

Abstract
Genetic analyses of patients with neurodegenerative disorders have identified multiple genes that need to be investigated for the presence of damaging variants. However, mutation analysis by Sanger sequencing is costly and time consuming. We tested the utility of a recently designed semi-custom genome-wide array (NeuroX; Illumina, Inc) tailored to study neurodegenerative diseases (e.g. mutation screening). We investigated 192 patients with four different neurodegenerative disorders for the presence of rare damaging variations in 77 genes implicated in these diseases. Several causative mutations were identified and confirmed by Sanger sequencing including PSEN1 p.M233T responsible for Alzheimer’s disease in a large Italian family, as well as SOD1 p.A4V and p.I113T in patients with Amyotrophic Lateral Sclerosis. In total, we identified 78 potentially damaging rare variants (frequency <1%), including ABCA7 p.L400V in a family with Alzheimer’s disease and LRRK2 p.R1514Q in 6 out 98 patients with Parkinson’s Disease (6.1%). In conclusion, NeuroX appears to be helpful for rapid and accurate mutation screening, although further development may be still required to improve some current caveats.
Keywords: NeuroX, mutation, neurodegenerative disorder
1. Introduction
Alzheimer’s disease (AD) together with other common neurodegenerative disorders, such as Parkinson’s disease (PD), Frontotemporal Dementia (FTD) and Amyotrophic Lateral Sclerosis (ALS) have major socio-economic effects in different societies. The world-wide prevalence of dementia doubles every 20 years and the health-care cost for people with dementia is increasing almost exponentially, costing up to US$604 billion in 2010 (World Health Organization, 2012). Knowledge of genetic factors causing or modifying risk of neurodegenerative disorders could help manage the challenges of aging populations.
Rare causal mutations are highly penetrant, while common variants with >5% minor allele frequency (MAF), mainly assessed by Genome Wide Association Studies (GWASs), only modestly influence disease risk. For instance, analyses of autosomal dominant families with early-onset AD (<65 years) have found causal mutations in APP, PSEN1 and PSEN2; while GWASs identified or confirmed >20 loci associated with late-onset AD (APOE, ABCA7, BIN1, CASS4, CD2AP, CD33, CELF1, CLU, CR1, DSG2, EPHA1, FERMT2, HLA-DRB1, HLA-DRB5, INPP5D, MEF2C, MS4A4A, MS4A6A, NME8, PICALM, PTK2B, SLC24A4/RIN3, ZCWPW1 and SORL1) (Ghani and Rogaeva, 2014). Importantly, GWAS loci could harbour rare damaging variants with a greater risk-effect vs. common variants within the same gene. For example, both rare coding and common non-coding CLU variants are independently associated with AD (Bettens, et al., 2012). However, the role of copy number variations (CNVs) and recessive inheritance (e.g. estimated based on runs of homozygosity) need further exploration (Ghani, et al., 2012, Ghani, et al., 2013a).
The genetics of other neurodegenerative disorders is also complex. GWAS and family-studies have implicated at least 24 loci for PD (ACMSD, ATP13A2, BST1, FBXO7, FGF20, GAK, GBA, GIGYF2, GPNMB, HIP1R, HLA-DRB5, LAMP3, LRRK2, MAPT, PARK2, PARK7, PINK1, PLA2G6, SNCA, STBD1, STK39, STX1B, SYT11 and VPS35) (International Parkinson's Disease Genomics Consortium (IPDGC); Wellcome Trust Case Control Consortium 2 (WTCCC2), 2011,Nalls, et al., 2011,Singleton, et al., 2013,Valente, et al., 2004,van Duijn, et al., 2001). Similarly, ALS and FTD might be explained by mutations in several often overlapping genes (Hardy and Rogaeva, 2013). To date 26 genes have been identified for FTD and/or ALS (ALS2, ANG, ATXN2, C9orf72, CHMP2B, DAO, EWSR1, FIG4, FUS, GRN, HNRNPA1, HNRNPA2B1, MAPT, OPTN, PFN1, SETX, SIGMAR1, SOD1, SPG11, SQSTM1, TAF15, TARDBP, TMEM106B, UBQLN2, VAPB and VCP) (Ling, et al., 2013).
There are many similarities that connect different neurodegenerative disorders. For instance, rare TREM2 variants (e.g. p.R47H) are associated with AD (Guerreiro, et al., 2013), while the p.R47H has been reported as a risk for PD, FTD and ALS (Benitez and Cruchaga, 2013,Cady, et al., 2014,Rayaprolu, et al., 2013). Hence, the overlap between different genes implicated in neurodegenerative disorders has to be systematically explored using novel genotyping technologies that could be crucial for the fast, reliable and concurrent screening of known genes, instead of costly and time-consuming gene-by-gene Sanger sequencing (Guerreiro, et al., 2014).
Here, we tested the utility of a recently designed semi-custom genome-wide array (NeuroX; Illumina, Inc) tailored to investigate neurodegenerative diseases. It has the standard content of the Illumina Exome BeadChip (~240,000 coding variants) and ~24,000 custom variants, including reported mutations causing neurodegenerative disorders, as well as risk variants found by GWASs (Nalls, et al., 2014). Hence, NeuroX is suitable for both mutation screening and association studies (Kara, et al., 2014). We evaluated 192 patients affected by AD, PD, FTD or ALS and catalogued the coding variations detected in a subset of 77 genes implicated in these disorders.
2. Method
2.1 Human Samples
Informed consent was obtained from all participants in accordance with the respective ethical review board. Study participants were mainly Canadian of North European origin, recruited from hospitals specialized in neurodegenerative disorders. Cases with known pathological mutations were excluded from the study as described previously (Xi, et al., 2012), with the exception of cases (n=9) that were used as internal controls to evaluate the reliability of the NeuroX array.
We investigated 192 subjects, including 98 PD, 43 ALS, 37 AD and 14 FTD patients prioritized for having an early age of onset and/or family history of neurodegenerative disease (Table 1). Most of the samples were unrelated (n=179), however for 13 familial patients we included a second affected family member to prioritise variants for segregation analysis. Family members of FLO25, FLO61 and FLO66 were genotyped by Sanger sequencing for the PSEN1, ABCA7 or TREM2 mutations detected by NeuroX (assays are available upon request).
Table 1.
Samples characteristics in different disease categories
|
| |||
| Diagnosis | N cases | Mean Age at onset (SD) years | Familial cases |
|---|---|---|---|
|
| |||
| AD | 37 | 64 (14) | 100% |
|
| |||
| ALS | 42 | 55 (11.2) | 57% |
|
| |||
| FTD | 14 | 58 (10.5) | 100% |
|
| |||
| PD | 98 | 41 (7.9) | 49% |
|
| |||
2.2 Analysis of NeuroX array
The NeuroX data was loaded to GenomeStudio (Illumina Inc.). All markers were clustered using the default Gen Call threshold (0.15); 97.3% of markers had Gen Train score >0.7, indicating high genotype quality. Genotypes were then converted to PLINK input files and allele frequencies were calculated. ANNOVAR annotation of the 267,151 single nucleotide polymorphisms (SNPs) on NeuroX revealed that this array can assess 230,921 exonic, 2,068 intronic- and 63 exonic-splicing SNPs (2 bp from an exon/intron boundary). The coding SNPs consisted of 213,892 non-synonymous, 9,639 synonymous, 5,242 stop-gain, and 219 stop-loss variations. The average call rate was 99.7% among all samples. Only one FTD sample was removed from the study due to poor DNA quality as indicated by the extensive chromosomal abnormalities revealed by cnvPartition.
Markers within 77 genes implicated in AD, PD, FTD and/or ALS (Supplemental Table 1) were extracted (n=4,441), including 1,945 non-synonymous, 163 synonymous, 95 stop-gain, 36 splicing variants and 1 stop-loss variant. Non-polymorphic markers (n=2,359) and markers missing genotypes in >5% of the samples (n=99) were excluded. We also removed non-coding (n=1,654), synonymous (n=43) and common variants with MAF>1% at the1000Genomes (1000g2012apr_all) and Exome Variant Server (esp6500si_all) databases (n=138). Finally, variants overlapping segmental duplications were excluded due to possible genotyping error (Ghani, et al., 2013b). The remaining variants were filtered to those predicted to have a probable damaging effect on protein function, according to either PolyPhen (LJB2_PP2_HDIV_Pred) or SIFT (LJB2_SIFT) analyses implemented in ANNOVAR. Cases with damaging variants were extracted using PLINK.
3. Results
3.1 Analysis of internal controls
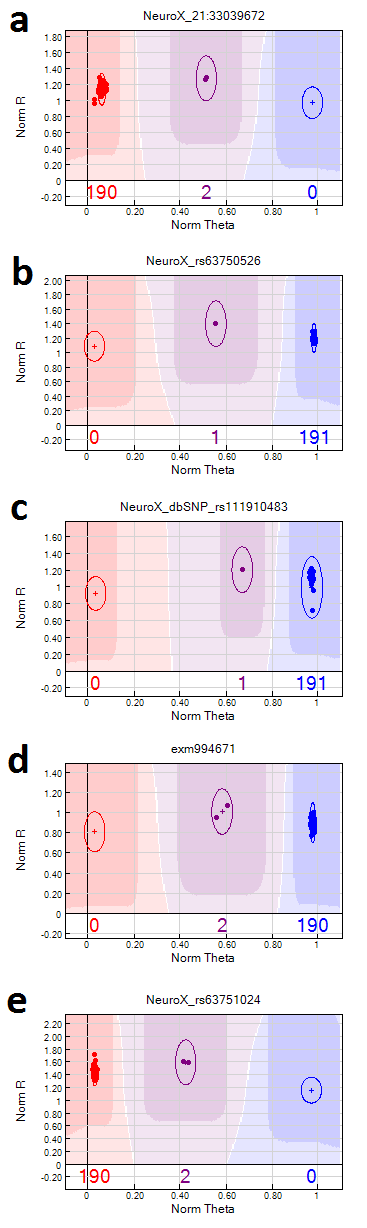
Five samples with heterozygous missense mutations previously detected by Sanger sequencing were all accurately recognized by NeuroX (Supplemental Figure 1a-d): an ALS case with SOD1 p.I114T (NeuroX_21:33039672), an AD case with PSEN1 p.A246E (NeuroX_rs63750526), a PD case with LRRK2 p.L1795F (NeuroX_rs111910483) and two PD cases with LRRK2 p.G2019S (exm994671). Also, a pair of previously reported monozygotic twins discordant for ALS (Xi Z, 2014) revealed identical genotypes for all NeuroX markers (n=267,607), except one in the TTC30B gene (exm244263). Sanger sequencing did not confirm this discrepancy, which was likely caused by a segmental duplication affecting the locus.
Four PD samples with known PARK2 CNVs were examined using cnvPartition and GenomeStudio (Supplemental Figure 2a-d). CnvPartition detected the heterozygous deletion affecting exons 3-6 in sample 6045 with CNV-values of 1 for 31 markers spanning the locus Chr6:162394342-162687778 (hg19); however it did not recognize the heterozygous duplications of exons 7 and 9 (sample 4932), exon 5 (sample 4033) or homozygous deletion of exon 2 (sample 6140). GenomeStudio also did not detect an increase in signal intensity for the duplication regions (likely due to the low density of NeuroX markers); however in sample 6140 a homozygous deletion is indicated by reduced signal intensity and an absence of genotype calls for 10 out of 15 NeuroX markers in exon 2. Notably, the 5 false-positive genotype calls included damaging variations (e.g. InDel Asp53Stop and p.Q34R), suggesting the need for careful validation of some NeuroX calls.
3.2 Discovery of new cases with Mendelian pathological mutations
A PSEN1 p.M233T mutation was detected in an Italian autosomal dominant FLO25 family with six members affected by AD (Figure 1a). The p.M233T substitution was found by heterozygosity of marker NeuroX_rs63751024 in two AD cases (8994 and 8995) (Supplemental Figure 1e). It was confirmed by Sanger sequencing, which revealed 8 carriers of the p.M233T among family members (Figure 1a). This pathogenic mutation was reported in several early onset AD families (Campion, et al., 1999,Guerreiro, et al., 2010, Kwok, et al.,1997, Park, et al.,2008, Raux, et al., 2005).
Figure 1.

Diagrams of three Italian families affected by Alzheimer’s Disease, in which mutations were identified by NeuroX. Genotypes are shown below the ID number with the mutant allele highlighted in bold font. Where available age at the time of examination is shown in the upper right corner and age of onset is indicated below the ID number. The gender of family members is masked to protect patient confidentiality. a) FLO25 with the PSEN1 p.M233T mutation (T>C). b) FLO61 with the rare ABCA7 p.L400V variant (C>G). c) FLO66 with the TREM2 p.D87N (G>A).
In addition to the internal control with the SOD1 p.I114T, we found the same mutation in patient 9108 with familial ALS (onset at age 50; disease duration 6 months). Sanger sequencing also detected the p.I114T in a currently unaffected sibling of patient 9108. Moreover, two unrelated familial ALS patients were heterozygous for SOD1 p.A5V (NeuroX_21:33032096): patient 9572 had onset at age 42 and a rapid ALS course (6 months), while the disease in patient 9432 was less severe with onset at age 58 and one year duration. Sanger sequencing confirmed the mutation and revealed the p.A5V in a currently asymptomatic sibling of patient 9432. The p.A5V is a common SOD1 mutation in the USA (also known as p.A4V, since the start methionine is post-translationally cleaved), while p.I114T is common in the UK (Battistini, et al., 2010,Niemann, et al., 2004).
3.4 Rare coding variants in 77 genes related to neurodegenerative disorders
A description of the variants identified in genes associated with four neurodegenerative disorders, their frequency in public databases, and predicted consequence on protein function are presented in Supplemental Table 2. In total, we detected 78 rare damaging SNPs observed in 98 out of 192 genotyped patients, including subjects with three (n=13) and two (n=22) concurrent variants. Of note, 211 InDel markers can be assessed by NeuroX, including 42 markers within the 77 genes of interest, of which only two were polymorphic, but not confirmed by Sanger sequencing.
3.5. Rare variants among PD patients
Several rare variants appeared to be relatively common (MAF >1%) in our PD cohort (Supplemental Table 2). For instance, the LRRK2 p.R1514Q variation (rs35507033 with MAF=0.2% in 1000Genomes) was heterozygous in 6 out of 98 unrelated PD samples (6.1%). In a previous study the p.R1514Q substitution was not significantly associated with sporadic late onset PD (>60 years). In datasets of different European origins, the MAF of p.R1514Q ranged from 1.0-3.3% in cases and 0-3.8% in controls (Toft, et al., 2007). The relatively high MAF of p.R1514Q in our study suggests that pathogenicity of the p.R1514Q substitution has to be further investigated in early onset PD dataset, since mean age at onset in our PD patients was only 41 years (Table 1).
We also identified several other very rare damaging variants, including TREM2 p.A105V in PD patient 5908 with age at onset of 46 (absent in 1000Genomes; MAF=0.022% at ESP); and APP p.E468K PD in patient 6892 with age at onset of 38 (MAF=0.074% at dbSNP138). Of note, AD mutations in APP are limited to duplications of the gene or missense mutations in exons 16 and 17, near the secretase processing sites (678-724 residues) (Ghani and Rogaeva, 2014), however the E468K variant is located in the soluble secreted part of the APP protein (sAPPβ). Interestingly, sAPPβ is reported to regulate Transthyretin (TTR), a potential biomarker for preclinical diagnosis of PD (Arguelles, et al., 2010,Li, et al., 2010). Also, two rare variants in exon 26 of GIGYF2 (PD-associated gene) were detected in familial patient 6171 with onset at age 30 (p.H1165R) and patient 8563 with onset at age 39 (p.P1149T).
In addition, we detected two potentially damaging PARK2 variants: p.A46T in patient 7932 (age at onset of 30) and p.R253C in patient 7031 (age at onset of 41). Both patients had family history of PD, however heterozygous mutations in this recessive PD gene have questionable pathological significance (Marras, et al., 2010). It is possible that disease manifestation in such cases is influenced by concurrent damaging variants. For instance, in PD patient 4033 with known heterozygous duplication of PARK2 exon 5 (age of onset 40) we also detected MAPT p.P140S, ABCA7 p.E316K and TREM2 p.H157Y. However, the risk associated with several variants is challenging to address. A large dataset would be required to determine whether genetic interactions manifest specific cellular/neuropathological phenotypes or lead to an earlier age of onset.
3.6 Rare variants among AD patients
We detected several mutations in genes reported by AD GWAS (ABCA7, ZCWPW1, SORL1, DSG2 and TREM2) (Lambert, et al., 2013). Two AD cases from Italian FLO61 family had an extremely rare heterozygous ABCA7 p.L400V variant absent in the ESP and 1000Genomes databases. Sanger sequencing confirmed the variant and detected another carrier that was unaffected at age 48 (Figure 1b). Both patients were also heterozygous for a rare ZCWPW1 p.H351R variant. Furthermore, two AD affected siblings of the Canadian TOR146 family (3515 and 3592), revealed the SORL1 p.H1813Q variant (MAF=0.003 in 1000Genomes), which was recently reported to be associated with AD in the analysis of probands from Caribbean Hispanic families (Vardarajan, et al., 2014). Notably, the same individuals in TOR146 are carriers of the DSG2 p.V56M variant.
First cousins with AD from Italian FLO66 family (8136 and 8145) were heterozygous for the extremely rare TREM2 p.D87N variant (MAF=0.074% in dbSNP138) affecting a residue conserved in all mammals. Sanger sequencing revealed that the p.D87N does not completely segregate with AD, since it was found in 4 out of 5 affected family members and in a subject unaffected by AD at age 80 (Figure 1c). However, p.D87N is likely a pathological variant with incomplete penetrance, as it was previously observed in 6 out of 1091 unrelated AD cases but not in 1105 controls (p=0.02) (Guerreiro, et al., 2013).
3.7 Rare variants among ALS patients
Heterozygous ATXN2 variants were detected in a familial ALS patient 9655 (p.M986V) and in an autopsy confirmed TDP43-positive ALS case 8861 (p.P954S). In another TDP43-positive ALS case 8851, we identified an extremely rare heterozygous p.D543N substitution at a highly conserved residue of ALS2, a known recessive gene causing the Juvenile form of ALS (Hadano, et al., 2001). However, the pathological significance of heterozygous ALS2 mutations remain to be investigated.
In ALS patient 8805 (age of onset 55) we observed two rare heterozygous variants (MAPT p.T17M and SETX p.C1554G). Currently it is unclear whether both variants contribute to the phenotype of this patient. Thus far, only three heterozygous SETX mutations were implicated in the Juvenile form of ALS (p.T3I, p.L389S and p.R2136H) (Chen, et al., 2004), and MAPT has not been associated with ALS.
4. Discussion
NeuroX is quite useful in the rapid screening of known mutations associated with neurodegenerative diseases. We identified several mutations reported to cause AD, PD or ALS. For instance, we detected the PSEN1 p.M233T mutation responsible for AD in a large family, which could be of clinical value, since carriers of such mutations might be enrolled in the longitudinal clinical AD trial of Dominantly Inherited Alzheimer Network (DIAN; http://www.dian-info.org/).
NeuroX allows the reliable concurrent investigation of rare coding variants in multiple genes for each subject, which is crucial for large studies investigating genes that could interact in the same network. For instance, rare variants in two AD-related genes (ABCA7 p.L400V and ZCWPW1 p.H351R) were observed in two related AD patients. NeuroX was also useful in detecting extremely rare variants. For example, we identified the very rare ALS2 p.D543N variant in an ALS patient. Several homozygous deletions in ALS2 were described in a young onset form of ALS (MIM #606352). Two affected sisters with Infantile-onset Ascending Hereditary Spastic Paralysis are the only cases described with a homozygous ALS2 missense mutation (p.C156Y) affecting a conserved residue in the Regulator of Chromatin Condensation (RCC1)-like domain and leading to down-regulation of the mutant protein (Eymard-Pierre, et al., 2006). Notably, the p.D543N mutation is also located in the same domain and its carrier needs to be investigated for the level of ALS2 protein and compound heterozygosity of ALS2.
Nevertheless, NeuroX has some limitations similar to other genome-wide arrays. For instance, the APOE alleles defined by SNPs rs429358 and rs7412 are not reliably detected (e.g. NeuroX markers for rs429358 were non-polymorphic in our study), since the APOE locus has high GC-content and its genotyping requires special conditions (Addya, et al., 1997). Also, NeuroX cannot evaluate repeat expansions (e.g. in C9orf72) known to be associated with several neurological disorders. In addition, CNVs, composing >12% of the human genome (Stankiewicz and Lupski, 2010) and implicated in many neuropsychiatric disorders (Cook and Scherer, 2008), are not reliably identified by NeuroX. However, the enriched probe density of NeuroX at a few loci (e.g. SNCA) could allow CNVs to be estimated based on signal intensities.
Furthermore, NeuroX contains only 211 InDels markers, while several million InDels are found throughout the human genome and many of them map to functionally important sites (Mullaney, et al., 2010). Also, careful consideration may be required when a NeuroX InDel marker appears polymorphic. For instance, an InDel marker in GRN (NeuroX_rs63750768) was found to be heterozygous in several of our samples but not confirmed by Sanger sequencing, because this 4-bp InDel variation overlaps a common SNP (rs25646) that interferes with the result of the InDel marker. Moreover, SNP probes on this array might occasionally have design problems producing misleading results. For example, the marker NeuroX_15:44856873 (rs80338869) was wrongly reported to be a premature stop codon in SPG11, however rs80338869 has three alleles and the array results reflect the common synonymous minor allele (MAF ~2%).
In summary, NeuroX provides rapid and accurate detection of coding variations within genes associated with neurodegenerative disorders, except a few loci and could be the first step in mutation analysis. For clinical diagnostic purposes, it can be supplemented with standard accredited genotyping methods to address its limitations.
Supplementary Material
Supplemental Table 1. List of known genes (and their transcripts) associated with four different neurodegenerative disorders. Locus positions are given based on hg19.
{kind=link}
Supplemental Table 2. Annotation details of the rare damaging variants identified among the 192 investigated patients with four different neurodegenerative disorders.
Supplemental Figure 1. Examples of heterozygous NeuroX marker genotypes in selected cases. a) SOD1 p.A5V (NeuroX_21:33039672) in two ALS cases; b) PSEN1 p.A246E (NeuroX_rs63750526) in an AD case; c) LRRK2 p.L1795F (NeuroX_dbSNP_rs111910483) in PD cases; d) LRRK2 p.G2019S (exm994671) in two PD patients; e) PSEN1 p.M233T (NeuroX_rs63751024) in two AD cases from FLO25 family.
Supplemental Figure 2. Log R ratio and B allele frequency graphs at the PARK2 locus for a) PD sample 6140 b) PD sample 6045 c) PD sample 4033 d) PD sample 4932.
Utility of novel genome-wide NeuroX array tailored to neurodegenerative diseases was tested.
We evaluated 192 patients for damaging variations in neurodegenerative disease genes.
78 potentially damaging rare variants and causative mutations were identified.
NeuroX is helpful for rapid and accurate mutation screening.
Further development may be still required to improve some current NeuroX caveats.
Acknowledgements
This work was supported by grants from the Canadian Institutes of Health Research, Ontario Research Fund, the W. Garfield Weston Foundation (ER, PSH); the Intramural Research Program of the National Institute on Aging, National Institutes of Health (NIH), part of the Department of Health and Human Services; project number ZO1 AG000958-11 (AS).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure Statement
None of the authors have conflict of interest, including financial interests in the results described.
References
- Arguelles S, Venero JL, Garcia-Rodriguez S, Tomas-Camardiel M, Ayala A, Cano J, Machado A. Use of haptoglobin and transthyretin as potential biomarkers for the preclinical diagnosis of Parkinson's disease. Neurochem Int. 2010;57(3):227–34. doi: 10.1016/j.neuint.2010.05.014. doi:10.1016/j.neuint.2010.05.014. [DOI] [PubMed] [Google Scholar]
- Battistini S, Ricci C, Giannini F, Calzavara S, Greco G, Del Corona A, Mancuso M, Battistini N, Siciliano G, Carrera P. G41S SOD1 mutation: A common ancestor for six ALS Italian families with an aggressive phenotype. Amyotroph Lateral Scler. 2010;11(1-2):210–5. doi: 10.3109/17482960902995592. doi:10.3109/17482960902995592. [DOI] [PubMed] [Google Scholar]
- Benitez BA, Cruchaga C. TREM2 and neurodegenerative disease. N Engl J Med. 2013;369(16):1567–8. doi: 10.1056/NEJMc1306509#SA4. doi:10.1056/NEJMc1306509#SA4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettens K, Brouwers N, Engelborghs S, Lambert JC, Rogaeva E, Vandenberghe R, Le Bastard N, Pasquier F, Vermeulen S, Van Dongen J, Mattheijssens M, Peeters K, Mayeux R, St George-Hyslop P, Amouyel P, De Deyn PP, Sleegers K, Van Broeckhoven C. Both common variations and rare non-synonymous substitutions and small insertion/deletions in CLU are associated with increased Alzheimer risk. Mol Neurodegener. 2012;7:3. doi: 10.1186/1750-1326-7-3. doi:10.1186/1750-1326-7-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cady J, Koval ED, Benitez BA, Zaidman C, Jockel-Balsarotti J, Allred P, Baloh RH, Ravits J, Simpson E, Appel SH, Pestronk A, Goate AM, Miller TM, Cruchaga C, Harms MB. TREM2 Variant p.R47H as a Risk Factor for Sporadic Amyotrophic Lateral Sclerosis. JAMA Neurol. 2014 doi: 10.1001/jamaneurol.2013.6237. doi:10.1001/jamaneurol.2013.6237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campion D, Dumanchin C, Hannequin D, Dubois B, Belliard S, Puel M, Thomas-Anterion C, Michon A, Martin C, Charbonnier F, Raux G, Camuzat A, Penet C, Mesnage V, Martinez M, Clerget-Darpoux F, Brice A, Frebourg T. Early-onset autosomal dominant Alzheimer disease: prevalence, genetic heterogeneity, and mutation spectrum. Am J Hum Genet. 1999;65(3):664–70. doi: 10.1086/302553. doi:10.1086/302553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YZ, Bennett CL, Huynh HM, Blair IP, Puls I, Irobi J, Dierick I, Abel A, Kennerson ML, Rabin BA, Nicholson GA, Auer-Grumbach M, Wagner K, De Jonghe P, Griffin JW, Fischbeck KH, Timmerman V, Cornblath DR, Chance PF. DNA/RNA helicase gene mutations in a form of juvenile amyotrophic lateral sclerosis (ALS4) Am J Hum Genet. 2004;74(6):1128–35. doi: 10.1086/421054. doi:10.1086/421054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook EH, Jr., Scherer SW. Copy-number variations associated with neuropsychiatric conditions. Nature. 2008;455(7215):919–23. doi: 10.1038/nature07458. doi:10.1038/nature07458. [DOI] [PubMed] [Google Scholar]
- Eymard-Pierre E, Yamanaka K, Haeussler M, Kress W, Gauthier-Barichard F, Combes P, Cleveland DW, Boespflug-Tanguy O. Novel missense mutation in ALS2 gene results in infantile ascending hereditary spastic paralysis. Ann Neurol. 2006;59(6):976–80. doi: 10.1002/ana.20879. doi:10.1002/ana.20879. [DOI] [PubMed] [Google Scholar]
- Ghani M, Pinto D, Lee JH, Grinberg Y, Sato C, Moreno D, Scherer SW, Mayeux R, St George-Hyslop P, Rogaeva E. Genome-wide survey of large rare copy number variants in Alzheimer's disease among Caribbean hispanics. G3 (Bethesda) 2012;2(1):71–8. doi: 10.1534/g3.111.000869. doi:10.1534/g3.111.000869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghani M, Rogaeva E. Autosomal Dominant Alzheimer’s Disease: Underlying Causes. In: Galimberti D, Scarpini E, editors. Neurodegenerative Diseases. Springer; London: 2014. pp. 27–47. [Google Scholar]
- Ghani M, Sato C, Lee JH, Reitz C, Moreno D, Mayeux R, St George-Hyslop P, Rogaeva E. Evidence of Recessive Alzheimer Disease Loci in a Caribbean Hispanic Data Set: Genome-wide Survey of Runs of Homozygosity. JAMA Neurol. 2013a doi: 10.1001/jamaneurol.2013.3545. doi:10.1001/jamaneurol.2013.3545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghani M, Sato C, Rogaeva E. Segmental duplications in genome-wide significant loci and housekeeping genes; warning for GAPDH and ACTB. Neurobiol Aging. 2013b;34(6):1710. doi: 10.1016/j.neurobiolaging.2012.11.006. e1-4. doi:10.1016/j.neurobiolaging.2012.11.006. [DOI] [PubMed] [Google Scholar]
- Guerreiro R, Bras J, Hardy J, Singleton A. Next generation sequencing techniques in neurological diseases: redefining clinical and molecular associations. Hum Mol Genet. 2014 doi: 10.1093/hmg/ddu203. doi:10.1093/hmg/ddu203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, Cruchaga C, Sassi C, Kauwe JS, Younkin S, Hazrati L, Collinge J, Pocock J, Lashley T, Williams J, Lambert JC, Amouyel P, Goate A, Rademakers R, Morgan K, Powell J, St George-Hyslop P, Singleton A, Hardy J. TREM2 variants in Alzheimer's disease. N Engl J Med. 2013;368(2):117–27. doi: 10.1056/NEJMoa1211851. doi:10.1056/NEJMoa1211851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerreiro RJ, Baquero M, Blesa R, Boada M, Bras JM, Bullido MJ, Calado A, Crook R, Ferreira C, Frank A, Gomez-Isla T, Hernandez I, Lleo A, Machado A, Martinez-Lage P, Masdeu J, Molina-Porcel L, Molinuevo JL, Pastor P, Perez-Tur J, Relvas R, Oliveira CR, Ribeiro MH, Rogaeva E, Sa A, Samaranch L, Sanchez-Valle R, Santana I, Tarraga L, Valdivieso F, Singleton A, Hardy J, Clarimon J. Genetic screening of Alzheimer's disease genes in Iberian and African samples yields novel mutations in presenilins and APP. Neurobiol Aging. 2010;31(5):725–31. doi: 10.1016/j.neurobiolaging.2008.06.012. doi:10.1016/j.neurobiolaging.2008.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadano S, Hand CK, Osuga H, Yanagisawa Y, Otomo A, Devon RS, Miyamoto N, Showguchi-Miyata J, Okada Y, Singaraja R, Figlewicz DA, Kwiatkowski T, Hosler BA, Sagie T, Skaug J, Nasir J, Brown RH, Jr., Scherer SW, Rouleau GA, Hayden MR, Ikeda JE. A gene encoding a putative GTPase regulator is mutated in familial amyotrophic lateral sclerosis 2. Nat Genet. 2001;29(2):166–73. doi: 10.1038/ng1001-166. doi:10.1038/ng1001-166. [DOI] [PubMed] [Google Scholar]
- Hardy J, Rogaeva E. Motor neuron disease and frontotemporal dementia: sometimes related, sometimes not. Exp Neurol. 2013 doi: 10.1016/j.expneurol.2013.11.006. doi:10.1016/j.expneurol.2013.11.006. [DOI] [PubMed] [Google Scholar]
- International Parkinson's Disease Genomics Consortium (IPDGC); Wellcome Trust Case Control Consortium 2 (WTCCC2) A two-stage meta-analysis identifies several new loci for Parkinson's disease. PLoS Genet. 2011;7(6):e1002142. doi: 10.1371/journal.pgen.1002142. doi:10.1371/journal.pgen.1002142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kara E, Xiromerisiou G, Spanaki C, Bozi M, Koutsis G, Panas M, Dardiotis E, Ralli S, Bras J, Letson C, Edsall C, Pliner H, Arepalli S, Kalinderi K, Fidani L, Bostantjopoulou S, Keller MF, Wood NW, Hardy J, Houlden H, Stefanis L, Plaitakis A, Hernandez D, Hadjigeorgiou GM, Nalls MA, Singleton AB. Assessment of Parkinson's disease risk loci in Greece. Neurobiol Aging. 2014;35(2):442. doi: 10.1016/j.neurobiolaging.2013.07.011. e9-e16. doi:10.1016/j.neurobiolaging.2013.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwok JB, Taddei K, Hallupp M, Fisher C, Brooks WS, Broe GA, Hardy J, Fulham MJ, Nicholson GA, Stell R, St George Hyslop PH, Fraser PE, Kakulas B, Clarnette R, Relkin N, Gandy SE, Schofield PR, Martins RN. Two novel (M233T and R278T) presenilin-1 mutations in early-onset Alzheimer's disease pedigrees and preliminary evidence for association of presenilin-1 mutations with a novel phenotype. Neuroreport. 1997;8(6):1537–42. doi: 10.1097/00001756-199704140-00043. [DOI] [PubMed] [Google Scholar]
- Lambert JC, Ibrahim-Verbaas CA, Harold D, Naj AC, Sims R, Bellenguez C, DeStafano AL, Bis JC, Beecham GW, Grenier-Boley B, Russo G, Thorton-Wells TA, Jones N, Smith AV, Chouraki V, Thomas C, Ikram MA, Zelenika D, Vardarajan BN, Kamatani Y, Lin CF, Gerrish A, Schmidt H, Kunkle B, Dunstan ML, Ruiz A, Bihoreau MT, Choi SH, Reitz C, Pasquier F, Cruchaga C, Craig D, Amin N, Berr C, Lopez OL, De Jager PL, Deramecourt V, Johnston JA, Evans D, Lovestone S, Letenneur L, Moron FJ, Rubinsztein DC, Eiriksdottir G, Sleegers K, Goate AM, Fievet N, Huentelman MW, Gill M, Brown K, Kamboh MI, Keller L, Barberger-Gateau P, McGuiness B, Larson EB, Green R, Myers AJ, Dufouil C, Todd S, Wallon D, Love S, Rogaeva E, Gallacher J, St George-Hyslop P, Clarimon J, Lleo A, Bayer A, Tsuang DW, Yu L, Tsolaki M, Bossu P, Spalletta G, Proitsi P, Collinge J, Sorbi S, Sanchez-Garcia F, Fox NC, Hardy J, Deniz Naranjo MC, Bosco P, Clarke R, Brayne C, Galimberti D, Mancuso M, Matthews F, Moebus S, Mecocci P, Del Zompo M, Maier W, Hampel H, Pilotto A, Bullido M, Panza F, Caffarra P, Nacmias B, Gilbert JR, Mayhaus M, Lannefelt L, Hakonarson H, Pichler S, Carrasquillo MM, Ingelsson M, Beekly D, Alvarez V, Zou F, Valladares O, Younkin SG, Coto E, Hamilton-Nelson KL, Gu W, Razquin C, Pastor P, Mateo I, Owen MJ, Faber KM, Jonsson PV, Combarros O, O'Donovan MC, Cantwell LB, Soininen H, Blacker D, Mead S, Mosley TH, Jr., Bennett DA, Harris TB, Fratiglioni L, Holmes C, de Bruijn RF, Passmore P, Montine TJ, Bettens K, Rotter JI, Brice A, Morgan K, Foroud TM, Kukull WA, Hannequin D, Powell JF, Nalls MA, Ritchie K, Lunetta KL, Kauwe JS, Boerwinkle E, Riemenschneider M, Boada M, Hiltuenen M, Martin ER, Schmidt R, Rujescu D, Wang LS, Dartigues JF, Mayeux R, Tzourio C, Hofman A, Nothen MM, Graff C, Psaty BM, Jones L, Haines JL, Holmans PA, Lathrop M, Pericak-Vance MA, Launer LJ, Farrer LA, van Duijn CM, Van Broeckhoven C, Moskvina V, Seshadri S, Williams J, Schellenberg GD, Amouyel P. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer's disease. Nat Genet. 2013;45(12):1452–8. doi: 10.1038/ng.2802. doi:10.1038/ng.2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Wang B, Wang Z, Guo Q, Tabuchi K, Hammer RE, Sudhof TC, Zheng H. Soluble amyloid precursor protein (APP) regulates transthyretin and Klotho gene expression without rescuing the essential function of APP. Proc Natl Acad Sci U S A. 2010;107(40):17362–7. doi: 10.1073/pnas.1012568107. doi:10.1073/pnas.1012568107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling SC, Polymenidou M, Cleveland DW. Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron. 2013;79(3):416–38. doi: 10.1016/j.neuron.2013.07.033. doi:10.1016/j.neuron.2013.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marras C, Klein C, Lang AE, Wakutani Y, Moreno D, Sato C, Yip E, Munhoz RP, Lohmann K, Djarmati A, Bi A, Rogaeva E. LRRK2 and Parkin mutations in a family with parkinsonism-Lack of genotype-phenotype correlation. Neurobiol Aging. 2010;31(4):721–2. doi: 10.1016/j.neurobiolaging.2008.05.030. doi:10.1016/j.neurobiolaging.2008.05.030. [DOI] [PubMed] [Google Scholar]
- Mullaney JM, Mills RE, Pittard WS, Devine SE. Small insertions and deletions (INDELs) in human genomes. Hum Mol Genet. 2010;19(R2):R131–6. doi: 10.1093/hmg/ddq400. doi:10.1093/hmg/ddq400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalls MA, Plagnol V, Hernandez DG, Sharma M, Sheerin UM, Saad M, Simon-Sanchez J, Schulte C, Lesage S, Sveinbjornsdottir S, Stefansson K, Martinez M, Hardy J, Heutink P, Brice A, Gasser T, Singleton AB, Wood NW. Imputation of sequence variants for identification of genetic risks for Parkinson's disease: a meta-analysis of genome-wide association studies. Lancet. 2011;377(9766):641–9. doi: 10.1016/S0140-6736(10)62345-8. doi:10.1016/S0140-6736(10)62345-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalls MAJB, Hernandez Dena G., Keller Margaux F., Majounie Elisa, Alan E, Renton MS, Jansen Iris, Guerreiro Rita, Lubbe Steven, Vincent Plagnol J, Raphael Gibbs CS, Pankratz Nathan, Sutherland Margaret, Bertram Lars, Christina Lill ALD, Faroud Tatiana, Eriksson Nicholas, Tung Joyce Y., Noah, Nichols JB, Arepalli Sampath, Pliner Hannah, Letson Chris, Heutink Peter, Maria, Martinez TG, Traynor Bryan J., Wood Nick, Hardy John, Singleton Andrew B., o.b.o.t.I.P.s.D.G.C.I.a., consortium., P.s.D.m.-a. NeuroX, a Fast and Efficient Genotyping Platform for Investigation of Neurodegenerative Diseases. Neurobiology of Aging. 2014 doi: 10.1016/j.neurobiolaging.2014.07.028. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niemann S, Joos H, Meyer T, Vielhaber S, Reuner U, Gleichmann M, Dengler R, Muller U. Familial ALS in Germany: origin of the R115G SOD1 mutation by a founder effect. J Neurol Neurosurg Psychiatry. 2004;75(8):1186–8. doi: 10.1136/jnnp.2003.028324. doi:10.1136/jnnp.2003.028324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park HK, Na DL, Lee JH, Kim JW, Ki CS. Identification of PSEN1 and APP gene mutations in Korean patients with early-onset Alzheimer's disease. J Korean Med Sci. 2008;23(2):213–7. doi: 10.3346/jkms.2008.23.2.213. doi:10.3346/jkms.2008.23.2.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raux G, Guyant-Marechal L, Martin C, Bou J, Penet C, Brice A, Hannequin D, Frebourg T, Campion D. Molecular diagnosis of autosomal dominant early onset Alzheimer's disease: an update. J Med Genet. 2005;42(10):793–5. doi: 10.1136/jmg.2005.033456. doi:10.1136/jmg.2005.033456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rayaprolu S, Mullen B, Baker M, Lynch T, Finger E, Seeley WW, Hatanpaa KJ, Lomen-Hoerth C, Kertesz A, Bigio EH, Lippa C, Josephs KA, Knopman DS, White CL, Caselli R, Mackenzie IR, Miller BL, Boczarska-Jedynak M, Opala G, Krygowska-Wajs A, Barcikowska M, Younkin SG, Petersen RC, Ertekin-Taner N, Uitti RJ, Meschia JF, Boylan KB, Boeve BF, Graff-Radford NR, Wszolek ZK, Dickson DW, Rademakers R, Ross OA. TREM2 in neurodegeneration: evidence for association of the p.R47H variant with frontotemporal dementia and Parkinson's disease. Mol Neurodegener. (3rd) 2013;8:19. doi: 10.1186/1750-1326-8-19. doi:10.1186/1750-1326-8-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singleton AB, Farrer MJ, Bonifati V. The genetics of Parkinson's disease: progress and therapeutic implications. Mov Disord. 2013;28(1):14–23. doi: 10.1002/mds.25249. doi:10.1002/mds.25249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stankiewicz P, Lupski JR. Structural variation in the human genome and its role in disease. Annu Rev Med. 2010;61:437–55. doi: 10.1146/annurev-med-100708-204735. doi:10.1146/annurev-med-100708-204735. [DOI] [PubMed] [Google Scholar]
- Toft M, Mata IF, Ross OA, Kachergus J, Hulihan MM, Haugarvoll K, Stone JT, Blazquez M, Gibson JM, Aasly JO, White LR, Lynch T, Adler CH, Gwinn-Hardy K, Farrer MJ. Pathogenicity of the Lrrk2 R1514Q substitution in Parkinson's disease. Mov Disord. 2007;22(3):389–92. doi: 10.1002/mds.21217. doi:10.1002/mds.21217. [DOI] [PubMed] [Google Scholar]
- Valente EM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S, Ali Z, Del Turco D, Bentivoglio AR, Healy DG, Albanese A, Nussbaum R, Gonzalez-Maldonado R, Deller T, Salvi S, Cortelli P, Gilks WP, Latchman DS, Harvey RJ, Dallapiccola B, Auburger G, Wood NW. Hereditary early-onset Parkinson's disease caused by mutations in PINK1. Science. 2004;304(5674):1158–60. doi: 10.1126/science.1096284. doi:10.1126/science.1096284. [DOI] [PubMed] [Google Scholar]
- van Duijn CM, Dekker MC, Bonifati V, Galjaard RJ, Houwing-Duistermaat JJ, Snijders PJ, Testers L, Breedveld GJ, Horstink M, Sandkuijl LA, van Swieten JC, Oostra BA, Heutink P. Park7, a novel locus for autosomal recessive early-onset parkinsonism, on chromosome 1p36. Am J Hum Genet. 2001;69(3):629–34. doi: 10.1086/322996. doi:10.1086/322996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vardarajan BN, Zhang Y, Lee JH, Cheng R, Bohm C, Ghani M, Reitz C, Reyes-Dumeyer D, Shen Y, Rogaeva E, St George-Hyslop P, Mayeux R. Coding mutations in SORL1 implicate alternate APP processing mechanisms in Alzheimer’s disease. Annals of Neurology. 2014 doi: 10.1002/ana.24305. (Submitted) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi Z YY, van Blitterswijk M, Dib S, Ghani M, Moreno D, Sato C, Liang Y, Singleton A, Robertson J, Rademakers R, Zinman L, Rogaeva E. Identical twins with the C9orf72 repeat expansion are discordant for ALS. Neurology. 2014 doi: 10.1212/WNL.0000000000000886. (In Press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi Z, Zinman L, Grinberg Y, Moreno D, Sato C, Bilbao JM, Ghani M, Hernandez I, Ruiz A, Boada M, Moron FJ, Lang AE, Marras C, Bruni A, Colao R, Maletta RG, Puccio G, Rainero I, Pinessi L, Galimberti D, Morrison KE, Moorby C, Stockton JD, Masellis M, Black SE, Hazrati LN, Liang Y, van Haersma de With J, Fornazzari L, Villagra R, Rojas-Garcia R, Clarimon J, Mayeux R, Robertson J, St George-Hyslop P, Rogaeva E. Investigation of c9orf72 in 4 neurodegenerative disorders. Arch Neurol. 2012;69(12):1583–90. doi: 10.1001/archneurol.2012.2016. doi:10.1001/archneurol.2012.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table 1. List of known genes (and their transcripts) associated with four different neurodegenerative disorders. Locus positions are given based on hg19.
Supplemental Table 2. Annotation details of the rare damaging variants identified among the 192 investigated patients with four different neurodegenerative disorders.
Supplemental Figure 1. Examples of heterozygous NeuroX marker genotypes in selected cases. a) SOD1 p.A5V (NeuroX_21:33039672) in two ALS cases; b) PSEN1 p.A246E (NeuroX_rs63750526) in an AD case; c) LRRK2 p.L1795F (NeuroX_dbSNP_rs111910483) in PD cases; d) LRRK2 p.G2019S (exm994671) in two PD patients; e) PSEN1 p.M233T (NeuroX_rs63751024) in two AD cases from FLO25 family.
Supplemental Figure 2. Log R ratio and B allele frequency graphs at the PARK2 locus for a) PD sample 6140 b) PD sample 6045 c) PD sample 4033 d) PD sample 4932.