

Abstract
Background
Proper migration of neurons is essential for the formation and normal functioning of the nervous system. Defects in neuronal migration underlie a number of neurologic diseases in humans. Although cell migration is crucial for neural development, molecular mechanisms guiding neuronal migration remain to be elucidated fully. Newborn neurons from the embryonic medial ganglionic eminence (MGE) migrate a long distance dorsally in the developing brain, giving rise to several types of interneurons in the neocortex.
New Method
In this study, we developed an immunocytochemistry (ICC) protocol to stain neurons migrating out of the MGE explant embedded in Matrigel. We also established a protocol to efficiently transfect cells in MGE explants, achieving a transduction efficiency of more than 30%.
Comparison with Existing Method
In addition, we developed microfluidic chambers for explants that allow visualization of the vectorial migration of individual neurons from mouse embryonic MGE explants. Our microfluidic system allows monitoring of the distribution of cellular organelles (e.g. Golgi) within migrating neurons which have been stained with commercial molecular dyes or transfected with adeno-associated virus (AAV) expressing reporter proteins.
Conclusion
These methods provide new paradigms to study neuronal migration in real-time.
Keywords: neuron migration, MGE explant, microfluidic device, transduction
1. Introduction
Neurological syndromes caused by disruption of neuronal migration can be devastating human disorders, which are mostly due to genetic causes (for an extensive review see reviews Reiner et al., 2013; Valiente and Marın, 2010). Studies have shown that disruption of cytoskeleton underlies a large spectrum of neuronal migration disorders (Métin et al., 2008). Migration defects of cortical interneurons can cause abnormal inhibitory responses in the developing cortex (Valiente and Marin, 2010). Ganglionic eminence (GE) neurons move tangentially and across the plane of the glial fiber system during brain development (Marín and Rubenstein, 2001). These GE-derived neurons contribute significantly to the GABAergic cortical cell population (Hernández-Miranda et al., 2010). The gamma amino butyric acid (GABA) interneurons must be produced and deployed to the cortex during fetal life in order for the neural circuits to function properly in the mature brain. Many GABA neurons migrate tangentially to reach their final destination. Tangential migration of GABA neurons is widespread in the embryonic rodent brain (Parnavelas et al., 2000), and three major routes of such migration have been recently characterized in detail: (1) from the medial GE (MGE) to the cortex; (2) from anterior entopeduncular area (AEP) to the neocortex and hippocampus; and (3) from the lateral GE (LGE) to the olfactory bulb.. This latter migratory route persists into adulthood as the rostral migratory stream. In this study we have focused on MGE neurons because they have a unique migratory potential, and may have applications in cell replacement therapies in certain neurodegenerative diseases. MGE neurons are the only primary neuronal precursors known to be able to disperse when grafted into the adult brain (Wichterle et al., 1999).
Here, we describe novel methods to study MGE migration, including a microfluidic chamber to culture MGE explants. Our methods may open new avenues for informative neurobiological investigations. An advantage of the neurotechnology system we have developed is that, once validated, it could be used for screening of drugs, which can affect neuronal migration. In addition, immunocytochemical (ICC) staining methods can be used to detect changes in the distribution of proteins thought to contribute to migration defects, such as DISC1, Lis1, and Ndel1 (Richter et al., 1999), and be used to express mutant proteins involved in neurological diseases instead of using transgenic animal models.
2. Materials and methods
2.1. Fabrication of the microfluidic microchambers
To probe the migration of neurons from a MGE explant towards a naturally chemotrophic cortical explant, we designed a microfluidic device with two compartments connected by an array a small channels. The two compartments, in the form of circular wells of 1.5 mm diameter and 3 mm depth were filled with gel matrix. Also filled with gel was the parallel array of microchannels of 25 µm width, 10 µm height, and average 150 µm length, connecting the two compartments. The cross section of the channels was chosen such that the migration and mechanical orientation of individual neurons could be recorded by time lapse microscopy. The device was fabricated by directly bonding polydimethylsiloxane (PDMS - Dow Corning Midland, MI) on glass coverslips, inside glass-bottom Petri-dishes (MatTek, Ashland, MA) after exposing the surfaces to be bonded to oxygen plasma for 30 seconds. The PDMS piece was cast on a microfabricated silicon wafer, to produce a replica of the photolithographic features on the wafer, baked for 12 hours at 65°C and punched using a 1.5mm puncher (Harris Uni-Core, Ted Pella, Reading, CA) to define the two tissue compartments. The geometry of the array of microchannels and their connection to the two compartments was precisely defined using photolithography techniques, in a 10 µm thick layer of SU-8 photoresist (Microchem, Newton, MA). Immediately after fabrication, devices are kept on ice and filled with the liquid Matrigel (BD Biosciences, Franklin Lakes, NJ), which is then allowed to gel at room temperature, and covered with Neurobasal medium (BD Biosciences).
2.2. Animals
C57BL/6 breeding pairs were obtained from Charles River Laboratories (Wilmington, MA). C57BL/6 matings were set up and the females monitored for vaginal plugs. The day vaginal plug was observed was considered embryonic day 0 (E0), and the male was separated from the female. The pregnant dams were housed in a temperature- and humidity-controlled environment on a 12-hour light/dark cycle with food and water available ad libitum.
2.3. Embryo collection and processing
For collection of embryos, the dams were anesthetized by intraperitoneal (i.p.) injections of a mixture of ketamine (50 mg/kg body weight) and xylazine (10 mg/kg body weight) and the embryos were removed by hysterotomy. The embryos were collected at E15 and decapitated immediately upon removal from the dam, as described previously (McCarthy et al., 2012).
2.4. Analysis of neuronal migration in MGE explants
Slice preparations were prepared from brains of E15 embryos as described with modifications (Crandall et al., 2007; McCarthy et al., 2011; McCarthy et al., 2012). Briefly, embryonic brains were removed and placed in 8% agarose (type VII; Sigma-Aldrich, St. Louis, MO). Coronal sections were cut at a thickness of 250 µm using a Vibratome. A sterile biopsy punch (Miltex Inc., York, PA) was used to collect disks of MGE from the vibratome sections (McCarthy et al., 2011). The majority of the neurons produced in the MGE are migratory neurons destined to become cortical GABA neurons (Butt et al., 2005; Rallu et al., 2002; Anderson et al., 2001). The disk explants from MGE additional punch-out from the cortical region were collected from the same embryonic slice. For staining experiments, MGE explants were cultured in 50 µl Matrigel covered with Neurobasal medium containing 2% B27 supplement, 0.25% glutamine, 0.0125% glutamate, 1× penicillin/streptomycin in glass bottom culture dishes (BD Biosciences). Following 48 hours in culture, the explants were fixed with 4% paraformaldehyde (PFA) for 1 hour, and stored in 1× Phosphate Buffered Saline (PBS) buffer at 4°C until ICC was performed. For microfluidic devices experiments, both wells were filled with Matrigel and covered with Neurobasal medium, described above. MGE explant was placed in one of the wells and a cortical explant placed in the other well.
2.5. Immunocytochemistry (ICC) of MGE explants
PFA fixed explants were treated with warm (37°C) 1× PBS for 90 sec (three 30 second incubations with warmed PBS), followed by incubation with 0.1% Triton X-100, 10% goat serum (Jackson Immuno Research) in 1× PBS for 1 hour. After washing 3 times for 10 min with 1× PBS, the samples were incubated overnight at 4°C with primary antibodies to βIII-tubulin and pericentrin (βIII-tubulin mouse monoclonal antibody, MAB55441, 1:1,500; EMD Millipore, Billerica, MA; pericentrin rabbit polyclonal antibody, PRB-432C, 1:800; Covance, Dedham, MA) to reveal the processes and of the centrosome respectively in migrating cells. The next day, explants were washed three times with 1× PBS and incubated at room temperature for 1 hour with secondary antibodies - anti-mouse Alexa 488 (1:1,000) or anti-rabbit Alexa 555 (1:800) (Invitrogen/Life Technologies, Grand Island, NY). Nuclei were labeled with ToPro3 (1:1,000; Invitrogen/Life Technologies, Grand Island, NY) by incubation for 15 min.
2.6. Lentiviral (LV) and adeno-associated virus (AAV) vectors and MGE transfection
Lentiviral vectors [LV-CMV-GFP (green fluorescent protein); Sena-Esteves et al., 2004] and AAV vectors (AAV-CBA-GFP; Broekman et al., 2006) packaged with serotype 8, 9, or rh10 capsids were kindly provided by Dr. Sena-Esteves (UMass Medical School, Worcester, MA). Viral titers of 5 × 107 transducing units (t.u.)/ml for lentiviral vectors and 5 × 108 genome copies (g.c.)/ml for AAV vectors were used to infect explants. For transduction with viral vectors, MGE explants were placed in 96-well plates and treated with 50 µl lentivirus or 5 µl AAV in 200 µl Neurobasal media containing 10 mM HEPES buffer, pH 7 for 1 hour. To improve the infection efficiency, samples were centrifuged at 1000 × g for 30 minutes in the 96-well plates. After centrifugation, the explants were placed in Matrigel and cultured, as described above.
2.7. Cell organelles labeling
Viable MGE explants were stained with Hoechst 33258 and BODIPY FL C5-ceramide dyes (both from Molecular Probe Inc., Eugene, OR) according to the manufacturer’s instructions. BODIPY FL C5-ceramide localizes to the Golgi apparatus, and Hoechst 33258 dye stains the nuclei.
2.8. Microscopy
Confocal images were acquired using an Olympus FluoView 1000 confocal microscope with independent photostimulation and imaging channels. This microscope has a variety of lasers covering the visible and near infrared spectrum. It also has a tunable two-photon source for reduced photodynamic damage, as well as DIC optics and a photomultiplier collector for transmitted light imaging. There are two sets of objectives, both water immersions: one set corrected for a coverslip and one set for direct immersion. There is a culture chamber attachment that surrounds the stage, which maintains temperature and CO2 levels and the acquisition software can be programmed for time-lapse acquisition. The current confocal laser lines available are 405, 488, 514, 561, and 635 nm. The 2 photon is a chameleon hidden photons (HP), with a range of 690-1020 nm and peak power of 3 Watts.
3. Results and discussion
3.1. Immunocytochemistry (ICC) of non-dissociated MGE explants embedded in Matrigel
MGE explants were cultured in Matrigel and placed in: 1) glass bottom culture dishes, or 2) microfluidic chambers (described below). Neurons migrated out from MGE explants in a symmetrical fashion over 48 hours (Fig. 1 C–E). By fixing and co-staining the cells with the neuronal microtubular marker, βIII-tubulin, the centromere marker, pericentrin, and the nuclear dye, ToPro3, we were able to visualize the morphology of individual neurons (Fig. 1E). This ICC method of analyzing neurons, which have migrated out of the MGE explants can be used to monitor alterations in neuronal morphology and expression of endogenous proteins/or receptors in neurons. Also, this assay can be used as a tool to compare the distribution of the neurons as a function of distance from the edge of the explant, as related to different genotypes (McCarthy et al., 2012), or to record epileptiform discharges in organotypic cultures of cortex and hippocampus (Berdichevsky et al., 2010).
Fig. 1. Experimental models to study MGE neurons migration from the explants of mouse embryos.

A timed-pregnant female mice was sacrificed and brains of the E15 embryos isolated, as described in the methods (1 A). The MGE explants were dissected out from 250 µm coronal Vibratome slices of brains (1 B). MGE explants were placed in a glass bottom culture dish containing 10 µl of Matrigel (1 C). Neurons migrated out of the MGE explant in a symmetrical and radial way, shown here after 48 hours (1 D). The explants were fixed and neurons in Matrigel were co-stained with the neuronal marker βIII-tubulin (red), the centromere marker pericentrin (green) and the nuclear marker ToPro3 (blue), shown at low magnification 20× and high magnification 100×) (1 E). Based on the experimental model schematized on the right (2), an MGE explant was placed in one of the wells of a microfluidic device and a cortical explant placed in the other well (1 F). Both wells were filled with Matrigel and covered with Neurobasal medium. Neurons migrated away from MGE explant within microchannels filled with Matrigel towards the cortical explant in the opposite well (1 G–I). Images of a neuron that migrated out of the explant inside of the microfluidic chambers are shown (1 I). The direction of migration is indicated by the white arrow.
3.2. Loading and culturing MGE explants in the microfluidic chambers
MGE explants were isolated from brains of E15 mice embryos as previously described. MGE explants previously infected with AAV or lentivirus, or labeled with specific dyes, were placed in the micro-wells (Fig. 1F). Explants were allowed to attach to the Matrigel for 10 min at 37°C, after which Neurobasal medium containing 2% B-27 supplement, penicillin, streptomycin and glutamine, was added to the microfluidic system. Following 48 hours in culture, the neurons migrating out of the explant into the microchambers (Fig. 1 G–I) were examined under a microscope using phase-contrast optics and by time-lapse image.
3.3. Adeno-associated virus (AAV) transduce MGE explants more efficient than lentivirus
In this study, we developed a protocol to transduce MGE explants with high efficiency. We compared transduction efficacy of AAV serotypes (AAVrh10, AAV 8, and AAV 9) and LV. We packaged AAV-CBA-GFP vector in cells transfected with expression plasmids for the following capsids: AAVrh10 (Fig. 2 A–B), AAV 8 (Fig. 2C), or AAV 9 (Fig. 2D), and lentiviral vector (LV-CMV-GFP) with an expression construct for vesicular stomatitis virus glycoprotein (VSV-G) (Fig. 2C). All AAV and LV vectors demonstrated GFP expression by transduction of MGE explant within 72 hours, but with different expression levels. Highest expression was detected with AAVrh10 while lowest was with LV.
Fig. 2. Transduction and labeling of MGE explants.
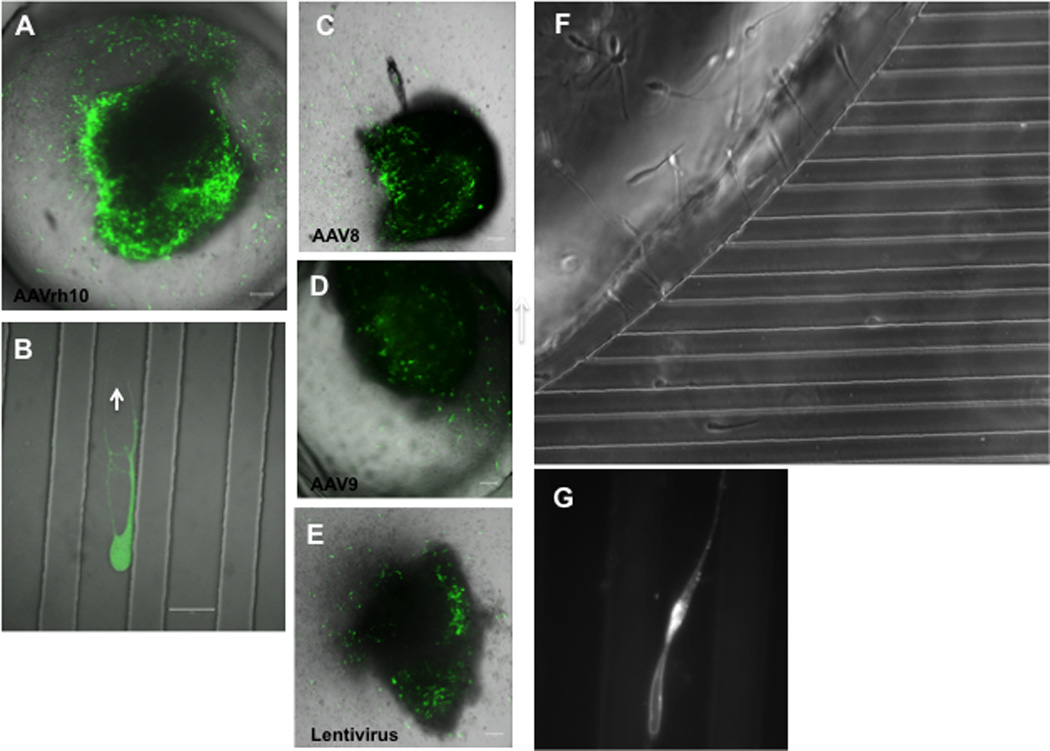
MGE explants from the brains of E15 mouse embryos were transfected with AAV or LV vectors expressing GFP and placed in microchambers filled with Matrigel and covered with Neurobasal medium. After 72 hours neurons expressing GFP migrated out of the explants. (A–E) MGE explants infected with either AAV serotyped with one of the following capsids: AAVrh10 (A–B), AAV 8 (C), and AAV 9 (D), or with lentivirus (E), showing infected neurons expressing GFP after 72 hours. In B, an individual neuron infected with AAVrh10 is shown inside of the microchannel (magnification bar = 20 µm). In addition, we monitored the migration in real-time of live neurons moving out of the explants (F), and labeled with BODIPY TR C5 (Golgi-tracker red, Molecular Probe; F–G). Forty-eight hours after placing MGE explant labeled with Golgi-tracker in a microfluidic system filed with Matrigel, individual neurons were observed inside of microchannels expressing the Golgi-tracker (G).
Further, MGE explants were stained with the Golgi marker BODIPY FL C5-ceramide (B–22650; Molecular Probes; Fig. 2 F–G). The BODIPY FL C5-ceramide dye was resuspended in sterile deionized water and diluted to 5 µM in 1 × Hank’s buffered salt solution (HBSS) immediately prior to use. This working dilution was added directly to the explants at a final concentration of 5 µM and incubated for 30 min at 4°C followed by several cold washes with 1×HBSS and 30 min room temperature incubation with explants in the microfluidic system filled with Matrigel. Live-cells microscopy showed neurons migrating out of explants inside of microchambers and labeled with the Golgi marker (Fig. 2G). All the neurons showed a similar staining pattern with red- emitting punctae for BODIPY FL C5-ceramide along the processes. In MGE cells, the centrosome and Golgi moved within the neurons a remarkably long distance away from the nucleus (up to 30 µm), and at the end of its forward migration, the centrosome and the Golgi apparatus folded into a single complex localized to a swelling distinct from the nuclear compartment, as previously observed (Métin et al., 2008)
4. Conclusions
Here we describe methods for culturing and staining neurons migrating out of MGE explants. Our ICC method allows co-staining of these with different intracellular probes. In addition, we also labeled neurons with live cell Golgi and nuclear trackers and imaged neurons in real time. Further, MGE explants showed differences in transduction efficiencies using AAV and lentiviral vectors. Most cells in the MGE explants were preferentially transduced with AAV vectors, serotype rh10 being the best and all better than with a lentiviral vector. Transduction efficiency could presumably be enhanced by increasing the titer of AAV vectors. In addition, our microfluidic devices can be used to culture MGE explants in different compartments and permits the visualization of the vectorial migration of individual neurons in culture chambers. The microfluidic platforms are amenable to high-resolution microscopy because they are constructed on glass coverslips. These attractive features of our staining techniques and microfluidic platforms make them ideal for studying neuronal migration at both a cellular and a molecular level.
Highlights.
-
♦
We established an immunocytochemistry (ICC) protocol to stain neurons migrating out of the MGE explant embedded in Matrigel.
-
♦
We also established a protocol to efficiently transfect cells in MGE explants
-
♦
We developed microfluidic chambers for explants that allow visualization of the vectorial migration of individual neurons from mouse embryonic MGE explants.
-
♦
Our microfluidic system allows monitoring of the distribution of cellular organelles (e.g. Golgi) within migrating neurons which have been stained with commercial molecular dyes or transfected with adeno-associated virus (AAV) expressing reporter proteins.
-
♦
Our methods provide new paradigms to study neuronal migration in real-time.
Acknowledgements
The authors thank Ms. Suzanne McDavitt for skilled editorial assistance. We also thank Igor Bagayev and Dr. Cristopher Bragg at MGH Neuroscience Center Confocal Microscope Facility. This work was supported by NIH/NINDS Grant P50 NS037409 (XOB and PB) and GM092804 (DI), and The Eleanor and Miles Shore 50th Anniversary Fellowship Program for Scholars in Medicine (FCN). Lentivirus and AAV vectors were kindly provided by Dr. Miguel Sena-Esteves (UMass Medical School, Worcester, MA). Microfabrication procedures were performed at the NIH/NIBIB BioMEMS Resource Center (EB002503).
Abbreviations
- AAV
adeno-associated virus
- AEP
anterior entopeduncular area
- GABA
gamma amino butyric acid
- GE
ganglionic eminence
- g.c.
genome copy
- GFP
green fluorescent protein
- HP
hidden photon
- ICC
immunocytochemistry
- i.p.
intraperitoneal
- LGE
lateral GE
- LV
lentivirus
- MGE
medial ganglionic eminence
- PFA
paraformaldehyde
- PBS
Phosphate Buffered Saline
- t.u.
transducing units
- VSV-G
vesicular stomatitis virus glycoprotein
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
Authors have no conflict of interest.
References
- Anderson SA, Marín O, Horn C, Jennings K, Rubenstein JL. Distinct cortical migrations from the medial and lateral ganglionic eminences. Development. 2001;128:353–363. doi: 10.1242/dev.128.3.353. [DOI] [PubMed] [Google Scholar]
- Berdichevsky Y, Stanley KJ, Yarmush ML. Building and manipulating neural pathways with microfluidics. Lab Chip. 2010;10:999–1004. doi: 10.1039/b922365g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broekman ML, Comer LA, Hyman BT, Sena-Esteves M. Adeno-associated virus vectors serotyped with AAV8 capsids are more efficient than AAV-1 or-2 serotypes for widespread gene delivery to the neonatal mouse brain. Neuroscience. 2006;138:501–510. doi: 10.1016/j.neuroscience.2005.11.057. [DOI] [PubMed] [Google Scholar]
- Butt SJ, Fuccillo M, Nery S, Noctor S, Kriegstein A, Corbin JG, Fishell G. The temporal and spatial origins of cortical interneurons predict their physiological subtype. Neuron. 2005;48:591–604. doi: 10.1016/j.neuron.2005.09.034. [DOI] [PubMed] [Google Scholar]
- Crandall JE, McCarthy DM, Araki KY, Sims JR, Ren J-Q, Bhide PG. Dopamine receptor activation modulates GABA neuron migration from the basal forebrain to the cerebral cortex. J Neurosci. 2007;27:3813–3822. doi: 10.1523/JNEUROSCI.5124-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernández-Miranda LR, Parnavelas JG, Chiara F. Molecules and mechanisms involved in the generation and migration of cortical interneurons. ASN Neuro. 2010;2:e00031. doi: 10.1042/AN20090053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marín O, Rubenstein JL. A long, remarkable journey: tangential migration in the telencephalon. Nat Rev Neurosci. 2001;2:780–790. doi: 10.1038/35097509. [DOI] [PubMed] [Google Scholar]
- McCarthy DM, Zhang X, Darnell SB, Sangrey GR, Yanagawa Y, Sadri-Vakili G, Bhide PG. Cocaine alters BDNF expression and neuronal migration in the embryonic mouse forebrain. J Neurosci. 2011;31:13400–13411. doi: 10.1523/JNEUROSCI.2944-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy DM, Gioioso V, Zhang X, Sharma N, Bhide PG. Neurogenesis and neuronal migration in the forebrain of the torsinA knockout mouse embryo. Dev Neurosci. 2012;34:366–378. doi: 10.1159/000342260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Métin C, Vallee RB, Rakic P, Bhide PG. Modes and mishaps of neuronal migration in the mammalian brain. J Neurosci. 2008;28:11746–11752. doi: 10.1523/JNEUROSCI.3860-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parnavelas JG. The origin and migration of cortical neurones: new vistas. Trends Neurosci. 2000;23:126–131. doi: 10.1016/s0166-2236(00)01553-8. [DOI] [PubMed] [Google Scholar]
- Rallu M, Corbin JG, Fishell G. Parsing the prosencephalon. Nat Rev Neurosci. 2002;3:943–951. doi: 10.1038/nrn989. [DOI] [PubMed] [Google Scholar]
- Reiner O. LIS1 and DCX: implications for brain development and human disease in relation to microtubules 2013. Scientifica. 2013 doi: 10.1155/2013/393975. Article ID 393975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter A, Loscher W. Gabapentin decreases the severity of dystonia at low doses in a genetic animal model of paroxysmal dystonic choreoathetosis. Eur J Pharmacol. 1999;369:335–338. doi: 10.1016/s0014-2999(99)00104-1. [DOI] [PubMed] [Google Scholar]
- Sena-Esteves M, Tebbets JC, Steffens S, Crombleholme T, Flake AW. Optimized large-scale production of high titer lentivirus vector pseudotypes. J Virol Methods. 2004;122:131–139. doi: 10.1016/j.jviromet.2004.08.017. [DOI] [PubMed] [Google Scholar]
- Valiente M, Marín O. Neuronal migration mechanisms in development and disease. Curr Opin Neurobiol. 2010;20:68–78. doi: 10.1016/j.conb.2009.12.003. [DOI] [PubMed] [Google Scholar]
- Wichterle H, Garcia-Verdugo JM, Herrera DG, Alvarez-Buylla A. Young neurons from medial ganglionic eminence disperse in adult and embryonic brain. Nat Neurosci. 1999;2:461–466. doi: 10.1038/8131. [DOI] [PubMed] [Google Scholar]