM, Chamberlain DW, Poutanen SM, et al. Pulmonary pathology of severe acute respiratory syndrome in Toronto. Mod Pathol 2005; 18: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nicholls J, Dong XP, Jiang G, et al. SARS: clinical virology and pathogenesis. Respirology 2003; 8(suppl): S6–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ding Y, Wang H, Shen H, et al. The clinical pathology of severe acute respiratory syndrome (SARS): a report from China. J Pathol 2003; 200: 282–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hsiao CH, Wu MZ, Chen CL, et al. Evolution of pulmonary pathology in severe acute respiratory syndrome. J Formos Med Assoc 2005; 104: 75–81. [PubMed] [Google Scholar]
- 24. Peiris JS, Chu CM, Cheng VC, et al. Clinical progression and viral load in a community outbreak of coronavirus‐associated SARS pneumonia: a prospective study. Lancet 2003; 361: 1767–1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zaki AM, van Boheemen S, Bestebroer TM, et al. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N Engl J Med 2012; 367: 1814–1820. [DOI] [PubMed] [Google Scholar]
- 26. Al‐Abdallat MM, Payne DC, Alqasrawi S, et al. Hospital‐associated outbreak of Middle East respiratory syndrome coronavirus: a serologic, epidemiologic, and clinical description. Clin Infect Dis 2014; 59: 1225–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Raj VS, Farag EA, Reusken CB, et al. Isolation of MERS coronavirus from a dromedary camel, Qatar, 2014. Emerg Infect Dis 2014; 20: 1339–1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Corman VM, Ithete NL, Richards LR, et al. Rooting the phylogenetic tree of MERS‐coronavirus by characterization of a conspecific virus from an African bat. J Virol 2014; 88: 11297–11303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Reusken CB, Haagmans BL, Muller MA, et al. Middle East respiratory syndrome coronavirus neutralising serum antibodies in dromedary camels: a comparative serological study. Lancet Infect Dis 2013; 13: 859–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Corman VM, Jores J, Meyer B, et al. Antibodies against MERS coronavirus in dromedary camels, Kenya, 1992–2013. Emerg Infect Dis 2014; 20: 1319–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fouchier RA, Kuiken T, Schutten M, et al. Aetiology: Koch's postulates fulfilled for SARS virus. Nature 2003; 423: 240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Haagmans BL, Kuiken T, Martina BE, et al. Pegylated interferon‐α protects type 1 pneumocytes against SARS coronavirus infection in macaques. Nat Med 2004; 10: 290–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. McAuliffe J, Vogel L, Roberts A, et al. Replication of SARS coronavirus administered into the respiratory tract of African green, rhesus and cynomolgus monkeys. Virology 2004; 330: 8–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Falzarano D, de Wit E, Feldmann F, et al. Infection with MERS‐CoV causes lethal pneumonia in the common marmoset. PLoS Pathog 2014; 10: e1004250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. de Wit E, Rasmussen AL, Falzarano D, et al. Middle East respiratory syndrome coronavirus (MERS‐CoV) causes transient lower respiratory tract infection in rhesus macaques. Proc Natl Acad Sci USA 2013; 110: 16598–16603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Subbarao K, McAuliffe J, Vogel L, et al. Prior infection and passive transfer of neutralizing antibody prevent replication of severe acute respiratory syndrome coronavirus in the respiratory tract of mice. J Virol 2004; 78: 3572–3577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Glass WG, Subbarao K, Murphy B, et al. Mechanisms of host defense following severe acute respiratory syndrome‐coronavirus (SARS‐CoV) pulmonary infection of mice. J Immunol 2004; 173: 4030–4039. [DOI] [PubMed] [Google Scholar]
- 38. Wentworth DE, Gillim‐Ross L, Espina N, et al. Mice susceptible to SARS coronavirus. Emerg Infect Dis 2004; 10: 1293–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Roberts A, Paddock C, Vogel L, et al. Aged BALB/c mice as a model for increased severity of severe acute respiratory syndrome in elderly humans. J Virol 2005; 79: 5833–5838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Roberts A, Deming D, Paddock CD, et al. A mouse‐adapted SARS‐coronavirus causes disease and mortality in BALB/c mice. PLoS Pathog 2007; 3: e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sheahan T, Whitmore A, Long K, et al. Successful vaccination strategies that protect aged mice from lethal challenge from influenza virus and heterologous severe acute respiratory syndrome coronavirus. J Virol; 85: 217–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Frieman M, Yount B, Agnihothram S, et al. Molecular determinants of severe acute respiratory syndrome coronavirus pathogenesis and virulence in young and aged mouse models of human disease. J Virol 2012; 86: 884–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gralinski LE, Bankhead A, 3rd , Jeng S, et al. Mechanisms of severe acute respiratory syndrome coronavirus‐induced acute lung injury. mBio 2013; 4: e00271–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhao J, Li K, Wohlford‐Lenane C, et al. Rapid generation of a mouse model for Middle East respiratory syndrome. Proc Natl Acad Sci USA; 111: 4970–4975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Collins SE, Mossman KL. Danger, diversity and priming in innate antiviral immunity. Cytokine Growth Factor Rev 2014, In press. [DOI] [PubMed] [Google Scholar]
- 46. Lancellotti M, Pereira RF, Cury GG, et al. Pathogenic and opportunistic respiratory bacteria‐induced apoptosis. Braz J Infect Dis 2009; 13: 226–231. [DOI] [PubMed] [Google Scholar]
- 47. DeDiego ML, Nieto‐Torres JL, Regla‐Nava JA, et al. Inhibition of NF‐κB‐mediated inflammation in severe acute respiratory syndrome coronavirus‐infected mice increases survival. J Virol 2014; 88: 913–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Frieman M, Ratia K, Johnston RE, et al. SARS‐CoV papain‐like protease ubiquitin‐like domain and catalytic domain regulate antagonism of IRF3 and NF‐κB signaling. J Virol 2009; 83: 6689–6705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. DeDiego ML, Nieto‐Torres JL, Jimenez‐Guardeno JM, et al. Severe acute respiratory syndrome coronavirus envelope protein regulates cell stress response and apoptosis. PLoS Pathog 2011; 7: e1002315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. DeDiego ML, Nieto‐Torres JL, Jimenez‐Guardeno JM, et al. Coronavirus virulence genes with main focus on SARS‐CoV envelope gene. Virus Res 2014; doi: 10.1016/j.virusres.2014.07.024. (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lamirande EW, DeDiego ML, Roberts A, et al. A live attenuated severe acute respiratory syndrome coronavirus is immunogenic and efficacious in golden Syrian hamsters. J Virol 2008; 82: 7721–7724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Fett C, DeDiego ML, Regla‐Nava JA, et al. Complete protection against severe acute respiratory syndrome coronavirus‐mediated lethal respiratory disease in aged mice by immunization with a mouse‐adapted virus lacking E protein. J Virol 2013; 87: 6551–6559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Almazan F, DeDiego ML, Sola I, et al. Engineering a replication‐competent, propagation‐defective Middle East respiratory syndrome coronavirus as a vaccine candidate. mBio 2013; 4: e00650–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Cinatl J, Morgenstern B, Bauer G, et al. Treatment of SARS with human interferons. Lancet 2003; 362: 293–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Strayer DR, Dickey R, Carter WA. Sensitivity of SARS/MERS CoV to interferons and other drugs based on achievable serum concentrations in humans. Infect Disord Drug Targets 2014; 14: 37–43. [DOI] [PubMed] [Google Scholar]
- 56. Frieman MB, Chen J, Morrison TE, et al. SARS‐CoV pathogenesis is regulated by a STAT1 dependent but a type I, II and III interferon receptor independent mechanism. PloS Pathog 2010; 6: e1000849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sheahan T, Morrison TE, Funkhouser W, et al. MyD88 is required for protection from lethal infection with a mouse‐adapted SARS‐CoV. PLoS Pathog 2008; 4 : e1000240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Frieman M, Heise M, Baric R. SARS coronavirus and innate immunity. Virus Res 2008; 133: 101–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kopecky‐Bromberg SA, Martínez‐Sobrido L, Frieman M, et al. Severe acute respiratory syndrome coronavirus open reading frame (ORF) 3b, ORF 6, and nucleocapsid proteins function as interferon antagonists. J Virol 2007; 81: 548–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Yang Y, Zhang L, Geng H, et al. The structural and accessory proteins M, ORF 4a, ORF 4b, and ORF 5 of Middle East respiratory syndrome coronavirus (MERS‐CoV) are potent interferon antagonists. Protein Cell 2013; 4: 951–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Matthews KL, Coleman CM, van der Meer Y, et al. The ORF4b‐encoded accessory proteins of Middle East respiratory syndrome coronavirus and two related bat coronaviruses localize to the nucleus and inhibit innate immune signalling. J Gen Virol 2014; 95: 874–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Totura AL, Baric RS. SARS coronavirus pathogenesis: host innate immune responses and viral antagonism of interferon. Curr Opin Virol 2012; 2: 264–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Menachery VD, Eisfeld AJ, Schafer A, et al. Pathogenic influenza viruses and coronaviruses utilize similar and contrasting approaches to control interferon‐stimulated gene responses. mBio 2014; 5: e01174–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. van Hemert MJ, van den Worm SH, Knoops K, et al. SARS‐coronavirus replication/transcription complexes are membrane‐protected and need a host factor for activity in vitro . PloS Pathog 2008; 4: e1000054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Knoops K, Kikkert M, Worm SH, et al. SARS‐coronavirus replication is supported by a reticulovesicular network of modified endoplasmic reticulum. PLoS Biol 2008; 6: e226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kato H, Takeuchi O, Mikamo‐Satoh E, et al. Length‐dependent recognition of double‐stranded ribonucleic acids by retinoic acid‐inducible gene‐I and melanoma differentiation‐associated gene 5. J Exp Med 2008; 205: 1601–1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Diamond MS, Farzan M. The broad‐spectrum antiviral functions of IFIT and IFITM proteins. Nat Rev Immunol 2013; 13: 46–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Daffis S, Szretter KJ, Schriewer J, et al. 2'‐O methylation of the viral mRNA cap evades host restriction by IFIT family members. Nature 2010; 468: 452–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Menachery VD, Yount BL, Jr. , Josset L, et al. Attenuation and restoration of severe acute respiratory syndrome coronavirus mutant lacking 2′‐O‐methyltransferase activity. J Virol 2014; 88: 4251–4264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Baas T, Taubenberger JK, Chong PY, et al. SARS‐CoV virus–host interactions and comparative etiologies of acute respiratory distress syndrome as determined by transcriptional and cytokine profiling of formalin‐fixed paraffin‐embedded tissues. J Interferon Cytokine Res 2006; 26: 309–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Frieman M, Yount B, Heise M, et al. Severe acute respiratory syndrome coronavirus ORF6 antagonizes STAT1 function by sequestering nuclear import factors on the rough endoplasmic reticulum/Golgi membrane. J Virol 2007; 81: 9812–9824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Zhao J, Falcon A, Zhou H, et al. Severe acute respiratory syndrome coronavirus protein 6 is required for optimal replication. J Virol 2009; 83: 2368–2373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. de Wilde AH, Raj VS, Oudshoorn D, et al. MERS‐coronavirus replication induces severe in vitro cytopathology and is strongly inhibited by cyclosporin A or interferon‐α treatment. J Gen Virol 2013; 94: 1749–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. de Lang A, Baas T, Teal T, et al. Functional genomics highlights differential induction of antiviral pathways in the lungs of SARS‐CoV‐infected macaques. PLoS Pathog 2007; 3: e112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Meraz MA, White JM, Sheehan KC, et al. Targeted disruption of the Stat1 gene in mice reveals unexpected physiologic specificity in the JAK–STAT signaling pathway. Cell 1996; 84: 431–442. [DOI] [PubMed] [Google Scholar]
- 76. Hogan RJ, Gao G, Rowe T, et al. Resolution of primary severe acute respiratory syndrome‐associated coronavirus infection requires Stat1 . J Virol 2004; 78: 11416–11421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Zornetzer GA, Frieman MB, Rosenzweig E, et al. Transcriptomic analysis reveals a mechanism for a prefibrotic phenotype in STAT1 knockout mice during severe acute respiratory syndrome coronavirus infection. J Virol 2010; 84: 11297–11309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Page C, Goicochea L, Matthews K, et al. Induction of alternatively activated macrophages enhances pathogenesis during severe acute respiratory syndrome coronavirus infection. J Virol 2012; 86: 13334–13349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Hamming I, Timens W, Bulthuis ML, et al. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J Pathol 2004; 203: 631–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Kuba K, Imai Y, Rao S, et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus‐induced lung injury. Nat Med 2005; 11: 875–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Dijkman R, Jebbink MF, Deijs M, et al. Replication‐dependent downregulation of cellular angiotensin‐converting enzyme 2 protein expression by human coronavirus NL63. J Gen Virol 2012; 93: 1924–1929. [DOI] [PubMed] [Google Scholar]
- 82. Imai Y, Kuba K, Rao S, et al. Angiotensin‐converting enzyme 2 protects from severe acute lung failure. Nature 2005; 436: 112–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Guy JL, Lambert DW, Warner FJ, et al. Membrane‐associated zinc peptidase families: comparing ACE and ACE2. Biochim Biophys Acta 2005; 1751: 2–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Kuba K, Imai Y, Penninger JM. Angiotensin‐converting enzyme 2 in lung diseases. Curr Opin Pharmacol 2006; 6: 271–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Arndt PG, Young SK, Poch KR, et al. Systemic inhibition of the angiotensin‐converting enzyme limits lipopolysaccharide‐induced lung neutrophil recruitment through both bradykinin and angiotensin II‐regulated pathways. J Immunol 2006; 177: 7233–7241. [DOI] [PubMed] [Google Scholar]
- 86. Gong MN. Genetic epidemiology of acute respiratory distress syndrome: implications for future prevention and treatment. Clin Chest Med 2006; 27: 705–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Malcolm KC, Kret JE, Young RL, et al. Bacteria‐specific neutrophil dysfunction associated with interferon‐stimulated gene expression in the acute respiratory distress syndrome. PLoS One 2011; 6: e21958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Cameron MJ, Ran L, Xu L, et al. Interferon‐mediated immunopathological events are associated with atypical innate and adaptive immune responses in patients with severe acute respiratory syndrome. J Virol 2007; 81: 8692–8706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Verma GP. Fundamentals of Histology. New Age International Pvt Ltd Publishers: New Delhi, India; 2001. [Google Scholar]
- 90. Fehrenbach H. Alveolar epithelial type II cell: defender of the alveolus revisited. Respir Res 2001; 2: 33–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Zhao J, Zhao J, Van Rooijen N, et al. Evasion by stealth. inefficient immune activation underlies poor T cell response and severe disease in SARS‐CoV infected mice. PloS Pathog 2009; 5: e1000636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Chambers RC, Scotton CJ. Coagulation cascade proteinases in lung injury and fibrosis. Proc Am Thorac Soc 2012; 9: 96–101. [DOI] [PubMed] [Google Scholar]
- 93. To KF, Tong JH, Chan PK, et al. Tissue and cellular tropism of the coronavirus associated with severe acute respiratory syndrome: an in‐situ hybridization study of fatal cases. J Pathol 2004; 202: 157–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Weinheimer VK, Becher A, Tonnies M, et al. Influenza A viruses target type II pneumocytes in the human lung. J Infect Dis 2012; 206: 1685–1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Gunther A, Ruppert C, Schmidt R, et al. Surfactant alteration and replacement in acute respiratory distress syndrome. Respir Res 2001; 2: 353–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Wong CK, Lam CW, Wu AK, et al. Plasma inflammatory cytokines and chemokines in severe acute respiratory syndrome. Clin Exp Immunol 2004; 136: 95–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Ng PC, Lam CW, Li AM, et al. Inflammatory cytokine profile in children with severe acute respiratory syndrome. Pediatrics 2004; 113: e7–14. [DOI] [PubMed] [Google Scholar]
- 98. Cheung OY, Chan JW, Ng CK, et al. The spectrum of pathological changes in severe acute respiratory syndrome (SARS). Histopathology 2004; 45: 119–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Booth CM, Matukas LM, Tomlinson GA, et al. Clinical features and short‐term outcomes of 144 patients with SARS in the greater Toronto area. J Am Med Assoc 2003; 289: 2801–2809. [DOI] [PubMed] [Google Scholar]
- 100. Haagmans BL, Osterhaus AD. Nonhuman primate models for SARS. PLoS Med 2006; 3: e194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Wygrecka M, Jablonska E, Guenther A, et al. Current view on alveolar coagulation and fibrinolysis in acute inflammatory and chronic interstitial lung diseases. Thromb Haemost 2008; 99: 494–501. [DOI] [PubMed] [Google Scholar]
- 102. Smits SL, van den Brand JM, de Lang A, et al. Distinct severe acute respiratory syndrome coronavirus‐induced acute lung injury pathways in two different nonhuman primate species. J Virol; 85: 4234–4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Adams RL, Bird RJ. Review article: coagulation cascade and therapeutics update: relevance to nephrology. Part 1: Overview of coagulation, thrombophilias and history of anticoagulants. Nephrology (Carlton) 2009; 14: 462–470. [DOI] [PubMed] [Google Scholar]
- 104. Cesarman‐Maus G, Hajjar KA. Molecular mechanisms of fibrinolysis. Br J Haematol 2005; 129: 307–321. [DOI] [PubMed] [Google Scholar]
- 105. Zhao X, Nicholls JM, Chen YG. Severe acute respiratory syndrome‐associated coronavirus nucleocapsid protein interacts with Smad3 and modulates transforming growth factor‐β signaling. J Biol Chem 2008; 283: 3272–3280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Wygrecka M, Markart P, Ruppert C, et al. Compartment‐ and cell‐specific expression of coagulation and fibrinolysis factors in the murine lung undergoing inhalational versus intravenous endotoxin application. Thromb Haemost 2004; 92: 529–540. [DOI] [PubMed] [Google Scholar]
- 107. Senoo T, Hattori N, Tanimoto T, et al. Suppression of plasminogen activator inhibitor‐1 by RNA interference attenuates pulmonary fibrosis. Thorax 2010; 65: 334–340. [DOI] [PubMed] [Google Scholar]
- 108. Sung JJ, Wu A, Joynt GM, et al. Severe acute respiratory syndrome: report of treatment and outcome after a major outbreak. Thorax 2004; 59: 414–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Guery B, Poissy J, el Mansouf L, et al. Clinical features and viral diagnosis of two cases of infection with Middle East respiratory syndrome coronavirus: a report of nosocomial transmission. Lancet 2013; 381: 2265–2272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Zhang Q, Lai K, Xie J, et al. Does unrestrained single‐chamber plethysmography provide a valid assessment of airway responsiveness in allergic BALB/c mice? Respir Res 2009; 10: 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Criee CP, Sorichter S, Smith HJ, et al. Body plethysmography – its principles and clinical use. Respir Med 2011; 105: 959–971. [DOI] [PubMed] [Google Scholar]
- 112. Thompson BT, Bernard GR. ARDS Network (NHLBI) studies: successes and challenges in ARDS clinical research. Crit Care Clin 2011; 27: 459–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Vlasak R, Luytjes W, Spaan W, et al. Human and bovine coronaviruses recognize sialic acid‐containing receptors similar to those of influenza C viruses. Proc Natl Acad Sci USA 1988; 85: 4526–4529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Collins AR. HLA class I antigen serves as a receptor for human coronavirus OC43. Immunol Invest 1993; 22: 95–103. [DOI] [PubMed] [Google Scholar]
- 115. Vijgen L, Keyaerts E, Moes E, et al. Complete genomic sequence of human coronavirus OC43: molecular clock analysis suggests a relatively recent zoonotic coronavirus transmission event. J Virol 2005; 79: 1595–1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Dijkman R, Jebbink MF, Koekkoek SM, et al. Isolation and characterization of current human coronavirus strains in primary human epithelial cell cultures reveal differences in target cell tropism. J Virol 2013; 87: 6081–6090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Collins AR. Human macrophages are susceptible to coronavirus OC43. Adv Exp Med Biol 1998; 440: 635–639. [DOI] [PubMed] [Google Scholar]
- 118. Bonavia A, Arbour N, Yong VW, et al. Infection of primary cultures of human neural cells by human coronaviruses 229E and OC43. J Virol 1997; 71: 800–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Vabret A, Mourez T, Gouarin S, et al. An outbreak of coronavirus OC43 respiratory infection in Normandy, France. Clin Infect Dis 2003; 36: 985–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Yeager CL, Ashmun RA, Williams RK, et al. Human aminopeptidase N is a receptor for human coronavirus 229E. Nature 1992; 357: 420–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Pfefferle S, Oppong S, Drexler JF, et al. Distant relatives of severe acute respiratory syndrome coronavirus and close relatives of human coronavirus 229E in bats, Ghana. Emerg Infect Dis 2009; 15: 1377–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Pyrc K, Dijkman R, Deng L, et al. Mosaic structure of human coronavirus NL63; one thousand years of evolution. J Mol Biol 2006; 364: 964–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Patterson S, Macnaughton MR. Replication of human respiratory coronavirus strain 229E in human macrophages. J Gen Virol 1982; 60: 307–314. [DOI] [PubMed] [Google Scholar]
- 124. Reed SE. The behaviour of recent isolates of human respiratory coronavirus in vitro and in volunteers: evidence of heterogeneity among 229E‐related strains. J Med Virol 1984; 13: 179–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Pene F, Merlat A, Vabret A, et al. Coronavirus 229E‐related pneumonia in immunocompromised patients. Clin Infect Dis 2003; 37: 929–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Hofmann H, Pyrc K, van der Hoek L, et al. Human coronavirus NL63 employs the severe acute respiratory syndrome coronavirus receptor for cellular entry. Proc Natl Acad Sci USA 2005; 102: 7988–7993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Huynh J, Li S, Yount B, et al. Evidence supporting a zoonotic origin of human coronavirus strain NL63. J Virol 2012; 86: 12816–12825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Arden KE, Nissen MD, Sloots TP, et al. New human coronavirus, HCoV‐NL63, associated with severe lower respiratory tract disease in Australia. J Med Virol 2005; 75: 455–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. van der Hoek L, Sure K, Ihorst G, et al. Croup is associated with the novel coronavirus NL63. PLoS Med 2005; 2: e240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Vabret A, Dina J, Gouarin S, et al. Detection of the new human coronavirus HKU1: a report of six cases. Clin Infect Dis 2006; 42: 634–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Risku M, Lappalainen S, Rasanen S, et al. Detection of human coronaviruses in children with acute gastroenteritis. J Clin Virol 2010; 48: 27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Yang ZY, Huang Y, Ganesh L, et al. pH‐dependent entry of severe acute respiratory syndrome coronavirus is mediated by the spike glycoprotein and enhanced by dendritic cell transfer through DC‐SIGN. J Virol 2004; 78: 5642–5650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Ksiazek TG, Erdman D, Goldsmith CS, et al. A novel coronavirus associated with severe acute respiratory syndrome. N Engl J Med 2003; 348: 1953–1966. [DOI] [PubMed] [Google Scholar]
- 134. Vijaykrishna D, Smith GJ, Zhang JX, et al. Evolutionary insights into the ecology of coronaviruses. J Virol 2007; 81: 4012–4020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Hon CC, Lam TY, Shi ZL, et al. Evidence of the recombinant origin of a bat severe acute respiratory syndrome (SARS)‐like coronavirus and its implications on the direct ancestor of SARS coronavirus. J Virol 2008; 82: 1819–1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Sims AC, Baric RS, Yount B, et al. Severe acute respiratory syndrome coronavirus infection of human ciliated airway epithelia: role of ciliated cells in viral spread in the conducting airways of the lungs. J Virol 2005; 79: 15511–15524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Nicholls JM, Butany J, Poon LL, et al. Time course and cellular localization of SARS‐CoV nucleoprotein and RNA in lungs from fatal cases of SARS. PLoS Med 2006; 3: e27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Nicholls JM, Poon LL, Lee KC, et al. Lung pathology of fatal severe acute respiratory syndrome. Lancet 2003; 361: 1773–1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Raj VS, Mou H, Smits SL, et al. Dipeptidyl peptidase 4 is a functional receptor for the emerging human coronavirus‐EMC. Nature; 495: 251–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Cotten M, Watson SJ, Zumla AI, et al. Spread, circulation, and evolution of the Middle East respiratory syndrome coronavirus. mBio 2014; 5: e01062–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Zielecki F, Weber M, Eickmann M, et al. Human cell tropism and innate immune system interactions of human respiratory coronavirus EMC compared to those of severe acute respiratory syndrome coronavirus. J Virol 2013; 87: 5300–5304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Eckerle I, Muller MA, Kallies S, et al. In‐vitro renal epithelial cell infection reveals a viral kidney tropism as a potential mechanism for acute renal failure during Middle East respiratory syndrome (MERS) coronavirus infection. Virol J 2013; 10: 359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Chu H, Zhou J, Wong BH, et al. Productive replication of Middle East respiratory syndrome coronavirus in monocyte‐derived dendritic cells modulates innate immune response. Virology 2014; 454–455: 197–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Memish ZA, Zumla AI, Al‐Hakeem RF, et al. Family cluster of Middle East respiratory syndrome coronavirus infections. N Engl J Med 2013; 368: 2487–2494. [DOI] [PubMed] [Google Scholar]
- 145. Roberts A, Vogel L, Guarner J, et al. Severe acute respiratory syndrome coronavirus infection of golden Syrian hamsters. J Virol 2005; 79: 503–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Martina BE, Haagmans BL, Kuiken T, et al. Virology: SARS virus infection of cats and ferrets. Nature 2003; 425: 915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Rowe T, Gao G, Hogan RJ, et al. Macaque model for severe acute respiratory syndrome. J Virol 2004; 78: 11401–11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Lawler JV, Endy TP, Hensley LE, et al. Cynomolgus macaque as an animal model for severe acute respiratory syndrome. PLoS Med 2006; 3: e149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Smits SL, de Lang A, van den Brand JM, et al. Exacerbated innate host response to SARS‐CoV in aged non‐human primates. PLoS Pathog 2010; 6: e1000756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. McCray PB, Jr. , Pewe L, Wohlford‐Lenane C, et al. Lethal infection of K18‐hACE2 mice infected with severe acute respiratory syndrome coronavirus. J Virol 2007; 81: 813–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Mantovani A, Biswas SK, Galdiero MR, Sica A, Locati M. Macrophage plasticity and polarization in tissue repair and remodelling. J Pathol 2013; 229: 176–185. [DOI] [PubMed] [Google Scholar]
- 152.van den Brand JMA, Smits SL. Haagmans BL. Pathogenesis of Middle East respiratory syndrome coronavirus. J Pathol 2015; 235: 175–184. [DOI] [PMC free article] [PubMed] [Google Scholar]