

Abstract
Motor deficits are present in cardiac arrest survivors and injury to cerebellar Purkinje cells (PCs) likely contribute to impairments in motor coordination and post-hypoxic myoclonus. NMDA receptor mediated excitotoxicity is a well-established mechanism of cell death in several brain regions, but the role of NMDA receptors in PC injury remains understudied. Emerging data in cortical and hippocampal neurons indicates that the GluN2A-containing NMDA receptors signal to improve cell survival and GluN2B-containing receptors contribute to neuronal injury. This study compared neuronal injury in the hippocampal CA1 region to that in PCs and investigated the role of NMDA receptors in PC injury in our mouse model of cardiac arrest and cardiopulmonary resuscitation (CA/CPR). Analysis of cell density demonstrated a 24% loss of PCs within 24 hours after 8 min CA/CPR and injury stabilized to 33% by 7 days. The subunit promiscuous NMDA receptor antagonist MK-801 protected both CA1 neurons and PCs from ischemic injury following CA/CPR, demonstrating a role for NMDA receptor activation in injury to both brain regions. In contrast, the GluN2B antagonist, Co 101244, had no effect on Purkinje cell loss while protecting against injury in the CA1 region. These data indicate that ischemic injury to cerebellar PCs progresses via different cell death mechanisms compared to hippocampal CA1 neurons.
Keywords: Cardiac arrest, excitotoxicity, global ischemia, Purkinje cells, NMDA
1. Introduction
Cardiac arrest is a leading cause of death and disability with approximately 600,000 cardiac arrests occurring annually in the United States (Go et al., 2013). Neurological injury resulting from cardiac arrest is the most common cause of mortality and morbidity among those who are successfully resuscitated (Laver et al., 2004; Lim et al., 2004). Transient global ischemia resulting from cardiac arrest and cardiopulmonary resuscitation (CA/CPR) results in death of selectively vulnerable populations of neurons (Ng et al., 1989; Horn & Schlote, 1992; Kofler et al., 2004). Rodent models of cerebral ischemia have been used extensively to examine injury mechanisms and neuroprotective therapies in cortex, striatum and hippocampus (Merchenthaler et al., 2003; Traystman, 2003; Kofler et al., 2004; Noppens et al., 2008). However, fewer studies have focused on the cerebellum following global ischemia, despite data indicating that cerebellar Purkinje cells are highly vulnerable to cerebral ischemia (Ng et al., 1989; Horn & Schlote, 1992; Welsh et al., 2002; Paine et al., 2012). Movement disorders are often observed in cardiac arrest survivors and are attributed primarily to striatal damage, but it is likely that cerebellar injury also contributes to motor deficits in post-cardiac arrest patients (Venkatesan & Frucht, 2006; Lu-Emerson & Khot, 2010). Purkinje neurons are the sole output of the cerebellar cortex and play a critical role in motor coordination, motor learning, and aspects of cognition (Gilbert & Thach, 1977; Llinás & Welsh, 1993). They receive and integrate excitatory inputs from sensory, vestibular and motor areas to allow for temporally and spatially precise movement (Thach, 1998; Bastian, 2006). There is evidence of Purkinje cell loss in post-mortem examinations of cardiac arrest victims and in rodent models of global cerebral ischemia however, the mechanism of injury to these cells remains understudied (Horn & Schlote, 1992; Welsh et al., 2002; Lim et al., 2004; Kelley et al., 2011).
Global ischemia results in neuronal death in the CA1 region of the hippocampus with a delayed onset of 2-3 days (Pulsinelli et al., 1982; Kofler et al., 2004). Mechanisms contributing to death of these neurons include excitotoxicity, oxidative stress and activation of apoptotic pathways (Martin et al., 1998; Saito et al., 2005; Hertz, 2008). Excitotoxicity resulting from over activation of glutamate receptors, particularly N-Methyl-D-aspartic acid (NMDA) receptors, is an early trigger for calcium dysregulation and cell death and has been the target of neuroprotective strategies (Newell et al., 1995; Arundine & Tymianski, 2004; Dhawan et al., 2011). Glutamate receptor antagonists can minimize injury of hippocampal CA1 and striatal neurons in vitro and in vivo mouse models of cerebral ischemia (Gill et al., 1987; Foster et al., 1988; Collins et al., 1989; Rothman & Olney, 1995), however, less data exists clarifying which receptors are responsible for glutamate toxicity in Purkinje cells following cerebral ischemia. There is evidence that glutamate excitotoxicity contributes to Purkinje cell death as AMPA receptor antagonists can protect against Purkinje cell loss following global cerebral ischemia (Balchen & Diemer, 1992; Brasko et al., 1995) and knockdown of glial glutamate transporter expression increases Purkinje cell loss following experimental cardiac arrest (Yamashita et al., 2006). Interestingly, recent studies using memantine, a non-competitive NMDA receptor antagonist have yielded conflicting results as to whether blocking NMDA activity prevents Purkinje cell injury following cardiac arrest (Tai & Truong, 2007; 2010; 2013). It is important to note that until recently, it was generally accepted that Purkinje cells lacked NMDA receptor expression, however recent work demonstrates a role for NMDA receptors in normal synaptic transmission and plasticity in Purkinje cells (Piochon et al., 2007; Renzi et al., 2007; Piochon et al., 2010; He et al., 2013) which leads us to hypothesize that NMDA receptor activation may be a major contributor to Purkinje cell death. The current study aims to increase our understanding of the role of NMDA receptors in Purkinje cell injury following global cerebral ischemia.
A subunit specific role for NMDA receptor contribution to excitotoxic neuronal death has been proposed based on in vitro and in vivo animal studies. Synaptic GluN2A-containing receptors can promote cell survival following an excitotoxic insult, while extrasynaptic GluN2B-containing receptors promote cell death (Lynch & Guttmann, 2002; Liu et al., 2007; Choo et al., 2012; Sanz-Clemente et al., 2013). While there is evidence of opposing roles in ischemic injury of GluN2A and GluN2B containing NMDA receptors in cortical and hippocampal neurons in vitro and in vivo, NMDA receptor subunit-dependent mechanisms have not been examined in the cerebellum.
In this study we compared NMDA receptor subunit contributions to ischemic injury in CA1 neurons and Purkinje cells and revealed regional differences in injury mechanisms following experimental CA/CPR.
2.0 Methods
For all experiments, 8-12 week male (20-25g) C57Bl/6 mice (Charles River laboratories, Portage, MI) were used. All experiments were approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Colorado, School of Medicine and were performed according to the guidelines from the National Institutes of Health. Mice were individually housed and allowed free access to food and water.
2.1 Cardiac arrest model
Cardiac arrest and cardiopulmonary resuscitation was performed as previously described (Kofler et al., 2004; Neigh et al., 2004) with slight modifications to head and body temperature. Mice were anesthetized initially with 3% isoflurane and maintained with 1.5-2% isoflurane in O2 enriched air via facemask (Isotec 5, Ohmeda, Reno, NV). Isoflurane exposure was consistent between groups (duration and dose) to ensure any preconditioning effects were uniform and not contributing to differences observed between treatment groups (Zheng & Zuo, 2003). Head and body temperatures were monitored using tympanic and rectal probes connected to separate automatic temperature controllers (Doric Instruments, San Diego, CA) to maintain 37±0.2°C during surgery using a head coil, heating pad and a heating lamp. For drug administration, a PE-10 catheter was inserted into the right jugular vein and flushed with heparinized 0.9% saline solution. Needle electrodes were placed subcutaneously on the chest for EKG monitoring (Medical Data Electronics, Arleta, CA) and mice were endotracheally intubated and connected to a mini ventilator (Harvard Apparatus, Holliston, MA). To induce cardiac arrest, 50 μl of 0.5M KCl was administered via the jugular vein. Cardiac arrest was confirmed by a flat line EKG and the endotracheal tube was then disconnected from the ventilator. During CA, a tympanic temperature of 37.5°C and rectal temperature of 35°C was maintained. Following 8 minutes of cardiac arrest, resuscitation was performed by mechanical ventilation (190 breaths/min), injections of epinephrine (200 μl, 16 μg/ml in 0.9% saline; maximal dose 1 ml) and simultaneous chest compressions (300 compressions/min). As soon as there was a return of spontaneous circulation (ROSC), compressions were terminated. When spontaneous breathing reached a rate of 30 breaths/min, mechanical ventilation was stopped and the endotracheal tube removed. Temperature probes and catheters were then removed, and the skin wounds were closed. Drug administration (saline, MK-801 and Co101244) was performed intravenously 30 minutes after resuscitation. Mice were returned to their home cages that were placed on a heated water blanket (35°C) for recovery and received soft chow and free access to water. At various times after resuscitation (3 hours-30 days), mice were transcardially perfused with 4% paraformaldehyde (PFA) and post-fixed in PFA at 4°C overnight. Cerebellums and hippocampi were paraffin embedded and 6 μm sections, at 100 μm intervals, were cut and collected and stained for histology and immunohistochemistry.
2.2 Histology
Hippocampus and cerebellum were analyzed by hematoxylin and eosin (H&E) and Fluoro-Jade B at 3 hrs, 1 and 3 days after CA/CPR. For pharmacological studies, neuronal injury in the hippocampus and cerebellum was analyzed 7 days after CA/CPR. Fluoro-Jade B staining (Millipore, Billerica, MA) was performed according to manufacturer’s instructions. Briefly, slides were deparaffinized and rehydrated, blocked with potassium permanganate solutions (0.06%) for 20 minutes, stained with Fluoro-Jade B (0.004%) for 15 minutes, dried and cover slipped with DPX mounting solution. Slices were visualized using bright field microscopy (Leica DM750, Buffalo Grove, IL) for H&E and with epifluorescence (Leica DM2000) for Fluoro-Jade B and imaged using QCapturePro software (QImaging, Surrey, Canada).
2.3 Immunohistochemistry
Immunohistochemistry was performed at 3 hrs, 1, 3, 7, and 30 days after CA/CPR on paraffin sections that were deparaffinized. Antigen retrieval was performed using sodium citrate buffer, pH 6.0 at 95°C for 30 minutes. Sections were blocked and permeabilized (10% normal donkey serum/0.5% Triton-X in PBS) for 2 hours and primary antibody incubation was performed overnight at 4°C. For colabeling experiments mouse anti-parvalbumin (Abcam, Cambridge, MA; 1:400)/(Abcam; 1:500), mouse anti-GluN1 (Phosphosolutions, Aurora, CO; 1:400)/rabbit anti-calbindin and rabbit GABA alpha1 (Millipore; 1:1000)/ mouse anti-calbindin (Santa Cruz Biotechnology, Santa Cruz, CA; 1:1000) were used in combination. Antibody labeling was detected using donkey anti-mouse or donkey anti-rabbit Alexa594 and Alexa488 secondary antibodies (Jackson ImmunoResearch, Westgrove, PA; 1:500). Fluorescent labeling was visualized using epifluorescence, and Qimaging monochromatic camera (Q Imaging). Image acquisition was performed using QCapture Pro (Q Imaging) with uniform imaging parameters across experimental groups (exposure time and gain). Analysis of calbindin labeling was performed using Image J software (NIH, Bethesda, MD). 6 sections containing lobules IV-VII in the vermis of the cerebellum were analyzed by counting calbindin positive cells per given length measured with the segmented line tool and was used to calculate cells/mm. These sections were selected as previous work indicated the greatest Purkinje cell injury following CA/CPR occurs within the vermis while the hemispheres are relatively spared (Welsh et al., 2002)
2.4 Reverse transcription polymerase chain reaction (RT-PCR)
Cerebellums from control animals were rapidly removed and snap frozen in OCT cryoprotective media and stored at -80°C until sectioning. Sections (8 μm) were sectioned using a cryostat, collected on uncharged slides and stored on dry ice. Laser capture microdissection (LCM) was performed on the same day as sectioning. Sections were ethanol fixed (70%), dehydrated in alcohol containing RNase inhibitor (Protect RNA 500x concentrate, Sigma-Aldrich, St. Louis, MO) and cleared with xylene. LCM was performed using an ArcturusXT microdissection system (Arcturus, New York, NY). Purkinje cells were captured with the spot tool to select individual cells to be collected on HS LCM microcaps using an infrared laser. Caps were pooled to yield 800-1000 cells from each animal. RNA extraction and isolation was performed using Arcturus PicoPure Kit. cDNA was generated from 100 ng RNA using iScript cDNA synthesis kit (BioRad, Hercules, CA). Quantitative RT-PCR was performed on CFX96 real-time PCR system (BioRad) using taqman primer/probes (Invitrogen, Grand Island, NY) for 18s, GluN1, GluN2A, GluN2A, GABAA α1 and GABAA α6 and SsoFast master mix (BioRad). Relative expression was determined using the 18s reference gene and the ΔΔCq method and data were normalized to GluN1 expression.
2.5 Whole cell electrophysiology
Acute cerebellar brain slices were prepared from adult male mice (8-12 wks.). Mice were placed under isoflurane anesthesia (3% in oxygen-enriched air) and transcardial perfusion of artificial cerebral spinal fluid (ACSF) was performed for 3 minutes. Mice were decapitated then brains were rapidly removed and sagittal sections (250 μm) were cut on a vibratome (Leica VT1000) in ice-cold ACSF. ACSF contained (in mM): 126 NaCl, 2.5 KCl, 2.5 CaCl2, 1.2 MgCl2, 1.2 NaH2PO4, 21.4 NaHCO3, and 11 D-glucose, bubbled with 95% O2/5% CO2 to maintain pH of 7.4. Slices were incubated in ACSF at 35°C for 30 minutes prior to transferring to a recording chamber on an upright microscope (Leica DMLFS) with differential interference contrast optics. Purkinje cells were identified by their location adjacent to the granule cell layer and large soma.
Whole-cell recordings were obtained using 2-4 MΩ pipettes fashioned from borosilicate glass on a P90 puller (Sutter Instruments, Novato, CA). Internal pipette solution contained (in mM): Kgluconate 135, NaCl 8, HEPES 10, EGTA 0.05, MgCl2 1, Mat 4, Na2GTP 0.3, pH to 7.3 with KOH. Recordings were performed at 32°C in magnesium-free ACSF containing picrotoxin (100 μM), glycine (25 μM), NBQX (10 μM) and TTX (250 NM) to allow for isolation of NMDA currents. Cells were held at -60 mV. Series resistance was compensated 70% and recordings were excluded from analysis if series resistance exceeded 20MΩ or changed >20%. NMDA currents were elicited by ejecting NMDA (10 mM) from an iontophoretic electrode (Kation Scientific, Minneapolis, MN) placed near the primary dendrite. Retention current (15 nA) was placed on the iontophoretic electrode to prevent leakage of NMDA between trials. NMDA was ejected by application of negative current pulses (150 nA, 4s) every minute. Drugs were perfused for 10 minutes and peak amplitude of the inward current elicited by NMDA iontophoresis was measured.
2.6 Drugs
For in vivo studies the investigator was blinded to treatment group and animals were randomized. MK-801 (1 mg/kg) and Co 101244 (3 mg/kg) (Tocris) were dissolved in saline vehicle. A single dose of drugs or vehicle was administered intravenously via jugular catheter 30 minutes after resuscitation. For electrophysiology, all drugs were obtained from Tocris and dissolved in water with the exception of picrotoxin which was dissolved in DMSO (0.1% final concentration).
2.7 Statistics
Time course of Purkinje cell injury was compared using one-way ANOVA, followed by Tukey’s multiple comparisons post-hoc analysis. Comparisons of MK-801, Co 101244 and vehicle treated groups were performed using one-way ANOVA followed by Dunnett’s post-hoc analysis to compare to vehicle group. Statistical analysis was performed using GraphPad Prism 6 (GraphPad, La Jolla, CA)
3.0 Results
3.1 Histological injury not observed in Purkinje cells after CA/CPR
Mice were subjected to 8 minutes of cardiac arrest followed by cardiopulmonary resuscitation. We first wanted to assess Purkinje cell injury using standard histological methods at 24 hours and 3 days after cardiac arrest. Ischemic injury to neurons is generally characterized by the presence of hypereosinophilic cytoplasm and dark pyknotic nuclei using H&E staining. While Purkinje cell morphology was not hypereosinophilic at 24 hours and 3 days after CA/CPR, some of the cell bodies did appear dark and shrunken and we did observe gaps in the Purkinje cell monolayer indicating the possibility of Purkinje cell loss (Figure 1A-C). Similarly, we observed very little injury with Fluoro-Jade B, a fluorescent marker of degenerating neurons. We observed fluorescently labeled Purkinje cells in 10.8±0.1% (n=4) of Purkinje cells at 3 days (Figure 1D-F). In contrast, ischemic injury was easily detectable in the CA1 region of the hippocampus (Figure 2B). Using H&E staining we observed injured neurons in 5.4±0.7% (n=4) of CA1 neurons 24 hours after CA/CPR and in 69.1±11% (n=5) of CA1 neurons at 3 days after (Figure 2A, B). A large injury was also detected using Fluoro-Jade B staining, where neurons in the CA1 were intensely labeled at 3 days but not at 24 hours (Figure 2C, D). The relative lack of injury observed in the CA1 at 24 hours after CA/CPR is consistent with previous studies demonstrating delayed neuronal death in this region (Pulsinelli et al., 1982; Kofler et al., 2004; Wang et al., 2013). TUNEL staining, which labels fragmented DNA, was also assessed in Purkinje cells and in the CA1 to determine whether apoptotic mechanisms were engaged in these brain regions. TUNEL positive cells were detected in the CA1 3 days after CA/CPR (Figure 3A), but were absent in the cerebellum (Figure 3B).
Figure 1.
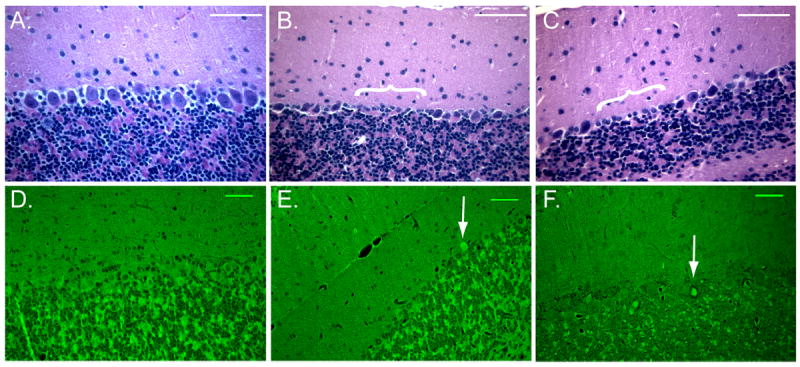
Histological analysis of cerebellum following CA/CPR fails to detect robust injury to Purkinje cells. Representative images of hematoxylin and eosin (H&E) staining in control (A.), 24 hours (B.) and 3 days (C.) after CA/CPR. Purkinje cell morphology is not dramatically altered by CA/CPR, however gaps in the Purkinje cell layer were observed (indicated by white bracket). Scale bars = 50 um. Fluoro-Jade B staining performed in control (D.), 24 hours (E.) and 3 days (F.) after CA/CPR labels a small portion of Purkinje cells (white arrows). N = 5-8. Scale bars = 50 um.
Figure 2.
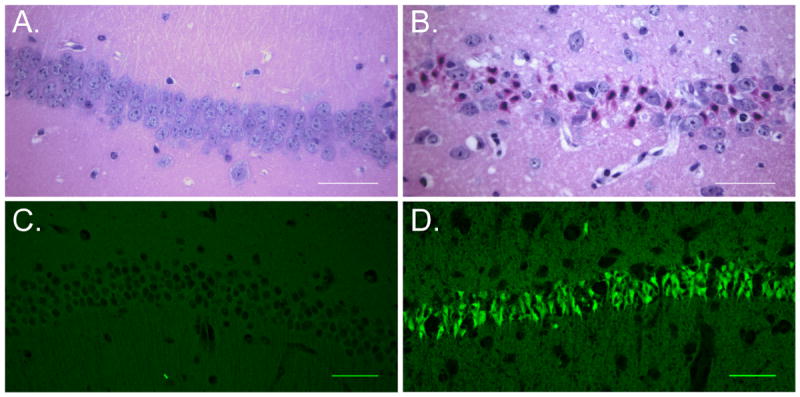
Histological analysis of CA1 region of the hippocampus demonstrates delayed neuronal death. Representative images of H&E demonstrate a lack of injury at 24 hours (A.) and large injury at 3 days (B.) as demonstrated by eosinophilic cytoplasm and pyknotic nuclei. Scale bar = 50um. Representative images of Fluoro-jade labeling is absent in the CA1 at 24 hours (C.) and intensely labeled at 3 days after CA/CPR. N=5-8. Scale bar = 50um
Figure 3.
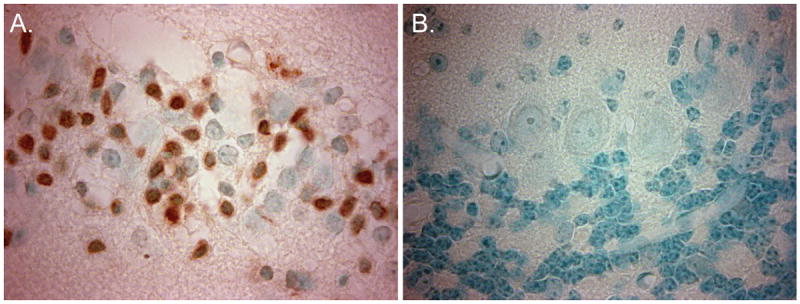
A marker of apoptosis is present in CA1 neurons but absent in Purkinje cells. (A.) Representative images of TUNEL staining in the CA1 region and (B.) in Purkinje cells. N = 3-4. Images captured using 100× oil objective.
3.2 Analysis of calbindin positive cell density in the cerebellum reveals Purkinje cell drop-out within 24 hours after CA/CPR
Despite the apparent lack of histological injury of Purkinje cells in cerebellar sections, H&E staining of the cerebellum revealed gaps in the Purkinje cell layer (Figure 1B and 1C) which led us to hypothesize Purkinje cell loss. To test whether differences in ischemic injury between CA1 and the cerebellum were due to a lack of injury to Purkinje cells or a failure of these methods to reliably measure injury in this cell type, we analyzed Purkinje density at various times after CA/CPR. Calbindin is a calcium binding protein which is highly expressed by Purkinje cells and is not present in other cell types within the cerebellum. Immunohistochemistry was performed using anti-calbindin antibody to specifically label Purkinje cells and linear cell density within the medial portions of the cerebellar vermis, which included lobules IV-VII, were analyzed. Cell density in sham controls was 31.2±1.0 cells/mm (n=5) (Figure 4A). Cell density measured at 3 hours after CA/CPR (28.3±1.6 cells/mm; n=5) was not different from sham controls (p=0.47). At 24 hours after CA/CPR, a 24% decrease of Purkinje cell density was observed (23.5±0.8 cells/mm; n=5; p=0.0006 compared to sham controls), indicating significant Purkinje cell death and removal (Figure 4B). A similar level of injury was observed at 3 days (24.74 ±0.5 cells/mm; p=0.0045 compared to sham controls), the time at which CA1 injury is analyzed. There was an additional decrease in cell density between 3 days and 7 days after CA/CPR to 20.9±0.9 cells/mm (n=6; p<0.0001 compared to sham and p=0.048 compared to 3 day). The injury was maximal at 7 days after CA/CPR (Figure 4C), as no further decrease in cell density was observed 30 days after CA/CPR compared to 7 days (21±1.3 cells/mm; n=6; p>0.99) (Figure 4D). To address the possibility that the decrease in cell density resulted from a decrease in calbindin immunoreactivity rather than Purkinje cell loss, other Purkinje cell markers were used. Parvalbumin colocalized with calbindin in the Purkinje cell layer and also labeled interneurons (Figure 5A and 5B) in the molecular layer. GluN1 and GABAA α1 were detected in Purkinje cells and in some neurons in the granule cell layer (Figure 5C and 5D). By co-labeling with these other Purkinje cell markers we confirmed a loss of Purkinje cell bodies in regions where calbindin labeling was absent. Thus, analysis of Purkinje cell density was a more robust measure of Purkinje cell injury in the cerebellum than other histological markers, and revealed a more rapid initiation of cell death in this region than was observed in the CA1 region.
Figure 4.
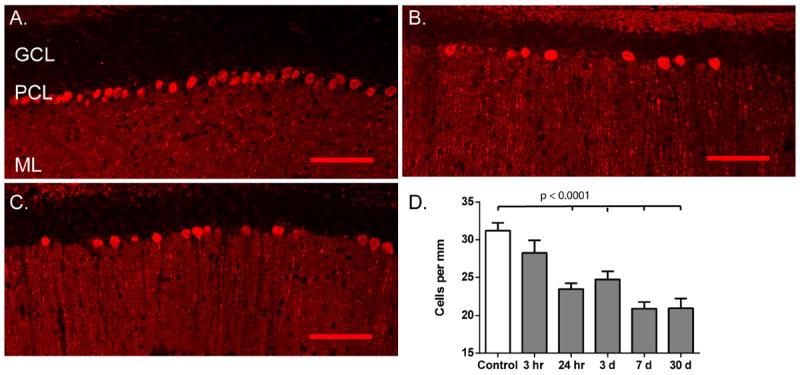
Calbindin immunohistochemistry demonstrates Purkinje cell death and loss at 24 hours. (A-C). Representative images showing labeling of Purkinje cells with anti-Calbindin D28K antibody. A continuous layer is observed in control (A.). Following cardiac arrest clear gaps, indicative of cell loss are observed at 24 hours (B.) and 7 days (C.) after CA/CPR. GCL: granule cell layer; PCL: Purkinje cell layer; ML: molecular layer. Scale bar = 50um. (D.) Quantification of Purkinje cell density analyzed at various times after CA/CPR. Loss of Purkinje cells is observed as early as 24 hours and is maximal at 7 days. Bars represent mean±SEM. N = 5-8. One-way ANOVA with Tukey’s multiple comparisons.
Figure 5.
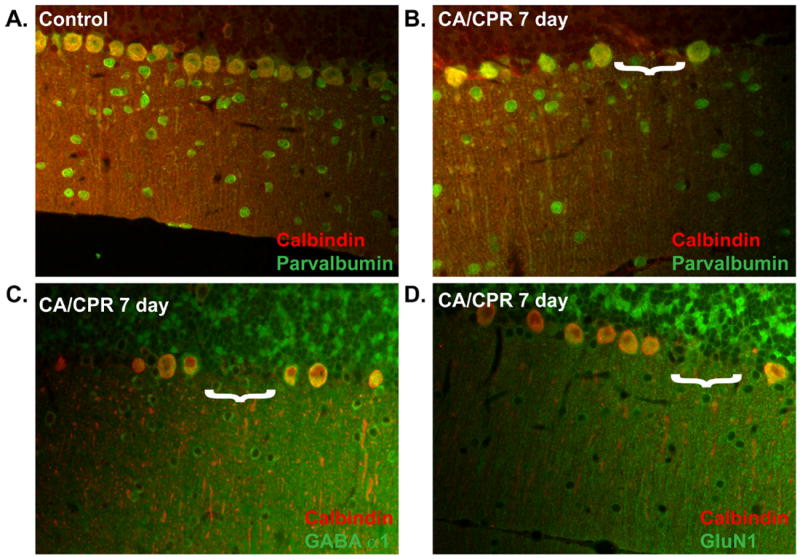
Confirmation of Purkinje cell loss using immunohistochemical markers. Dual labeling with anti-calbindin and anti-parvalbumin, GluN1 or GABAA α1 antibodies. Colocalization was observed with calbindin and parvalbumin in the Purkinje cell layer in controls (A) and 7 days after CA/CPR (B). Colocalization was observed in the Purkinje cell layer for calbindin with NR1 (C) and with GABAA α1 (D) 7 days after CA/CPR. Gaps in Purkinje cell layer are indicated by white bracket. N = 3-4.
3.3 NMDA receptor inhibition protects against Purkinje cell drop-out following CA/CPR
Next we tested whether NMDA receptor activation contributes to Purkinje cell degeneration. We first wanted to confirm the presence of functional NMDA receptor-mediated currents in adult mice and to determine the relative contribution of GluN2A and GluN2B containing receptors. Previous work suggests GluN2A/B containing receptors are expressed in Purkinje cells of adult mice (>6 wks.) (Piochon et al., 2007; Renzi et al., 2007). Laser capture microdissection was performed to isolate Purkinje cells for quantitative RT-PCR. Expression of GABAA α6, the subunit expressed in granule cells, was abundant in cerebellar sections but was observed at very low levels in RNA isolations from laser capture microdissections, demonstrating an enrichment of Purkinje cells (Figure 6B). GABAA α1 which is expressed by Purkinje cells was observed in both laser capture and whole cerebellar sections. RNA isolated from Purkinje cells was used to examine the presence of GluN1, GluN2A and GluN2B mRNA in Purkinje cells. GluN1, the requisite subunit of the NMDA receptor, was most abundant and used as our comparator for analyzing the relative expression of the other NMDA receptor subunits, 1.08±0.06 (n=4). GluN2A had a relative expression of 0.27±0.03 (n=4) and GluN2B relative expression was 0.04±0.01 (n=4) (Figure 6A). To assess NMDA receptor function we performed whole-cell electrophysiology on Purkinje cells in acute brain slices obtained from adult male C57Bl6 mice (8-12 wks.). Exogenous application of NMDA (10 mM, pH 8.0) near the primary dendrite of Purkinje cells using iontophoretic application (150nA, 4s) resulted in a large inward current, 362.8±72.11 pA (n=7) (Figure 6B). Application of the GluN2B selective antagonist ifenprodil (5 μM) inhibited a small fraction, approximately 10%, (33.2±10.4 pA; n=5) of the total NMDA receptor-mediated current. The remaining current was blocked by the non-selective NMDA receptor antagonist D-AP5 (5 μM) (Figure 6B). This confirms previous work demonstrating a predominately GluN2A-containing population of NMDA receptors on Purkinje cells (Renzi et al., 2007).
Figure 6.
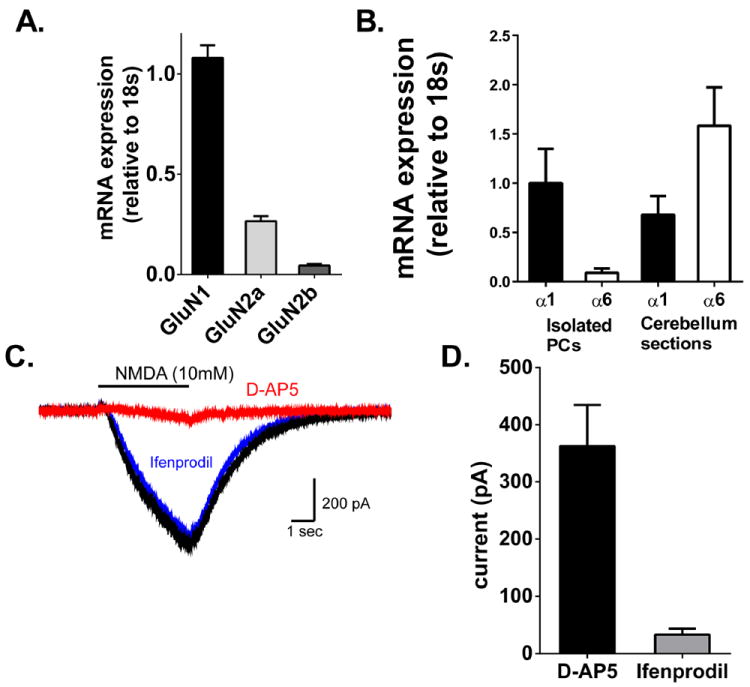
NMDA receptors in Purkinje cells are predominantly GluN1/GluN2A containing receptors. (A.) Relative expression of GluN1 subunits in Purkinje cells. mRNA expression in Purkinje cells isolated by laser capture microdissection were quantified relative to the housekeeping gene 18s and normalized to GluN1 expression. Bars represent mean±SEM (n=4) (B) GABAA α6 mRNA was expressed at very low levels in LCM samples, demonstrating an enrichment of Purkinje cells with few granule cells being collected. GABAA α1 mRNA was observed in both LCM and whole cerebellar sections. N=4. (C.) Representative traces of antagonist-sensitive current elicited by iontophoresis of NMDA (10 mM, 4s). Currents elicited in the presence of antagonist were subtracted from baseline currents to generate antagonist-sensitive current. Solid line indicates timing of NMDA iontophoresis. (D.) Summarized data of current (in pA) blocked by D-AP5 (n=7) and ifenprodil (n=5). Bars represent mean±SEM.
To determine whether NMDA receptor activation contributes to Purkinje cell degeneration following CA/CPR, the NMDA receptor antagonist MK-801 was administered to mice 30 minutes after resuscitation and Purkinje cell density was analyzed at 7 days after CA/CPR. Analysis at 7 days after CA/CPR was selected because the loss of Purkinje cells was maximal at this time point. Purkinje cell density in vehicle-treated mice at 7 days after CA/CPR was 23±.8 cells/mm (n=11) (Figure 7A). In mice that were administered MK-801 (1 mg/kg; iv) Purkinje cell density was 27.8±0.6 cells/mm (n=6; p=0.0019) compared to vehicle treated CA/CPR mice (Figure 7A). This represents remarkable neuroprotection as Purkinje cell density in MK-801 treated mice is not significantly different from sham controls. In contrast, cerebellar sections from mice that were administered the NR2B selective antagonist Co 101244 (3 mg/kg, iv) showed a reduction in Purkinje cell density (20.24±1.15; n=5) that was not different from vehicle-treated mice (p=0.77) (Figure 7A). This provides another contrast to CA1 hippocampal neurons, that demonstrated reduced neuronal injury following treatment with both MK-801 (12.6±3.4 % injured neurons; n=5, p=0.0126) and Co 101244 (14.1±2.4% injured neurons; n=5, p=0.015) compared to vehicle controls (22.7±2.2% injured neurons; n=8) (Figure 7B). Therefore, preventing NMDA receptor activation provides significant protection against Purkinje cell loss caused by CA/CPR that is independent of NR2B activation.
Figure 7.
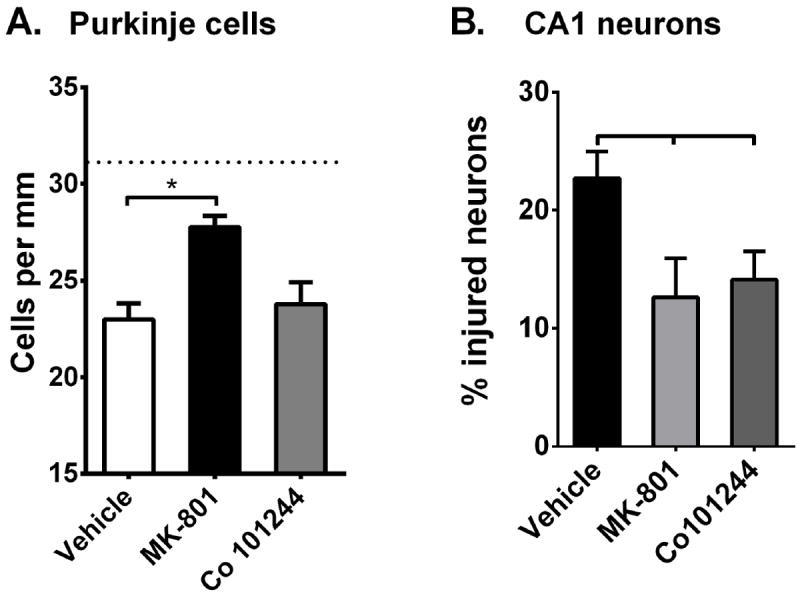
Region-specific neuroprotection observed with GluN2B antagonist Co 101244. (A.) Quantification of Purkinje cell density measured using calbindin antibody at 7 days after CA/CPR in vehicle (saline), MK-801 (1 mg/kg) and Co101244 (3 mg/kg) treated mice. Dotted line represents Purkinje cell density in sham controls. (B.) Quantification of injury to CA1 neurons at 7 days after CA/CPR in vehicle (saline), MK-801 (1 mg/kg) and Co101244 (3 mg/kg) treated mice. N= 5-12. Comparisons were made using One-way ANOVA with Dunnett’s post-hoc analysis (* p<0.05).
4. Discussion
Delayed neuronal death resulting from global cerebral ischemia has been demonstrated in select populations of cells including hippocampal CA1 neurons (Pulsinelli et al., 1982; Kofler et al., 2004). Immunohistochemical analysis of Purkinje cell density revealed a more rapid and robust level of injury than was detected using H&E and Fluoro-Jade staining. Our results indicate that while Purkinje cells and CA1 neurons are sensitive to injury caused by CA/CPR, the loss of Purkinje cells occurs with a more rapid time course than was observed in CA1 neurons. We also show NMDA receptor dependence to ischemic injury in both regions, but that there was a cell specific protection of CA1 neurons with the GluN2B selective antagonist. The differences in time course and mechanism of injury in these two ischemia sensitive brain regions could have significant impact on the therapeutic window and efficacy of therapies designed to prevent neuronal death after cardiac arrest.
The more rapid progression of degeneration in the cerebellum suggests differences in injury mechanisms between Purkinje cells and CA1 neurons. The time course of Purkinje cell loss obtained using our mouse model of CA/CPR is consistent with those observed in post mortem examinations of human cerebellums following cardiac arrest or other hypoxic events. Purkinje cell loss in cardiac arrest victims that were successfully resuscitated and survived between 1 to 186 hours after resuscitation occurred with a slightly delayed time course but was more rapid than CA1 (Horn & Schlote, 1992). Similarly, Purkinje cell death in drowning/asphyxia victims was observed in patients who died 5 hours after the event but not in those that suffered immediate death (Hausmann et al., 2007). Thus, our observation that death occurs between 3 and 24 hours suggests this model recapitulates aspects of ischemic cell death observed in the clinical population. However, the time course of Purkinje cell injury is somewhat conflicting with a previous study using a rat model of CA/CPR, which observed Fluoro-Jade B positive staining at 4 days after CA/CPR (Tai & Truong, 2013). One major difference however was that the simple lobule in the cerebellar hemisphere was analyzed while our study examined injury within the medial cerebellum. Additionally, methods used to detect and quantify injury were different between studies and species were different. Our data in mouse indicates that Purkinje cell removal (linear density) is a more reliable indicator of injury than Fluoro-Jade analysis used in previous Purkinje cell studies. Therefore, we are confident that a large portion of Purkinje cell death occurs rapidly. However, it is worth pointing out that we do see a modest increase in Purkinje cell death between 24 and 72 hrs, which may represent a population of Purkinje cells the exhibit delayed neuronal death as described previously (Tai & Truong, 2010).
Injury to CA1 neurons is not detectable at 24 hrs and is apparent at 3 and 7 days after CA/CPR. The accelerated loss of Purkinje cells observed following cardiac arrest is in contrast to many studies that demonstrate delayed neuronal injury in the CA1 region (Pulsinelli et al., 1982; Kirino et al., 1984; Kofler et al., 2004). Purkinje cells have several properties that are unique from CA1 neurons which may contribute to the more rapid onset of injury after CA/CPR. Purkinje cells fire action potentials spontaneously (30-50 Hz) and synaptic inputs can drive the Purkinje cells to fire at rates up to 300 Hz. Additionally, Purkinje cells receive large excitatory drive from climbing fiber and parallel fibers resulting in large calcium transients during synaptic activity (Eccles et al., 1966; Napper & Harvey, 1988; Perkel et al., 1990). This imposes a large metabolic demand due to high activity of ionic pumps on Purkinje cells which may contribute to their sensitivity to cerebral ischemia, as ischemia is a metabolic challenge resulting in nutrient and oxygen depletion and loss of ATP.
Another indication for unique injury mechanisms in Purkinje cells is the lack of morphological changes that are typical of ischemic injury in other neuronal populations. H&E staining of cerebellar sections showed a fading of cytoplasm and gaps in the Purkinje cell layer, rather than an eosinophilic cytoplasm and condensed pyknotic nuclei that are classically associated with ischemia induced neuronal injury. There are several studies reporting that AMPA receptor-mediated excitotoxicity or hypoxia can result in Purkinje cell dark cell degeneration, which is characterized by cytosolic and nuclear condensation and neuronal shrinkage that results in a dark purple H&E labeling. There were some cells that appeared dark and shrunken, but these morphological changes were inconsistent and therefore not ideal for quantification. Therefore, while H&E staining reveals alterations in PC morphology, cell death analysis with this method is not optimal. Similar to the fading cytoplasm observed by H&E, Fluoro-Jade B labeling was faint in Purkinje cells relative to CA1 neurons which were intensely labeled. Our failure to detect TUNEL labeling in Purkinje cells following cardiac arrest suggest that injury to these cells may not be mediated through classical apoptotic mechanisms. It remains unclear why standard histological markers fail to detect ischemic injury to Purkinje cells. Others have used Fluoro-Jade B to demonstrate Purkinje cell injury in rat models of cerebral ischemia (Tai and Truong) (Tai & Truong, 2007; 2010), however our data indicates that alternative methods are more robust and reliable. The presence of gaps in the Purkinje cell layer observed at 24hrs with H&E and calbindin immunohistochemistry suggest that Purkinje cells die and are cleared rapidly. Future studies are warranted to more clearly identify cell death mechanisms in Purkinje cells and to investigate the surprisingly rapid clearance of Purkinje cells following CA/CPR.
For decades NMDA receptors have been shown to contribute to neuronal injury following cerebral ischemia and excitotoxicity. Pharmacological antagonism of NMDA receptors has been shown to protect multiple neuronal populations and brain regions against experimental ischemia (Gill et al., 1987; Foster et al., 1988; Collins et al., 1989; Maier et al., 1995; Newell et al., 1995; Rothman & Olney, 1995; Dhawan et al., 2011). In vitro studies have implicated NMDA receptor activation as a trigger for neuronal death in cortical, striatal and hippocampal neurons. Despite the abundance of literature implicating the NMDA receptor as a mediator of cell death in multiple brain regions, the role of NMDA receptors in Purkinje cell injury, remains unclear. Excitotoxic and ischemic injury to Purkinje cells has been primarily attributed to AMPA receptor mediated mechanisms (Balchen & Diemer, 1992; Brasko et al., 1995). This is likely due to a previous dogma that Purkinje cells lacked expression of the NMDA receptor at their synapses (Konnerth et al., 1990; Perkel et al., 1990). This is true for Purkinje cells in juvenile rodents, however more recent work has demonstrated the presence of NMDA receptors on Purkinje cells at climbing fiber synapses in the adult mouse and rat (Piochon et al., 2007; Renzi et al., 2007). Our data with exogenous application of NMDA is consistent with the presence of functional NMDA receptors on Purkinje cells. The protection from Purkinje cell loss that we observed with administration of MK-801 supports a role for the NMDA receptor in glutamate transmission at Purkinje cell synapses and in mediating excitotoxicity following CA/CPR in adults. Previous studies using memantine, a non-competitive antagonist of NMDA receptors, have observed conflicting results, with one study demonstrating protection and two failing to observe a benefit to Purkinje cells with the NMDA receptor inhibitor memantine (Tai & Truong, 2007; 2010; 2013). In studies where there was a lack of protection, the authors argue a lack of NMDA receptor contribution to Purkinje cell death; however they also did not observe protection to CA1 neurons, indicating memantine may not sufficiently block NMDA receptors to prevent excitotoxic cell death. Differences with our results with MK-801 are may be due to memantine’s lower potency and reduced efficacy at depolarized potentials (Frankiewicz et al., 1996). While we interpret our findings to indicate a role of NMDA receptors in excitotoxic Purkinje cell injury, it is possible that the protection observed with MK-801 is due to a presynaptic inhibition of glutamate release onto on Purkinje cells. Unilateral ablation of the inferior olive input via the climbing fiber prevents Purkinje cell death following CA/CPR coupled with the observation that MK-801 can reduce olivary firing rate may suggest that MK-801 protection is indirect via altering climbing fiber glutamatergic transmission (Welsh et al., 2002). However, extensive data suggests that the dramatic elevation of glutamate observed during or immediately following prolonged hypoxia/ischemia occurs through action potential-independent spontaneous release from synaptic and extrasynaptic sites, reversal of glutamate transporters and release from glia (Krnjevic, 2008). Therefore, reductions in presynaptic firing of climbing fibers or parallel fibers with NMDA antagonists (MK-801) are not likely to prevent excitotoxic levels of glutamate onto Purkinje cells. Thus, antagonists of NMDA receptors and their downstream signaling may represent a viable therapeutic approach, particularly if subunit selective approaches can be used to minimize side effects.
Purkinje cells and CA1 neurons are both sensitive to NMDA receptor mediated ischemic injury, but excitotoxic mechanisms of injury may differ between these regions. The lack of Purkinje cell protection observed with the GluN2B antagonist contrasts with the protection observed in CA1 neurons. GluN2B has been implicated as a mediator of injury in response to focal and global ischemia (Gogas, 2006; Liu et al., 2007; Choo et al., 2012). Differences in subunit specificity of injury mechanisms are likely to have implications for signaling pathways engaged downstream of NMDA receptor activation. Indeed, multiple studies have suggested opposing roles for GluN2A and GluN2Bfor regulating cell survival with GluN2A engaging prosurvival mechanisms and GluN2B promoting cell death. Specifically, GluN2A couples to CREB phosphorylation and activation while GluN2B opposes CREB activation and signals to death-associated protein kinase (DAPK) (Hardingham et al., 2002; Tu et al., 2010). Our results with Co 101244 in the hippocampus are consistent with others demonstrating neuroprotection against injury from focal or global cerebral ischemia with GluN2B selective antagonists in the cortex, hippocampus and striatum (Gotti et al., 1988; Bath et al., 1996; Doğan et al., 1997; Picconi et al., 2006; Chen et al., 2008; Mishra et al., 2011). Genetic or pharmacological inhibition of GluN2A in vitro increases cell death, suggesting an opposing pro-survival role for this subunit in cortical and striatal neurons; however there is less evidence for GluN2A playing a protective role in vivo. Our results demonstrating a lack of protection with a GluN2B antagonist implicates GluN2A as mediating injury in Purkinje cells, however the current lack of in vivo pharmacological and genetic tools to specifically block GluN2A make this difficult to test directly. The excitotoxic link between GluN2A and injury implies fundamentally different cell signaling downstream of the NMDA receptor in Purkinje cells compared to other neuronal populations where GluN2A promotes cell survival. This would have implications not only to injury, but to normal physiological responses as well.
The lack of protection observed in vivo with Co 101244 was consistent with our quantitative RT-PCR and electrophysiology data showing relatively low mRNA expression of GluN2B and GluN2B activation mediates only about 10% of the total NMDA receptor mediated current in Purkinje cells. It is important to note that while NMDA receptor inhibition is neuroprotective in experimental studies, therapies targeting NMDA receptor activation in clinical trials have been unsuccessful (Ikonomidou & Turski, 2002). Clinical trials testing NMDA receptor antagonist following stroke have failed due to a narrow therapeutic window and negative consequences associated with blocking normal synaptic transmission through NMDA receptors. Experimental studies indicating a specific role for GluN2B in injury have led to proposals of specifically targeting this subunit as a therapy for ischemic brain injury (Wang & Shuaib, 2005; Gogas, 2006; Harraz et al., 2012). Our results indicate this strategy would be ineffective in preventing cerebellar injury. Further understanding of the pathways engaged downstream of NMDA receptor activation in Purkinje cells are required to help elucidate targets for neuroprotective therapies.
In summary, this work highlights the importance of examining ischemic injury to Purkinje cells in addition to other ischemia sensitive brain regions. Our data demonstrate that timing and mechanism of Purkinje cell injury necessitate a different method of histological analysis to reliably estimate injury following cerebral ischemia. The rapid injury observed in Purkinje cells is in contrast to other brain areas where injury progresses over 2-3 days, and likely has implication for therapeutic window which should be considered in pre-clinical studies. Our data also demonstrates that the subunit composition of NMDA receptors has significant implications for injury mechanisms and highlights an important difference between Purkinje cells and other ischemia sensitive brain regions.
Highlights.
Immunohistochemistry and cell density analysis of ischemic injury in cerebellum
Purkinje cell death rapid and not via apoptosis following CA/CPR
GluN2B antagonist selectively protects CA1 neurons
Acknowledgments
Project funded by AHA/Philips Resuscitation Fellowship 12POST11930031 (NQ), NIH NS080851 (PSH) and Walter S. and Lucienne Driskill Foundation grant (RJT). We wish to thank Nicole Spoestra from the Laser Capture Microdissection Core at University of Colorado Cancer Center (P30CA046934) for her technical assistance.
Abbreviations
- ACSF
artificial cerebral spinal fluid
- CA/CPR
cardiac arrest and cardiopulmonary resuscitation
- H&E
hematoxylin and eosin
- NMDA
N-Methyl-D-aspartic acid
Footnotes
Conflict of interest: The authors have no conflicts of interest to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Arundine M, Tymianski M. Molecular mechanisms of glutamate-dependent neurodegeneration in ischemia and traumatic brain injury. Cellular and Molecular Life Sciences (CMLS) 2004;61:657–668. doi: 10.1007/s00018-003-3319-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balchen T, Diemer NH. The AMPA antagonist, NBQX, protects against ischemia-induced loss of cerebellar Purkinje cells. Neuroreport. 1992;3:785–788. doi: 10.1097/00001756-199209000-00016. [DOI] [PubMed] [Google Scholar]
- Bastian AJ. Learning to predict the future: the cerebellum adapts feedforward movement control. Curr Opin Neurobiol. 2006;16:645–649. doi: 10.1016/j.conb.2006.08.016. [DOI] [PubMed] [Google Scholar]
- Bath CP, Farrell LN, Gilmore J, Ward MA, Hicks CA, O’Neill MJ, Bleakman D. The effects of ifenprodil and eliprodil on voltage-dependent Ca2+ channels and in gerbil global cerebral ischaemia. European journal of pharmacology. 1996;299:103–112. doi: 10.1016/0014-2999(95)00846-2. [DOI] [PubMed] [Google Scholar]
- Brasko J, Rai P, Sabol MK, Patrikios P, Ross DT. The AMPA antagonist NBQX provides partial protection of rat cerebellar Purkinje cells after cardiac arrest and resuscitation. Brain research. 1995;699:133–138. doi: 10.1016/0006-8993(95)01015-n. [DOI] [PubMed] [Google Scholar]
- Chen M, Lu T, Chen X, Zhou Y, Chen Q, Feng X, Xu L, Duan W, Xiong Z. Differential roles of NMDA receptor subtypes in ischemic neuronal cell death and ischemic tolerance. Stroke; a journal of cerebral circulation. 2008;39:3042–3048. doi: 10.1161/STROKEAHA.108.521898. [DOI] [PubMed] [Google Scholar]
- Choo AM, Geddes-Klein DM, Hockenberry A, Scarsella D, Mesfin MN, Singh P, Patel TP, Meaney DF. NR2A and NR2B subunits differentially mediate MAP kinase signaling and mitochondrial morphology following excitotoxic insult. Neurochemistry international. 2012;60:506–516. doi: 10.1016/j.neuint.2012.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins RC, Dobkin BH, Choi DW. Selective vulnerability of the brain: new insights into the pathophysiology of stroke. Ann Intern Med. 1989;110:992–1000. doi: 10.7326/0003-4819-110-12-992. [DOI] [PubMed] [Google Scholar]
- Dhawan J, Benveniste H, Luo Z, Nawrocky M, Smith S, Biegon A. A new look at glutamate and ischemia: NMDA agonist improves long-term functional outcome in a rat model of stroke. Future neurology. 2011;6:823–834. doi: 10.2217/fnl.11.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doğan A, Rao AM, Başkaya MK, Rao VL, Rastl J, Donaldson D, Dempsey RJ. Effects of ifenprodil, a polyamine site NMDA receptor antagonist, on reperfusion injury after transient focal cerebral ischemia. Journal of neurosurgery. 1997;87:921–926. doi: 10.3171/jns.1997.87.6.0921. [DOI] [PubMed] [Google Scholar]
- Eccles JC, Llinás R, Sasaki K. The excitatory synaptic action of climbing fibres on the purinje cells of the cerebellum. J Physiol (Lond) 1966:268–296. doi: 10.1113/jphysiol.1966.sp007824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster AC, Gill R, Woodruff GN. Neuroprotective effects of MK-801 in vivo: selectivity and evidence for delayed degeneration mediated by NMDA receptor activation. J Neurosci. 1988:4745–4754. doi: 10.1523/JNEUROSCI.08-12-04745.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankiewicz T, Potier B, Bashir ZI, Collingridge GL, Parsons CG. Effects of memantine and MK-801 on NMDA-induced currents in cultured neurones and on synaptic transmission and LTP in area CA1 of rat hippocampal slices. British journal of pharmacology. 1996;117:689–697. doi: 10.1111/j.1476-5381.1996.tb15245.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert PF, Thach WT. Purkinje cell activity during motor learning. Brain research. 1977;128:309–328. doi: 10.1016/0006-8993(77)90997-0. [DOI] [PubMed] [Google Scholar]
- Gill R, Foster AC, Woodruff GN. Systemic administration of MK-801 protects against ischemia-induced hippocampal neurodegeneration in the gerbil. J Neurosci. 1987:3343–3349. doi: 10.1523/JNEUROSCI.07-10-03343.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Borden WB, Bravata DM, Dai S, Ford ES, Fox CS, Franco S, Fullerton HJ, Gillespie C, Hailpern SM, Heit JA, Howard VJ, Huffman MD, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Magid D, Marcus GM, Marelli A, Matchar DB, McGuire DK, Mohler ER, Moy CS, Mussolino ME, Nichol G, Paynter NP, Schreiner PJ, Sorlie PD, Stein J, Turan TN, Virani SS, Wong ND, Woo D, Turner MB. Executive summary: heart disease and stroke statistics--2013 update: a report from the American Heart Association. Circulation. 2013;127:143–152. doi: 10.1161/CIR.0b013e318282ab8f. [DOI] [PubMed] [Google Scholar]
- Gogas KR. Glutamate-based therapeutic approaches: NR2B receptor antagonists. Current opinion in pharmacology. 2006;6:68–74. doi: 10.1016/j.coph.2005.11.001. [DOI] [PubMed] [Google Scholar]
- Gotti B, Duverger D, Bertin J, Carter C, Dupont R, Frost J, Gaudilliere B, MacKenzie ET, Rousseau J, Scatton B. Ifenprodil and SL 82.0715 as cerebral anti-ischemic agents. I. Evidence for efficacy in models of focal cerebral ischemia. The Journal of pharmacology and experimental therapeutics. 1988;247:1211–1221. [PubMed] [Google Scholar]
- Hardingham GE, Fukunaga Y, Bading H. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nature neuroscience. 2002;5:405–414. doi: 10.1038/nn835. [DOI] [PubMed] [Google Scholar]
- Harraz MM, Eacker SM, Wang X, Dawson TM, Dawson VL. MicroRNA-223 is neuroprotective by targeting glutamate receptors. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:18962–18967. doi: 10.1073/pnas.1121288109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hausmann R, Seidl S, Betz P. Hypoxic changes in Purkinje cells of the human cerebellum. Int J Legal Med. 2007:175–183. doi: 10.1007/s00414-006-0122-x. [DOI] [PubMed] [Google Scholar]
- He Q, Titley H, Grasselli G, Piochon C, Hansel C. Ethanol affects NMDA receptor signaling at climbing fiber-Purkinje cell synapses in mice and impairs cerebellar LTD. Journal of neurophysiology. 2013;109:1333–1342. doi: 10.1152/jn.00350.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertz L. Bioenergetics of cerebral ischemia: a cellular perspective. Neuropharmacology. 2008;55:289–309. doi: 10.1016/j.neuropharm.2008.05.023. [DOI] [PubMed] [Google Scholar]
- Horn M, Schlote W. Delayed neuronal death and delayed neuronal recovery in the human brain following global ischemia. Acta Neuropathologica. 1992;85:79–87. doi: 10.1007/BF00304636. [DOI] [PubMed] [Google Scholar]
- Ikonomidou C, Turski L. Why did NMDA receptor antagonists fail clinical trials for stroke and traumatic brain injury? Lancet neurology. 2002;1:383–386. doi: 10.1016/s1474-4422(02)00164-3. [DOI] [PubMed] [Google Scholar]
- Kelley MH, Kuroiwa M, Taguchi N, Herson PS. Sex difference in sensitivity to allopregnanolone neuroprotection in mice correlates with effect on spontaneous inhibitory post synaptic currents. Neuropharmacology. 2011;61:724–729. doi: 10.1016/j.neuropharm.2011.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirino T, Tamura A, Sano K. Delayed neuronal death in the rat hippocampus following transient forebrain ischemia. Acta neuropathologica. 1984;64:139–147. doi: 10.1007/BF00695577. [DOI] [PubMed] [Google Scholar]
- Kofler J, Hattori K, Sawada M, DeVries A, Martin L, Hurn P, Traystman R. Histopathological and behavioral characterization of a novel model of cardiac arrest and cardiopulmonary resuscitation in mice. Journal of neuroscience methods. 2004;136:33–44. doi: 10.1016/j.jneumeth.2003.12.024. [DOI] [PubMed] [Google Scholar]
- Konnerth A, Llano I, Armstrong CM. Synaptic currents in cerebellar Purkinje cells. Proceedings of the National Academy of Sciences of the United States of America. 1990;87:2662–2665. doi: 10.1073/pnas.87.7.2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krnjevic K. Electrophysiology of cerebral ischemia. Neuropharmacology. 2008;55:319–333. doi: 10.1016/j.neuropharm.2008.01.002. [DOI] [PubMed] [Google Scholar]
- Laver S, Farrow C, Turner D, Nolan J. Mode of death after admission to an intensive care unit following cardiac arrest. Intensive care medicine. 2004;30:2126–2128. doi: 10.1007/s00134-004-2425-z. [DOI] [PubMed] [Google Scholar]
- Lim C, Alexander MP, LaFleche G, Schnyer DM, Verfaellie M. The neurological and cognitive sequelae of cardiac arrest. Neurology. 2004;63:1774–1778. doi: 10.1212/01.wnl.0000144189.83077.8e. [DOI] [PubMed] [Google Scholar]
- Liu Y, Wong TP, Aarts M, Rooyakkers A, Liu L, Lai TW, Wu DC, Lu J, Tymianski M, Craig AM, Wang YT. NMDA receptor subunits have differential roles in mediating excitotoxic neuronal death both in vitro and in vivo. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2007;27:2846–2857. doi: 10.1523/JNEUROSCI.0116-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llinás R, Welsh JP. On the cerebellum and motor learning. Curr Opin Neurobiol. 1993:958–965. doi: 10.1016/0959-4388(93)90168-x. [DOI] [PubMed] [Google Scholar]
- Lu-Emerson C, Khot S. Neurological sequelae of hypoxic-ischemic brain injury. NeuroRehabilitation. 2010;26:35–45. doi: 10.3233/NRE-2010-0534. [DOI] [PubMed] [Google Scholar]
- Lynch DR, Guttmann RP. Excitotoxicity: perspectives based on N-methyl-D-aspartate receptor subtypes. The Journal of pharmacology and experimental therapeutics. 2002;300:717–723. doi: 10.1124/jpet.300.3.717. [DOI] [PubMed] [Google Scholar]
- Maier CM, Sun GH, Kunis DM, Giffard RG, Steinberg GK. Neuroprotection by the N-methyl-D-aspartate receptor antagonist CGP 40116: in vivo and in vitro studies. Journal of neurochemistry. 1995;65:652–659. doi: 10.1046/j.1471-4159.1995.65020652.x. [DOI] [PubMed] [Google Scholar]
- Martin LJ, Al-Abdulla NA, Brambrink AM, Kirsch JR, Sieber FE, Portera-Cailliau C. Neurodegeneration in excitotoxicity, global cerebral ischemia, and target deprivation: A perspective on the contributions of apoptosis and necrosis. Brain research bulletin. 1998;46:281–309. doi: 10.1016/s0361-9230(98)00024-0. [DOI] [PubMed] [Google Scholar]
- Merchenthaler I, Dellovade TL, Shughrue PJ. Neuroprotection by estrogen in animal models of global and focal ischemia. Annals of the New York Academy of Sciences. 2003;1007:89–100. doi: 10.1196/annals.1286.009. [DOI] [PubMed] [Google Scholar]
- Mishra V, Verma R, Singh N, Raghubir R. The neuroprotective effects of NMDAR antagonist, ifenprodil and ASIC1a inhibitor, flurbiprofen on post-ischemic cerebral injury. Brain research. 2011;1389:152–160. doi: 10.1016/j.brainres.2011.03.011. [DOI] [PubMed] [Google Scholar]
- Napper RM, Harvey RJ. Number of parallel fiber synapses on an individual Purkinje cell in the cerebellum of the rat. J Comp Neurol. 1988:168–177. doi: 10.1002/cne.902740204. [DOI] [PubMed] [Google Scholar]
- Neigh GN, Kofler J, Meyers JL, Bergdall V, Perle KM, Traystman RJ, DeVries AC. Cardiac arrest/cardiopulmonary resuscitation increases anxiety-like behavior and decreases social interaction. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism. 2004;24:372–382. doi: 10.1097/01.WCB.0000112323.75217.B4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newell DW, Barth A, Papermaster V, Malouf AT. Glutamate and non-glutamate receptor mediated toxicity caused by oxygen and glucose deprivation in organotypic hippocampal cultures. The Journal of neuroscience : the official journal of the Society for Neuroscience. 1995;15:7702–7711. doi: 10.1523/JNEUROSCI.15-11-07702.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng T, Graham DI, Adams JH, Ford I. Changes in the hippocampus and the cerebellum resulting from hypoxic insults: frequency and distribution. Acta neuropathologica. 1989;78:438–443. doi: 10.1007/BF00688181. [DOI] [PubMed] [Google Scholar]
- Noppens RR, Kofler J, Grafe MR, Hurn PD, Traystman RJ. Estradiol after cardiac arrest and cardiopulmonary resuscitation is neuroprotective and mediated through estrogen receptor-β. Journal of Cerebral Blood Flow & Metabolism. 2008;29:277–286. doi: 10.1038/jcbfm.2008.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paine MG, Che D, Li L, Neumar RW. Cerebellar Purkinje cell neurodegeneration after cardiac arrest: Effect of therapeutic hypothermia. Resuscitation. 2012;83:1511–1516. doi: 10.1016/j.resuscitation.2012.05.022. [DOI] [PubMed] [Google Scholar]
- Perkel DJ, Hestrin S, Sah P, Nicoll RA. Excitatory synaptic currents in Purkinje cells. Proc R Soc Lond. 1990;241:116–121. doi: 10.1098/rspb.1990.0074. [DOI] [PubMed] [Google Scholar]
- Picconi B, Tortiglione A, Barone I, Centonze D, Gardoni F, Gubellini P, Bonsi P, Pisani A, Bernardi G, Luca M, Calabresi P. NR2B Subunit Exerts a Critical Role in Postischemic Synaptic Plasticity. Stroke; a journal of cerebral circulation. 2006;37:1895–1901. doi: 10.1161/01.STR.0000226981.57777.b0. [DOI] [PubMed] [Google Scholar]
- Piochon C, Irinopoulou T, Brusciano D, Bailly Y, Mariani J, Levenes C. NMDA receptor contribution to the climbing fiber response in the adult mouse Purkinje cell. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2007;27:10797–10809. doi: 10.1523/JNEUROSCI.2422-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piochon C, Levenes C, Ohtsuki G, Hansel C. Purkinje cell NMDA receptors assume a key role in synaptic gain control in the mature cerebellum. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2010;30:15330–15335. doi: 10.1523/JNEUROSCI.4344-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulsinelli WA, Brierley JB, Plum F. Temporal profile of neuronal damage in a model of transient forebrain ischemia. Annals of neurology. 1982;11:491–498. doi: 10.1002/ana.410110509. [DOI] [PubMed] [Google Scholar]
- Renzi M, Farrant M, Cull-Candy SG. Climbing-fibre activation of NMDA receptors in Purkinje cells of adult mice. The Journal of physiology. 2007;585:91–101. doi: 10.1113/jphysiol.2007.141531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothman SM, Olney JW. Excitotoxicity and the NMDA receptor--still lethal after eight years. Trends in neurosciences. 1995;18:57–58. doi: 10.1016/0166-2236(95)93869-y. [DOI] [PubMed] [Google Scholar]
- Saito A, Maier CM, Narasimhan P, Nishi T, Song YS, Yu F, Liu J, Lee YS, Nito C, Kamada H, Dodd RL, Hsieh LB, Hassid B, Kim EE, Gonzalez M, Chan PH. Oxidative stress and neuronal death/survival signaling in cerebral ischemia. Mol Neurobiol. 2005;31:105–116. doi: 10.1385/MN:31:1-3:105. [DOI] [PubMed] [Google Scholar]
- Sanz-Clemente A, Nicoll RA, Roche KW. Diversity in NMDA receptor composition: many regulators, many consequences. The Neuroscientist : a review journal bringing neurobiology, neurology and psychiatry. 2013;19:62–75. doi: 10.1177/1073858411435129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai KK, Truong DD. NMDA receptor-mediated excitotoxicity contributes to the cerebral hypoxic injury of a rat model of posthypoxic myoclonus. Brain research. 2007;1133:209–215. doi: 10.1016/j.brainres.2006.11.076. [DOI] [PubMed] [Google Scholar]
- Tai KK, Truong DD. Memantine exacerbates myoclonic jerks in a rat model of posthypoxic myoclonus. Brain research. 2010;1343:194–198. doi: 10.1016/j.brainres.2010.04.058. [DOI] [PubMed] [Google Scholar]
- Tai KK, Truong DD. Amiloride but not memantine reduces neurodegeneration, seizures and myoclonic jerks in rats with cardiac arrest-induced global cerebral hypoxia and reperfusion. PloS one. 2013;8:e60309. doi: 10.1371/journal.pone.0060309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thach WT. A role for the cerebellum in learning movement coordination. Neurobiology of learning and memory. 1998;70:177–188. doi: 10.1006/nlme.1998.3846. [DOI] [PubMed] [Google Scholar]
- Traystman RJ. Animal models of focal and global cerebral ischemia. ILAR journal / National Research Council, Institute of Laboratory Animal Resources. 2003;44:85–95. doi: 10.1093/ilar.44.2.85. [DOI] [PubMed] [Google Scholar]
- Tu W, Xu X, Peng L, Zhong X, Zhang W, Soundarap MM, Soundarapandian MM, Balel C, Wang M, Jia N, Zhang W, Lew F, Chan SL, Chen Y, Lu Y. DAPK1 interaction with NMDA receptor NR2B subunits mediates brain damage in stroke. Cell. 2010;140:222–234. doi: 10.1016/j.cell.2009.12.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatesan A, Frucht S. Movement disorders after resuscitation from cardiac arrest. Neurologic clinics. 2006;24:123–132. doi: 10.1016/j.ncl.2005.11.001. [DOI] [PubMed] [Google Scholar]
- Wang CX, Shuaib A. NMDA/NR2B selective antagonists in the treatment of ischemic brain injury. Current drug targets CNS and neurological disorders. 2005;4:143–151. doi: 10.2174/1568007053544183. [DOI] [PubMed] [Google Scholar]
- Wang J, Fujiyoshi T, Kosaka Y, Raybuck JD, Lattal KM, Ikeda M, Herson PS, Koerner IP. Inhibition of soluble epoxide hydrolase after cardiac arrest/cardiopulmonary resuscitation induces a neuroprotective phenotype in activated microglia and improves neuronal survival. Journal of Cerebral Blood Flow & Metabolism. 2013;33:1574–1581. doi: 10.1038/jcbfm.2013.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsh J, Yuen G, Placantonakis D, Vu T, Haiss F, O’Hearn E, Molliver M, Aicher S. Why do Purkinje cells die so easily after global brain ischemia? Aldolase C, EAAT4, and the cerebellar contribution to posthypoxic myoclonus. Advances in neurology. 2002;89:331–359. [PubMed] [Google Scholar]
- Yamashita A, Makita K, Kuroiwa T, Tanaka K. Glutamate transporters GLAST and EAAT4 regulate postischemic Purkinje cell death: An in vivo study using a cardiac arrest model in mice lacking GLAST or EAAT4. Neuroscience research. 2006;55:264–270. doi: 10.1016/j.neures.2006.03.007. [DOI] [PubMed] [Google Scholar]
- Zheng S, Zuo Z. Isoflurane preconditioning reduces purkinje cell death in an in vitro model of rat cerebellar ischemia. Neuroscience. 2003;118:99–106. doi: 10.1016/s0306-4522(02)00767-4. [DOI] [PubMed] [Google Scholar]