

Abstract
Effective combined antiretroviral therapy (cART) in HIV infected patients has made HIV a treatable condition; however, debilitating HIV-associated neurocognitive disorders (HAND) can still affect up to 50% of HIV infected individuals even under cART. While cART has greatly reduced the prevalence of the most severe form of HAND, milder forms have increased in prevalence, leaving a the total proportion of HIV-infected individuals suffering from HAND relatively unchanged. In this study an in vitro drug screen identified fluconazole and paroxetine as protective compounds against HIV gp120 and Tat neurotoxicity. Using an accelerated, consistent SIV/macaque model of HIV-associated CNS disease, we tested the in vivo neuroprotective capabilities of combination fluconazole/paroxetine (FluPar) treatment. FluPar treatment protected macaques from SIV-induced neurodegeneration, as measured by neurofilament light chain in the CSF, APP accumulation in the axons, and CaMKIIα in the frontal cortex, but did not significantly reduce markers of neuroinflammation or plasma or CNS viral loads. Since HIV and SIV neurodegeneration is often attributed to accompanying neuroinflammation, this study provides proof of concept that neuroprotection can be achieved even in the face of ongoing neuroinflammation and viral replication.
Keywords: HIV, SIV, fluconazole, paroxetine, selective serotonin reuptake inhibitor, CNS
Introduction
HIV-associated neurocognitive disorders (HAND) are a significant, life-altering complication of HIV infection that can persist even when viremia is suppressed by combination antiretroviral therapy (cART) (Simioni et al, 2010). HAND encompasses a spectrum of disorders ranging from asymptomatic neurocognitive impairment (ANI), which does not affect the patient’s daily function, to mild neurocognitive disorder (MND), which affects the patient’s daily function at work, school, or home, to HIV-associated dementia (HAD), which is a highly debilitating, frank dementia (Antinori et al, 2007). Although modern cART regimens have decreased the prevalence of HAD, the prevalence of the more minor conditions (ANI and MND) has increased, thus maintaining the prevalence of HAND around 50% in the HIV-infected population (Ances and Ellis, 2007; Heaton et al, 2010). Despite many years of investigation and many clinical trials, no treatment for HAND exists.
Given the need for neuroprotective compounds to treat HAND, an in vitro drug screen was used to identify compounds that would protect mixed hippocampal cultures from HIV gp120 and Tat neurotoxicity (Nath et al, 2012). Fluconazole, an antifungal, and paroxetine, a selective serotonin reuptake inhibitor (SSRI), proved neuroprotective in this screen. These drugs were attractive neuroprotective agents because they are already approved for long-term use in humans and are well-tolerated for prolonged use.
Paroxetine was particularly appealing for HAND treatment because of a study by Letendre and colleagues, who demonstrated that patients taking selective serotonin reuptake inhibitors were less likely to have detectable viral loads in the CSF and performed better in neuropsychological testing (Letendre et al, 2007). Additionally, SSRIs or serotonin alone, have been neuroprotective in animal models of Parkinson’s disease, Alzheimer’s disease, and Huntington’s disease (Chiou et al, 2006; Chung et al, 2010; Chung et al, 2011; Mattson et al, 2004; Nelson et al, 2007; Peng et al, 2008; Shibui et al, 2009). Most relevant to this study, Lee and colleagues showed that paroxetine reversed the loss of amplifying neuroprogenitor cells in the hippocampus of gp120 transgenic mice (Lee et al, 2011). Furthermore, SSRIs or serotonin alone inhibited HIV replication in vitro in both cell lines and primary monocyte derived macrophages (Benton et al, 2010; Kristiansen and Hansen, 2000; Maneglier et al, 2008), which belong to the same family of cells as perivascular macrophages and microglia, the productively infected cells in the CNS.
The in vitro drug screen data together with the literature on SSRI neuroprotection and inhibition of HIV replication inspired both the preclinical evaluation of potential protective effects of fluconazole and paroxetine in an accelerated, consistent SIV/pigtailed macaque model of HIV-associated CNS disease and a Phase I/II human clinical trial, which is still enrolling participants. Using the SIV model of HIV-associated CNS disease, we found FluPar treatment to be protective against neurodegeneration without blunting neuroinflammation or viral replication in the CNS or plasma.
Methods
Cell culture
Neuronal cultures from rodent hippocampus were prepared from embryonic day 18 Sprague–Dawley rats using methods similar to those described previously (Haughey et al, 2004). Tissues were dissociated by gentle trituration with a firepolished glass pipette in calcium-free Hank’s balanced salt solution. The single cell suspension was centrifuged at 1000 g and re-suspended in minimal essential medium containing 10% heat-inactivated fetal bovine serum and 1% (v/v) antibiotic and antimycotic solution (penicillin G 104 units/mL, streptomycin 10 mg/mL and amphotericin B 25 mg/mL; Sigma; St. Louis, MO). Cells were allowed to attach for 3 h before the media was replaced with serum-free neurobasal medium containing 2% (v/v) B-27 supplement (Gibco; Rockville, MD) and 1% (v/v) antibiotic and antimycotic mix (Sigma). Rat mixed hippocampal neurons were generated from freshly cultured rat hippocampi in neurobasal media containing 5% (v/v) fetal bovine serum and 2% (v/v) B27 supplement. Hippocampal neurons were plated in 96-well plates at a density of 4 × 105 cells per milliliter for neurotoxicity studies.
Compound screening
The Microsource Discovery Spectrum Collection of compounds contains 2000 compounds, of which about half are FDA approved drugs and the remaining compounds are natural products or other compounds with some prior human exposure and safety testing data. The compound collection is dispensed and maintained on 96 well plates at a concentration of 10 mM in 100% DMSO and stored at −80°C. The compound mother plates were thawed one time in order to make 4 sets of daughter plates, and one set of daughter plates was used for these screening assays.
Previously, a total of 2000 compounds were screened using the oxidative stressor 3-nitropropionic acid (3-NP) neurotoxicity assay in rat mixed hippocampal cultures, with screening at 10uM drug concentration against the toxic effects of 3mM 3-NP for 24 h (Nath et al, 2012). From these screening assays, 256 compounds showed > 50% protection. Additionally, of these “hits,” 146 compounds displayed complete (100% protection) and 53 demonstrated > 200% protection. We then tested the efficacy of these compounds for neuroprotection against HIV gp120 and Tat by measuring cell survival using the MTT assay as described previously (Nath et al, 2012).
Cell survival assay
Neuronal cell viability was assessed with 3-(4,5-dimethylthiazol-2-yl)- 2,5-diphenyltetrazolium bromide (MTT) assay. This assay (Mosmann, 1983) is based on the ability of a mitochondrial dehydrogenase enzyme from viable cells to cleave the tetrazolium rings of the pale yellow MTT and form dark blue formazan crystals. These crystals are largely impermeable to cell membranes, and thus, accumulate within healthy cells. The resultant formazan precipitates are solubilized with dimethyl sulfoxide (DMSO) and read on a multiwell scanning spectrophotometer (SpectraMAX M5e, Molecular Devices). The number of surviving cells is directly proportional to the level of the formazan product created.
Animal infection and treatment
Pigtailed macaques (Macaca nemestrina) were used for these studies. Prior to inoculation, blood and CSF were collected at least three times, at least two weeks apart, under ketamine anesthesia, to establish baseline levels of several parameters. Six macaques were mock-inoculated procedural controls. Twenty-one macaques were co-inoculated intravenously with an immunosuppressive swarm (SIV/DeltaB670) and a neurovirulent clone (SIV/17E-Fr) of SIV (Zink et al, 1999). Fifteen SIV-infected macaques remained untreated and 6 were treated orally with a combination of fluconazole (12.5 mg, once daily) and paroxetine (5 mg once daily) beginning at 12 days postinoculation (dpi) and continuing until euthanasia. SIV-infected macaques were scheduled for euthanasia at approximately 84 dpi, the time at which all animals have AIDS and most have developed encephalitis in this model (Clements et al, 2008; Zink et al, 1999). If an animal developed clinical symptoms, as described previously (Weed et al, 2003), prior to the scheduled euthanasia date, the animal was humanely euthanized. Blood and CSF were collected at days 7, 10, 14, 21, and 28 dpi, every two weeks thereafter, and at euthanasia. At euthanasia, the macaques were perfused with PBS under barbiturate anesthesia to remove blood from the vasculature. The brain was sliced in 0.5 cm coronal sections. Portions of the brain and other tissues were snap frozen then stored at −80°C until use, while other portions were fixed and paraffin embedded. The Johns Hopkins Animal Care and Use Committee approved all animal studies. Animals were treated humanely in accordance with federal guidelines.
SIV RNA measurement in plasma, CSF, and brain by qRT-PCR
SIV RNA was measured in plasma, CSF, brain and spleen by qRT-PCR as described previously (Meulendyke et al, 2012). SIV RNA levels were expressed as copy equivalents per mL of plasma/CSF or per μg tissue RNA. Statistical analyses of viral loads were performed on log-transformed values.
CCL2 and IL-6 measurement by ELISA
CCL2 and IL-6 were measured by ELISA in CSF collected at various points throughout infection from SIV-infected, untreated and FluPar-treated macaques as described previously (Mankowski et al, 2004).
Immunohistochemistry neuronal damage and neuroinflammatory markers
Amyloid precursor protein (APP) accumulates in damaged axons and therefore was quantified in the long white matter tracts of the corpus callosum. Immunohistochemical staining and quantitation of APP was performed as described previously (Mankowski et al, 2002b). Quantitative immunohistochemistry for the neuroinflammatory markers CD68 (marker of macrophage/microglial activation) and MHC class II (marker of macrophage/microglial and endothelial cell activation) was performed on deep white matter, adjacent to the basal ganglia as described previously (Clements et al, 2002; Zink et al, 2001). Quantitation of all these markers was performed as described previously, with the exception that the microscope, camera and software were Nikon Eclipse 90i (microscope) with motorized stage, Nikon DS-Ri1 (camera), NIS Elements AR (software; all microscopy tools from Nikon; Melville, NY).
Neurofilament light chain measurement by ELISA
The presence of neurofilament light chain in the CSF indicates neuronal damage in several neurodegenerative diseases, including HIV (Abdulle et al, 2007; Jessen Krut et al, 2014; Norgren et al, 2003; Rosengren et al, 1996). Neurofilament light chain was measured in CSF from SIV-infected, untreated and SIV-infected, FluPar-treated macaques using a highly sensitive, commercially available ELISA (IBL International; Toronto, ON). Data were expressed as fold change in CSF sampled prior to euthanasia compared to preinoculation levels.
CaMKIIα detection by Western blotting
Samples from frontal cortex gray matter of uninfected, SIV-infected, untreated and SIV-infected, FluPar-treated macaques were homogenized in 0.5% NP-40 lysis buffer containing protease inhibitor cocktail (Sigma), insoluble debris was removed by centrifugation, and protein was quantitated using the BioRad Protein Assay Kit (BioRad; Hercules, CA). Twenty-five micrograms of protein per sample were resolved under reducing conditions using 10% bis-tris gels and MOPS buffer and then transferred to PVDF membranes. Membranes were blocked in TBST (20 mM Tris, 137 mM NaCl, pH 7.5, 0.1% Tween-20) containing 5% nonfat dry milk then probed using rabbit anti-CaMKII (pan) primary antibodies (1:1000, Cell Signaling Technology; Danvers, MA) diluted in TBST containing 5% BSA for 2 days at 4°C. Membranes were washed with TBST then probed with goat-anti-rabbit-HRP antibodies (1:10,000; Dako; Glostrup, Denmark) diluted in 5% BSA-TBST, washed, then finally developed with Super Signal West Dura Substrate (ThermoFisher Scientific; Waltham, MA). The CaMKα isoform was distinguished from other isoforms by molecular weight (~50 kDa). Membranes were then probed using mouse-anti-ß-actin primary antibodies (1:5000; Sigma-Aldrich) diluted in 5% BSA-TBST. Membranes were washed with TBST then probed with goat-anti-mouse-HRP antibodies (1:10,000; Dako) diluted in 5% BSA-TBST, washed, then finally developed with Super Signal West Pico Substrate (ThermoFisher Scientific). Images of the films were digitally scanned then quantitated using ImageQuant TL 7.0 software (Amersham; Piscataway, NJ). To normalize each sample for protein loading, CaMKIIα was normalized to ß-actin. The experiment was performed in triplicate. To account for blot-to-blot variation, the CaMKIIα/ß-actin ratio of each sample was normalized to one of the uninfected controls and the final quantitation represented the average of the triplicates.
Striatal serotonin measurement by ELISA
Striatal samples were weighed, diluted 1/20 (weight/volume) in 0.1% ascorbic acid, and homogenized by sonication. Insoluble debris was removed by centrifugation and serotonin was measured using an ultrasensitive ELISA according to the manufacturer’s protocol (Eagle Biosciences; Nashua, NH). Data were expressed as ng serotonin per mg tissue.
Neopterin measurement by ELISA
Neopterin, a marker of macrophage/microglial activation, was measured in CSF from SIV-infected, untreated and SIV-infected, FluPar-treated macaques using a commercially available ELISA (IBL International; Toronto, ON). Data were expressed as fold change in CSF sampled prior to euthanasia compared to preinoculation levels.
IDO mRNA measurement by qRT-PCR
Total RNA was isolated from macaque parietal cortex and IDO mRNA and 18S RNA were measured by multiplexed qRT-PCR as described previously (Zink et al, 2010) using a Rotor-Gene Q thermocycler (Qiagen). IDO mRNA was normalized to 18S RNA and expressed as fold change compared to the median SIV-infected, untreated, using the ∆∆Ct method. For samples with undetectable levels of IDO mRNA, the IDO Ct was set to the highest detectable Ct + 3.57 cycles, which would be approximately 10-fold lower level of IDO mRNA than the lowest detectable level. The ∆∆Ct for all samples with undetectable IDO was set to lowest undetectable ∆∆Ct.
Statistical analyses
For in vitro drug screening assays, all data are represented as mean +/− SEM and analyzed by one-way analysis of variance (ANOVA). Group-wise post hoc comparisons were assessed by Newman-Keuls multiple comparison tests.
For in vivo data, group vs. group (SIV-infected, untreated vs. SIV-infected, FluPar-treated) statistical comparisons were made using the Mann-Whitney test, the nonparametric equivalent of the Student’s t test. For measurements taken longitudinally throughout SIV infection (i.e.- plasma/CSF viral loads, CCL2 CSF levels, and IL-6 CSF levels) 3 statistical questions were asked. 1) Were the two groups (SIV-infected, untreated and SIV-infected, FluPar-treated) different just prior to FluPar treatment (initiated at 12 dpi)? For this group vs. group comparison, a Mann-Whitney test was performed on the data at 10 dpi (log10 transformed values for viral load or, fold change at 10 dpi compared to preinoculation for CCL2 and IL-6 levels in the CSF). 2) Did FluPar treatment have a rapid impact on the variable (change from 10 dpi to 21 dpi, the end of the acute phase of disease and the beginning of the asymptomatic/chronic phase of disease)? The fold change from 21 dpi vs. 10 dpi was calculated for each macaque and for comparison of groups the Mann-Whitney test was performed. 3) Did FluPar impact the variable during the chronic phase of the disease? Change over time from 21 dpi until euthanasia was measured by a linear regression model for longitudinal data (repeated measurements over time), whereby group vs. group comparisons tested the differences in slopes (coefficients of the “dpi” variable). For viral loads, log10 transformed values were used. For CCL2 and IL-6 levels in the CSF the fold change compared to preinoculation level was used. For all statistical analyses, the significance level was set at p < 0.05.
Results
Fluconazole and paroxetine were neuroprotective against HIV gp120 and Tat toxicity
The Microsource Discovery Spectrum collection of compounds contains 2000 FDA approved drugs and other naturally occurring compounds. We screened the entire library at a single concentration of 10 M of each compound, and found that more than 256 compounds demonstrated > 50% protection (Nath et al, 2012). The antidepressant paroxetine and the antifungal fluconazole were among these active neuroprotective compounds, with neuroprotection against oxidative stress confirmed in a secondary assay. In order to utilize these compounds as potential adjunctive neuroprotective therapies against HAND, we evaluated the effects of both compounds as protectants against the HIV viral toxic proteins gp120 and Tat (Fig. 1). Both paroxetine and fluconazole displayed concentration dependent neuroprotective actions against both viral proteins, with paroxetine protective at 100–500 nM, while fluconazole had significant neuroprotection at 1–5 uM concentrations. These are drug concentrations that have been found in the CNS of drug treated patients (Henry et al, 2000; Thaler et al, 1995).
Fig. 1.
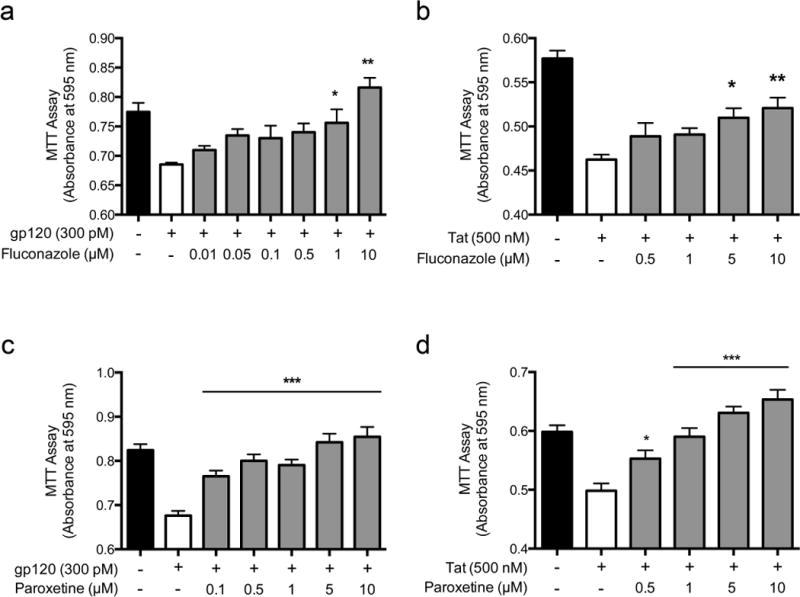
Fluconazole (a & b) and paroxetine (c & d) were neuroprotective against HIV gp120 and Tat toxicity in a drug screen using primary rat mixed hippocampal cultures. Mixed hippocampal cultures were pretreated with varying doses of fluconazole (a & b) or paroxetine (c & d) for 1 h prior to 18 h incubation with either HIV gp120 (a & c) or Tat (b & d), then assayed for cell viability using the MTT assay. Data are represented as mean +/-SEM and analyzed by one-way analysis of variance (ANOVA). Group-wise post hoc comparisons were assessed by Newman-Keuls multiple comparison tests. * p < 0.05, ** p < 0.01, *** p < 0.001
FluPar treatment reduced SIV-induced neurodegeneration
Given the protective effects of fluconazole and paroxetine on HIV gp120 and Tat neurotoxicity in vitro, we tested the neuroprotective effects of combination fluconazole/paroxetine (FluPar) treatment in a SIV model of HIV-associated CNS disease. Pigtailed macaques were dually inoculated with an immunosuppressive swarm (SIV/DeltaB670) and a neurovirulent clone (SIV/17E-Fr) of SIV, then either left untreated or treated with a combination fluconazole and paroxetine (FluPar) beginning at 12 dpi until euthanasia, approximately 84 dpi.
To assess the neuroprotective effect of FluPar treatment, we examined 3 indicators of neurodegeneration: CSF neurofilament light chain levels, APP accumulation in axons, and tissue levels of CaMKIIα. The presence of neurofilament light chain in the CSF is indicative of neuronal damage, particularly axonal damage, in several neurodegenerative diseases, including HIV (Abdulle et al, 2007; Jessen Krut et al, 2014; Norgren et al, 2003; Rosengren et al, 1996). FluPar-treated macaques had significantly lower levels of neurofilament light chain in the CSF compared to SIV-infected, untreated macaques (p = 0.023; Fig. 2a).
Fig. 2.
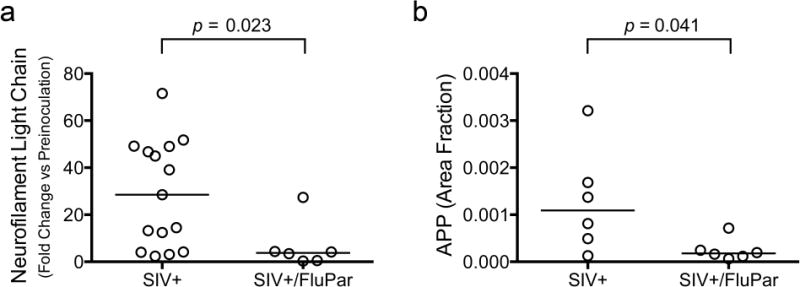
FluPar treatment reduced neuronal damage. Neurofilament light chain was measured in the CSF preinoculation and just prior to euthanasia by ELISA. Data are expressed as fold change at euthanasia compared to preinoculation levels. FluPar-treated macaques had significantly lower levels of neurofilament light chain in the CSF at euthanasia compared to controls (p = 0.023; a). APP was measured in the corpus callosum using quantitative immunohistochemistry as a marker of axonal degeneration. FluPar-treated macaques had significantly lower levels of APP than untreated controls (p = 0.041, b). Bars represent medians. Statistical comparisons were made using the Mann-Whitney test.
We previously demonstrated axonal accumulation of APP in our SIV macaque model, which was highly correlative with behavioral deficits (Mankowski et al, 2002b; Weed et al, 2003). We found that FluPar-treated macaques had significantly lower levels of APP accumulation than SIV-infected, untreated macaques (p = 0.041; Fig. 2b).
CaMKIIα is a synaptodendritic kinase important for learning and memory (Coultrap and Bayer, 2012; Frankland et al, 2001; Lisman et al, 2012). We previously showed CaMKII dysregulation in the hippocampus and frontal cortex of SIV-infected rhesus macaques (Macaca mulatta) (Gupta et al, 2010). In collaboration with the Jordan-Sciutto lab, we recently showed lower levels of CaMKIIα in the frontal cortex of SIV-infected macaques treated combination antiretroviral therapy (cART) compared to both uninfected and SIV-infected, untreated controls (Akay et al, 2014). In this study, Western blotting demonstrated that FluPar-treated macaques had higher levels of CaMKIIα than the untreated controls (p = 0.004; Fig. 3). Together, these data indicate that FluPar treatment protected macaques from SIV-induced neurodegeneration based on several indicators of neuronal integrity.
Fig. 3.
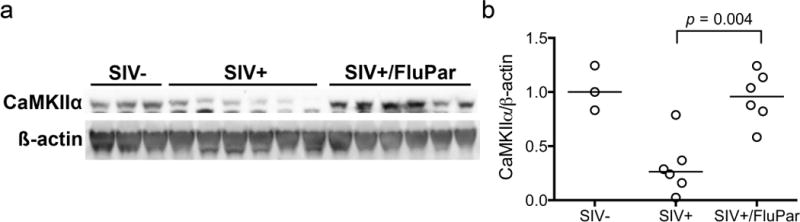
FluPar treatment prevented SIV-induced CaMKIIα loss in the frontal cortex. CaMKIIα was measured in frontal cortex homogenates by Western blotting and normalized to ß-actin to control for protein loading. FluPar-treated macaques had significantly higher levels of CaMKIIα in the frontal cortex than untreated controls (p = 0.004). Bars in b represent medians. Statistical comparisons were made using the Mann-Whitney test.
FluPar treatment did not affect plasma or CSF viral loads
SIV viral loads were measured throughout infection in the plasma and CSF of SIV-infected macaques that were either untreated or treated with FluPar beginning at 12 dpi. FluPar treatment did not significantly impact plasma or CSF viral loads either soon after treatment initiation (fold change of 21 dpi compared to peak at 10 dpi) or during the chronic phase of disease from 21 dpi until euthanasia (Fig. 4). Untreated macaques had slightly higher plasma and CSF viral loads at 10 dpi than FluPar-treated macaques, but the slope of viral RNA levels for the two groups did not differ after treatment initiation.
Fig. 4.
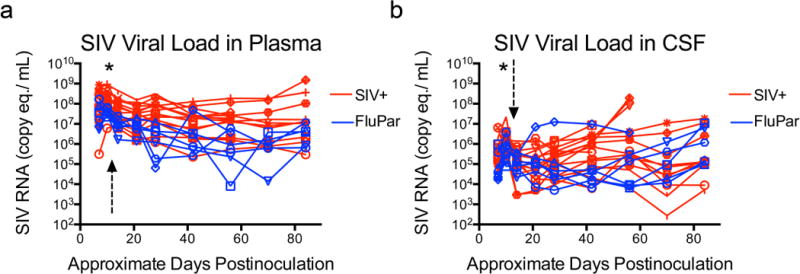
FluPar treatment had no effect on plasma or CSF viral loads. Viral RNA in plasma (a) and CSF (b) was quantitated in samples collected throughout infection by qRT-PCR. Arrow indicates time of treatment initiation (12 dpi). * indicates p < 0.05, Mann-Whitney comparison. Other statistical comparisons made using these longitudinally measured data are described in detail in the methods.
FluPar treatment did not affect SIV replication in the brain
SIV replication in the brain was measured by qRT-PCR in both the basal ganglia and parietal cortex. Median viral loads in the basal ganglia and parietal cortex were lower in FluPar-treated macaques than uninfected controls, but the differences were not statistically significant (p = 0.299 and p = 0.777, respectively; Fig. 5). If there were small differences in brain viral loads, a larger sample size would be needed to detect the difference.
Fig. 5.
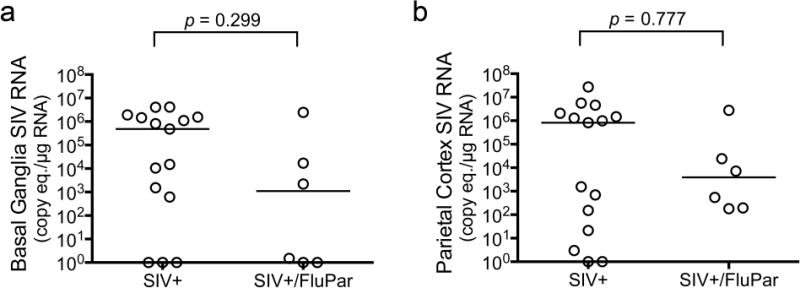
FluPar treatment did not reduce viral load in the brain. Viral RNA was quantitated in the basal ganglia (a) and parietal cortex (b) by qRT-PCR. Bars represent medians. Statistical comparisons were made using the Mann-Whitney test.
Effect of FluPar treatment on brain serotonin levels
Both SIV and HIV infection have been shown to decrease levels of serotonin in the striatum (Drewes et al, submitted; Reynolds and Sardar, 1996). The paroxetine component of FluPar treatment is an SSRI. Therefore we expected FluPar-treated macaques to have higher levels of serotonin than untreated macaques. FluPar-treated macaques tended to have higher levels of striatal serotonin than untreated controls, however this did not quite reach statistical significance (p = 0.076; Fig. 6).
Fig. 6.
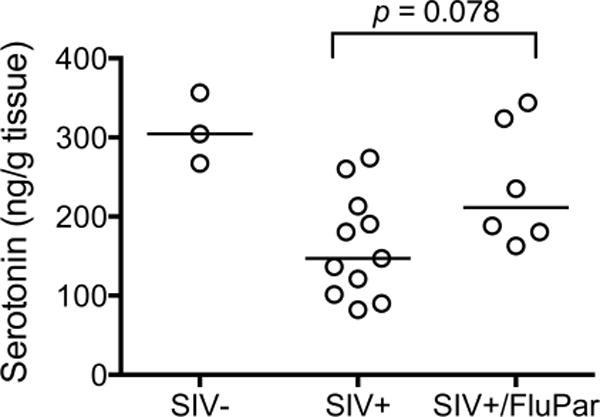
SIV-infected, FluPar-treated macaques had higher median levels of striatal serotonin than SIV-infected, untreated macaques (p = 0.078). Serotonin in the striatum was measured by ELISA. Bars represent medians. Statistical comparisons were made using the Mann-Whitney test.
FluPar treatment did not affect SIV-induced neuroinflammation
SIV encephalitis is a pathological hallmark of SIV infection of the CNS that closely reflects HIV CNS infection (Mankowski et al, 2002a). The encephalitis is accompanied and/or mediated by a predictable, well-documented neuroinflammatory cascade. FluPar’s effect on SIV-induced neuroinflammation was measured using several methods. CCL2 and IL-6 levels in the CSF are early predictive indicators of which macaques will develop encephalitis (Mankowski et al, 2004; Zink et al, 2001). CSF was collected periodically throughout infection and levels of CCL2 and IL-6 were measured by ELISA. FluPar treatment did not significantly impact levels of CCL2 or IL-6 in the CSF either soon after treatment initiation (fold change of 21 dpi compared to peak at 10 dpi) or the chronic progression from 21 dpi until euthanasia (Fig. 7a, b). To examine neuroinflammation in brain tissue, we used quantitative immunohistochemistry to measure markers of activated macrophages/microglia (CD68 and MHC class II) in the subcortical white matter adjacent to the basal ganglia. Although the median level of CD68 and MHC class II staining was lower in the FluPar treated macaques than untreated controls, the difference was not statistically significant (p = 0.197 and p = 0.387, respectively; Fig. 7c, d). Macrophage activation was also assessed by measuring the small molecule, neopterin, in the CSF. Neopterin is produced by macrophages subjected to proinflammatory stimuli, such as interferon gamma, and is elevated in the CSF of HIV-infected individuals, even in the setting of cART (Hagberg et al, 2010). FluPar-treated macaques did not have significantly different levels of neopterin in the CSF than their untreated counterparts (p = 0.788; Fig. 7e). Indoleamine 2,3-dioxygenase (IDO) is tryptophan-catabolizing enzyme induced by Type I and II interferons, which can contribute to immune dysregulation, oxidative stress, and neuronal dysfunction. IDO is elevated in both SIV and HIV CNS disease (Burudi et al, 2002; Drewes et al, submitted; Sardar and Reynolds, 1995). We measured IDO mRNA in the parietal cortex of both SIV-infected, untreated macaques and SIV-infected, FluPar treated macaques and found that FluPar treatment had no significant impact (p = 0.603; Fig. 7f). Taken together these data demonstrate that FluPar treatment had no significant effect on SIV-induced neuroinflammation in this model.
Fig. 7.
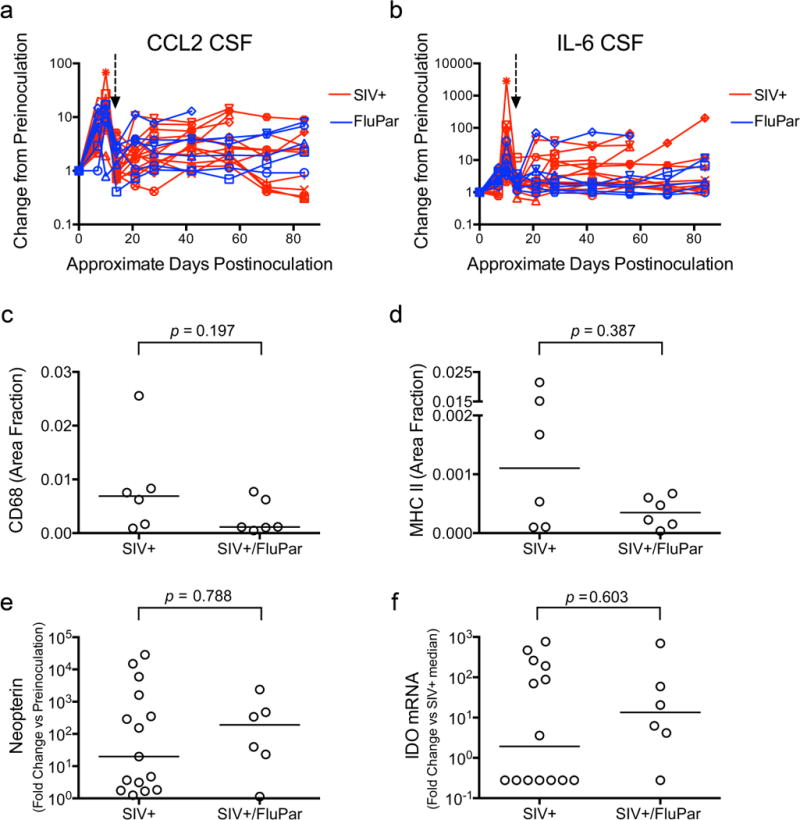
FluPar treatment did not resolve neuroinflammation. CCL2 (a) and IL-6 (b) in CSF were quantitated in samples collected throughout infection by ELISA. Arrow indicates time of treatment initiation (12 dpi). CD68 (c) and MHC class II (d) were measured in deep white matter, adjacent to the basal ganglia by quantitative immunohistochemistry. Neopterin levels in the CSF were measured by ELISA (e). IDO mRNA in the parietal cortex was measured by qRT-PCR (f). Bars in c-f represent medians. Statistical comparisons made using longitudinally measured data in a and b are described in detail in the methods. Statistical comparisons in c-f were made using the Mann-Whitney test.
Discussion
Combination fluconazole/paroxetine (FluPar) treatment prevented neurodegeneration in an accelerated, consistent SIV/macaque model of HIV-associated CNS disease without significantly impacting neuroinflammation or viral replication, which is a unique mode of protection for HAND. The drugs were selected based on results from a unique drug screen designed to measure protection against HIV gp120 and Tat neurotoxicity in a primary mixed hippocampal cell culture system, which inspired not only this study, but also an ongoing clinical trial.
In our SIV/macaque model of HIV-associated CNS disease, FluPar treatment protected against neurodegeneration as determined by three separate measures. First, FluPar treatment reduced the level neurofilament light chain in the CSF. Neurofilament light chain is an important structural component of axons and is elevated in the CSF of patients with neurodegenerative diseases with wide ranging etiologies, such as Alzheimer’s disease, amyotrophic lateral sclerosis, cerebral infarction, multiple sclerosis, Parkinson’s disease, and HIV (Abdulle et al, 2007; Jessen Krut et al, 2014; Norgren et al, 2003; Rosengren et al, 1996). Second, FluPar treatment prevented axonal APP accumulation, supporting the neurofilament light chain data indicating protection from axonal damage. APP is most commonly recognized as the precursor to Aß, a major component of senile plaques characteristic of Alzheimer’s disease. APP accumulates in damaged axons and has been shown to accumulate both in the brains of SIV-infected macaques and HIV-infected humans (Giometto et al, 1997; Mankowski et al, 2002b). Finally, FluPar treatment prevented CaMKIIα loss, indicating protection from synaptodendritic damage. CaMKIIα is important for learning and memory (Coultrap and Bayer, 2012; Frankland et al, 2001; Lisman et al, 2012). Therefore any changes in levels or activation could have significant cognitive consequences. We previously showed alterations in CaMKIIα in both the pigtailed macaque (Macaca nemestrina) model used in this study and the less pathogenic rhesus macaque (Macaca mulatta) model (Akay et al, 2014; Gupta et al, 2010). In contrast to the pigtailed macaque model, total levels of CaMKIIα were unchanged while CaMKIIα activation was decreased in SIV-infected rhesus macaques, indicating that SIV-induced disruption of CaMKIIα could be species-, model-, and disease stage-specific. CaMKIIα alterations have not yet been reported in human brains, however rat hippocampal cultures exposed to supernatants from HIV-infected human monocyte-derived macrophages had decreased CaMKIIα activation (Gupta et al, 2010), indicating that HIV can cause CaMKIIα dysregulation.
Further studies will be needed to parse out the individual neuroprotective contributions of fluconazole and paroxetine and to dissect the molecular mechanisms of protection against SIV/HIV-induced neurodegeneration. There are currently no published reports on the neuroprotective capabilities of fluconazole, so we cannot speculate on its mechanism of neuroprotection, although there is evidence that it may reach therapeutic levels in the brain (Thaler et al, 1995). In contrast, paroxetine and other SSRIs have proven neuroprotective in several animal models of neurodegenerative diseases (Chiou et al, 2006; Chung et al, 2010; Chung et al, 2011; Lim et al, 2009; Mattson et al, 2004; Nelson et al, 2007; Peng et al, 2008; Shibui et al, 2009). SSRI neuroprotection has been attributed to inhibition of neuroinflammation and oxidative stress (Chiou et al, 2006; Chung et al, 2010; Chung et al, 2011; Lim et al, 2009; Zhang et al, 2012), inhibition of apoptosis and promotion of neuroprotective BDNF signaling (Mattson et al, 2004), rescue or enhancement of neurogenesis (Jiang et al, 2014; Klomp et al, 2014; Lee et al, 2011; Stagni et al, 2013), and even inhibition of glutamate excitotoxicity (Vizi et al, 2013) in both animal models and cell culture, all of which have been implicated in the development of HAND (Lindl et al, 2010; Mocchetti et al, 2014; Potter et al, 2013; Steiner et al, 2006). In this study, we found FluPar treatment prevented neurodegeneration without significantly inhibiting multiple measures of neuroinflammation, indicating further investigation into the mechanism of action is needed.
It is unclear whether paroxetine’s neuroprotective effects in this SIV model of HIV-associated CNS disease could be due to its effects on serotonin or other novel mechanisms of action. In this study, FluPar-treated macaques showed a trend toward increased striatal serotonin levels that did not quite reach statistical significance (Fig. 6). Because we measured total tissue levels of serotonin, it is possible that FluPar treatment could have elevated synaptic serotonin, thereby facilitating neuroprotection. Alternatively, FluPar neuroprotection could be afforded by a here-to-fore unidentified mechanism. In a MPTP mouse model of Parkinson’s disease, the SSRI fluoxetine prevented neuroinflammation and dopaminergic neuron loss without affecting striatal serotonin levels, indicating that tissue serotonin levels may not correspond with neuroprotection (Chung et al, 2011).
Neuroinflammation and viral replication are important mediators in HIV/SIV CNS disease (Gonzalez-Scarano and Martin-Garcia, 2005). Paroxetine, other SSRIs, and serotonin itself have been shown to inhibit HIV replication in vitro in both cell lines and primary monocyte-derived macrophages (Benton et al, 2010; Kristiansen and Hansen, 2000; Maneglier et al, 2008). Furthermore, Letendre and colleagues showed HIV-infected patients taking SSRIs were less likely to have detectable viral loads in the CSF (Letendre et al, 2007), so we were disappointed that FluPar treatment did not improve plasma, CSF, or brain tissue SIV viral loads. However, it was important to find that viral loads were not elevated by FluPar treatment, which would contraindicate its use for HIV, regardless of neuroprotection.
The neuroprotective results of this SIV/macaque study heighten enthusiasm for the ongoing clinical trial testing the efficacy of combination fluconazole/paroxetine treatment in patients with HAND, which was inspired by the same in vitro drug screen data in Fig. 1. There are important differences in study design, outcome measures, and benefits vs. limitations between the SIV/macaque study presented here and the human clinical trial underway. In this SIV/macaque study, macaques were administered FluPar daily beginning at 12 dpi, at the end of the acute phase of disease. In contrast, patients enrolled in the human clinical trial have already developed HAND, and therefore are in a more advanced stage of disease. In our study, it is likely that FluPar-treatment prevented neurodegeneration because it was administered early in infection, and it is not known whether FluPar will reverse neurodegeneration in patients that have already developed HAND. A study using the gp120 transgenic mouse model found that paroxetine rescued gp120-induced deficiencies in hippocampal neurogenesis, indicating paroxetine has potential to reverse some kinds of neurological damage (Lee et al, 2011). In this SIV/macaque study, we also measured different disease outcomes than are planned for the human clinical trial. The SIV/macaque model has the advantage of extensive tissue sampling, so in addition to measuring markers of disease progression in plasma and CSF, we were able to measure these markers directly in tissues, which is not possible in human patients. Of the 3 neurodegenerative parameters which were protected by FluPar treatment, only neurofilament light chain in the CSF can be measured in human patients, and we would recommend the investigators involved in the clinical trial do so, if sample availability permits. The human clinical trial has the advantage of measuring neurocognitive outcomes, which is cost-prohibitive in the SIV/macaque model. Another benefit of the human clinical trial is its placebo-controlled 2×2 factorial design whereby patients will receive 1.) placebo, 2.) fluconazole alone, 3.) paroxetine alone, or 4.) combination fluconazole/paroxetine, which will help discern individual drug effects. For these reasons, we are optimistically awaiting the outcome of the human clinical trial.
The finding that FluPar treatment protected SIV-infected macaques from neurodegeneration, despite ongoing neuroinflammation and viral replication has important implications in light of emerging latency reversing HIV eradication strategies. In these strategies, HIV infected reservoirs are purged by reactivating latently infected cells, which are subsequently killed by cytotoxic T cells or cytopathic effects while cART prevents new rounds of infection (Margolis, 2014; Xing and Siliciano, 2013). In addition to inducing viral replication, reversing latency will likely trigger inflammatory cascades. Because many antiretroviral drugs have low blood brain barrier penetration (Letendre et al, 2008), viral replication and neuroinflammation after latency reversal could smolder, causing neurodegeneration. There may be a need for adjunct therapies, such as FluPar, that could prevent neurodegeneration even in the face of viral replication and neuroinflammation.
Finally, these studies highlight the importance of collaborative efforts to merge drug discovery and preclinical testing facilitated by initiatives such as the NIMH Center for Novel Therapeutics of HIV-associated Cognitive Disorders. Through these collaborative initiatives, we are hopeful that more potential therapies can be brought through the pipeline of discovery, to preclinical testing, and finally to the clinic to treat HAND as the disease currently stands and in the emerging frontier of HIV eradication.
Acknowledgments
These studies were funded by the National Institutes of Health (grants P30 MH075673, R01 MH087233), R01 MH085554, P01 MH070306, and P40 OD013117).
The authors thank Dr. Robert J. Adams for overseeing the health and care of the animals. We also thank Dr. Lucio Gama for assistance with the IDO qRT-PCR assay and Dr. Zhaohao Liao for assistance with the serotonin ELISA.
Footnotes
Conflicts of interest
The authors have no conflicts of interest.
References
- Abdulle S, Mellgren A, Brew BJ, Cinque P, Hagberg L, Price RW, Rosengren L, Gisslen M. CSF neurofilament protein (NFL) – a marker of active HIV-related neurodegeneration. J Neurol. 2007;254:1026–32. doi: 10.1007/s00415-006-0481-8. [DOI] [PubMed] [Google Scholar]
- Akay C, Cooper M, Odeleye A, Jensen BK, White MG, Vassoler F, Gannon PJ, Mankowski J, Dorsey JL, Buch AM, Cross SA, Cook DR, Pena MM, Andersen ES, Christofidou-Solomidou M, Lindl KA, Zink MC, Clements J, Pierce RC, Kolson DL, Jordan-Sciutto KL. Antiretroviral drugs induce oxidative stress and neuronal damage in the central nervous system. J Neurovirol. 2014;20:39–53. doi: 10.1007/s13365-013-0227-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ances BM, Ellis RJ. Dementia and neurocognitive disorders due to HIV-1 infection. Semin Neurol. 2007;27:86–92. doi: 10.1055/s-2006-956759. [DOI] [PubMed] [Google Scholar]
- Antinori A, Arendt G, Becker JT, Brew BJ, Byrd DA, Cherner M, Clifford DB, Cinque P, Epstein LG, Goodkin K, Gisslen M, Grant I, Heaton RK, Joseph J, Marder K, Marra CM, McArthur JC, Nunn M, Price RW, Pulliam L, Robertson KR, Sacktor N, Valcour V, Wojna VE. Updated research nosology for HIV-associated neurocognitive disorders. Neurology. 2007;69:1789–99. doi: 10.1212/01.WNL.0000287431.88658.8b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benton T, Lynch K, Dube B, Gettes DR, Tustin NB, Ping Lai J, Metzger DS, Blume J, Douglas SD, Evans DL. Selective serotonin reuptake inhibitor suppression of HIV infectivity and replication. Psychosom Med. 2010;72:925–32. doi: 10.1097/PSY.0b013e3181f883ce. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burudi EM, Marcondes MC, Watry DD, Zandonatti M, Taffe MA, Fox HS. Regulation of indoleamine 2,3-dioxygenase expression in simian immunodeficiency virus-infected monkey brains. J Virol. 2002;76:12233–41. doi: 10.1128/JVI.76.23.12233-12241.2002. doi: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiou SH, Chen SJ, Peng CH, Chang YL, Ku HH, Hsu WM, Ho LL, Lee CH. Fluoxetine up-regulates expression of cellular FLICE-inhibitory protein and inhibits LPS-induced apoptosis in hippocampus-derived neural stem cell. Biochem Biophys Res Commun. 2006;343:391–400. doi: 10.1016/j.bbrc.2006.02.180. [DOI] [PubMed] [Google Scholar]
- Chung YC, Kim SR, Jin BK. Paroxetine prevents loss of nigrostriatal dopaminergic neurons by inhibiting brain inflammation and oxidative stress in an experimental model of Parkinson’s disease. J Immunol. 2010;185:1230–7. doi: 10.4049/jimmunol.1000208. [DOI] [PubMed] [Google Scholar]
- Chung YC, Kim SR, Park JY, Chung ES, Park KW, Won SY, Bok E, Jin M, Park ES, Yoon SH, Ko HW, Kim YS, Jin BK. Fluoxetine prevents MPTP-induced loss of dopaminergic neurons by inhibiting microglial activation. Neuropharmacology. 2011;60:963–74. doi: 10.1016/j.neuropharm.2011.01.043. [DOI] [PubMed] [Google Scholar]
- Clements JE, Babas T, Mankowski JL, Suryanarayana K, Piatak M, Jr, Tarwater PM, Lifson JD, Zink MC. The central nervous system as a reservoir for simian immunodeficiency virus (SIV): steady-state levels of SIV DNA in brain from acute through asymptomatic infection. J Infect Dis. 2002;186:905–13. doi: 10.1086/343768. [DOI] [PubMed] [Google Scholar]
- Clements JE, Mankowski JL, Gama L, Zink MC. The accelerated simian immunodeficiency virus macaque model of human immunodeficiency virus-associated neurological disease: from mechanism to treatment. J Neurovirol. 2008;14:309–17. doi: 10.1080/13550280802132832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coultrap SJ, Bayer KU. CaMKII regulation in information processing and storage. Trends Neurosci. 2012;35:607–18. doi: 10.1016/j.tins.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drewes JL, Meulendyke KA, Liao Z, Witwer KW, Gama L, Ubaida Mohien C, Li M, Notarangelo FM, Tarwater PM, Schwarcz R, Graham DR, Zink MC. Quinolinic acid/tryptophan ratios are an early predictor of SIV encephalitis in macaques and remain elevated in the brain under cART. doi: 10.1007/s13365-015-0334-2. (submitted) doi: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankland PW, O’Brien C, Ohno M, Kirkwood A, Silva AJ. Alpha-CaMKII-dependent plasticity in the cortex is required for permanent memory. Nature. 2001;411:309–13. doi: 10.1038/35077089. [DOI] [PubMed] [Google Scholar]
- Giometto B, An SF, Groves M, Scaravilli T, Geddes JF, Miller R, Tavolato B, Beckett AA, Scaravilli F. Accumulation of beta-amyloid precursor protein in HIV encephalitis: relationship with neuropsychological abnormalities. Ann Neurol. 1997;42:34–40. doi: 10.1002/ana.410420108. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Scarano F, Martin-Garcia J. The neuropathogenesis of AIDS. Nat Rev Immunol. 2005;5:69–81. doi: 10.1038/nri1527. [DOI] [PubMed] [Google Scholar]
- Gupta RG, Kelly KM, Helke KL, Queen SE, Karper JM, Dorsey JL, Brice AK, Adams RJ, Tarwater PM, Kolson DL, Mankowski JL. HIV and SIV induce alterations in CNS CaMKII expression and activation: a potential mechanism for cognitive impairment. Am J Pathol. 2010;176:2776–84. doi: 10.2353/ajpath.2010.090809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagberg L, Cinque P, Gisslen M, Brew BJ, Spudich S, Bestetti A, Price RW, Fuchs D. Cerebrospinal fluid neopterin: an informative biomarker of central nervous system immune activation in HIV-1 infection. AIDS Res Ther. 2010;7:15. doi: 10.1186/1742-6405-7-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haughey NJ, Cutler RG, Tamara A, McArthur JC, Vargas DL, Pardo CA, Turchan J, Nath A, Mattson MP. Perturbation of sphingolipid metabolism and ceramide production in HIV-dementia. Ann Neurol. 2004;55:257–67. doi: 10.1002/ana.10828. [DOI] [PubMed] [Google Scholar]
- Heaton RK, Clifford DB, Franklin DR, Jr, Woods SP, Ake C, Vaida F, Ellis RJ, Letendre SL, Marcotte TD, Atkinson JH, Rivera-Mindt M, Vigil OR, Taylor MJ, Collier AC, Marra CM, Gelman BB, McArthur JC, Morgello S, Simpson DM, McCutchan JA, Abramson I, Gamst A, Fennema-Notestine C, Jernigan TL, Wong J, Grant I. HIV-associated neurocognitive disorders persist in the era of potent antiretroviral therapy: CHARTER Study. Neurology. 2010;75:2087–96. doi: 10.1212/WNL.0b013e318200d727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry ME, Moore CM, Kaufman MJ, Michelson D, Schmidt ME, Stoddard E, Vuckevic AJ, Berreira PJ, Cohen BM, Renshaw PF. Brain kinetics of paroxetine and fluoxetine on the third day of placebo substitution: a fluorine MRS study. Am J Psychiatry. 2000;157:1506–8. doi: 10.1176/appi.ajp.157.9.1506. [DOI] [PubMed] [Google Scholar]
- Jessen Krut J, Mellberg T, Price RW, Hagberg L, Fuchs D, Rosengren L, Nilsson S, Zetterberg H, Gisslen M. Biomarker evidence of axonal injury in neuroasymptomatic HIV-1 patients. PLoS One. 2014;9:e88591. doi: 10.1371/journal.pone.0088591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang L, Liu C, Tong J, Mao R, Chen D, Wang H, Huang J, Li L. Fluoxetine pretreatment promotes neuronal survival and maturation after auditory fear conditioning in the rat amygdala. PLoS One. 2014;9:e89147. doi: 10.1371/journal.pone.0089147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klomp A, Vaclavu L, Meerhoff GF, Reneman L, Lucassen PJ. Effects of chronic fluoxetine treatment on neurogenesis and tryptophan hydroxylase expression in adolescent and adult rats. PLoS One. 2014;9:e97603. doi: 10.1371/journal.pone.0097603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristiansen JE, Hansen JB. Inhibition of HIV replication by neuroleptic agents and their potential use in HIV infected patients with AIDS related dementia. Int J Antimicrob Agents. 2000;14:209–13. doi: 10.1016/S0924-8579(99)00157-0. [DOI] [PubMed] [Google Scholar]
- Lee MH, Wang T, Jang MH, Steiner J, Haughey N, Ming GL, Song H, Nath A, Venkatesan A. Rescue of adult hippocampal neurogenesis in a mouse model of HIV neurologic disease. Neurobiol Dis. 2011;41:678–87. doi: 10.1016/j.nbd.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letendre S, Marquie-Beck J, Capparelli E, Best B, Clifford D, Collier AC, Gelman BB, McArthur JC, McCutchan JA, Morgello S, Simpson D, Grant I, Ellis RJ. Validation of the CNS Penetration-Effectiveness rank for quantifying antiretroviral penetration into the central nervous system. Arch Neurol. 2008;65:65–70. doi: 10.1001/archneurol.2007.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letendre SL, Marquie-Beck J, Ellis RJ, Woods SP, Best B, Clifford DB, Collier AC, Gelman BB, Marra C, McArthur JC, McCutchan JA, Morgello S, Simpson D, Alexander TJ, Durelle J, Heaton R, Grant I. The role of cohort studies in drug development: clinical evidence of antiviral activity of serotonin reuptake inhibitors and HMG-CoA reductase inhibitors in the central nervous system. J Neuroimmune Pharmacol. 2007;2:120–7. doi: 10.1007/s11481-006-9054-y. [DOI] [PubMed] [Google Scholar]
- Lim CM, Kim SW, Park JY, Kim C, Yoon SH, Lee JK. Fluoxetine affords robust neuroprotection in the postischemic brain via its anti-inflammatory effect. J Neurosci Res. 2009;87:1037–45. doi: 10.1002/jnr.21899. [DOI] [PubMed] [Google Scholar]
- Lindl KA, Marks DR, Kolson DL, Jordan-Sciutto KL. HIV-associated neurocognitive disorder: pathogenesis and therapeutic opportunities. J Neuroimmune Pharmacol. 2010;5:294–309. doi: 10.1007/s11481-010-9205-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisman J, Yasuda R, Raghavachari S. Mechanisms of CaMKII action in long-term potentiation. Nat Rev Neurosci. 2012;13:169–82. doi: 10.1038/nrn3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maneglier B, Guillemin GJ, Clayette P, Rogez-Kreuz C, Brew BJ, Dormont D, Advenier C, Therond P, Spreux-Varoquaux O. Serotonin decreases HIV-1 replication in primary cultures of human macrophages through 5-HT(1A) receptors. Br J Pharmacol. 2008;154:174–82. doi: 10.1038/bjp.2008.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mankowski JL, Clements JE, Zink MC. Searching for clues: tracking the pathogenesis of human immunodeficiency virus central nervous system disease by use of an accelerated, consistent simian immunodeficiency virus macaque model. J Infect Dis. 2002a;186(Suppl 2):S199–208. doi: 10.1086/344938. [DOI] [PubMed] [Google Scholar]
- Mankowski JL, Queen SE, Clements JE, Zink MC. Cerebrospinal fluid markers that predict SIV CNS disease. J Neuroimmunol. 2004;157:66–70. doi: 10.1016/j.jneuroim.2004.08.031. [DOI] [PubMed] [Google Scholar]
- Mankowski JL, Queen SE, Tarwater PM, Fox KJ, Perry VH. Accumulation of beta-amyloid precursor protein in axons correlates with CNS expression of SIV gp41. J Neuropathol Exp Neurol. 2002b;61:85–90. doi: 10.1093/jnen/61.1.85. doi: [DOI] [PubMed] [Google Scholar]
- Margolis DM. How Might We Cure HIV? Curr Infect Dis Rep. 2014;16:392. doi: 10.1007/s11908-014-0392-2. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Maudsley S, Martin B. BDNF and 5-HT: a dynamic duo in age-related neuronal plasticity and neurodegenerative disorders. Trends Neurosci. 2004;27:589–94. doi: 10.1016/j.tins.2004.08.001. [DOI] [PubMed] [Google Scholar]
- Meulendyke KA, Pletnikov MV, Engle EL, Tarwater PM, Graham DR, Zink MC. Early minocycline treatment prevents a decrease in striatal dopamine in an SIV model of HIV-associated neurological disease. J Neuroimmune Pharmacol. 2012;7:454–64. doi: 10.1007/s11481-011-9332-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mocchetti I, Bachis A, Campbell LA, Avdoshina V. Implementing neuronal plasticity in NeuroAIDS: the experience of brain-derived neurotrophic factor and other neurotrophic factors. J Neuroimmune Pharmacol. 2014;9:80–91. doi: 10.1007/s11481-013-9488-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- Nath S, Bachani M, Harshavardhana D, Steiner JP. Catechins protect neurons against mitochondrial toxins and HIV proteins via activation of the BDNF pathway. J Neurovirol. 2012;18:445–55. doi: 10.1007/s13365-012-0122-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson RL, Guo Z, Halagappa VM, Pearson M, Gray AJ, Matsuoka Y, Brown M, Martin B, Iyun T, Maudsley S, Clark RF, Mattson MP. Prophylactic treatment with paroxetine ameliorates behavioral deficits and retards the development of amyloid and tau pathologies in 3xTgAD mice. Exp Neurol. 2007;205:166–76. doi: 10.1016/j.expneurol.2007.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norgren N, Rosengren L, Stigbrand T. Elevated neurofilament levels in neurological diseases. Brain Res. 2003;987:25–31. doi: 10.1016/S0006-8993(03)03219-0. [DOI] [PubMed] [Google Scholar]
- Peng Q, Masuda N, Jiang M, Li Q, Zhao M, Ross CA, Duan W. The antidepressant sertraline improves the phenotype, promotes neurogenesis and increases BDNF levels in the R6/2 Huntington’s disease mouse model. Exp Neurol. 2008;210:154–63. doi: 10.1016/j.expneurol.2007.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potter MC, Figuera-Losada M, Rojas C, Slusher BS. Targeting the glutamatergic system for the treatment of HIV-associated neurocognitive disorders. J Neuroimmune Pharmacol. 2013;8:594–607. doi: 10.1007/s11481-013-9442-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds GP, Sardar AM. 5-Hydroxytryptamine deficits in the caudate nucleus in AIDS. AIDS. 1996;10:1303–4. doi: 10.1097/00002030-199609000-00026. doi: [DOI] [PubMed] [Google Scholar]
- Rosengren LE, Karlsson JE, Karlsson JO, Persson LI, Wikkelso C. Patients with amyotrophic lateral sclerosis and other neurodegenerative diseases have increased levels of neurofilament protein in CSF. J Neurochem. 1996;67:2013–8. doi: 10.1046/j.1471-4159.1996.67052013.x. [DOI] [PubMed] [Google Scholar]
- Sardar AM, Reynolds GP. Frontal cortex indoleamine-2,3-dioxygenase activity is increased in HIV-1-associated dementia. Neurosci Lett. 1995;187:9–12. doi: 10.1016/0304-3940(95)11324-P. [DOI] [PubMed] [Google Scholar]
- Shibui Y, He XJ, Uchida K, Nakayama H. MPTP-induced neuroblast apoptosis in the subventricular zone is not regulated by dopamine or other monoamine transporters. Neurotoxicology. 2009;30:1036–44. doi: 10.1016/j.neuro.2009.07.003. [DOI] [PubMed] [Google Scholar]
- Simioni S, Cavassini M, Annoni JM, Rimbault Abraham A, Bourquin I, Schiffer V, Calmy A, Chave JP, Giacobini E, Hirschel B, Du Pasquier RA. Cognitive dysfunction in HIV patients despite long-standing suppression of viremia. AIDS. 2010;24:1243–50. doi: 10.1097/QAD.0b013e3283354a7b. [DOI] [PubMed] [Google Scholar]
- Stagni F, Magistretti J, Guidi S, Ciani E, Mangano C, Calza L, Bartesaghi R. Pharmacotherapy with fluoxetine restores functional connectivity from the dentate gyrus to field CA3 in the Ts65Dn mouse model of down syndrome. PLoS One. 2013;8:e61689. doi: 10.1371/journal.pone.0061689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner J, Haughey N, Li W, Venkatesan A, Anderson C, Reid R, Malpica T, Pocernich C, Butterfield DA, Nath A. Oxidative stress and therapeutic approaches in HIV dementia. Antioxid Redox Signal. 2006;8:2089–100. doi: 10.1089/ars.2006.8.2089. [DOI] [PubMed] [Google Scholar]
- Thaler F, Bernard B, Tod M, Jedynak CP, Petitjean O, Derome P, Loirat P. Fluconazole penetration in cerebral parenchyma in humans at steady state. Antimicrob Agents Chemother. 1995;39:1154–6. doi: 10.1128/AAC.39.5.1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vizi ES, Kisfali M, Lorincz T. Role of nonsynaptic GluN2B-containing NMDA receptors in excitotoxicity: evidence that fluoxetine selectively inhibits these receptors and may have neuroprotective effects. Brain Res Bull. 2013;93:32–8. doi: 10.1016/j.brainresbull.2012.10.005. [DOI] [PubMed] [Google Scholar]
- Weed MR, Hienz RD, Brady JV, Adams RJ, Mankowski JL, Clements JE, Zink MC. Central nervous system correlates of behavioral deficits following simian immunodeficiency virus infection. J Neurovirol. 2003;9:452–64. doi: 10.1080/13550280390218751. [DOI] [PubMed] [Google Scholar]
- Xing S, Siliciano RF. Targeting HIV latency: pharmacologic strategies toward eradication. Drug Discov Today. 2013;18:541–51. doi: 10.1016/j.drudis.2012.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Zhou H, Wilson BC, Shi JS, Hong JS, Gao HM. Fluoxetine protects neurons against microglial activation-mediated neurotoxicity. Parkinsonism Relat Disord. 2012;18(Suppl 1):S213–7. doi: 10.1016/S1353-8020(11)70066-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zink MC, Brice AK, Kelly KM, Queen SE, Gama L, Li M, Adams RJ, Bartizal C, Varrone J, Rabi SA, Graham DR, Tarwater PM, Mankowski JL, Clements JE. Simian immunodeficiency virus-infected macaques treated with highly active antiretroviral therapy have reduced central nervous system viral replication and inflammation but persistence of viral DNA. J Infect Dis. 2010;202:161–70. doi: 10.1086/653213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zink MC, Coleman GD, Mankowski JL, Adams RJ, Tarwater PM, Fox K, Clements JE. Increased macrophage chemoattractant protein-1 in cerebrospinal fluid precedes and predicts simian immunodeficiency virus encephalitis. J Infect Dis. 2001;184:1015–21. doi: 10.1086/323478. [DOI] [PubMed] [Google Scholar]
- Zink MC, Suryanarayana K, Mankowski JL, Shen A, Piatak M, Jr, Spelman JP, Carter DL, Adams RJ, Lifson JD, Clements JE. High viral load in the cerebrospinal fluid and brain correlates with severity of simian immunodeficiency virus encephalitis. J Virol. 1999;73:10480–8. doi: 10.1128/jvi.73.12.10480-10488.1999. doi: [DOI] [PMC free article] [PubMed] [Google Scholar]