

Abstract
BACKGROUND
Autosomal recessive polycystic kidney disease (ARPKD) is a genetic disorder characterized by the development of renal cysts of the tubular epithelial cell origin. Epithelial Na+ channel (ENaC) is responsible for sodium reabsorption in the aldosterone-sensitive distal nephron. Here we investigated the ENaC expression and activity in cystic tissue taken from rats with ARPKD.
METHODS
PCK rats were treated with the selective ENaC inhibitor benzamil given in the drinking water, and after 4 or 12 weeks the severity of morphological malformations in the kidneys was assessed. ENaC and AQP2 expression and ENaC activity were tested with immunohistochemistry and patch clamp electrophysiology, respectively.
RESULTS
Treatment with benzamil exacerbated development of cysts compared to the vehicle-treated animals. In contrast, the 12 weeks treatment with a loop diuretic furosemide had no effect on cystogenesis. Single channel patch clamp analysis revealed that ENaC activity in the freshly isolated cystic epithelium was significantly lower than in the non-cystic collecting ducts isolated from PCK or normal Sprague-Dawley rats. Immunohistochemical analysis confirmed that β-ENaC and AQP2 expressions in cysts are decreased compared to non-dilated tubules from PCK rat kidneys.
CONCLUSION
We demonstrated that cystic epithelium exhibits low ENaC activity and this phenomenon can contribute to cyst progression.
INTRODUCTION
Polycystic kidney diseases (PKD) are a group of inherited nephropathies marked with progressive formation of fluid-filled cysts along the nephron. Autosomal recessive form of PKD (ARPKD) has an incidence of 1:20,000 to 1:40,000 and is characterized by a high mortality rate in early life (1). The cysts in ARPKD are formed by dilated collecting ducts (CD) that remain contiguous with the remaining nephron, allowing urine to continue to flow through the collecting system (2). Within this segment, Na+ is transported from the luminal space into principal cells through the apical epithelial Na+ channels (ENaC), and is extruded through the basolateral membrane in exchange for uptake of K+ by Na/K-ATPase. Proper ENaC function in the CD is the rate-limiting step for sodium reabsorption, which ultimately regulates renal sodium homeostasis and blood pressure (3,4). Disturbances of transtubular electrolyte and fluid transport are the central contributors to the process of cystogenesis (5). ENaC dysfunction associated with improper handling of NaCl has been demonstrated in renal epithelial cells from animal and human ARPKD cell lines (6–9). However, conflicting data have been reported, and it is still unclear how ENaC-mediated sodium transport in the CDs contributes to the development of cysts. In this study we focused on the role of ENaC-mediated sodium reabsorption in the orthologous PCK rat model of ARPKD (PCK/CrljCrl-Pkhd1pck/CRL) (10). A combination of electrophysiological, histochemical and chronic studies in vivo and ex vivo was used here to investigate how ENaC contributes to the development of ARPKD.
RESULTS
Role of ENaC-mediated sodium transport in the development of cysts
Amiloride and its derivatives (e.g. benzamil) have been long used as potassium-sparing diuretics that block sodium reabsorption through ENaC, and thus have potent antihypertensive effects, especially in such states of ENaC activation as in Liddle's syndrome (4). Here we used PCK rats to investigate the effects of benzamil at different stages of the disease to determine whether ENaC inhibitors modulate cyst formation. Benzamil (15 mg/L) was administered in drinking water to 4 week old PCK rats for either 4 or 12 weeks. As demonstrated by our data, benzamil aggravates cyst formation in PCK rats (Figure 1). After 4 weeks of treatment, kidney/body weight ratio was not different between the groups, whereas by week 12, benzamil-treated rats developed severe renal hypertrophy as measured by kidneys/total body weight ratio. Cyst progression in benzamil-treated rats was significantly higher than in the control groups after both 4 and 12 weeks of treatment. Therefore, we conclude that ENaC-mediated sodium absorption plays a critical role in cystogenesis in ARPKD.
Figure 1.

Inhibition of ENaC results in the progression of cyst formation in PCK rats. (a) Morphology of the kidneys collected from PCK rats after 4 or 12 weeks treatment with benzamil (benza) or vehicle (control). Scale bar is 5 mm. (b) Kidney weight/total body weight (TBW) ratios were calculated and graphed as a percentage. (c) Cysts area, % of total slice. Kidney sections were morphometrically analyzed as described in Materials and Methods to determine the surface area of the remaining kidney tissue relative to the cyst space. N = 6 rats in each group. *p<0.05 vs vehicle-control.
Furosemide, a loop diuretic which acts by inhibiting NKCC2 activity in the thick ascending limb, was used to test if the enlargement of cysts was specific to ENaC inhibition. In this work performed in independent groups of animals we observed relatively higher cyst size in control groups compared to the benzamil experiment control group. Most likely, this difference is due to variability of phenotype between breeder families. However, the animals were distributed between control and experimental groups randomly and statistically, control cyst area values in benzamil and furosemide experiments were homogeneous. Since we did not compare them, we decided the effect of furosemide can be evaluated as an independent experiment, although we realize that individual background of these litters could affect sensitivity to furosemide. As shown in Figure 2, treatment with furosemide (15 mg/L) given in drinking water did not produce any changes in the rate of cyst progression, nor did it change the kidney/body weight ratio. Therefore, we showed that this effect is specific to inhibition of ENaC, supporting the idea that this channel is an important participant of the CD dilation mechanism in the ARPKD.
Figure 2.

Treatment with furosemide has no effect on cyst formation in PCK rats. (a) Morphology of the kidneys collected from PCK rats after 12 weeks treatment with furosemide (furo). Scale bar is 5 mm. (b) Kidneys weight % of total body weight. (c) Cysts area, % of total slice. N = 6 rats in each group.
ENaC and AQP2 abundance in PCK rat kidneys
To test if there is abnormal salt and water reabsorption in cystic epithelium we initially characterized expression of β-ENaC subunit and aquaporin-2 (AQP2) water channel in the PCK rats. Immunohistochemical analysis was carried out on the 16 week old PCK rats to show protein localization and distribution in the kidney. The experiments revealed that the expression of β-ENaC and AQP2 was significantly lower in cysts compared to non-cystic tubules (Figure 3). Also, intracellular distribution of signal in the cystic epithelium was more diffused, polarity of the protein localization was disturbed, visual difference between primary and intercalated cells was indistinct and the epithelial layer seemed dedifferentiated. Thus we hypothesized from these data that in the cysts ENaC and AQP2 expression and activity are abnormally downregulated, and this subsequently leads to fluid accumulation. An additional study revealed that the 12-week benzamil treatment did not change the expression and distribution of ENaC only blocking ion permeation through the channel (Figure 4).
Figure 3.

ENaC and AQP2 abundance in PCK rat kidneys. Images of the representative immunohistochemical stainings for β-ENaC subunit (a) and AQP2 (b) in a 16-wks old PCK rat kidney. Magnification – 40x; scale bar is 50 μm. CD –collecting ducts. Summary graphs demonstrate individual and average signal intensities in the analyzed tubules and cysts. Circles, crosses and triangles denote three rats used for the study. **p<0.001.
Figure 4.
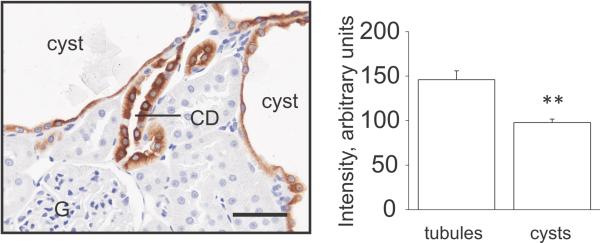
Lack of an effect of benzamil treatment on β-ENaC abundance and distribution. After the experiment with 12-weeks benzamil treatment immunohistochemical analysis of renal β-ENaC expression was performed. Magnification – 40x; scale bar is 50 μm. CD – collecting duct, G-glomerulus. **p<0.001.
ENaC activity in cystic epithelia
Next, evaluation of ENaC activity was performed with single channel patch-clamp analysis of CDs epithelium freshly isolated from 16 week old PCK and Sprague-Dawley (SD) rats. PCK strain was originally derived from a spontaneous mutation in SD rats (10); therefore, SD rats represent an appropriate additional control for comparison to PCK rats. Cystic epithelial layers and non-cystic collecting ducts were manually isolated from the kidneys and split open so that the apical membrane of the principal cells was available for cell-attached seals on the membrane. This approach enabled us to directly monitor ENaC single channel activity in real time. Figure 5a shows single channel ENaC recordings in the CDs isolated from SD rats and non-dilated and cystic tubules of PCK rats. Data summarized in Figure 5b clearly indicates that ENaC activity is much lower in the cyst cells compared to the non-dilated tubules of PCK rats and CDs isolated from SD rats. Similarly, Veizis et al. showed decreased ENaC-mediated sodium reabsorption in the collecting duct principal cells isolated from BPK mice (a model for ARPKD), and hypothesized that such reduction in sodium transport contributes to accumulation of luminal fluid in PKD cysts (7).
Figure 5.

ENaC activity in cystic and non-dilated tubules. (a) Representative current traces for ENaC activity measured in cell attached patches in freshly isolated non-cystic CDs and cysts of 16 week old SD or PCK rats. These patches were held at a test potential of Vh = –Vp = −40 mV. Inward Li+ currents are downward. Dashed lines indicate the respective current state with a “c” and “on” denoting the closed and open states. (b) Summary graph of observed ENaC activity (NPo). N = 6–8, recorded from 3–4 tubules or cysts isolated from different rats per group; number of patches acquired from an animal was 1–4. *P<0.05 versus PCK tubules; † P<0.05 versus SD tubules.
DISCUSSION
Processes that trigger formation of PKD cysts remain in the focus of many researchers. Cyst initiation and expansion is a complex process characterized by abnormalities in tubular cell proliferation, fluid secretion, extracellular matrix formation, apical or basolateral membrane-specific distribution of various proteins, and imbalance between cellular proliferation and apoptosis (1). We investigated impaired sodium reabsorption in the dilated collecting ducts, a new mechanism that contributes to cyst growth.. Earlier Rohatgi and colleagues (11) analyzed cyst fluid composition from ARPKD nephrectomy specimens at the time of renal transplantation revealing a low Na+ concentration and osmolality similar to that of serum. The authors hypothesize that ARPKD cysts are not secretory epithelia and the low Na+ concentration could be explained by enhanced collecting duct Na+ absorption or intact Na+ absorptive pathways upstream of the dilated collecting duct. The current study investigated if ENaC-mediated sodium reabsorption in the cyst wall is altered and involved in cyst progression. Our experiments revealed that dilated tubules exhibit low activity and abundance of such important proteins for proper reabsorption as ENaC that it leads to fluid accumulation in the cysts. Aquaporin-2 staining data presented on Figure 3 serve as additional indirect evidence of impaired fluid reabsorption in the cystic epithelium; however further studies are required to speculate on the specific mechanism. Treatment with benzamil confirms that blockade of ENaC-mediated sodium reuptake in the nephron aggravates cyst formation. Furosemide, a loop diuretic inhibiting NKCC2 transporters, had no effect on cyst expansion indicating that improper sodium reabsorption, specifically via ENaC, is critical for these CD malformations.
Molecular mechanisms involved in the reduced ENaC activity in cystic epithelium require extensive studies to determine potential therapeutic targets. A large panel of research is focused on identification of epidermal growth factor (EGF) family proteins as important signaling molecules in the development of ARPKD (9,12–17) and other childhood diseases (18). It was recently shown that TRPP2 and TRPV4 form an EGF activated calcium permeable channel at the apical membrane of renal collecting duct cells and this pathway may play a role in polycystic kidney disease (19). Previous data reveal that EGF chronically downregulates ENaC activity in vitro (20,21) and in vivo (22). EGF receptors' signaling is abnormal in the cystic CDs, and this could potentially contribute to the ENaC-dependent cysts progression in ARPKD (12). Another mechanism that might potentially lower ENaC activity in dilated collecting ducts is mediated via purinergic signaling. It is well established that ATP decreases ENaC activity in CD principal cells via P2Y2 receptors (23). Extracellularly, PKD cysts accumulate up to 10 μM of ATP, (24,25) which is unusually high compared to healthy kidney interstitial ATP levels (400 nM) (26). Growing evidence suggests that autocrine and paracrine effects of purinergic signaling via P2 receptors could detrimentally contribute to cyst expansion (27–29); P2 receptors antagonists were shown to have therapeutic potential in PKD (27).
Findings reported here are of clear clinical significance, as the deteriorative effects of benzamil on cyst formation should be taken into consideration during selection of diuretics for ARPKD treatment. However, it is unclear whether this mechanism is specific for ARPKD or common for other forms of PKD. Thus, role of ENaC in ADPKD requires further studies. Furthermore, understanding the linkage between electrolytes and water excretion is important in evaluating any potential medication for PKD treatment. For example, vasopressin receptor antagonists demonstrated high promise in the treatment of PKD (30), and it is known that vasopressin signaling is critical for control of water electrolyte homeostasis including ENaC-mediated sodium absorption. For instance, it was recently shown that adenylyl cyclase VI, which is a key mediator of cyst formation and renal injury (31), mediates vasopressin-stimulated ENaC activity (32). In summary, our study provides the evidence demonstrating critical role of ENaC-mediated signaling mechanisms in the development of cysts in ARPKD. Thus, agonists of ENaC or signaling molecules involved in appropriate regulation of ENaC-mediated sodium balance could be potential targets for treatment of PKD and other possible proliferative diseases.
MATERIALS AND METHODS
Animals
PCK and SD rats were purchased from Charles River. At the age of 4 weeks (right after weaning) PCK rats were given 15 mg/L benzamil (Sigma-Aldrich, St. Louis, MO), 15 mg/L furosemide (Sigma-Aldrich) or vehicle in drinking water (ad libitum). After 4 or 12 week treatments the rats were sacrificed and their kidneys were collected to perform histological and patch-clamp analysis. Animal use and welfare procedures adhered to the NIH Guide for the Care and Use of Laboratory Animals following protocols reviewed and approved by the Medical College of Wisconsin Institutional Animal Care and Use Committee.
Histological and Immunohistochemical Analysis
Isolated kidneys were fixed in 40% Formalin and routinely embedded, cut at 4 μm slices, dried and deparaffinized for subsequent histochemistry. Hematoxylin and Eosin (H&E) staining was used to assess kidney morphology. The kidney slices were scanned with Nikon (Melville, NY) Super CoolScan 9000 interfaced with NikonScan 4 software. Further analysis of cyst area was performed using color threshholding method using Metamorph (Molecular Devices, Sunnyvale, CA) software as previously described (33). For immunohistochemistry, tissue sections were incubated with anti-β-ENaC antibodies (StressMarq, Victoria, Canada) or anti-AQP2 antibodies (Santa Cruz Biotechnology, Dallas, TX). Secondary detection was performed with goat anti-rabbit biotinylated IgG (Biocare, Concord, CA) followed by streptavidin horseradish peroxidase (Biocare) and visualized with DAB (DAKO). All slides were counterstained with a Mayer hematoxylin (DAKO, Carpinteria, CA), dehydrated, and mounted with permanent mounting media (Sakura, Torrance, CA).
Captured images were analyzed with ImageJ software (NIH, Bethesda, MD) to measure signal intensities (arbitrary units, ranged from 0 to 255) in tubules and cysts. Intensities from 7–18 individual tubules or cysts were collected each animal; total number of animals per group was three.
Electrophysiology
Patch clamp analysis was used to assess ENaC activity in isolated split-open cystic epithelia or non-dilated CD tubules. CDs from Sprague-Dawley rats, cystic epithelia and non-cystic CDs from PCK rats were manually isolated with forceps in ice-cold saline under a stereomicroscope. CD-derived cyst monolayers were gently teased from open cyst cavities in kidney slices from PCK rats and further mechanically separated from surrounding tissue. Isolated tissues were attached to 5×5 mm coverglass coated with poly-L-lysine (Sigma-Aldrich). CDs were split-opened with two sharpened micropipettes, and controlled with micromanipulators to gain access to the apical membrane. Single-channel data were acquired and subsequently analyzed with Axopatch 200B or 700B amplifiers interfaced via a Digidata 1440A to a PC running the pClamp 10.2 suite of software (Molecular Devices). When signals are acquired with an Axopatch 200B amplifier currents were filtered with an 8-pole, low pass Bessel filter LPF-8 (Warner Inst., Hamden, CT) at 0.2 kHz. The pipette was pulled with a P-97 puller (Sutter, Novato, CA). The resistance of the pipette in the corresponding bath medium was 7–12 megohms. Bath solution consisted of (in mM): 150 NaCl, 1 CaCl2, 2 MgCl2, 10 HEPES (pH 7.4). Pipette solution for cell attached configuration contained (in mM): 140 LiCl, 2 MgCl2 and 10 HEPES (pH 7.4). Gap-free single channel current data from gigaohm seals in principal cells were acquired and subsequently analyzed. The channel events were analyzed by Clampfit 10.2 software (Molecular Devices) as previously described (22). Channel activity was calculated as NPo where N refers to the number of channels in the patch and Po characterizes their open-probability.
Statistical Analysis
All data are presented as mean ± SEM. The data were analyzed using a t-test and Mann–Whitney U-test. P<0.05 was considered to indicate a statistically significant difference.
Acknowledgments
Statement of Financial Support: This study was supported by the National Heart, Lung, and Blood Institute grants R01 HL108880 (to AS) and K99 HL116603 (to TSP), National Kidney Foundation IG1724 and MCW RAC Pilot Grant (to TSP), and the Ben J. Lipps Research Fellowship from the American Society of Nephrology (to DVI).
Footnotes
DISCLOSURES There are no competing financial, personal, or professional interests for any of the authors.
REFERENCES
- 1.Sweeney WE, Jr., Avner ED. Pathophysiology of childhood polycystic kidney diseases: new insights into disease-specific therapy. Pediatr Res. 2014;75:148–57. doi: 10.1038/pr.2013.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wilson PD. Polycystic kidney disease. N Engl J Med. 2004;350:151–64. doi: 10.1056/NEJMra022161. [DOI] [PubMed] [Google Scholar]
- 3.Staruschenko A. Regulation of transport in the connecting tubule and cortical collecting duct. Compreh Physiol. 2012;2:1541–84. doi: 10.1002/cphy.c110052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rossier BC, Staub O, Hummler E. Genetic dissection of sodium and potassium transport along the aldosterone-sensitive distal nephron: Importance in the control of blood pressure and hypertension. FEBS Letters. 2013;587:1929–41. doi: 10.1016/j.febslet.2013.05.013. [DOI] [PubMed] [Google Scholar]
- 5.Avner ED, Sweeney WE, Jr., Nelson WJ. Abnormal sodium pump distribution during renal tubulogenesis in congenital murine polycystic kidney disease. Proc Natl Acad Sci USA. 1992;89:7447–51. doi: 10.1073/pnas.89.16.7447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Olteanu D, Yoder BK, Liu W, et al. Heightened epithelial Na+ channel-mediated Na+ absorption in a murine polycystic kidney disease model epithelium lacking apical monocilia. Am J Physiol Cell Physiol. 2006;290:C952–C63. doi: 10.1152/ajpcell.00339.2005. [DOI] [PubMed] [Google Scholar]
- 7.Veizis EI, Carlin CR, Cotton CU. Decreased amiloride-sensitive Na+ absorption in collecting duct principal cells isolated from BPK ARPKD mice. Am J Physiol Renal Physiol. 2004;286:F244–F54. doi: 10.1152/ajprenal.00169.2003. [DOI] [PubMed] [Google Scholar]
- 8.Rohatgi R, Greenberg A, Burrow CR, Wilson PD, Satlin LM. Na transport in autosomal recessive polycystic kidney disease (ARPKD) cyst lining epithelial cells. J Am Soc Nephrol. 2003;14:827–36. doi: 10.1097/01.asn.0000056481.66379.b2. [DOI] [PubMed] [Google Scholar]
- 9.Veizis IE, Cotton CU. Abnormal EGF-dependent regulation of sodium absorption in ARPKD collecting duct cells. Am J Physiol Renal Physiol. 2005;288:F474–F82. doi: 10.1152/ajprenal.00227.2004. [DOI] [PubMed] [Google Scholar]
- 10.Katsuyama M, Masuyama T, Komura I, Hibino T, Takahashi H. Characterization of a novel polycystic kidney rat model with accompanying polycystic liver. ExpAnim. 2000;49:51–5. doi: 10.1538/expanim.49.51. [DOI] [PubMed] [Google Scholar]
- 11.Rohatgi R, Zavilowitz B, Vergara M, Woda C, Kim P, Satlin LM. Cyst fluid composition in human autosomal recessive polycystic kidney disease. Pediatr Nephrol. 2005;20:552–3. doi: 10.1007/s00467-004-1728-1. [DOI] [PubMed] [Google Scholar]
- 12.Zheleznova NN, Wilson PD, Staruschenko A. Epidermal growth factor-mediated proliferation and sodium transport in normal and PKD epithelial cells. Biochim Biophys Acta. 2011;1812:1301–13. doi: 10.1016/j.bbadis.2010.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ryan S, Verghese S, Cianciola NL, Cotton CU, Carlin CR. Autosomal recessive polycystic kidney disease epithelial cell model reveals multiple basolateral epidermal growth factor receptor sorting pathways. Mol Biol Cell. 2010;21:2732–45. doi: 10.1091/mbc.E09-12-1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wilson SJ, Amsler K, Hyink DP, et al. Inhibition of HER-2(neu/ErbB2) restores normal function and structure to polycystic kidney disease (PKD) epithelia. Biochim Biophys Acta. 2006;1762:647–55. doi: 10.1016/j.bbadis.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 15.Wilson PD. Apico-basal polarity in polycystic kidney disease epithelia. Biochim Biophys Acta. 2011;1812:1239–48. doi: 10.1016/j.bbadis.2011.05.008. [DOI] [PubMed] [Google Scholar]
- 16.MacRae DK, Nemo R, Sweeney WE, Jr., Avner ED. EGF-related growth factors in the pathogenesis of murine ARPKD. Kidney Int. 2004;65:2018–29. doi: 10.1111/j.1523-1755.2004.00623.x. [DOI] [PubMed] [Google Scholar]
- 17.Nemo R, Murcia N, Dell KM. Transforming growth factor alpha (TGF-alpha) and other targets of tumor necrosis factor-alpha converting enzyme (TACE) in murine polycystic kidney disease. Pediatr Res. 2005;57:732–7. doi: 10.1203/01.PDR.0000159513.51898.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Frey MR, Brent Polk D. ErbB receptors and their growth factor ligands in pediatric intestinal inflammation. Pediatr Res. 2014;75:127–32. doi: 10.1038/pr.2013.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang ZR, Chu WF, Song B, et al. TRPP2 and TRPV4 form an EGF-activated calcium permeable channel at the apical membrane of renal collecting duct cells. PLoS ONE. 2013;8:e73424. doi: 10.1371/journal.pone.0073424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Levchenko V, Zheleznova NN, Pavlov TS, Vandewalle A, Wilson PD, Staruschenko A. EGF and its related growth factors mediate sodium transport in mpkCCDc14 cells via ErbB2 (neu/HER-2) receptor. J Cell Physiol. 2010;223:252–9. doi: 10.1002/jcp.22033. [DOI] [PubMed] [Google Scholar]
- 21.Liu L, Duke BJ, Malik B, Yue Q, Eaton DC. Biphasic regulation of ENaC by TGF-{alpha} and EGF in renal epithelial cells. Am J Physiol Renal Physiol. 2009;296:F1417–F27. doi: 10.1152/ajprenal.90337.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pavlov TS, Levchenko V, O'Connor PM, et al. Deficiency of renal cortical EGF increases ENaC activity and contributes to salt-sensitive hypertension. J Am Soc Nephrol. 2013;24:1053–62. doi: 10.1681/ASN.2012080839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pochynyuk O, Bugaj V, Rieg T, et al. Paracrine regulation of the epithelial Na+ channel in the mammalian collecting duct by purinergic P2Y2 receptor tone. J Biol Chem. 2008;283:36599–607. doi: 10.1074/jbc.M807129200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wilson PD, Hovater JS, Casey CC, Fortenberry JA, Schwiebert EM. ATP release mechanisms in primary cultures of epithelia derived from the cysts of polycystic kidneys. J Am Soc Nephrol. 1999;10:218–29. doi: 10.1681/ASN.V102218. [DOI] [PubMed] [Google Scholar]
- 25.Schwiebert EM, Wallace DP, Braunstein GM, et al. Autocrine extracellular purinergic signaling in epithelial cells derived from polycystic kidneys. Am J Physiol Renal Physiol. 2002;282:F763–75. doi: 10.1152/ajprenal.0337.2000. [DOI] [PubMed] [Google Scholar]
- 26.Palygin O, Levchenko V, Ilatovskaya DV, et al. Real-time electrochemical detection of ATP and H2O2 release in freshly isolated kidneys. Am J Physiol Renal Physiol. 2013;305:F134–41. doi: 10.1152/ajprenal.00129.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chang MY, Lu JK, Tian YC, et al. Inhibition of the P2×7 receptor reduces cystogenesis in PKD. J Am Soc Nephrol. 2011;22:1696–706. doi: 10.1681/ASN.2010070728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schwiebert EM. ATP release mechanisms, ATP receptors and purinergic signalling along the nephron. Clin Exp Pharmacol Physiol. 2001;28:340–50. doi: 10.1046/j.1440-1681.2001.03451.x. [DOI] [PubMed] [Google Scholar]
- 29.Schwiebert EM, Kishore BK. Extracellular nucleotide signaling along the renal epithelium. Am J Physiol Renal Physiol. 2001;280:F945–F63. doi: 10.1152/ajprenal.2001.280.6.F945. [DOI] [PubMed] [Google Scholar]
- 30.Torres VE, Harris PC. Strategies Targeting cAMP Signaling in the Treatment of Polycystic Kidney Disease. J Am Soc Nephrol. 2014;25:18–32. doi: 10.1681/ASN.2013040398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rees S, Kittikulsuth W, Roos K, Strait KA, Van Hoek A, Kohan DE. Adenylyl Cyclase 6 Deficiency Ameliorates Polycystic Kidney Disease. J Am Soc Nephrol. 2014;25:232–7. doi: 10.1681/ASN.2013010077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Roos KP, Bugaj V, Mironova E, et al. Adenylyl Cyclase VI Mediates Vasopressin-Stimulated ENaC Activity. J Am Soc Nephrol. 2013;24:218–27. doi: 10.1681/ASN.2012050449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kwon M, Pavlov TS, Nozu K, et al. G-protein signaling modulator 1 deficiency accelerates cystic disease in an orthologous mouse model of autosomal dominant polycystic kidney disease. Proc Natl Acad Sci U S A. 2012;109:21462–7. doi: 10.1073/pnas.1216830110. [DOI] [PMC free article] [PubMed] [Google Scholar]