

Abstract
Regional depolarization of the mitochondrial network can alter cellular electrical excitability and increase the propensity for reentry, in part, through the opening of sarcolemmal KATP channels. Mitochondrial inner membrane potential (ΔΨm) instability or oscillation can be induced in myocytes by exposure to reactive oxygen species (ROS), laser excitation, or glutathione depletion, and is thought to be a major factor in arrhythmogenesis during ischemia-reperfusion. Nevertheless, the correlation between ΔΨm recovery kinetics and reperfusion-induced arrhythmias has been difficult to demonstrate experimentally. Here, we investigate the relationship between subcellular changes in ΔΨm, cellular glutathione redox potential, electrical excitability, and wave propagation during coverslip-induced ischemia-reperfusion (IR) in neonatal rat ventricular myocyte (NRVM) monolayers. Ischemia led to decreased action potential amplitude and duration followed by electrical inexcitability after ~ 15 min of ischemia. ΔΨm depolarization occurred in two phases during ischemia: in phase 1 (< 30 min ischemia), mitochondrial clusters within individual NRVMs depolarized, while phase 2 ΔΨm depolarization (30–60 min) was characterized by global functional collapse of the mitochondrial network across the whole ischemic region of the monolayer, typically involving a propagating metabolic wave. Oxidation of the glutathione (GSSG:GSH) redox potential occurred during ischemia, followed by recovery upon reperfusion (i.e., lifting the coverslip). ΔΨm recovered in the mitochondria of individual myocytes quite rapidly upon reperfusion (< 5 min), but was highly unstable, characterized by subcellular oscillations or flickering of clusters of mitochondria in NRVMs across the reperfused region. Electrical excitability also recovered in a heterogeneous manner, providing an arrhythmogenic substrate which led to formation of sustained reentry. Treatment with 4′-chlorodiazepam, a peripheral benzodiazepine receptor ligand, prevented ΔΨm oscillation, improved GSH recovery rate, and prevented reentry during reperfusion, indicating that stabilization of mitochondrial network dynamics is an important component of preventing post-ischemic arrhythmias.
Keywords: Ischemia-reperfusion, energy metabolism, ventricular arrhythmias, fibrillation, mitochondria, benzodiazepine
1 Introduction
Although the relative rate of mortality from cardiovascular disease has declined, coronary heart disease remains a leading cause of death, responsible for ~ 1 of every 6 deaths in the United States [1]. Ischemia caused by coronary occlusion, thrombosis, or spontaneous spasm leads to a variety of changes in ion homeostasis, energetics and oxidative processes that contribute to mitochondrial and cellular injury. Reperfusion of the ischemic area is the first line therapy to decrease ischemic damage and prevent cell death; however, reperfusion itself can lead to a more severe damage, a phenomenon known as reperfusion injury [2–4]. Reperfusion is also associated with an increased risk of potentially fatal arrhythmias [5–7].
Several arrhythmogenic mechanisms have been proposed to be involved in ischemia-reperfusion (IR)-induced arrhythmias. In pathological situations such as IR, abnormal automaticity and triggered activity [8] arising from ectopic pacemakers in the myocardium, particularly near the border zone between well-perfused and ischemic or infarcted tissue, can contribute to increased risk of arrhythmias [9]. Heterogeneous alterations in gap junctional conductance, resting membrane potential, excitability, and action potential duration (APD) together promote reentrant arrhythmias by increasing dispersion of refractoriness, slowing conduction velocity (CV), and/or creating regional excitation block (for a detailed review see ref. [10]).
The ability of the mitochondrial network of the cardiomyocyte to restore energy production, control reactive oxygen species (ROS), restore proton and ion gradients, and limit necrotic and apoptotic cell death is a key determinant of survival after IR. Mitochondrial failure can scale to affect cardiac cell function, and ultimately to the organ level, causing electrical and contractile dysfunction [11]. Dynamic instabilities of mitochondrial inner membrane potential (ΔΨm) are known to occur as the level of oxidative stress on the mitochondria increases. If a number of mitochondria in the network approach a threshold of redox stress, a state called mitochondrial criticality is reached, rendering the mitochondria hypersensitive to small perturbations. In this state, depolarization of only a few mitochondria can lead to a propagated wave of depolarization, complete collapse, or oscillation of ΔΨm in the whole network of a cardiac myocyte [12–14]. Coupling between mitochondria occurs through the autocatalytic mechanism called ROS-induced ROS-release (RIRR) [15–17]. RIRR was originally demonstrated by increased ROS generation via photodynamically-induced mitochondrial depolarization [15]. However, an increase in mitochondrial ROS generation due to inhibition of scavenging systems can also lead to a similar critical state [18]. In its initial description, activation of the permeability transition pore (PTP) was suggested as the main mechanism of RIRR [15], as evidenced by inhibition with the adenine nucleotide translocase inhibitor bongkrekic acid and by an increase in inner membrane calcein permeability; however, cyclosporine A (CsA) could not prevent RIRR induced by laser excitation [15]. Our previous studies revealed that RIRR also underlies self-sustained cell-wide mitochondrial oscillations in adult cardiomyocytes triggered either by highly localized laser excitation [19], or glutathione depletion [18]. These oscillations were also insensitive to CsA and independent of [Ca2+]c, but do not involve a large change in membrane permeability. Ligands of the mitochondrial benzodiazepine receptor, such as 4′-chlorodiazepam (4′-Cl-DZP) or PK-11195, were shown to stabilize the stressed mitochondria and prevent depolarization and oscillation of ΔΨm [19]. These effects were interpreted to suggest that activation of the ROS-sensitive energy-dissipating inner membrane anion channel (IMAC) [20, 21] was the main mechanism involved in RIRR and the mitochondrial instability resulting from oxidative stress [19].
With regard to the effects of ΔΨm loss or oscillation on cellular electrophysiology, decreasing cellular ATP/ADP ratio during ΔΨm depolarization can activate sarcolemmal KATP channels and profoundly alter cellular electrical excitability and APD [22, 23]. In isolated cardiomyocytes subjected to metabolic stress, IK,ATP oscillated in synchrony with ΔΨm depolarization, and induced APD oscillations [24]. We proposed that the resulting heterogeneous spatiotemporal instability of ΔΨm and action potentials (AP) would increase the dispersion of repolarization in the myocardium, increasing the vulnerability to cardiac arrhythmias. This was supported by experiments in intact perfused hearts subjected to antioxidant depletion or IR, whereby 4′-Cl-DZP suppressed arrhythmias and preserved cardiac contractile function [11, 25–27]. However, cellular oscillations in ΔΨm following IR were not demonstrated in the intact heart, in part due to technical limitations.
Recently, using computational and experimental methods, we explored the impact of regional mitochondrial depolarization, i.e., the formation of a metabolic current sink, on electrical propagation and arrhythmogenesis in monolayers of cardiomyocytes [28]. Induction of metabolic sinks, through regional chemical uncoupling of mitochondria in NRVM monolayers, decreased AP amplitude (APA) and caused slowing of conduction velocity (CV), shortening of APD and wavelength, and finally, inexcitability of the sink area. Using glibenclamide to inhibit KATP channels, we provided evidence that the opening of KATP channels was a major factor in producing these effects, as well as in causing reentry. Since IR induces much more complex changes in ion balance, ROS, and energetics than simple chemical uncoupling of oxidative phosphorylation, it remains to be determined if similar arrhythmogenic mechanisms are involved in the response to IR. To explore this, here we utilize a coverslip-induced IR model [29, 30] to study the electrophysiological effects, cytosolic GSH/GSSG redox and ΔΨm responses to IR in NRVM monolayers. The findings reveal complex patterns of ΔΨm loss and recovery during ischemia and reperfusion, which strongly influence the occurrence of post-ischemic reentrant arrhythmias.
2 Materials and Methods
2.1 NRVM Monolayers
Ventricles of 2-day old neonatal Sprague-Dawley rats (Harlan Laboratories) were excised, chopped into small pieces, washed with HBSS (Invitrogen) and then trypsinized overnight at 4°C. All animal protocols conformed to the guidelines of the National Institutes of Health [31]. The next day, cardiomyocytes were isolated using collagenase and two rounds of preplating were done to reduce the fibroblast concentration. 106 cells were plated on each fibronectin-coated (25 μg/ml) circular cover glass (D = 22 mm) and cultured in Medium 199 (Invitrogen) containing 10% heat-inactivated bovine serum (Invitrogen). The medium was changed daily. Experiments were performed on beating and confluent monolayers on the 3rd to 5th day of culture.
2.2 Inducing Ischemia and Reperfusion
Experiments started by equilibration of the monolayers with a modified Tyrode’s solution consisting of (in mmol/l): 135 NaCl, 5.4 KCl, 1.8 CaCl2, 1 MgCl2, 0.33 NaH2PO4, 5 HEPES, and 5 glucose. The monolayer was paced by application of voltage pulses at 1 Hz at the edge of the monolayer using bipolar point electrodes. A 15 mm circular glass coverslip (#1, Fisher Scientific) was placed on the center of the monolayer to reduce availability of nutrients and O2 and create a restricted extracellular space, hence inducing ischemic conditions. Reperfusion was performed by removing the coverslip after 1 hour of ischemia.
2.3 Measuring ΔΨm
To study the effect of IR on mitochondrial function, the fluorescent potentiometric indicator tetramethylrhodamine methyl ester (TMRM) was used to record ΔΨm. Monolayers were loaded with 50 or 100 nmol/l TMRM for 1 hour in the 37°C incubator. They were then washed and put in normal Tyrode’s solution in the heated (37°C) chamber of an inverted microscope (Eclipse TE2000-E, Nikon). Excitation light of 545 ± 12 nm was used and the emitted fluorescence (605 ± 35 nm) was recorded with an EMCCD camera (Cascade II, Photometrics, Tucson, AZ) using Micro-Manager (Vale Lab, UCSF, CA) or custom software developed in LabVIEW (Texas Instruments, Dallas, TX). Mitochondrial depolarization leads to loss of TMRM from the matrix and into the cytoplasm, which causes a decrease in the spatial dispersion of TMRM fluorescence. Spatial dispersion was quantified by calculating the coefficient of variation of the image fluorescence intensity, defined as the ratio of standard deviation to the mean (SD/mean), a dimensionless measure that provides a superior indicator for mitochondrial polarization than fluorescence intensity alone [32, 33], avoiding artifacts related to changes in dye load, illumination, bleaching, etc. (See Supplemental Figure 1). The data was analyzed using ImageJ, MATLAB (MathWorks, Natick, MA), and Origin (OriginLab, Northampton, MA).
2.4 Measuring oxidative Stress
Cyto-Grx1-roGFP2 [34] adenovirus was added to the media on the second day of culture. Experiments were done on the 4th–5th day of culture. Images were taken using a spinning disk confocal microscope (Andor Revolution XD, Olympus) at 10× magnification and analyzed using ImageJ and MATLAB. For each data point, two images were taken using laser excitation at 405 nm and 488 nm and recording the emission at 525 ± 25 nm. The excitation ratio (405 nm / 488 nm) indicates the redox status of the GSH/GSSG couple. Cyto-Grx1-roGFP2 ratios were normalized to between 0–1, with 1 and 0 representing the maxim and minimum levels of oxidation, respectively, during the ischemic or reperfusion phases.
2.5 Recording Sarcolemmal Membrane Voltage
To record the sarcolemmal membrane voltage and propagation of the excitation wave through the monolayer, the optical mapping technique was used. Monolayers were stained with 5 μmol/l di-4-ANEPPS (voltage-sensitive fluorescent dye; Invitrogen) for 30 minutes in the 37°C incubator, and then washed and put in the custom-built chamber of the optical mapping setup. The chamber was maintained at 37°C during the experiment. Excitation light of 480 ± 15 nm was used and emitted fluorescence was recorded at 500 Hz, with a 464-element photodiode array (WuTech), after passing through a red dye-coated glass filter (605 nm; long-pass). After filtering and digitization, data was analyzed with software developed using LabVIEW and MATLAB. Basic processing included zero-phase application of low-pass and high-pass butterworth filters, median filtering, detrending, normalization and application of a spatial filter.
2.6 Statistics
Data are presented as mean ± standard error of the mean (SEM) unless otherwise indicated. Statistical analyses were performed using MATLAB and Origin. Comparison between groups of data was performed using Student’s t-test or the χ2 contingency test (for the incidence of arrhythmias in Table 1). A P value of < 0.05 was used as the criterion for statistical significance.
Table 1.
Incidence of reentry: monolayers paced at 1 Hz.
| Control (N=10) | 4′-Cl-DZP (N=10) | Glibenclamide (N=8) | |
|---|---|---|---|
| Ischemia | 0 | 0 | 0 |
| Reperfusion | 70% | 10% | 87.5% |
3 Results
3.1 Loss and recovery of mitochondrial function during ischemia-reperfusion
Initially, cells under normoxic conditions were paced at 1 Hz with voltage pulse stimulation applied at the edge of the monolayer. Mitochondria were polarized and ΔΨm was stable (Figure 1A). After the coverslip was lowered onto the central part of the monolayer, ΔΨm declined over time in the ischemic region (Supplemental Figure 2). A border zone of about 2 mm was observed between the edge of the coverslip and the ischemic zone, due to continued availability of oxygen and nutrients through diffusion into this region. Loss of ΔΨm in the central ischemic zone, but not the border zone, was evident after 1 hour of ischemia (Figure 1B). Reperfusion by coverslip removal initiated a general repolarization of ΔΨm in the formerly ischemic region (Figure 1C–D).
Figure 1. Effect of 1 hour ischemia and the following reperfusion on the ΔΨm.
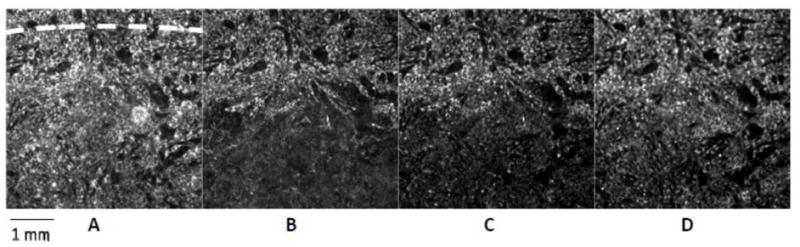
A: Start of ischemia; dashed line indicates the edge of the coverslip; the small area above the dashed line is not covered. B: Start of reperfusion; mitochondria in the ischemic area are depolarized. Mitochondrial ΔΨm recovers upon reperfusion. Recovery is shown at 45 minutes (C) and 2 hours (D) of reperfusion. 10× objective lens.
When observed at a higher resolution, ΔΨm was lost in two phases during ischemia: an initial slow and partial phase followed by a rapid global decline (Movie 1; Figure 2). During the initial 30 minutes of ischemia, individual clusters of mitochondria became depolarized (Figure 2A), followed by the second phase, which started, on average, after 37.6 ± 2.93 minutes of ischemia (N = 17), whereupon the mitochondrial network of the myocytes rapidly became depolarized and cells underwent contracture (Figure 2B). Figure 2C shows the decrease in spatial dispersion (i.e., ΔΨm depolarization) during these two phases over 60 minutes of ischemia for one monolayer and the subsequent recovery of ΔΨm in the same region after reperfusion. During ischemia, a wave of ΔΨm loss started from the center of the ischemic region and extended outwards (Movie 2). However, mitochondria in the uncovered area and the border zone were stable, apart from occasional ΔΨm oscillation (flickering) of a few individual mitochondria near the border zone. Observing the monolayer during a long period of ischemia showed that the mitochondria in the border zone could stay polarized even after 17 hours of ischemia followed by subsequent reperfusion (data not shown). Supplemental Figure 3A shows loss and recovery of ΔΨm in several monolayers during IR protocol to illustrate the extent of variability of the response. Reperfusion during the first phase of mitochondrial depolarization (up to 30 minutes of ischemia) resulted in stable recovery of ΔΨm; however, extending the ischemic period to 1 hour of ischemia resulted in an unstable recovery of ΔΨm following reperfusion (Movie 3). Sporadic episodes of ΔΨm loss and recovery (frequency < 20 mHz), evident as random large amplitude ΔΨm oscillations of separate clusters of mitochondria (Figure 3) were observed throughout the entire field of view in the post-ischemic zone. Early after reperfusion, adjacent clusters showed highly synchronized ΔΨm oscillations within individual myocytes, consistent with RIRR as a coupling factor; however, later in the reperfusion period, the correlation between ΔΨm oscillations in adjacent mitochondrial clusters declined (Figure 3B).
Figure 2. Change of ΔΨm during ischemia and reperfusion.

A: TMRM fluorescence images of NRVMs in the central region of the monolayer at 10 (left) and 20 min (right) of ischemia. During phase 1 ΔΨm depolarization, individual clusters of mitochondria become depolarized (indicated by white arrows; fluorescence intensity scaled as indicated by the lookup table; 40× objective lens). B: Two phases of ΔΨm loss during long ischemia show sparse (phase 1; < 30 min ischemia), then pronounced (phase 2; 30–60 min ischemia), redistribution of TMRM fluorescence to the cytoplasm, with ΔΨm recovering quickly upon reperfusion (same field of view as in panel A; selected frames from Supplemental Movies 1 and 3; durations of ischemia or reperfusion shown below the frames in min). C: Spatial TMRM dispersion during IR reflects timing of ΔΨm depolarization and repolarization independent of artifacts due to total fluorescence intensity variation (see Supplemental Figure 1 and ref. [33]). ΔΨm instability limits the recovery rate of mitochondrial membrane potential averaged over the entire image.
Figure 3. Oscillations of ΔΨm upon reperfusion.

A: Subcellular ΔΨm oscillations in mitochondrial clusters within NRVMs in the monolayer (25 × 25 μm2 zoomed region from the 40× image sequence; frame interval 1 min for minutes 50–55 of reperfusion). B: Two upper and left lower panel: intensity plots of TMRM signals during reperfusion for mitochondria at positions A, B, and C, as indicated on panel A, reveals sustained ΔΨm oscillation with variable periodicity throughout the reperfusion period. Lower right panel: Oscillations of adjacent clusters A and B are more synchronized during early reperfusion but are less synchronized at longer reperfusion times.
ΔΨm oscillations after reperfusion were generally sustained for the whole period of recording (up to 120 minutes). However, in some cases, the mitochondrial network of an individual myocyte or a few myocytes depolarized abruptly. This ΔΨm depolarization affected the neighboring myocytes and a wave of depolarization propagated through the monolayer (Figure 4A). This type of depolarization, which was irreversible, occurred with variable onset after reperfusion. Such events could start as late as 2 hours after the onset of reperfusion (Movie 4), or as shown in the dispersion plot in Supplemental Figure 4 (black line), it could start within the first 15 minutes of reperfusion.
Figure 4. Representative traces of IR-induced changes in TMRM intensity in the presence and absence of 4′-Cl-DZP.

A: Large amplitude ΔΨm oscillations of a mitochondrial cluster upon reperfusion are shown relative to a global irreversible ΔΨm depolarization wave occurring later in the same experiment. B: Stable recovery of mitochondrial energetics in the presence of 4′-Cl-DZP. C: Number of transient ΔΨm depolarizations in the first 30 minutes of reperfusion after 1 hour ischemia for 40 randomly selected mitochondrial clusters in each of 4 separate control (4.49 ± 0.2 oscillations/30 min) and 4′-Cl-DZP treatment experiments (0.55 ± 0.09 oscillations/30 min; P < 0.0001). Image field analyzed: 150 μm2 in the reperfused zone; small dots show individual data points. Box plot depicts mean (small filled squares) and standard deviation (vertical lines) with median and standard error represented by the open box height and inclusive horizontal line, respectively.
3.2 Effect of 4′-Cl-DZP on ΔΨm oscillations and glutathione redox potential
4′-Cl-DZP treatment was previously shown to stabilize ΔΨm in adult cardiomyocytes displaying RIRR-mediated mitochondrial ΔΨm oscillations [11], and to prevent the loss of ΔΨm in hearts subjected to glutathione depletion [25]. Interestingly, 4′-Cl-DZP slowed the rate of oxidation of the cytosolic GSH pool during ischemia (Figure 5A), and tended to delay phase 2 ΔΨm depolarization, which started at 43.57 ± 2.54 minutes (N = 7; not statistically significant). Supplemental Figure 3B shows loss and recovery of ΔΨm in several monolayers during IR in the presence of 4′-Cl-DZP. When 4′-Cl-DZP (16 μmol/l) was present throughout the IR protocol, GSH/GSSG redox state recovered faster upon reperfusion (Figure 5B), and ΔΨm stably repolarized (Figure 4B). Only a few clusters showed oscillations and these oscillations were very sparse (Figure 4C).
Figure 5. GSH/GSSG dynamics during coverslip-induced IR and effects of 4′-Cl-DZP.
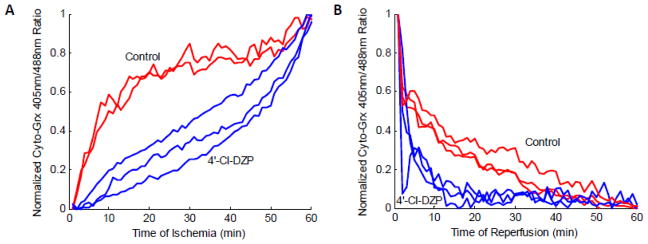
A: Ischemia-induced oxidation of the cytoplasmic GSH pool, followed by net GSH reduction on reperfusion. GSSG:GSH increased rapidly during early ischemia (< 10 min) in control monolayers, but in those treated with 4′-Cl-DZP, GSH oxidation was blunted, accelerating only after ~ 45 min of ischemia. B: 4′-Cl-DZP enhanced the recovery kinetics of the GSH pool during reperfusion.
We also tested for the possible involvement of PTP in IR-induced mitochondrial oscillations in separate experiments. The inhibitor of PTP, CsA (1 μmol/l), present during the entire IR protocol, did not prevent ischemia-induced loss of ΔΨm, nor did it suppress oscillations of ΔΨm after reperfusion, arguing against a role for PTP in the reperfusion-induced mitochondrial oscillations. On the other hand, the global loss of ΔΨm, at longer reperfusion times, was not observed in the presence of CsA (Supplemental Figure 4), suggesting a role for PTP in mitochondrial ΔΨm depolarization in late reperfusion injury. This is consistent with previous work demonstrating that there is sequential activation of the IMAC (4′-Cl-DZP-sensitive) and PTP (CsA-sensitive) pores upon oxidative stress [18].
3.3 Arrhythmias
Under normoxic control conditions, the excitation wave propagated through the monolayer uniformly (Movie 5) with a CV = 15.5 ± 6.1 (SD) cm/s (N = 10), evoking action potentials with APD50 = 138 ± 54 (SD) msec (N = 15). After placing the coverslip over the center part of the monolayer, APA and APD50 decreased to 77 ± 25 (SD)% and 57.9 ± 30.8 (SD)%, respectively, of initial values after 5 min of ischemia (Supplemental Figure 5), while conduction velocity (CV) slowed to 8.3 ± 4.4 (SD) cm/s. Therefore, the wavelength shortened in the ischemic area over time (Movie 6). Cells in the ischemic region stopped contracting approximately 6 minutes after coverslip placement. After 15.4 ± 5.4 (SD) min (N = 13) of ischemia, the area under the coverslip became completely inexcitable (Figure 6). At this time, the activation wave passed around the ischemic area, exciting only the cells in the non-ischemic region and the border zone (Movie 7). At the normal pacing rate of 1 Hz, reentry did not occur; however, when the monolayer was paced at 3 Hz, in 3 of 18 monolayers a heterogeneous decrease in excitability beneath the coverslip caused a unidirectional block and led to reentry. Overall, reentry was not a typical event during ischemia.
Figure 6. Electrical propagation during ischemia/reperfusion.
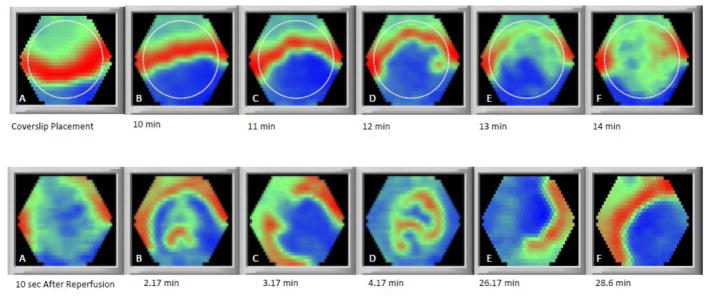
Upper panels: Coverslip-induced ischemia induced inexcitability and prevented the propagation of the excitation wave through the ischemic region. The dashed line indicates the edge of the coverslip. Lower panels: Sequelae of abnormal electrical propagation and reentry upon reperfusion. A: The excitation wave passes around the reperfused area. B: After a short time, wavelets form in the reperfused area. C: Formation of two reentrant waves with rotors located on the boundary of the border zone and reperfused zone. D: Three reentrant waves underlying fibrillatory activity. E: Small reentrant waves coalesce and form a single reentrant spiral wave. F: After 28 min of reperfusion, the reentrant wave ceases and near normal propagation is observed; however, CV in the reperfused area remains slow through the end of the experiment (38 minutes of reperfusion; not shown).
Excitability recovered shortly after reperfusion, but not homogeneously (Figure 6). The recovery started 1.1 ± 0.6 (SD) minutes (N = 12) after the onset of reperfusion. The excitation wave entered the recovered regions forming several slowly propagating stray wavelets in the post-ischemic zone, which reentered into the non-ischemic area (Movie 8). Due to the low CV and short wavelength in this region, this phase was soon (ranging from a few seconds up to 3 minutes) replaced by micro-reentries and fibrillatory activity (Movie 9), which later evolved into sustained reentry, anchored on the boundary between the border zone and ischemic zone, in 7 of 10 monolayers (Movie 10). The reentrant spiral wave remained anchored near the border between the reperfused and non-ischemic zones, either staying at the same point or migrating to other points on the boundary or, occasionally, inside the reperfused area.
4′-Cl-DZP had no significant effects on APD or CV at baseline (Supplementary Table). Consistent with its protective effect against the oxidative stress, 4′-Cl-DZP preserved CV early during ischemia (CV = 14.16 ± 6.74 (SD) after 5 min of ischemia). This is in line with the findings that oxidative stress could severely affect gap junction conduction [35]. While 4′-Cl-DZP was shown to blunt APD shortening and prolong electrical excitability in intact perfused hearts during ischemia [11], in the present experiments, 4′-Cl-DZP did not significantly affect the AP failure and inexcitability induced by coverslip-induced ischemia in the NRVM monolayers. Recovery of excitability upon reperfusion was, however, somewhat delayed, similar to its effects in intact hearts. For several minutes after the initial recovery of AP generation, the monolayers did not respond to the stimulation pulses in a 1:1 fashion. However, by 10–20 minutes after reperfusion started, normal excitability was restored. As shown in Figure 7, while 4′-Cl-DZP treatment delayed recovery after ischemia, it prevented reperfusion-induced reentry (χ2 test: P < 0.01 compared with control; Table 1) and facilitated a near complete recovery of APD to the pre-ischemic values within 2–4 minutes (Movie 11). By the time excitability recovered, CV and wavelength also recovered to the point that prevented formation of stray wavelets in the post-ischemic region, which had caused reentry in the control. Spontaneous activity elicited on the border zone was also suppressed in the presence of 4′-Cl-DZP, occurring only in 1 out of 10 monolayers; for this singular event, reentry occurred 20 minutes after the onset of reperfusion (late reperfusion arrhythmia).
Figure 7. 4′-Cl-DZP stabilizes electrical activity during reperfusion.

Changes in optical action potential amplitude and duration during IR in the absence (A) or presence (B) of 4′-Cl-DZP (16 μmol/l). 4′-Cl-DZP did not prevent ischemia-induced loss of electrical excitability, but enhanced APD recovery to pre-ischemic values after reperfusion. Ischemia rendered the monolayer inexcitable with (C) or without 4′-Cl-DZP, but completely prevented reentry upon reperfusion (D). See text and Table 1 for analysis of arrhythmia incidence. Dashed line in panel C indicates the edge of the coverslip.
4 Discussion
The main contributions of this work are that 1) oscillations of mitochondrial ΔΨm are consistently observed upon reperfusion in monolayers of cardiac myocytes after a long period of ischemia, 2) ΔΨm depolarization occurs in two phases during coverslip-mediated ischemia in NRVM monolayers; localized ΔΨm loss in a limited number of cells for ischemic times of 30 min or less, and global ΔΨm depolarization of the network between 30 min and 1 hour of ischemia, 3) reentrant arrhythmias develop spontaneously upon reperfusion after 1 hour ischemia during the course of ΔΨm recovery, 4) ΔΨm instability/oscillation is prevalent in the post-ischemic zone after reperfusion and correlates with the window of vulnerability for tachyarrhythmias, 5) 4′-Cl-DZP enhances ΔΨm recovery on reperfusion by preventing ΔΨm oscillations while stabilizing electrical excitability, and 6) inhibition of the PTP with CsA does not impact early ΔΨm oscillations on reperfusion, but may prevent later widespread ΔΨm collapse. Together, the findings suggest that ΔΨm stabilization during reperfusion is an important goal when developing antiarrhythmic strategies.
Although first recognized almost 80 years ago [36], the mechanisms underlying reperfusion arrhythmias remain poorly understood, in part due to the multifactorial changes that are induced by ischemia [6]. Ideally, in vitro models of IR should preserve as many of the essential features of myocardial IR injury while reducing the complexity of the system enough to measure relevant variables and modulate potential therapeutic targets. The coverslip IR model in the NRVM monolayer, originally developed by Pitts and Toombs [29], meets these criteria in several important ways: it creates a regional ischemic microenvironment that not only limits cellular access to oxygen and substrates, but also results in a restricted extracellular space, within which ischemic metabolites (e.g., lactate) and ions (H+, K+) accumulate, just as in the ischemic heart. Moreover, the ability to simulate reperfusion and study the interface between the ischemic and non-ischemic cells in the monolayer allows one to examine the effects of heterogeneity in the excitable medium, essential for understanding the origin of arrhythmias, as demonstrated in previous optical mapping studies [30, 37]. An additional advantage realized in the present work is that the functional impact of IR at the macroscopic (whole monolayer) and microscopic (subcellular) level can be investigated. The platform also permitted the ready introduction of a genetically-encoded fluorescent probe for measuring GSH/GSSG redox potential, a key indicator of the antioxidant response.
Reperfusion after 1 hour of ischemia markedly destabilized the mitochondrial network of the cells in the NRVM monolayer. This occurred in phases - early reperfusion was characterized by asynchronous ΔΨm oscillations in subcellular clusters of mitochondria, while late reperfusion was more likely to display widespread, irreversible ΔΨm collapse happening occasionally anywhere between 15 minutes to 2 hours after the onset of reperfusion. Notably, in some experiments, the reperfusion-induced mitochondrial instability was observed to spread beyond the post-ischemic zone. The normoxic regions of the monolayer underwent sporadic and dispersed loss of ΔΨm, even though they had not been affected during ischemia. This suggests that toxic metabolites can diffuse out of the ischemic zone to destabilize mitochondria in otherwise healthy cells. NRVMs at the outer edge of the coverslip did not depolarize during ischemia and tended to resist ΔΨm loss during reperfusion, suggesting that they were preconditioned by their proximity to the ischemic zone.
Within the reperfused zone, recovery was characterized by random ΔΨm oscillations or flickering, demonstrating that spatiotemporal heterogeneity of energetics spans from the subcellular to the syncytial level. 4′-Cl-DZP prevented mitochondrial destabilization and facilitated AP recovery upon reperfusion while preventing post-ischemic arrhythmias. It also significantly decreased the rate of GSH oxidation during ischemia and hastened recovery of the GSH pool during reperfusion, consistent with its presumed role in inhibiting mitochondrial RIRR. In contrast, CsA was ineffective in preventing early ΔΨm flickers, although later widespread ΔΨm loss appeared to be prevented (Supplemental Figure 4), supporting a role for PTP opening in late reperfusion injury. Due to the irregularity of this late mitochondrial depolarization, statistical verification of the CsA effect on PTP was not possible; however, these observations comport with the idea that there is a hierarchy of mitochondrial channels activated by RIRR during IR or oxidative stress; the first sensitive to inhibition by benzodiazepines and the second sensitive to CsA [18]. While the molecular entities responsible for this pharmacological distinction are incompletely resolved, we have attributed the antiarrhythmic and ΔΨm stabilization effects of 4′-Cl-DZP to inhibition of IMAC, and the latter CsA-sensitive ΔΨm depolarization to the PTP [18]. 4′-Cl-DZP does not inhibit the Ca2+-dependent activation of PTP in isolated mitochondria (Supplemental Figure 6), but an interaction between a benzodiazepine binding site and the PTP structure involving the ATP synthase has recently been proposed [38]. Regardless of whether or not the two inner membrane pores physically interact, a functional interaction is likely, since ROS bursts evoked during RIRR would be expected to sensitize the PTP to Ca2+-dependent activation [18].
The association between ΔΨm heterogeneity and arrhythmias upon reperfusion, and the sensitivity of both to inhibition by 4′-Cl-DZP, supports the linkage between mitochondrial and electrical instability, but the details of how they are linked bears further discussion. The activity of many ion channels and transporters may be affected by the pathophysiological changes induced by IR, including acidification, ROS accumulation, decreased ATP availability, etc. With respect to RIRR, a major consequence of ΔΨm loss or oscillation is the opening of KATP channels, secondary to ATP hydrolysis, which profoundly affects the action potential duration and excitability. We proposed that KATP current activation, occurring heterogeneously in regions of the tissue or in the cell monolayer could underlie increased vulnerability to reentrant arrhythmias by increasing dispersion of AP repolarization [11]. In a recent study, such metabolic current sinks, generated regionally by local perfusion of the monolayer with a mitochondrial uncoupler, increased the incidence of reentry in NRVM monolayers, as well as in a 2D computer simulation of mitochondrial oscillation in an integrated guinea pig heart cell model [28]. The arrhythmias were prevented by blocking KATP current with glibenclamide; however, the evidence also pointed to a contribution of other mechanisms, including effects on conduction velocity, presumably related to inhibition of gap junctional conductance [28].
In the present study, we found that the mechanism of IR arrhythmias differed from that of simple chemical uncoupling of oxidative phosphorylation. Blocking KATP current using glibenclamide or glimepiride had no effect on ΔΨm depolarization during coverslip-induced ischemia or mitochondrial oscillations during reperfusion (Supplemental Figure 7). Glibenclamide slightly decreased the loss of APD during ischemia (Supplemental Figure 8), and somewhat unexpectedly, it did not significantly decrease the incidence of arrhythmias upon reperfusion; reentry occurred in 7 out of 8 monolayers (χ2 test: N.S. compared with control; Table 1). Clearly, IR induced a much more complex set of cellular changes that were not reversed by inhibition of a single ion channel target. Unlike agents that prevent mitochondrial instability (e.g., 4′-Cl-DZP), KATP channel inhibition would not be expected to fix the global defects in energy supply and ROS imbalance that accompanies IR. Moreover, as these KATP channel inhibitors are not specific to sarcolemmal KATP channels, the detrimental actions of these compounds could be the result of blocking mitochondrial KATP channels. Mitochondrial KATP channels were shown previously to be critical in ischemic preconditioning [39–41]; hence blocking them might prevent their protective effects. Glibenclamide has been shown to be partially effective in preventing reperfusion arrhythmias in perfused whole heart experiments [11] and suppresses reperfusion arrhythmias in dogs [42, 43], but others have observed adverse effects on post-ischemic arrhythmias in sheep [44]. Species differences in the electrophysiology of the cardiomyocytes could partly account for differences in the antiarrhythmic action of glibenclamide in these studies. For example, in NRVMs, AP repolarization is more dependent on the transient outward K+ current Ito [45], unlike in guinea pig, large animal, or human heart cells, and the differences in the electrophysiology of NRVMs in the monolayer culture should not be ignored. For example, as shown by den Haan et al. [46], cultured NRVM monolayers have a less polarized resting membrane potential (~ −60 mV) than intact adult or neonatal rat hearts (~−80 mV), which could alter the sensitivity of the cultured cells′ electrophysiology to ion channel modulators. In addition, differences in metabolism in adult versus neonatal hearts are well known [47, 48], so while the NRVM monolayer is a practical and useful model to study electrical activity and mitochondrial function during IR, extrapolation of the findings to arrhythmogenic mechanisms in adult hearts should be done with caution. While KATP current can contribute to arrhythmogenesis in NRVM monolayers after chemical uncoupling of ΔΨm [28], the present findings suggests that IR induces a unique set of alterations in the electrophysiological substrate, underscoring the utility of the coverslip IR model for mechanistic investigation. This has been illustrated nicely in an earlier study by de Diego et al. [30], who showed that increased dephosphorylation of Cx43 during ischemia contributes to CV slowing. Slow recovery of Cx43 phosphorylation was considered to be a major arrhythmogenic factor during early reperfusion. Later, Wang et al. showed the protective effects of nutrition restriction preconditioning against IR using the same model [37]; post-ischemic mitochondrial depolarization was prevented and IR-induced arrhythmias were decreased, although the subcellular dynamics of ΔΨm were not investigated. Our high resolution observations of the mitochondrial dynamics during IR, in the present work, strongly support the hypothesis that mitochondrial ΔΨm instability plays a major role in post-ischemic arrhythmias. The similar protection afforded by 4′-Cl-DZP on ΔΨm, mitochondrial GSH recovery and post-ischemic arrhythmias suggests mitochondria as a major potential target for antiarrhythmic interventions.
Supplementary Material
Highlights.
Simulating ischemia-reperfusion (IR) in vitro is challenging; many methods do not reproduce conditions in the intact heart
Coverslip-induced IR in beating myocyte monolayers enables studies of mitochondrial dysfunction and arrhythmias in vitro
Mitochondrial potential (ΔΨm) is lost in 2 phases during ischemia; spatiotemporal instability of ΔΨm occurs upon reperfusion
Reperfusion-induced reentry correlates with ΔΨm instability; 4′-chlorodiazepam stabilizes ΔΨm and prevents arrhythmias
Mitochondrial ΔΨm stabilization during reperfusion is the key to preventing arrhythmias during ischemia-reperfusion injury
Acknowledgments
This work was supported by NIH grants R01HL105216 and R37HL54598 (B.O’R.).
Footnotes
Disclosures/Conflicts of Interest: None declared.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Blaha MJ, et al. Executive summary: Heart disease and stroke statistics--2014 update: A report from the american heart association. Circulation. 2014 Jan 21;129(3):399–410. doi: 10.1161/01.cir.0000442015.53336.12. [DOI] [PubMed] [Google Scholar]
- 2.Maxwell SRJ, Lip GYH. Reperfusion injury: A review of the pathophysiology, clinical manifestations and therapeutic options. Int J Cardiol. 1997 Jan 31;58(2):95–117. doi: 10.1016/s0167-5273(96)02854-9. [DOI] [PubMed] [Google Scholar]
- 3.Zweier JL, Flaherty JT, Weisfeldt ML. Direct measurement of free radical generation following reperfusion of ischemic myocardium. Proceedings of the National Academy of Sciences. 1987;84(5):1404–7. doi: 10.1073/pnas.84.5.1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carry MM, Mrak RE, Murphy ML, Peng CF, Straub KD, Fody EP. Reperfusion injury in ischemic myocardium: Protective effects of ruthenium red and of nitroprusside. Am J Cardiovasc Pathol. 1989;2(4):335–44. [PubMed] [Google Scholar]
- 5.Woodward B, Zakaria MN. Effect of some free radical scavengers on reperfusion induced arrhythmias in the isolated rat heart. J Mol Cell Cardiol. 1985;17(5):485–93. doi: 10.1016/s0022-2828(85)80053-5. [DOI] [PubMed] [Google Scholar]
- 6.Manning AS, Hearse DJ. Reperfusion-induced arrhythmias: Mechanisms and prevention. Journal of molecular and cellular cardiology. 1984 Jun;16(6):497–518. doi: 10.1016/s0022-2828(84)80638-0. [DOI] [PubMed] [Google Scholar]
- 7.Pepe M, Zanna D, Quagliara D, Caiati C, Marzullo A, Palmiotto AI, et al. Sudden cardiac death secondary to demonstrated reperfusion ventricular fibrillation in a woman with takotsubo cardiomyopathy. Cardiovascular Pathology. 2011;20(4):254–7. doi: 10.1016/j.carpath.2010.06.006. [DOI] [PubMed] [Google Scholar]
- 8.Hauer RN, de Bakker JM, de Wilde AA, Straks W, Vermeulen JT, Janse MJ. Ventricular tachycardia after in vivo DC shock ablation in dogs. electrophysiologic and histologic correlation. Circulation. 1991 Jul;84(1):267–78. doi: 10.1161/01.cir.84.1.267. [DOI] [PubMed] [Google Scholar]
- 9.Hume J, Katzung BG. Physiological role of endogenous amines in the modulation of ventricular automaticity in the guinea-pig. The Journal of physiology. 1980 Dec;309:275–86. doi: 10.1113/jphysiol.1980.sp013508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zaitsev AV, Warren MD. Mechanisms of ischemic ventricular fibrillation. In: Zipes DP, Jalife J, editors. Cardiac Electrophysiology: From Cell to Bedside. 5. Philadelphia, PA: Saunders; 2009. [Google Scholar]
- 11.Akar FG, Aon MA, Tomaselli GF, O’Rourke B. The mitochondrial origin of postischemic arrhythmias. J Clin Invest. 2005 Dec;115(12):3527–35. doi: 10.1172/JCI25371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aon MA, Cortassa S, O’Rourke B. Percolation and criticality in a mitochondrial network. Proc Natl Acad Sci U S A. 2004 Mar 30;101(13):4447–52. doi: 10.1073/pnas.0307156101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhou L, Aon MA, Almas T, Cortassa S, Winslow RL, O’Rourke B. A reaction-diffusion model of ROS-induced ROS release in a mitochondrial network. PLoS Comput Biol. 2010;6(1) doi: 10.1371/journal.pcbi.1000657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Aon MA, Cortassa S, O’Rourke B. The fundamental organization of cardiac mitochondria as a network of coupled oscillators. Biophys J. 2006;91(11):4317–27. doi: 10.1529/biophysj.106.087817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zorov DB, Filburn CR, Klotz LO, Zweier JL, Sollott SJ. Reactive oxygen species (ROS)-induced ROS release: A new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J Exp Med. 2000 Oct 2;192(7):1001–14. doi: 10.1084/jem.192.7.1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial ROS-induced ROS release: An update and review. Biochimica et Biophysica Acta (BBA)-Bioenergetics. 2006;1757(5):509–17. doi: 10.1016/j.bbabio.2006.04.029. [DOI] [PubMed] [Google Scholar]
- 17.Brady NR, Hamacher-Brady A, Westerhoff HV, Gottlieb RA. A wave of reactive oxygen species (ROS)-induced ROS release in a sea of excitable mitochondria. Antioxidants & redox signaling. 2006;8(9–10):1651–65. doi: 10.1089/ars.2006.8.1651. [DOI] [PubMed] [Google Scholar]
- 18.Aon MA, Cortassa S, Maack C, O’Rourke B. Sequential opening of mitochondrial ion channels as a function of glutathione redox thiol status. The Journal of biological chemistry. 2007 Jul 27;282(30):21889–900. doi: 10.1074/jbc.M702841200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aon MA, Cortassa S, Marban E, O’Rourke B. Synchronized whole-cell oscillations in mitochondrial metabolism triggered by a local release of reactive oxygen species in cardiac myocytes. J Biol Chem. 2003 Aug 20;278:44735–44. doi: 10.1074/jbc.M302673200. [DOI] [PubMed] [Google Scholar]
- 20.Beavis AD. On the inhibition of the mitochondrial inner membrane anion uniporter by cationic amphiphiles and other drugs. J Biol Chem. 1989 Jan 25;264(3):1508–15. [PubMed] [Google Scholar]
- 21.Beavis AD. Properties of the inner membrane anion channel in intact mitochondria. J Bioenerg Biomembr. 1992 Feb;24(1):77–90. doi: 10.1007/BF00769534. [DOI] [PubMed] [Google Scholar]
- 22.Lederer WJ, Nichols CG, Smith GL. The mechanism of early contractile failure of isolated rat ventricular myocytes subjected to complete metabolic inhibition. The Journal of physiology. 1989 Jun;413:329–49. doi: 10.1113/jphysiol.1989.sp017657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sasaki N, Sato T, Marban E, O’Rourke B. ATP consumption by uncoupled mitochondria activates sarcolemmal K(ATP) channels in cardiac myocytes. Am J Physiol Heart Circ Physiol. 2001;280(4):H1882–8. doi: 10.1152/ajpheart.2001.280.4.H1882. [DOI] [PubMed] [Google Scholar]
- 24.O’Rourke B, Ramza BM, Marban E. Oscillations of membrane current and excitability driven by metabolic oscillations in heart cells. Science. 1994 Aug 12;265(5174):962–6. doi: 10.1126/science.8052856. [DOI] [PubMed] [Google Scholar]
- 25.Brown D, Aon M, Akar F, O’Rourke B. A ligand to the mitochondrial benzodiazepine receptor prevents ventricular arrhythmias and LV dysfunction after ischemia or glutathione depletion. FASEB J. 2008;22:747.7. [Google Scholar]
- 26.Brown DA, Aon MA, Akar FG, Liu T, Sorarrain N, O’Rourke B. Effects of 4′-chlorodiazepam on cellular excitation-contraction coupling and ischaemia-reperfusion injury in rabbit heart. Cardiovascular research. 2008 Jul 1;79(1):141–9. doi: 10.1093/cvr/cvn053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brown DA, Aon MA, Frasier CR, Sloan RC, Maloney AH, Anderson EJ, et al. Cardiac arrhythmias induced by glutathione oxidation can be inhibited by preventing mitochondrial depolarization. J Mol Cell Cardiol. 2010;48(4):673–9. doi: 10.1016/j.yjmcc.2009.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhou L, Solhjoo S, Millare B, Plank G, Abraham MR, Cortassa S, et al. Effects of regional mitochondrial depolarization on electrical propagation: Implications for arrhythmogenesis. Circ Arrhythm Electrophysiol. 2014;7:143–51. doi: 10.1161/CIRCEP.113.000600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pitts KR, Toombs CF. Coverslip hypoxia: A novel method for studying cardiac myocyte hypoxia and ischemia in vitro. American Journal of Physiology-Heart and Circulatory Physiology. 2004;287(4):H1801–12. doi: 10.1152/ajpheart.00232.2004. [DOI] [PubMed] [Google Scholar]
- 30.de Diego C, Pai RK, Chen F, Xie LH, De Leeuw J, Weiss JN, et al. Electrophysiological consequences of acute regional ischemia/reperfusion in neonatal rat ventricular myocyte monolayers. Circulation. 2008;118(23):2330–7. doi: 10.1161/CIRCULATIONAHA.108.789149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.National Research Council. Guide for the care and use of laboratory animals. 8. Washington, DC: The National Academies Press; 2011. [Google Scholar]
- 32.Romashko DN, Marban E, O’Rourke B. Subcellular metabolic transients and mitochondrial redox waves in heart cells. Proc Natl Acad Sci U S A. 1998;95(4):1618–23. doi: 10.1073/pnas.95.4.1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Duchen MR, Surin A, Jacobson J. Imaging mitochondrial function in intact cells. Meth Enzymol. 2003;361:353–89. doi: 10.1016/s0076-6879(03)61019-0. [DOI] [PubMed] [Google Scholar]
- 34.Albrecht SC, Barata AG, Großhans J, Teleman AA, Dick TP. In vivo mapping of hydrogen peroxide and oxidized glutathione reveals chemical and regional specificity of redox homeostasis. Cell metabolism. 2011;14(6):819–29. doi: 10.1016/j.cmet.2011.10.010. [DOI] [PubMed] [Google Scholar]
- 35.Sovari AA, Rutledge CA, Jeong EM, Dolmatova E, Arasu D, Liu H, et al. Mitochondria oxidative stress, connexin43 remodeling, and sudden arrhythmic death. Circ Arrhythm Electrophysiol. 2013 Jun;6(3):623–31. doi: 10.1161/CIRCEP.112.976787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tennant R, Wiggers CJ. The effect of coronary occlusion on myocardial contraction. American Journal of Physiology--Legacy Content. 1935;112(2):351–61. [Google Scholar]
- 37.Wang S, Chen J, Valderrábano M. Nutrient restriction preserves calcium cycling and mitochondrial function in cardiac myocytes during ischemia and reperfusion. Cell Calcium. 2012;51(6):445–51. doi: 10.1016/j.ceca.2012.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Giorgio V, von Stockum S, Antoniel M, Fabbro A, Fogolari F, Forte M, et al. Dimers of mitochondrial ATP synthase form the permeability transition pore. Proc Natl Acad Sci U S A. 2013 Apr 9;110(15):5887–92. doi: 10.1073/pnas.1217823110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu Y, Sato T, O’Rourke B, Marban E. Mitochondrial ATP-dependent potassium channels : Novel effectors of cardioprotection? Circulation. 1998 Jun 23;97(24):2463–9. doi: 10.1161/01.cir.97.24.2463. [DOI] [PubMed] [Google Scholar]
- 40.Akao M, Ohler A, O’Rourke B, Marban E. Mitochondrial ATP-sensitive potassium channels inhibit apoptosis induced by oxidative stress in cardiac cells. Circ Res. 2001 Jun 22;88(12):1267–75. doi: 10.1161/hh1201.092094. [DOI] [PubMed] [Google Scholar]
- 41.O’Rourke B. Evidence for mitochondrial K+ channels and their role in cardioprotection. Circ Res. 2004 Mar 5;94(4):420–32. doi: 10.1161/01.RES.0000117583.66950.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Billman GE, Avendano CE, Halliwill JR, Burroughs JM. The effects of the ATP-dependent potassium channel antagonist, glyburide, on coronary blood flow and susceptibility to ventricular fibrillation in unanesthetized dogs. Journal of Cardiovascular Pharmacology. 1993;21:197–204. doi: 10.1097/00005344-199302000-00003. [DOI] [PubMed] [Google Scholar]
- 43.Billman GE. The cardiac sarcolemmal ATP-sensitive potassium channel as a novel target for anti-arrhythmic therapy. Pharmacology & therapeutics. 2008 Oct;120(1):54–70. doi: 10.1016/j.pharmthera.2008.07.004. [DOI] [PubMed] [Google Scholar]
- 44.del Valle HF, Lascano EC, Negroni JA, Crottogini AJ. Glibenclamide effects on reperfusion-induced malignant arrhythmias and left ventricular mechanical recovery from stunning in conscious sheep. Cardiovasc Res. 2001 Jun;50(3):474–85. doi: 10.1016/s0008-6363(01)00209-7. [DOI] [PubMed] [Google Scholar]
- 45.Kassiri Z, Zobel C, Molkentin JD, Backx PH. Reduction of ito causes hypertrophy in neonatal rat ventricular myocytes. Circ Res. 2002;90(5):578–85. doi: 10.1161/01.res.0000012223.86441.a1. [DOI] [PubMed] [Google Scholar]
- 46.den Haan AD, Veldkamp MW, Bakker D, Boink GJ, Janssen RB, de Bakker JM, et al. Organ explant culture of neonatal rat ventricles: A new model to study gene and cell therapy. PloS one. 2013;8(3):e59290. doi: 10.1371/journal.pone.0059290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wittels B, Bressler R. Lipid metabolism in the newborn heart. J Clin Invest. 1965;44(10):1639–46. doi: 10.1172/JCI105270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rogers C. Fatty acids and phospholipids of adult and newborn rat hearts and of cultured, beating neonatal rat myocardial cells. Lipids. 1974;9(8):541–7. doi: 10.1007/BF02532502. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.