

Abstract
The blood-brain barrier (BBB) remains a formidable obstacle in medicine, preventing efficient penetration of chemotherapeutic and diagnostic agents to malignant gliomas. Here, we demonstrate that a transactivator of transcription (TAT) peptide-modified gold nanoparticle platform (TAT-Au NP) with a 5 nm core size is capable of crossing the BBB efficiently and delivering cargoes such as the anticancer drug doxorubicin (Dox) and Gd3+ contrast agents to brain tumor tissues. Treatment of mice bearing intracranial glioma xenografts with pH-sensitive Dox-conjugated TAT-Au NPs via a single intravenous administration leads to significant survival benefit when compared to the free Dox. Furthermore, we demonstrate that TAT-Au NPs are capable of delivering Gd3+ chelates for enhanced brain tumor imaging with a prolonged retention time of Gd3+ when compared to the free Gd3+ chelates. Collectively, these results show promising applications of the TAT-Au NPs for enhanced malignant brain tumor therapy and non-invasive imaging.
Keywords: blood-brain barrier, gold nanoparticle, malignant glioma
Introduction
Malignant gliomas are primary brain tumors derived from glial origin and account for ∼70% of primary brain cancer diagnoses with overall low survival rates.[1] Glioblastoma (GBM), a grade IV astrocytoma, is the most predominate form of malignant glioma with patients displaying a 5-year survival rate of less than 9.8% and median survival time of 14.6 months after treatment.[2,3] Chemotherapy, a standard-of-care adjuvant for the treatment of patients with GBM, achieves very modest clinical benefits due to the impermeable nature of the blood-brain barrier (BBB) to most anti-cancer drugs,[4] which is maintained by the tight junctions between endothelial cells, surrounding pericytes, as well as the end feet of astrocytes.[5] Many potent therapeutic agents have been shown to kill glioma cells effectively in vitro, but could not affect tumor progression when administered systemically in vivo due to inaccessibility imposed by the BBB.[5] In addition, the poor solubility and short half-lives of many therapeutic drugs in the circulation further limit BBB penetration.
Another important aspect on malignant glioma is imaging, which is essential for preoperative planning, intraoperative removal of tumors, and therapeutic follow-up. Magnetic resonance imaging (MRI), a non-invasive imaging modality, is the primary method for detecting a brain tumor in clinic. Gadolinium (Gd3+) chelates such as Gd-DTPA and gadodiamine (Omniscan) are the typical contrast agents used for tumor imaging enhancement.[6] Such molecules show contrast in the brain tumor by relying on accumulation in the extracellular space where the BBB is compromised.[6] Due to the infiltrative nature of the disease and the great spatial variability in vascular disruption,[7] it is difficult to delineate the margins of the malignant glioma with high sensitivity. Therefore, there is a critical need for advances in delivery platforms to improve the effectiveness in the context of glioma diagnositics and treatment.
Searching for ideal platfroms that are capable of delivering therapeutic payloads across the BBB is currently under extensive investigation.[8] Currently, nanoparticle-based delivery systems are considered to lead to breakthroughs in maglignant glioma treatment. Anchoring antibodies and peptides onto nanoparticles (NPs) have been shown to improve BBB penetration via interactions with transporters or receptors on endothelial cells.[9] In addition, adsorptive-mediated transcytosis, triggered by electrostatic interactions between cationic targeting components and the negatively-charged membrane of brain capillary endothelial cells, has been demonstrated as an effective pathway for drug delivery to the brain.[9] Transactivator of transcription (TAT) peptide (sequence = YGRKKRRQRRR), derived from HIV, has been used to deliver a wide range of cargoes across lipophilic barriers.[10-14] On the other hand, it has been estabilished that as brain tumor progress, the BBB becomes compromised due to vascular leakiness. As a result, NPs in the size range of 10-100 nm can discriminate between normal brain and tumor tissues by accumulating within the tumor via the “enhanced permeability and retention effect” (EPR effect), leading to prolonged retention time of these particles.[15,16]
Among the various types of nanomaterials, gold nanoparticles (Au NPs) possess many features such as tunable size, a large surface area-to-volume ratio, flexibility to undergo versatile surface modifications, a high degree of biocompatibility and so on, making them good candidates as carriers for delivering cargoes across the BBB and targeting brain tumors.[17] The size of Au NPs plays a critical role in the ability of Au NPs to permeate through the brain microvasculature.[18-20] Using an in vitro BBB model, it has been predicted that Au NPs possessing a small core size and coated with low molecular weight polyethylene glycol (PEG) would achieve the optimal penetration of the brain microvasculature.[18] Recent animal studies suggest that Au NPs in the sub-100 nm range could enter the brain parenchyma after systemic administration.[19,20]
Based on these findings, we reasoned that incorporating the BBB-permeable TAT peptide onto the surface of small core-sized Au NPs would enhance endothelial cell membrane interaction and improve their penetration into brain tumors. Here, we present that a multifunctional 5 nm Au NPs platform with a short PEG (molecular weight 2000) and TAT peptide modified surface (TAT-Au NPs) is able to efficiently deliver both therapeutic and diagnostic agents across the BBB to reach the tumor tissues to enhance their effects on brain tumor treatment and imaging (Scheme 1). As a proof of concept, we first demonstrate that TAT-Au NPs without a cargo could cross the BBB efficiently and accumulate in brain tumors after systemic administration. We then show that such a platform could be used to deliver anticancer drugs to cross the BBB, improving overall survival in an orthotopic murine model. Furthermore, we demonstrate that gadolinium conjugated TAT-Au NPs show not only efficient BBB permeability and selective accumulation within brain tumors, but also improved MRI sensitivity, which could be used for monitoring malignant progression and therapeutic response, in vivo.
Scheme 1. Schematic illustrating the mechanism of Au NP-mediated therapeutic and diagnostic efficacy in brain tumors.

The chemotherapeutic drug, Dox, is conjugated to TAT-Au NPs via a pH sensitive linker (upper left panel) or the MRI contrast agent Gd3+ is chelated on the TAT-Au NPs (upper right panel) that circulates in the blood and selectively accumulates in the tumor via the EPR effect and adsorptive-mediated endocytosis.
Results
BBB penetration and tumor targeting of TAT-modified and non-modified PEGylated Au NPs
Non-modified PEGylated Au NPs (Nm-Au NPs) and TAT-modified PEGylated Au NPs (TAT-Au NPs) were synthesized and characterized as depicted in Fig. 1. Nm-Au NPs were synthesized by coating Au NP precursors with neutral HS-PEG-OCH3 (MW 2000 Da). The core size of Nm-Au NPs was 5.9 ± 2.1 nm as determined by transmission electron microscopy (TEM) (Fig. 1b) with a hydrodynamic diameter of 20.9 ± 0.5 nm and a polydispersity index of 10.7 ± 1.2 (Fig. 1d). TAT-Au NPs were prepared by ligand exchange reaction on the Au NPs with HS-PEG-OCH3 and HS-PEG-COOH (MW 1000 Da) followed by coupling with the amino group of lysines on the TAT peptide (Fig. S1). The core size of TAT-Au NPs was determined as 4.7±1.7 nm (Fig. 1c) with a hydrodynamic diameter of 21.4±0.9 nm and a polydispersity index of 9.1±0.7 (Fig. 1d). TAT-Au NPs were well dispersed in aqueous solution to form a homogenous solution as determined by TEM, and possessed an associated surface plasmon resonance at 520 nm (Fig. S1). Agarose gel electrophoresis was used to monitor peptide conjugation, which altered the nanoparticle size and surface charge density.[21-23] TAT-Au NPs were visualized as a distinct band that migrated more towards the negative electrode, which was different from Nm-Au NP due to the attachment of positively charged TAT peptide (Fig. S1).
Figure 1. The BBB penetration and tumor targeting studies of non-modified PEGylated (Nm-Au NP) and TAT-Au NPs.

(a) Schematic representation of Nm-Au NPs and TAT-Au NPs. (b) TEM images of Nm-Au NP and (c) TAT-Au NP. (d) Table of the core size (based on TEM) and hydrodynamic diameter (based on DLS) of TAT-Au NPs and Nm-Au NPs. (e) Histological sections demonstrating Au NPs overcome the BBB and penetrate into normal brain tissue. Brain tissue was collected 24 hours after injection of Nm-Au NPs or TAT-Au NPs and sections were stained with H&E. Au NPs were visualized by silver enhancement staining (particles indicated by black dots). Brain vasculature is indicated by yellow arrows. (f) Accumulation of Au NPs was enhanced in the brain tumor tissues, especially the TAT- Au NPs. (g-h) Quantification of the gold density and overall accumulation in the brain from the glioma bearing mice via ICP-MS (n=3).
To assess the BBB penetrating capabilities of Au NPs in vivo, the distribution of particles within the brain of glioma bearing mice was analyzed 24 hours after intravenous (I.V.) administration of Nm-Au NPs and TAT-Au NPs, respectively. Au NPs in the brain tissues were visualized as black aggregates by silver enhancement staining, a technique based on the preferential reduction of silver ions on Au NPs.[24] As shown in Fig. 1e, both Nm-Au NPs and TAT-Au NPs penetrated the brain microvasculature and were localized within the brain parenchyma. Au NPs primarily accumulated within endothelial cells of the microvasculature and diffused into the parenchyma within a ∼20 μm radius from the site of the endothelial cells (Fig. 1e), suggesting that our Au NPs could passively penetrate the BBB. Importantly, our TAT-Au NPs showed a higher level of accumulation within endothelial cells and disseminated ∼40 μm from the microvasculature.
Next, we compared the tumor-targeting ability of Nm-Au NPs and TAT-Au NPs. Since it has been previously reported that the brain tumor vasculature is compromised, allowing for the passive accumulation of nanoparticles within solid tumors.[16] This disruption of the BBB likely contributed to the increased accumulation of Nm-Au NPs in the brain tumor after 24 hours of I.V. injection (Fig. 1f). However, the TAT-Au NPs were enriched to an even higher extent in the tumor when compared to normal tissue in the contralateral brain (Fig. 1f). In Nm-Au NP treated-mice, gold content was 5.81±2.25 μg per gram of brain tissue and 0.6±0.27% of the injected dose (ID) accumulated in the brain tissues as quantified by ICP-MS (Fig. 1g and 1h). In contrast, the gold content was 22.34±5.03 μg per gram of brain tissue and 2.9±0.72% ID in the TAT-Au NPs treated mice, which represented a ∼4.8-fold increase in the accumulation of Au NPs in the brain when compared to Nm-Au NPs, suggesting that TAT peptides promote the penetration of Au NPs across the BBB and may be used to deliver therapeutic or diagnostic agents to brain tumor tissues.
Synthesis and drug-release dynamics of pH-sensitive TAT-Au NP-Dox conjugates
After demonstrating that TAT-Au NPs were able to cross the BBB efficiently and accumulate at the tumor site, we next considered how to make use of it to deliver therapeutic agents to intracranial glioma. For this purpose, we chose doxorubicin (Dox) as a model drug. Dox is a DNA intercalating agent that is unable to penetrate the BBB by itself, yet has been shown to possess potent cytotoxicity when cultured with glioma cells.[25] We chose to conjugate Dox to Au NPs through an acid-labile hydrazone linker, hypothesizing that the acidic tumor microenvironment caused by the overproduction of lactic acid via fast metabolic rates of cancer cells would allow for a triggered and controlled drug-release.[26] In addition, intracellular compartments like endosomes and lysosomes may also trigger Dox release from conjugates due to their acidic environment (pH = 4.5-6.0).[27]
To generate the hydrazone linkage, Au NP precursors were modified with methyl thioglycolate, HS-PEG-OCH3, HS-PEG-COOH and subsequently treated with hydrazine (Fig. 2a). The ketone group of Dox was conjugated to the hydrazine-modified Au NPs.[28] Conjugates were well dispersed in aqueous solution with an average core size of 5 nm (Fig. S2a). The number of Dox molecules per Au NP was determined by UV-Vis spectrometry based on the extinction coefficient of Dox (1.15 ×104 M-1 cm-1) and Au NPs (1.5 ×107 M-1 cm-1) (Fig. S2b).[29,30] On average, each Au NP was conjugated with ∼150 Dox molecules without causing precipitation of NPs. The presence of Dox on the Au NPs was further confirmed by [1]H NMR spectrum, which showed the characteristic resonances of Dox at 3.9, 4.5, 4.8, 4.9, 5.2, 5.4, 7.6, and 7.9 ppm (Fig. S2c). Compared to TAT-Au NP, the TAT-Au NP-Dox showed a band shift more toward the negative electrode in gel electrophoresis due to protonation of the free amine in Dox (Fig. S2d). Conjugations of TAT peptide and Dox onto Au NPs were further confirmed by conjugation of FITC-labeled TAT peptide identified by the fluorescence of FITC at 515 nm, and characteristic absorbance of Dox at 590 nm in the fluorescence spectrum. (Fig. S2e and S2f). An in vitro release study of Dox from TAT-Au NP-Dox conjugates by UV-Vis spectrometry confirmed that drug release from Au NPs was both pH- and time-dependent. At pH 7.4, the conjugates were stable and thus allowed for their systemic transportation; while at pH 4.7, over 90% of the Dox was released from the Au NPs within 6 hours (Fig. 2b).
Figure 2. pH-sensitive TAT-Au NP-Dox conjugates induce enhanced cytotoxicity towards glioma cells.

(a) TAT-Au NP-Dox conjugates release the free Dox under acidic conditions. (b) Time-dependent drug release at pH 7.4 and 4.7. (c) Quantification of the drug uptake of U87 cells treated with free Dox, Au NP-Dox and TAT-Au NP-Dox, respectively, by monitoring the Dox fluorescence. (d) Light microscopy images of glioma cells treated with the TAT-Au NPs, Dox, Au NP-Dox and TAT-Au NP-Dox conjugates at Dox concentration of 0.5 μM, respectively. (e) MTT quantification of cytotoxicity by Dox, Au NP-Dox and TAT-Au NP-Dox conjugates to U87, U251 and GBM 43 human glioma cell lines, respectively.
Enhanced in vitro therapeutic effect of TAT-Au NP-Dox for glioma therapy
To examine the intracellular uptake of the TAT-Au NP-Dox conjugates, flow cytometry was used to assess the proportion of glioma cells that have uptaken the conjugates in vitro. After 24 hours incubation with the conjugates, 100% of U87 glioma cells showed uptake of Dox (Fig. S3). A 2-fold increase in the mean Dox fluorescence intensity of the TAT-Au NP-Dox treated cells was observed when compared to those treated with the free Dox or the Au NP-Dox (Fig. 2c). To evaluate whether the covalent conjugation affected the cytotoxicity, we incubated U87 glioma cells with the same amount of TAT-Au NP, Dox, Au NP-Dox and TAT-Au NP-Dox, and found that the incubation with TAT-Au NP-Dox led to enhanced cytotoxicity than free Dox and Au NP-Dox based on cell morphology (Fig. 2d). The cytotoxicity of TAT-Au NP-Dox became apparent after 4 hours incubation and increased with time (Fig. S4). The IC50 of TAT-Au NP-Dox (56 nM) was 3 to 4-fold lower than free Dox (180 nM) and Au NP-Dox (220 nM) for U87 cell lines (Fig. 2e). Similar enhanced cytotoxicity using TAT-Au NP-Dox was also observed in other human glioma cell lines. Compared to free Dox, a 14- and 3.5-fold decrease in the IC50 was observed in U251 and GBM43 cell lines, respectively (Fig. 2e). Similar to free Dox, TAT-Au NP-Dox treated cells were also positive for cleaved capase-3, a key marker of apoptosis (Fig. S5), suggesting that TAT-Au NP-Dox may release the free Dox to kill cancer cells.
In vitro studies demonstrated that TAT-Au NPs were stable and biocompatible. U87 cells continuously incubated with these particles for 72 hours showed no detectable differences in cell morphology (Fig. 2d) and quantitative studies examining the viability of these treated cells were similar to the untreated group as evaluated by an MTT assay (Fig. S6). Although TAT peptide is generally known to be cytotoxic, our results indicated that the toxic property of TAT peptide was dramatically altered once attached to the Au NPs in a manner that prevented cell-mediated harm.
To examine the drug release pathway, intracellular localization of Dox was monitored by confocal microscopy. As shown in Fig. 3a, TAT-Au NP-Dox conjugates were efficiently taken up by U87 cells, and primarily localized to the lysosomes within 4 hours, where the acidic environment likely triggered the Dox release. Consistent with the primary mechanism of Dox cytotoxicity as a DNA intercalator and inhibitor of topoisomerase II,[31] we observed Dox accumulation in the perinuclear region and cell nuclei after 24 hours incubation with TAT-Au NP-Dox (Fig. 3b). The intracellular distribution of Au NPs, as observed by TEM, demonstrated Au NP localization within cellular lysosomes, some of which were close to the perinuclear region (Fig. 3c and 3d). In addition, TAT-Au NP-Dox conjugates were also observed binding to the cellular membrane, suggesting that there exists a strong interaction between the TAT peptide and the lipid bilayer (Fig. 3c). Previous work has shown that TAT-Au NPs can interact with membrane barriers in cancer cells and be taken up by cell vesicles, which then fuse with lysosomes as part of a transport pathway that is in agreement with the behavior of our TAT peptide-modified nanoparticles.[11,13] In contrast, no Au NPs were found inside the nucleus (Fig. 3d), further confirming that the observed cytotoxicity was due to the action of released Dox from TAT-Au NPs but not due to the platform itself. Moreover, TAT-Au NP-Dox conjugates showed an enhanced level of cellular uptake when compared to Nm-Au NP-Dox conjugates by TEM, suggesting that TAT peptide improved cellular penetration (Fig. S7). Taken together, our results strongly suggest that the overall transportation pathway includes cellular uptake of TAT-Au-Dox conjugates, release of the free Dox inside lysosomes, and nuclear translocation of free Dox.
Figure 3. In vitro studies of Au-NP-mediated Dox release in glioma cell lysosomes and cell nuclei.

(a) Co-localization of Dox in lysosomes after 4 hours of incubation as assessed by confocal microscopy. (b) Dox accumulation was observed in cell nuclei at 24 hours. (c) TEM images of glioma cells after 24 hours incubation with TAT-Au NP-Dox revealed that Au NPs were distributed around the cell membrane and within the cell lysosomes. (d) Au NPs were localized in the perinuclear region but could not enter nuclei. Red arrows indicate Au NPs.
Therapeutic efficacy of TAT-Au NP-Dox in an intracranial U87 mouse model
With the encouraging finding that our TAT-Au NPs are able to efficiently cross the BBB and TAT-Au NP-Dox conjugates show enhanced cytotoxicity to the glioma cells, we next addressed whether TAT-Au NP-Dox conjugates could cross the BBB and deliver Dox to an orthotopic U87 glioma mouse model after I.V. administration (Fig. 4). Consistent with the report that Dox is excluded from the brain parenchyma,[25] confocal microscopy showed that Dox itself could not accumulate in the tumor tissues within the first 24 hours (Fig. 4a). In contrast, injection of same molar dose of TAT-Au NP-Dox showed obvious Dox accumulation within brain tumor tissue (Fig. 4a). Consistent with this observation, a similar accumulation pattern of Au NPs was observed in adjacent brain sections using silver enhancement staining (Fig. 4b). The gold content quantified in the brain tissue by ICP-MS (4.5±1.1 μg per gram) (Fig. 4c) further confirmed that the TAT-Au NP-Dox conjugates penetrated through the BBB and reached tumor tissue. In addition, the gold content in urine samples was quantified to be 2.6±0.21 μg per gram (Fig. 4c), suggesting the Au NPs were small enough to undergo renal clearance. We also found that the conjugates had relatively long circulation of Au NPs in the blood. 8.1±0.34% ID of Au NPs were found in the blood after 24 hours post injection (Fig. 4d).
Figure 4. Therapeutic efficacy of a pH-sensitive TAT-Au NP-Dox delivery system in an intracranial U87 glioma mouse model.
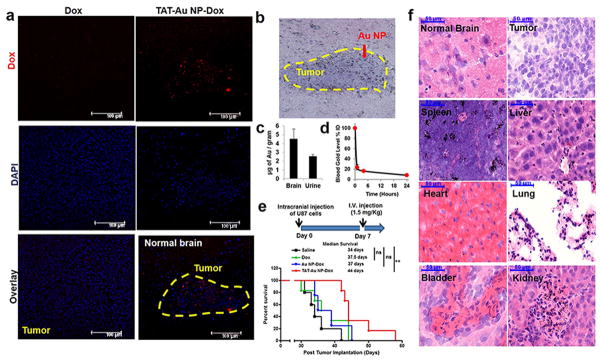
(a) Confocal images of mouse brain tissues 24 hours after I.V. injection showed Dox accumulation in a mouse after TAT-Au-NP administration. No Dox accumulation was observed when the drug alone was administered (b) Brain tissue from a TAT-Au NP-Dox injected mouse stained by H&E and silver enhancement confirming the localization of TAT-Au NP on the brain tumor tissue. (c) Quantification of the gold contents in the brain and urine samples from TAT-Au NP-Dox treated mice (n=5). (d) Quantification of the gold in the blood samples from TAT-Au NP-Dox injected mice. (e) Kaplan-Meier survival curves of intracranial U87 glioma tumor bearing mice I.V. injected with normal saline (n=5), Dox (n=6), Au NP-Dox (n=4) and (f) TAT-Au NP-Dox (n=6) at Dox concentration of 1.5 mg/kg. (f) Histology images of tissue samples with H&E and silver enhancement staining from the TAT-Au NP-Dox-treated mouse post 6 weeks of injection.
We next assessed whether administration of TAT-Au NP-Dox via a single-dose I.V. injection was able to improve survival of glioma bearing mice (Fig. 4e). U87 glioma-bearing mice were divided into four groups and injected with TAT-Au NP-Dox, Au NP-Dox, Dox, and normal saline at a single dose of 1.5 mg/kg, respectively. The median overall survival of Dox-treated mice (37.5 days) and Au NP-Dox-treated mice (37 days) showed only modest benefit compared to the saline-treated group (34 days) (Fig. 4e). In contrast, the median survival of the TAT-Au NP-Dox-treated mice (44 days) was significantly longer than that of saline (p<0.01) or Dox-treated mice (p<0.05) (Fig. 4e). Importantly, we found that the I.V. administered TAT-Au NPs did not show significant systemic toxicity. Normal mice (n=4) injected with TAT-Au NPs survived more than 10 months (Fig. S8), and also no premature animal death and body weight loss were observed over 30 days after the tumor implantation for the TAT-Au NP-Dox treated mice (Fig. S9). To evaluate whether the TAT-Au NP-Dox conjugates may do harm to non-diseased organs, we analyzed the cell morphology and Au NPs' accumulation in organs of brain, spleen, liver, heart, lung, bladder and kidney post 6 weeks of TAT-Au NP-Dox injection (Fig. 4f), and found that the treatment of TAT-Au NP-Dox did not cause any morphological change when compared to saline-treated mice (Fig. S10), suggesting that Au NPs were not toxic to these organs. Besides, no Au NPs were found in the normal brain or the tumor area 6 weeks post injection, suggesting that the NPs were completely cleared from the brain. Similar to other NP carriers, Au NPs showed more accumulation in the spleen and liver than in other organs, suggesting that their clearance was through the reticuloendothelial system. Interestingly, it has been reported that the major side effect of Dox is cardiotoxicity,[32] however, only negligible accumulation of Au NPs was observed in the heart, and little change to the myocardial tissue was observed after administration of TAT-Au NP-Dox, suggesting that our delivery system may help to overcome the toxicity to the heart caused by Dox. Furthermore, we noticed that the Au NPs were localized to the glomeruli of the kidneys and stroma of the bladder, suggesting that renal excretion may be the major clearance pathway for Au NPs, which is consistent with the amount of Au NPs found in the urine samples.
Gd3+-conjugated TAT-Au NPs as a non-invasive and highly sensitive tool for brain tumor imaging
With the encouraging finding that TAT-Au NPs were capable of efficiently delivering therapeutic agent such as Dox to brain tumor and enhance its therapeutic effects, we attempted to extend this platform to delivering other cargos such as MRI contrast agents. At present, the prevalent contrast agents used in clinic are Gd3+ chelates, which are unable to cross the BBB efficiently and retain in brain tissues for enough time by undergoing both rapid extravasation and renal clearance. We reasoned that our Au NPs platforms could facilitate the delivery of Gd3+ chelates across both BBB and disrupted BBB and increase the retention time. Therefore, we designed to synthesize the Gd3+ conjugated TAT-Au NPs (TAT-Au NP-Gd) (Fig. 5). The synthesis of TAT-Au NP-Gd conjugates entailed the preparation of the thiol-modified diethylenetriaminepentaacetic acid (DTPA) (Fig. 5a and Fig. S11) and its attachment to the surface of TAT-Au NPs via an Au-S bond for Gd3+ chelation. Using this method, a stable TAT-Au NP-Gd conjugate was obtained in aqueous solution with the loading of approximately 120 Gd3+ molecules on each Au NP as quantified by ICP-OES. TEM revealed that the TAT-Au NP-Gd conjugates were mono-dispersed and uniform (Fig. S12). The average core size of Au NPs was 5.2 nm (Fig. S12) with a hydrodynamic diameter of ∼22 nm as determined by DLS. The Gd3+ chelate modification on the Au NPs surface was further confirmed by via gel electrophoresis (Fig. S12).
Figure 5. TAT-Au NP-Gd conjugates for enhanced malignant glioma imaging.

(a) Construction of the TAT-Au NP-Gd conjugate. (b) T1-weighted MRI of TAT-Au NP-Gd water solutions with increasing amount of Gd3+ concentration and corresponding T1 map. (c) T1- weighted MRI and T1 map of U87 cells treated with growth medium alone (control), gadodiamide, or TAT-Au NP-Gd for 24 hours at [Gd3+] = 1.5 μM at 9.4 T (400 MHz). (d) Quantification of Gd3+ in U87 cells treated with the TAT-Au NP-Gd at [Gd3+] = 75 μM for 24 hours, cells treated with gadodiamide with the same concentration and growth medium (control), respectively. (e) MRI T1 map of the brain in a xenograft U87 glioma bearing mice before and after injection of TAT-Au NP-Gd conjugates (0.02 mmol/kg of Gd3+) at 9.4 T (400 MHz). (f) T2 of the mouse brain before injection. (g) T2 of the mouse brain 24 hours after injection. Displayed image FOV is 12 × 8 mm. The brain tumor is indicated by the red dashed circle. (h) Time-dependent R1 for the brain tumor.
Before we assess whether the TAT-Au NP-Gd conjugates can enhance the contrast in vivo, we first evaluate its sensitivity in vitro. We found that our conjugates were able to generate much stronger T1-weighted contrast enhancement signals with increasing conjugate concentrations compared to the control (water alone) (Fig. 5b). Remarkably, the corresponding T1 of protons was significantly shortened with increasing concentrations of the conjugate and the longitudinal relaxivity (r1) of the conjugates was determined as 7.4 mM-1 S-1 by taking the slope of 1/T1 as a function of Gd3+ concentration. This was 2.2-fold as high as the gadodiamide (3.4 mM-1 S-1), suggesting that conjugation to Au NPs significantly increased the sensitivity of the Gd3+ contrast agent.
To assess the feasibility for malignant glioma imaging, the MRI properties of TAT-Au NP-Gd conjugate treated glioma cells were investigated by compared to the clinical contrast agent gadodiamide. Due to the inability of gadodiamide to enter cells, gadodiamide-treated cells did not show signal enhancement (T1=2.44±0.08s) when compared to untreated cells (T1=2.43±0.05s) (Fig. 5c). In contrast, cells treated with TAT-Au NP-Gd conjugates at 1.5 μM of Gd3+ showed remarkable signal enhancement in T1-weighted MRI as well as shorter T1 (2.16±0.03s), suggesting that Gd3+ was efficiently taken up by the glioma cells. The cellular uptake of Gd3+ in the glioma cells was further confirmed by ICP-OES (Fig. 5d). Compared to gadodiamide, TAT-Au NP-Gd showed 82-fold increase in intracellular Gd3+ concentration, suggesting that the TAT-Au NP-Gd was able to penetrate the cell membrane efficiently and facilitate the uptake of the Gd3+ chelate by glioma cells. It is worth to note that no cytotoxicity was observed in U87 cells incubated with the TAT-Au NP-Gd conjugates over a 24-hour incubation (Fig. S13). The T1 of the conjugate treated cells decreased as conjugate concentration increased, as shown in the T1 map (Fig. S13). Up to 33% of T1 reduction at 150 μM was observed in the T1 map. Consistent with this, the longitudinal relaxation rate (R1) of water protons in the treated cells increased in a concentration dependent manner (Fig. S13).
The in vivo delivery of the conjugates to the brain tumor was investigated on the U87 mouse model. TAT-Au NP-Gd conjugates were injected intravenously into the mice at a Gd3+ dose of 0.02 mmol/kg, and the T1 maps of the mouse brain were obtained noninvasively using MRI at different time intervals after injection (Fig. 5e). 15 minutes after injection, the R1 value for the tumor was increased (shorter T1) by 17.7% from 0.36 to 0.42 s-1 and the corresponding Gd3+ concentration at the tumor was estimated to be 8.6 μM based on the longitudinal relaxivity r1 of the conjugates (7.4 mM-1 S-1). At 1.5 hours post-injection, the R1 value increased by 40% to 0.5 s-1 and the Gd3+ concentration in the tumor reached a peak of 19.3 μM. Later on, the intensity gradually decreased; but even after 24 hours, the intensity of R1 was still enhanced by 17.1% compared to the pre-injection time point, suggesting the TAT-Au NP-Gd conjugates had a longer retention time at the tumor site. Meanwhile, the conjugates also showed up to 19.6% R1 enhancement in the normal brain area 1.5 hours after injection (Fig. S14), indicating the conjugates were also permeable to the normal BBB. In contrast, when the mouse was injected with gadodiamine, R1 at the tumor area was only enhanced to 6% after 15 mins post-injection and underwent fast decay in 45 minutes (Fig. S15), suggesting that gadodiamine was rapidly cleared from the tumor site. In addition, low MRI intensity enhancement was observed in the non-tumor area, consistent with the impermeable nature of the gadodiamine to the BBB. T1-weighted images of the mice injected with the conjugates showed obvious contrast enhancement in the brain tumor area (Fig. 6a), which was in line with the T1 maps showing the increased R1 for the tumor.
Figure 6. Biodistribution of the TAT-Au NP-Gd conjugates in brain tissues from the TAT-Au NP-Gd treated mouse.

(a) T2 and the corresponding T1-weighted MRI horizontal section of the brain in a glioma bearing mouse post 24 hours of injection of the TAT-Au NP-Gd conjugates. The brain tumor is indicated by the red dashed circle. (b) and (c) Histology studies of the brain tissue from the TAT-Au NP-Gd treated mouse post 24 hours of injection. The brain tissue was stained by H&E staining to visualize the tissue structure and silver enhancement staining for Au NP. Au NPs were enhanced as black dots in the brain.
While the conjugates showed the enhanced brain tumor imaging via MRI, they also allowed for histology diagnosis via silver enhancement staining (Fig. 6b and 6c). Au NP distribution in brain tissue overlapped very well with signal enhancement in T1-weighted MRI (Fig. 6a and Fig. 6b), suggesting the successful delivery of TAT-Au NP-Gd conjugates to the brain tumor. The distribution of Au NPs in the tumor was relatively homogenous, further confirming the enhanced MRI at the tumor site. In contrast, the Au NPs in the non-tumor area, were localized at the vasculature and the surrounding parenchyma. Importantly, the Au NP conjugates showed significantly higher accumulation within the tumor than in normal brain tissue (Fig. 6c), suggesting excellent delivering selectivity of our platform. Consistent with this observation, there was a clear difference in Au NP distribution between the cancerous tissue and normal brain tissue at the tumor border.
Discussion
Searching for effective treatments for GBM has been painstakingly slow due to the impermeability of the BBB, which only selectively allows small lipophilic molecules, electro-neutral compounds, and/or nutrients under 400-600 Daltons to passively diffuse from the blood into the brain. Few chemotherapeutic agents can readily cross the BBB via systemic administration.[4] For example, one of the drug candidates for brain tumor is Dox, which is almost two thousand-fold more potent for glioma cells in vitro than the current standard-of-care agent, temozolomide (TMZ).[25] Unfortunately, Dox itself cannot cross the BBB readily, and it only possesses a short plasma half-life. Therefore, it is not used for glioma therapy so far. One possible solution is to administer anticancer drugs locally, immediately after surgical resection of the tumor.[33,34] However, the depth of drug penetration relies on diffusion and multiple dosing is limited. An alternative strategy is to incorporate anticancer drugs into a delivery system that is capable of crossing the BBB. After the conjugates reach the brain tumor tissue selectively, the release of free Dox can kill cancer cells and inhibit tumor growth. This strategy possesses an advantage for targeted and repetitive treatment of brain tumors. Seeking BBB-permeable platforms to deliver these potent agents across the BBB via systemic delivery is under extensive investigation.
Engineered nanoparticles with customizable size and surface properties hold the promise to overcome the BBB limitation that currently restricts many systemically-administrated agents. Previous studies have shown that only ∼0.3% of the injected Au NPs with a 10 nm size reach the brain tissues and the percentage decreases dramatically with an increase in NP size.[35,36] For example, less than 0.08% of the 15 nm Au NPs were localized in the brain while Au NPs larger than 50 nm were impermeable to the BBB.[35,36] Although the BBB of a brain tumor is compromised, the tumor microvessels retain some of the properties of the normal BBB,[37-39] limiting agents based on size. Therefore, we chose to synthesize Au NPs with smaller size and found that our Nm-Au NPs with a core diameter of 5 nm were able to passively accumulate inside the tumor bed. In addition to size, the TAT peptide has been reported to facilitate the BBB transportation of proteins as well as nanoparticles including liposomes, micelles and quantum dots in mice.[10,40-42] It has also been shown that TAT peptide labeling increased internalization efficiency of magnetic nanoparticles into progenitor cells for in vivo tracking and recovering.[43] However, TAT peptide modified Au NPs for in vivo BBB transportation has not been explored before. Our results indicated that TAT-Au NPs led to a significant increase of their accumulation within the brain tumor after systemic administration (from 0.6% to∼2.9 % of ID), suggesting the TAT modified Au NPs has much better delivery capacity than Au NPs alone. Our studies showed that TAT-Au NPs targeted the intracranial brain tumor by the EPR effect with an enhancement in penetration likely due to TAT peptide-mediated absorptive-mediated transcytosis across the disrupted BBB and into the tumor area.
To assess the TAT-Au NP delivery system for glioma therapy, we synthesized TAT-Au NP-Dox conjugates with a pH-dependent reversible hydrazone linkage. In our in vitro experiments, we found that intracellular drug uptake was mediated by the TAT-Au NPs, as shown by a 2-fold increase of Dox in the glioma cells when compared to cells treated with Dox. Moreover, TAT-Au NP-Dox conjugates showed 3 to 14-fold enhancement in cytotoxicity to several glioma cell lines when compared with Dox, which might allow for effectively controlling brain tumor growth at lower drug dosages. Although several in vitro studies have shown that TAT-Au NPs can enhance cellular uptake,[13,14] no in vivo efficacy studies in the context of brain tumors have demonstrated this before. Our in vivo experiments showed that the TAT peptide selectively improved the cell uptake of Dox at the brain tumor site. Consistent with these observations, animal survival experiements showed that a single regimen of the TAT-Au NP-Dox conjugates led to a obvious overall survival improvement when compared to both saline- and Dox-treated mice, respectively.
To improve the brain tumor imaging and monitor the disease progression after treatment, delineation of glioma margins with high sensitivity is a major challenge in MRI using gadolinium chelates as contrast agents. These contrast agents do not cross the BBB and cannot be taken up by cancer cells efficiently,[6,7] which cause the difficulty to precisely monitor disease progression. Compared to the Gd3+ chelate, TAT-Au NP-Gd conjugates showed a 2.2-fold higher relaxivity and 82-fold enhancement in Gd3+ cellular uptake, which allowed for sensitive detection of the cancer cells via MRI. Previously, it was reported that a Gd3+concentration of millimolar was needed to achieve 107-109 Gd3+ per cell for contrast enhancement in cells.[44] Using the TAT-Au NP-Gd conjugates, adequate cell MRI contrast was achieved at 1.5 μM concentration, which is 3 times lower than that of the Gd3+ enriched by DNA-Au NPs and 150 times lower than that of the cell penetrating peptide-coupled Gd3+ chelates.[45-46] More importantly, our in vivo MRI studies showed that TAT-Au NP-Gd conjugates crossed both intact BBB and disrupted BBB in the tumor area and had a longer retention time, suggesting the promising applications of these conjugates in glioma imaging. Meanwhile, the corresponding Gd3+ concentration in brain tumor could be estimated based on the T1 map, providing a non-invasive way to quantify the NP concentration in the tumor area with the fixed Gd3+/NP ratio and thus insuring that the appropriate concentration range of a therapeutic agent can be achieved. While the TAT-Au NP-Gd allows non-invasive imaging of conjugate uptake in the brain, it could also serve as a contrast agent for both time-dependent MRI and histological determination of the tumor margins.
Since TAT-Au NPs loaded with drug or contrast agent have been shown to penetrate into an intracranial tumor effectively, a multifunctional platform may be further developed to achieve simultaneous drug-delivery and contrast enhancement for glioma. Furthermore, using imaging techniques such as MRI to monitor drug delivery in tissues non-invasively may allow us to predict tumor responses in a precise and three-dimensional manner. Moving forward, the loading ratio of both drug molecules and contrast agents on this platform needs to be balanced in order to achieve desirable drug concentrations and adequate imaging signal at the site of a brain tumor.
In addition, our TAT-Au NP platform provides a high degree of both biocompatibility and stability in vitro and in vivo. The Au NP structure is physically smaller and chemically more stable when compared to organic-based nanocarriers such as liposomes and micelles, ensuring the integrity of the nanocarrier structure. Hydrophilic PEG coating was applied to increase the steric hindrance, preventing aggregation in aqueous solution and reducing recognition by the reticuloendothelial system.[47] More importantly, TAT-Au NP did not cause significant long-term systemic toxicity. At 6 weeks after injection, few Au NPs were detected in the tumor area and non-tumor tissue in the brain, suggesting that Au NPs were mostly cleared from this site. In addition, no alterations in cell morphology were detected in the major organs after injection with TAT-Au NP-Dox conjugates. While the Au NPs were large enough to accumulate in the brain tumor via the EPR effect, they were also small enough to be cleared via renal excretion, a main pathway of clearance out of the body.
In summary, we have demonstrated that the TAT-Au NP platform was able to cross the BBB and deliver the BBB-impermeable Dox to brain tumor tissue and thus improved the survival of mice bearing an intracranial U87 glioma. We also demonstrated that this system was able to efficiently deliver Gd3+ contrast agent across the BBB to reach brain tumors and thus improved the brain tumor imaging with MRI. Together, we have shown that a TAT-Au NP is a powerful delivery platform for enhanced glioma therapy and imaging with MRI. To the best of our knowledge, our work represents the first example of in vivo targeted therapy using TAT-Au NPs to overcome the BBB for controlled drug delivery and imaging in an intracranial malignant glioma model after systemic administration. Besides Dox and Gd3+, this platform may also be suitable for delivering other potent but BBB-impermeable anticancer drugs and contrast agents for enhanced glioma therapy and imiging. Recently, it was reported that some other targeting moieties such as tranferrin, cyclic [RGDyK] peptide, and angiopep-2 peptides incresed the penetration across the BBB or specifically bind to the receptors overexpressed on the tumor vasculature and cancer cell membrane.[19,37,48] Incorporating these targeting reagents into our Au NP platform may further improve its specificity. Our results showed that the system is a promising delivery platform for crossing the BBB and will find wide applications in malignant glioma therapy and diagnosis.
Methods
Materials
Gold (III) chloride (HAuCl4), tetraoctylammonium bromide (TOAB), sodium borohydride (NaBH4), dodecylamine (DDA), methyl thioglycolate (MTG), hydrazine, triethylamine, toluene, chloroform, and ethanol were purchased from Sigma-Aldrich (St. Louis, MO, USA). Biofunctional HOOC-PEG-SH (MW 1000 Da) and monofunctional PEG-SH (MW 2000 Da) were purchased from Laysan Bio (Arab, AL, USA). Doxorubicin hydrochloride was purchased from Ontario Chemicals (Ontario, Canada). 1-Ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC) and N-hydroxysulfosuccinimide (sulfo-NHS) were purchased from Pierce. S-2-(4-aminobenzyl)-diethylenetriamine pentaacetic acid•4TFA (pNH2-Bn-DTPA) was purchased from Green Chempharm, Inc. N-Succinimidyl 3-(2-Pyridyldithio)propionate (SPDP) was purchased from TCI Chemicals. TAT peptides with the sequence containing YGRKKRRQRRR and FITC-LC-YGRKKRRQRRR were purchased from Anaspec (Fremont, CA, USA).
Cell culture and animals
Glioma cell lines, U87, U251, GBM43 and GL261 were cultured in DMEM (Dulbecco's Modification of Eagle's Medium) (Mediatech Inc., Manassas, VA, USA), containing 2% penicillin and streptomycin antibiotic (Cellgro, Mediatech, Inc., Manassas, VA, USA) and 10% fetal bovine serum (FBS; Atlanta Biologicals, Lawrenceville, GA, USA). 6-8 week-old male athymic nude mice were purchased from Charles River Laboratory (Wilmington, MA, USA) and C57BL/6 mice were obtained from Jackson Laboratories. Brain tumor bearing mice were established by intracranially injecting glioma cells into the right hemisphere of the brain. A burr hole centered 2 mm lateral to the sagittal suture and 2 mm posterior to the coronal suture was drilled into the right side of the skull. After positioning the animals in a stereotactic frame, 4×105 glioma cells in 2.5 μL PBS were injected at a depth of approximately 3 mm into the brain. Animals were cared for according to an animal protocol approved by Institutional Animal Care and Use Committee at the University of Chicago.
Synthesis of TAT-Au NPs and Nm-Au NPs
Dodecylamine coated Au NPs were synthesized based on the reduction of HAuCl4 by NaBH4 in toluene.[48] To synthesize TAT-Au NPs, Au NPs in chloroform (4×10-8 mol) were treated with a mixture of biofunctional HOOC-PEG-SH (MW 1000 Daltons, 8×10-6 mol) and monofunctional CH3O-PEG-SH (MW 2000 Daltons, 3.2×10-5 mol) for 24 hours. After purification by centrifugation, the -COOH on the Au NPs was activated by EDC and sulfo-NHS for 15 minutes in MES buffer and coupled with the -NH2 groups on the TAT peptide. The Nm-Au NP was synthesized by treatment of Au NPs with CH3O-PEG-SH (MW 2000 Daltons) and purified by centrifugation.
Synthesis of pH sensitive TAT-Au NP-Dox conjugates
To synthesize TAT-Au NP-Dox, methyl thioglycolate (2.4×10-5 mol), biofunctional HOOC-PEG-SH (1.12×10-5 mol) and monofunctional PEG-SH (4.48×10-5 mol) were mixed with Au NPs (4×10-8 mol) in chloroform for 24 hours to achieve functionality and stability in aqueous solution. After purification, 400 μL of 10% hydrazine solution was added to the above Au NP solution and the mixture was incubated overnight at room temperature. The hydrazine modified Au NPs were then incubated with Dox (2.4×10-5 mol) for 24 hours. The -COOH on the Au NPs was activated by 0.6 mg of EDC and 1 mg of sulfo-NHS to conjugate with the -NH2 groups on the TAT peptide. Similarly, Au NP-Dox with the same drug loading was synthesized. Each step of the synthesis was monitored by transmission electron microscopy (TEM), dynamic light scattering (DLS), gel electrophoresis, and UV-Vis spectrometry. The final Dox and TAT peptide concentration in the TAT-Au NP-Dox solution were determined by UV-Vis and fluorescence spectrometry.
Synthesis of TAT-Au NP-Gd conjugates
To synthesize the conjugates, pNH2-Bn-DTPA (99.5 mg, 0.1 mmol) was added to a flask (50 mL) containing PBS buffer (5.2 mL, 40 mM, pH 7.4) to give a clear solution. Then SPDP (31.2 mg, 0.1 mmol) in anhydrous DMF (5.2 mL) was added dropwisely and the mixture was stirred at room temperature for 2 hours. The solvents were removed by evaporation in high vacuum and the residue was dissolved in deionized water. The DTPA-SS was purified by C18 RP HPLC eluting with 0-50% acetonitrile in TEAA (0.1 M, pH 7.0). The Maldi-TOF MS of the product gave the expected peaks at [MH]+ = 696 and [MNa]+ = 718. DTPA-SS (2.25×10-6 mol) was reacted with 8.6 ×10-6 mol TECP in water for 5 minutes to break the disulfide bond to generate DTPA-SH. TAT-Au NPs (1.5 ×10-8 mol) were added to the above mixture and stirred for 24 hours. The TAT-Au NP-DTPA conjugates were purified by centrifugation and dissolved in water, which was then treated with GdCl3 (1.125×10-5 mol) for 24 hours. The final TAT-Au NP-Gd conjugates were obtained and washed using the centrifugation tubes with 10,000 Da membrane until no free Gd3+ was detected by xylenol orange diodium. The conjugates were characterized by UV-Vis spectrometry, DLS and TEM. The Gd3+ ion per Au NP and cellular uptake were quantified using inductively coupled plasma-atomic emission spectroscopy (ICP-OES).
In vitro Dox release experiments
In vitro Dox release experiments were performed using PBS buffer (pH = 7.4,) and 2-(N-morpholino) ethanesulfonic acid (MES) buffered saline (pH = 4.7), respectively. For each release study, TAT-Au NP-Dox solution was incubated with 0.5 mL buffer solution at room temperature. Release media was taken out for analysis at specific time intervals by centrifuging at 13,000 rpm for 20 minutes. The released percentage of the Dox molecules in the supernatant was determined by UV-Vis spectrometry.
Flow cytometric assessment of TAT-Au NP-Dox treated glioma cells
2×105 U87 glioma cells were incubated for 24 and 48 hours with TAT-Au NP-Dox, Au NP-Dox, and Dox at 1 μM Dox, respectively. Cells were then washed 3 times with 10% FBS DMEM and fixed in BD fixation buffer. Cells were re-suspended in 200 μL FACS buffer (1% BSA and 0.01% NaN3). A BD Biosciences FACSCanto flow cytometer with a 488 nm Sapphire Laser was used to detect intracellular Dox fluorescence via the PE-A channel. 10,000 events for each condition were collected and analyzed. For apoptosis quantification, cleaved caspase-3 (Asp175) antibody (Cell Signaling Technology, Inc. Danvers, MA) was added to the cell suspension and incubated for 1 hour on ice. After washing, 1:500 diluted anti-rabbit Alexa Fluor® 647 conjugated secondary antibody was treated with the cells in dark for another hour. The apoptotic cells were detected via the APC-A channel.
Evaluation of TAT-Au NP-Dox cytotoxicity via MTT assays
Glioma cells were seeded on 96 well plates at 1×104 cells per well and incubated with TAT-Au NP-Dox, Au NP-Dox, Dox, and TAT-Au NP at 37 °C and 5% CO2 for 48 h, respectively. MTT labeling reagent 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide (Cell Proliferation Kit I (MTT), Roche Applied Sciences, Indianapolis) was added to the wells and incubated for 4 hours according to the manufacturer's protocol. 100 μL of 10% SDS solubilization solution was added to the wells to dissolve the purple formazan salt crystals. The plates were read on a GeneMate microplate reader. 4-6 replicates were included for each condition and the experiments were repeated twice.
Determination of the uptake of TAT-Au NP-Dox by confocal microscopy
5×104 U87 glioma cells were seeded into glass-bottom culture dishes. TAT-Au NP-Dox, Au NP-Dox and Dox in 10% FBS DMEM was added to the dishes at the Dox concentration of 1 μM, respectively. The treated cells were monitored at 4 and 24 hours post-incubation via confocal microscopy. To determine the location of intracellular lysosomes, the treated cells were washed 3 times with PBS and incubated with LysoTracker Green DND-26 (Invitrogen, USA) at 7.5×10-8 M for 30 minutes. Live cell images were taken by a Leica SP5 II STED-CW super-resolution laser scanning confocal microscope.
Determining Au NP localization in the glioma cells by TEM
1×106 U87 glioma cells were seeded into culture dishes. Cells were incubated with TAT-Au NP-Dox and Au NP-Dox at Au NP concentration of 2 μM for 24 hours, respectively. The cells were rinsed twice with serum-free DMEM and fixed in glutaraldehyde for 2 hours. The cell pellets were washed and fixed with 1% osmiumtetroxide in 0.1 M sodium cacodylate buffer for 1 hour. The pellets were washed with sodium cacodylate buffer, and 1% uranyl acetate was added for staining. After dehydration with a series of washes with increased ethanol concentration, propylene oxide (PO) was added at 2 : 1 PO : Spur resin, 1 : 1 PO : Spur resin, and 100% Spur resin. The samples were maintained overnight at 60 °C. The resin blocks were cut with an ultramicrotome at ∼100 nm thickness and transferred onto copper TEM grids. The images were taken on a FEI Tecnai F30 scanning transmission electron microscope with high performance CCD camera.
Biodistribution of Au NPs on the tissues
For histology studies, the brain, spleen, liver, heart, lung, kidney and bladder of mice were freshly frozen in Tissue-Tek® O.C.T. compound and were sectioned at 10 μm thickness. The tissue slides were co-stained by silver enhanced staining to visualize the NPs and hematoxylin and eosin (H&E) for structural morphology study of the brain. For quantification, the brains of mice were digested in concentrated nitric acid and the gold content was quantified by inductively coupled plasma-mass spectrometry (ICP-MS).
MRI experiments
All MRI experiments were performed on a 30-cm horizontal bore Bruker BioSpec 9.4 Tesla Small Animal MR System (Bruker-Biospin, Billerica, MA) with 11.6 cm inner diameter, actively shielded gradient coils (maximum constant gradient strength for all axes: 230 mT m−1) and a volume quad coil (Bruker BioSpin MRI GmbH Quad coil, OD/ID = 59/35mm, length = 38mm). Glioma-bearing mice were anesthetized during procedures with 1.5–2% isoflurane mixed with medical air to maintain a surgical plane of anesthesia. Temperature was monitored continuously by a thermometer and controlled with warm air. Heart rate, respiration rate, and blood pressure were monitored with an optical detection system from SA Instruments (Stony Brook, NY), developed for use in MRI. For animal experiments, high-resolution anatomic images were acquired first with multi-slice RARE (rapid acquisition with relaxation enhancement) sequences in both coronal and axial orientations (repetition time (TR)/echo time (TEeffective) = 4000/20.4 msec, array size = 256×256, field of view = 25.6 mm × 25.6 mm, slice thickness = 0.5 mm, RARE factor = 4, NEX (number of excitation) = 2). For the phantom experiments, slice thickness was 1 mm. Then, T1-weighted imaging and T1 measurement were performed for selected slices based on T2-weighted imaging. For gradient echo (a fast low-angle shot sequence), the following parameters were used: TR/TE = 700/5 msec, flip angle = 30°, slice thickness = 1 mm. All other parameters were as in the spin echo sequence. For T1 measurement, spin-lattice relaxation (T1) time was determined based on acquisitions with a saturation recovery sequence using the following parameters: TR = 14.1, 50, 100, 150, 200, 250, 300, 500, 750, 1000, 2000, 3000, 5000, 10,000, and 15,000 msec, TE = 10 msec, array size = 128×128, slice thickness = 1 – 3 mm, NEX = 1. The data was processed and analyzed using a computer program written in IDL (Exelis VIS, Inc., Boulder, CO).
Statistical analysis
All statistical analyses were performed using Graphpad Prism 4 (GraphPad Software Inc., San Diego CA), and numerical data were reported as a Mean±SEM. For comparisons between two groups, unpaired student t test was applied, and for comparisons among more than two groups, ANOVA with bonferroni's correction was used. The IC50 values were calculated using the sigmoidal dose-response formula implemented in Prism. Survival curves were generated by the Kaplan–Meier method, and the log-rank test was used to compare the distributions of survival times. All reported p values were two-sided and were considered to be statistically significant at * p<0.05, **p<0.01, ***p<0.001.
Supplementary Material
Acknowledgments
This work is supported by the National Institute of Neurological Disorders and Stroke U01NS069997 and R01NS077388 to MSL, K01 HG006699 to QD, and F32NS073366 and K99NS082381 to DAW. We thank Yimei Chen at the Electron Microscopy Core Facility of University of Chicago and Dr. Hisashi Fujioka at Case Western Reserve University for the assistance in TEM image analysis. We thank the MRI Core Facility at The University of Chicago for technical support.
Footnotes
Supporting Information is available on the WWW under http://www.small-journal.com or from the author.
Contributor Information
Dr. Yu Cheng, The Brain Tumor Center, The University of Chicago, Chicago, Illinois, USA, Fax: +1 773 834 2608.
Dr. Qing Dai, Department of Chemistry, Institute of Biophysics Dynamics and Howard Hughes Medical Institute, The University of Chicago, Chicago, Illinois, USA.
Ramin Morshed, The Brain Tumor Center, The University of Chicago, Chicago, Illinois, USA, Fax: +1 773 834 2608.
Dr. Xiaobing Fan, Department of Radiology, The University of Chicago, Chicago, Illinois, USA
Michelle L. Wegscheid, The Brain Tumor Center, The University of Chicago, Chicago, Illinois, USA, Fax: +1 773 834 2608
Dr. Derek A. Wainwright, The Brain Tumor Center, The University of Chicago, Chicago, Illinois, USA, Fax: +1 773 834 2608
Yu Han, The Brain Tumor Center, The University of Chicago, Chicago, Illinois, USA, Fax: +1 773 834 2608.
Lingjiao Zhang, The Brain Tumor Center, The University of Chicago, Chicago, Illinois, USA, Fax: +1 773 834 2608.
Dr. Brenda Auffinger, The Brain Tumor Center, The University of Chicago, Chicago, Illinois, USA, Fax: +1 773 834 2608
Alex L. Tobias, The Brain Tumor Center, The University of Chicago, Chicago, Illinois, USA, Fax: +1 773 834 2608
Dr. Esther Rincón, The Brain Tumor Center, The University of Chicago, Chicago, Illinois, USA, Fax: +1 773 834 2608
Dr. Bart Thaci, The Brain Tumor Center, The University of Chicago, Chicago, Illinois, USA, Fax: +1 773 834 2608
Dr. Atique U. Ahmed, The Brain Tumor Center, The University of Chicago, Chicago, Illinois, USA, Fax: +1 773 834 2608
Prof. Peter Warnke, Department of Surgery, The University of Chicago, Chicago, Illinois, USA
Prof. Chuan He, Department of Chemistry, Institute of Biophysics Dynamics and Howard Hughes Medical Institute, The University of Chicago, Chicago, Illinois, USA
Prof. Maciej S. Lesniak, Email: mlesniak@surgery.bsd.uchicago.edu, The Brain Tumor Center, The University of Chicago, Chicago, Illinois, USA, Fax: +1 773 834 2608.
References
- 1.Wen PY, Kesari S. N Engl J Med. 2008;359:492–507. doi: 10.1056/NEJMra0708126. [DOI] [PubMed] [Google Scholar]
- 2.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJB, Belanger K, Brandes AA, Marosi C, Bogdahn U, Curschmann J, et al. N Engl J Med. 2005;352:987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 3.Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, Ludwin SK, Allgeier A, Fisher B, Belanger K, Hau P, Brandes AA, Gijtenbeek J, Marosi C, Vecht CJ, Mokhtari K, Wesseling P, Villa S, Eisenhauer E, Gorlia T, Weller M, Lacombe D, Cairncross JG, Mirimanoff RO. Lancet Oncol. 2009;10:459–466. doi: 10.1016/S1470-2045(09)70025-7. [DOI] [PubMed] [Google Scholar]
- 4.Friedman HS, Kerby T, Calvert H. Clin Cancer Res. 2000;6:2585–2597. [PubMed] [Google Scholar]
- 5.Lesniak MS, Brem H. Nat Rev Drug Discover. 2004;3:499–508. doi: 10.1038/nrd1414. [DOI] [PubMed] [Google Scholar]
- 6.Yang CT, Chuang KH. Medchemcomm. 2012;3:552–565. [Google Scholar]
- 7.Jain RK, di Tomaso E, Duda DG, Loeffler JS, Sorensen AG, Batchelor TT. Nat Rev Neurosci. 2007;8:610–622. doi: 10.1038/nrn2175. [DOI] [PubMed] [Google Scholar]
- 8.Jain KK. Nanomedicine. 2012;7:1225–1233. doi: 10.2217/nnm.12.86. [DOI] [PubMed] [Google Scholar]
- 9.Beduneau A, Saulnier P, Benoit JP. Biomaterials. 2007;28:4947–4967. doi: 10.1016/j.biomaterials.2007.06.011. [DOI] [PubMed] [Google Scholar]
- 10.Schwarze SR, Ho A, Vocero-Akbani A, Dowdy SF. Science. 1999;285:1569–1572. doi: 10.1126/science.285.5433.1569. [DOI] [PubMed] [Google Scholar]
- 11.Ruan G, Agrawal A, Marcus AI, Nie S. J Am Chem Soc. 2007;129:14759–14766. doi: 10.1021/ja074936k. [DOI] [PubMed] [Google Scholar]
- 12.Liu L, Guo K, Lu J, Venkatraman SS, Luo D, Ng KC, Ling EA, Moochhala S, Yang YY. Biomaterials. 2008;29:1509–1517. doi: 10.1016/j.biomaterials.2007.11.014. [DOI] [PubMed] [Google Scholar]
- 13.Krpetic Z, Saleemi S, Prior IA, Sée V, Qureshi R, Brust M. ACS Nano. 2011;5:5195–5201. doi: 10.1021/nn201369k. [DOI] [PubMed] [Google Scholar]
- 14.Yuan H, Fales AM, Vo-Dinh T. J Am Chem Soc. 2012;134:11358–11361. doi: 10.1021/ja304180y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maeda H, Wu J, Sawa T, Matsumura Y, Hori K. J Control Release. 2000;65:271–284. doi: 10.1016/s0168-3659(99)00248-5. [DOI] [PubMed] [Google Scholar]
- 16.Jensen SA, Day ES, Ko CH, Hurley LA, Luciano JP, Kouri FM, Merkel TJ, Luthi AJ, Patel PC, Cutler JI, Daniel WL, Scott AW, Rotz MW, Meade TJ, Giljohann DA, Mirkin CA, Stegh AH. Sci Transl Med. 2013;5:209ra152. doi: 10.1126/scitranslmed.3006839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weintraub K. Nature. 2013;495:S14–16. doi: 10.1038/495S14a. [DOI] [PubMed] [Google Scholar]
- 18.Etame AB, Smith CA, Chan WC, Rutka JT. Nanomedicine. 2011;7:992–1000. doi: 10.1016/j.nano.2011.04.004. [DOI] [PubMed] [Google Scholar]
- 19.Wiley DT, Webster P, Gale A, Davis ME. Proc Natl Acad Sci U S A. 2013;110:8662–8667. doi: 10.1073/pnas.1307152110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Frigell J, Garcia I, Gomez-Vallejo V, Llop J, Penades S. J Am Chem Soc. 2013;136:449–457. doi: 10.1021/ja411096m. [DOI] [PubMed] [Google Scholar]
- 21.Hasenoehrl C, Alexander CM, Azzarelli NN, Dabrowiak JC. Electrophoresis. 2012;33:1251–1254. doi: 10.1002/elps.201100556. [DOI] [PubMed] [Google Scholar]
- 22.Doane TL, Cheng Y, Babar A, Hill RJ, Burda C. J Am Chem Soc. 2010;132:15624–15631. doi: 10.1021/ja1049093. [DOI] [PubMed] [Google Scholar]
- 23.Liu Y, Shipton MK, Ryan J, Kaufman ED, Franzen S, Feldheim DL. Anal Chem. 2007;79:2221–2229. doi: 10.1021/ac061578f. [DOI] [PubMed] [Google Scholar]
- 24.Perrault SD, Chan WC. Proc Natl Acad Sci U S A. 2010;107:11194–11199. doi: 10.1073/pnas.1001367107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lesniak MS, Upadhyay U, Goodwin R, Tyler B, Brem H. Anticancer Res. 2005;25:3825–3831. [PMC free article] [PubMed] [Google Scholar]
- 26.Webb BA, Chimenti M, Jacobson MP, Barber DL. Nat Rev Cancer. 2011;11:671–677. doi: 10.1038/nrc3110. [DOI] [PubMed] [Google Scholar]
- 27.Fehrenbacher N, Jaattela M. Cancer Res. 2005;65:2993–2995. doi: 10.1158/0008-5472.CAN-05-0476. [DOI] [PubMed] [Google Scholar]
- 28.Aryal S, Grailer JJ, Pilla S, Steeber DA, Gong SQ. J Mater Chem. 2009;19:7879–7884. [Google Scholar]
- 29.Liu X, Atwater M, Wang J, Huo Q. Colloids Surf B Biointerfaces. 2007;58:3–7. doi: 10.1016/j.colsurfb.2006.08.005. [DOI] [PubMed] [Google Scholar]
- 30.Lee CC, Gillies ER, Fox ME, Guillaudeu SJ, Fréchet JM, Dy EE, Szoka FC. Proc Natl Acad Sci U S A. 2006;103:16649–16654. doi: 10.1073/pnas.0607705103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Muller I, Jenner A, Bruchelt G, Niethammer D, Halliwell B. Biochem Biophys Res Commun. 1997;230:254–257. doi: 10.1006/bbrc.1996.5898. [DOI] [PubMed] [Google Scholar]
- 32.Arola OJ, Saraste A, Pulkki K, Kallajoki M, Parvinen M, Voipio-Pulkki LM. Cancer Res. 2000;60:1789–1792. [PubMed] [Google Scholar]
- 33.Raza SM, Pradilla G, Legnani FG, Thai QA, Olivi A, Weingart JD, Brem H. Expert Opin Biol Ther. 2005;5:477–494. doi: 10.1517/14712598.5.4.477. [DOI] [PubMed] [Google Scholar]
- 34.Guerin C, Olivi A, Weingart JD, Lawson HC, Brem H. Invest New Drugs. 2004;22:27–37. doi: 10.1023/b:drug.0000006172.65135.3e. [DOI] [PubMed] [Google Scholar]
- 35.De Jong WH, Hagens WI, Krystek P, Burger MC, Sips AJ, Geertsma RE. Biomaterials. 2008;29:1912–1919. doi: 10.1016/j.biomaterials.2007.12.037. [DOI] [PubMed] [Google Scholar]
- 36.Sonavane G, Tomoda K, Makino K. Colloids Surf B Biointerfaces. 2008;66:274–280. doi: 10.1016/j.colsurfb.2008.07.004. [DOI] [PubMed] [Google Scholar]
- 37.Yan H, Wang L, Wang J, Weng X, Lei H, Wang X, Jiang L, Zhu J, Lu W, Wei X, Li C. ACS Nano. 2012;6:410–420. doi: 10.1021/nn203749v. [DOI] [PubMed] [Google Scholar]
- 38.Hobbs SK, Monsky WL, Yuan F, Roberts WG, Griffith L, Torchilin VP, Jain RK. Proc Natl Acad Sci U S A. 1998;95:4607–4612. doi: 10.1073/pnas.95.8.4607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sarin H, Sanevsky KA, Wu H, Brimacombe KR, Fung SH, Sousa AA, Auh S, Wilson CM, Sharma K, Aronova MA, Leapman RD, Griffiths GL, Hall MD. J Transl Med. 2008;6:80. doi: 10.1186/1479-5876-6-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Santra, Yang SH, Stanley JT, Holloway PH, Moudgil BM, Walter G, Mericle RA. Chem Commun. 2005:3144–3146. doi: 10.1039/b503234b. [DOI] [PubMed] [Google Scholar]
- 41.Qin Y, Chen H, Zhang Q, Wang X, Yuan W, Kuai R, Tang J, Zhang L, Zhang Z, Zhang Q, Liu J, He Q. Int J Pharm. 2011;420:304–312. doi: 10.1016/j.ijpharm.2011.09.008. [DOI] [PubMed] [Google Scholar]
- 42.Liu L, Venkatraman SS, Yang YY, Guo K, Lu J, He B, Moochhala S, Kan L. Biopolymers. 2008;90:617–623. doi: 10.1002/bip.20998. [DOI] [PubMed] [Google Scholar]
- 43.Lewin M, Carlesso N, Tung CH, Tang XW, Cory D, Scadden DT, Weissleder R. Nat Biotechnol. 2000;18:410–414. doi: 10.1038/74464. [DOI] [PubMed] [Google Scholar]
- 44.Song Y, Xu X, MacRenaris KW, Zhang XQ, Mirkin CA, Meade TJ. Angew Chem Int Ed Engl. 2009;48:9143–9147. doi: 10.1002/anie.200904666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Prantner AM, Sharma V, Garbow JR, Piwnica-Worms D. Mol Imaging. 2003;2:333–341. doi: 10.1162/15353500200303106. [DOI] [PubMed] [Google Scholar]
- 46.Allen MJ, Meade TJ. J Biol Inorg Chem. 2003;8:746–750. doi: 10.1007/s00775-003-0475-2. [DOI] [PubMed] [Google Scholar]
- 47.Jokerst JV, Lobovkina T, Zare RN, Gambhir SS. Nanomedicine (Lond) 2011;6:715–28. doi: 10.2217/nnm.11.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cheng Y, Meyers JD, Agnes RS, Doane TL, Kenney ME, Broome AM, Burda C, Basilion JP. Small. 2011;7:2301–2306. doi: 10.1002/smll.201100628. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.