

Abstract
Notch activity regulates tumor biology in a context-dependent and complex manner. Notch may act as an oncogene or a tumor suppressor gene even within the same tumor type. Recently, Notch signaling has been implicated in cellular senescence. Yet, it remains unclear as to how cellular senescence checkpoint functions may interact with Notch-mediated oncogenic and tumor suppressor activities. Herein, we used genetically engineered human esophageal keratinocytes and esophageal squamous cell carcinoma cells to delineate the functional consequences of Notch activation and inhibition along with pharmacological intervention and RNA interference (RNAi) experiments. When expressed in a tetracycline-inducible manner, the ectopically expressed activated form of Notch1 (ICN1) displayed oncogene-like characteristics inducing cellular senescence corroborated by the induction of G0/G1 cell-cycle arrest, Rb dephosphorylation, flat and enlarged cell morphology and senescence-associated β-galactosidase activity. Notch-induced senescence involves canonical CSL/RBPJ-dependent transcriptional activity and the p16INK4A-Rb pathway. Loss of p16INK4A or the presence of human papilloma virus (HPV) E6/E7 oncogene products not only prevented ICN1 from inducing senescence, but permitted ICN1 to facilitate anchorage-independent colony formation and xenograft tumor growth with increased cell proliferation and reduced squamous-cell differentiation. Moreover, Notch1 appears to mediate replicative senescence as well as TGF-β-induced cellular senescence in non-transformed cells and that HPV E6/E7 targets Notch1 for inactivation to prevent senescence, revealing a tumor suppressor attribute of endogenous Notch1. In aggregate, cellular senescence checkpoint functions may influence dichotomous Notch activities in the neoplastic context.
Keywords: Notch, Rb, p16, HPV, E7, senescence, squamous cell carcinoma
Introduction
Esophageal squamous cell carcinoma (ESCC) is among the deadliest cancers known 1 and is a paradigm for the investigation of all types of squamous cell carcinomas (SCCs). Common genetic lesions associated with ESCC include p53 mutations, p16INK4A loss, cyclin D1 overexpression, EGFR overexpression and telomerase activation 2. Ectopically expressed telomerase (hTERT) or human papilloma virus (HPV) E6/E7 gene products immortalize human esophageal epithelial cells (keratinocytes) overcoming replicative senescence 3, 4. Oncogenes induce senescence in immortalized esophageal keratinocytes 5–7. Senescence serves as a failsafe mechanism to prevent oncogene-induced aberrant proliferation. In fact, malignant transformation of esophageal keratinocytes requires concurrent inactivation of the senescence checkpoint functions regulated by the p53 and Rb pathways to negate oncogene-induced senescence 5, 7–9.
The Notch pathway regulates cell fate and differentiation through cell-cell communication. The mammalian Notch family comprises four transmembrane receptor proteins (Notch1 to Notch4). Ligands (JAG1/2, DLL1, 3 and 4) bind Notch receptors through cell-cell contact to trigger γ-secretase-mediated proteolytic cleavage of Notch receptor proteins, resulting in nuclear translocation of the intracellular domain of Notch (ICN), the activated form of Notch. ICN of all Notch receptor paralogs forms a transcriptional activation complex containing a common transcription factor CSL (a.k.a. RBPJκ) and the coactivator Mastermind-like (MAML)10. Notch1 target genes include the HES/HEY family of transcription factors, Notch3 and IVL, a marker of squamous-cell differentiation. Squamous-cell differentiation is impaired by Notch1 loss, CSL loss or ectopic expression of dominant negative MAML1 (DNMAML1) in the skin and the esophagus in mice 11–13.
The highly context-dependent nature of Notch functions adds complexity to its roles in cancers. While Notch acts as an oncogene in T cell acute lymphoblastic leukemia, both oncogenic and tumor suppressor roles have been found in solid tumors even within identical tumor types 14. Notch1 may be activated in SCCs15, 16. The active form of Notch1 (i.e. ICN1) transforms keratinocytes in concert with HPV E6/E717, 18, although Notch1 may be downregulated to sustain E6/E7 expression at the late steps of malignant transformation 19. Multiple lines of evidence indicate a tumor suppressor role of Notch in SCCs. They include loss-of-function mutations identified in primary SCCs including ESCC 20–23 and tumor-prone phenotypes in genetically engineered mouse models targeting the Notch pathway 24–30. By maintaining epidermal integrity and barrier functions, Notch may prevent the tumor-promoting inflammatory microenvironment in the skin 30. It is unclear in what specific context Notch may act as an oncogene or a tumor suppressor in SCCs.
Notch1 is activated in vascular endothelial cells undergoing replicative senescence 31, 32. Although Notch1 has been implicated in cell-cycle arrest associated with squamous-cell differentiation 12, 33, it is unclear whether Notch1 induces or mediates senescence in cells of epithelial origin and how senescence may be linked to the either oncogenic or tumor suppressor attributes of Notch1. Herein we investigated the functional consequences of Notch1 activation and inhibition in esophageal keratinocytes and ESCC cells, revealing unique interactions between Notch1 and cellular senescence checkpoint functions via transforming growth factor (TGF)-β signaling which may influence dichotomous Notch1 functions in SCCs and other cancers.
Results
Notch1 is activated in human esophageal keratinocytes undergoing replicative senescence
The role of Notch1 in senescing epithelial cells remains unknown. We examined Notch1 in well-characterized primary human esophageal keratinocytes EPC2, which undergo replicative senescence by 40–44 population doublings (PDs)34 with an increased doubling time (Figure 1a and b). The activated form of Notch1 (ICN1Val1744) was upregulated at 43 PDs in cells with senescent characteristics corroborated by Rb dephosphorylation, upregulation of p53, p16INK4A and p21 (CDKN1A), flat and enlarged cell morphology and the increased senescenceassociated β-galactosidase (SABG) activity (Figure 1, c–e). Pharmacological Notch inhibition by a γ-secretase inhibitor (GSI) suppressed ICN1Val1744 and antagonized the above changes (Figure 1), suggesting that Notch1 may regulate replicative senescence in keratinocytes.
Figure 1. Notch1 is activated in EPC2 cells undergoing replicative senescence.
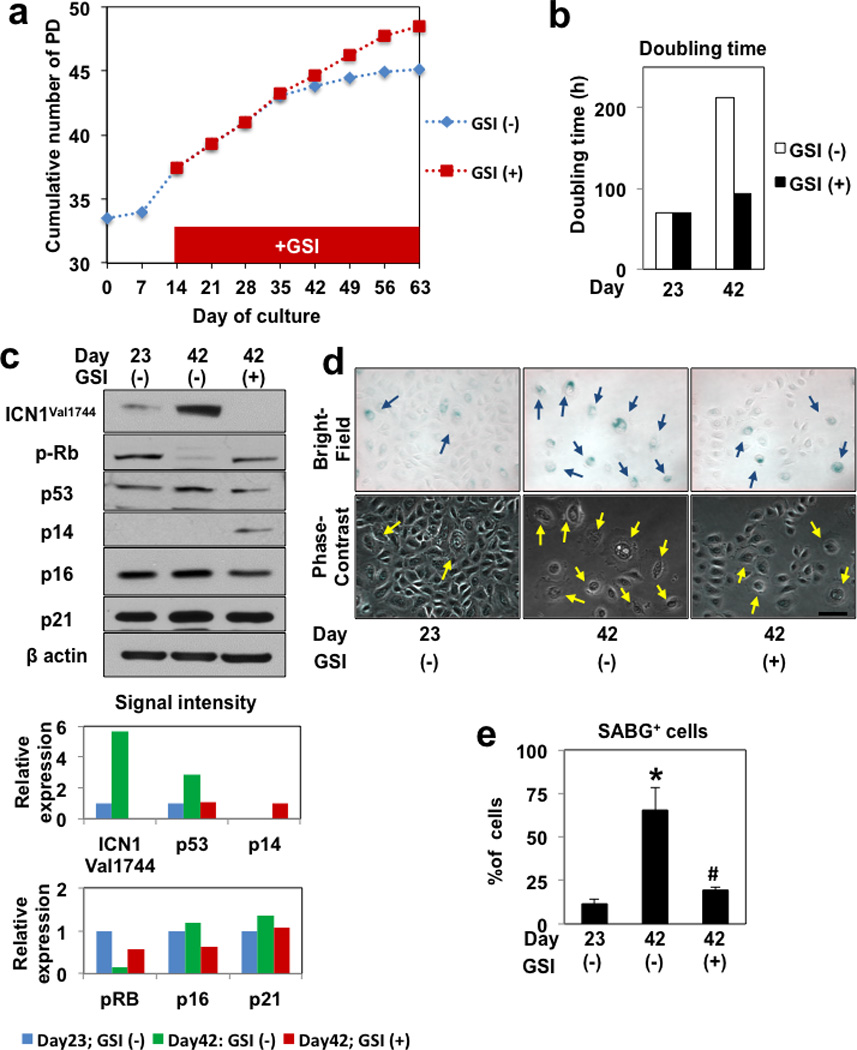
A frozen vial of primary culture of EPC2 cells (27.5 PD) was thawed and grown in the presence or absence of GSI for a period indicated in (a). Cells were harvested at indicated time points to determine population doubling (a) as well as doubling time (b), and subjected to Western blotting (c) and SABG assays (d and e). In (c), β-actin served as a loading control; ICN1Val1744, the activated form of Notch1; p-Rb, phospho-RbS780. In densitometry, the signal intensity for molecule of interest was calibrated by that of β-actin at each time point. In (d), representative bright-field and phase contrast images demonstrate SABG-positive cells and the corresponding cells with flat and enlarged cell morphology (arrows) as scored in (e); *, P <0.05 vs. Day 23 and GSI (−); #, P <0.05 vs. Day 42 and GSI (−); (n=6). Note a reduced cell density at day 42 (43 PD) without GSI. Note that GSI suppressed ICN1Val174 (c), preventing the extension of doubling time (b) as well as the induction of SABG positive cells in (d) and (e).
ICN1 induces senescence via canonical CSL-dependent transcription
To delineate the functional consequences of Notch1 activation, we used the tetracycline-inducible system to express ICN1 ectopically. Doxycycline (DOX) induced ICN1 within 24 h to activate its downstream molecules including HES5 and Notch3 in a dose-dependent manner in EPC2-hTERT, a telomerase-immortalized EPC2 derivative (Figure 2a and b; Supplementary Figure S1a). ICN1 induced p16INK4A, p21 and Rb dephosphorylation as a function of time to inhibit cell proliferation, leading to G0/G1 cell-cycle arrest (Figure 1b – d). Senescence was suggested by flat and enlarged cell morphology and DOX dose-dependent SABG induction (Figure 1e and f). ICN1 also induced its target genes and senescence in human esophageal keratinocytes EPC1 and its derivative EPC1-hTERT; however, p16INK4A was not detectable in the latter (Supplementary Figures S1b and S2), suggesting p16INK4A loss in EPC1 during telomerase-induced immortalization 35. Of note, DOX alone did not induce senescence in parental cell lines nor in those carrying a control vector (data not shown), indicating that DOX per se did not induce senescence.
Figure 2. ICN1 induces Notch target genes and senescence in EPC2-hTERT cells.
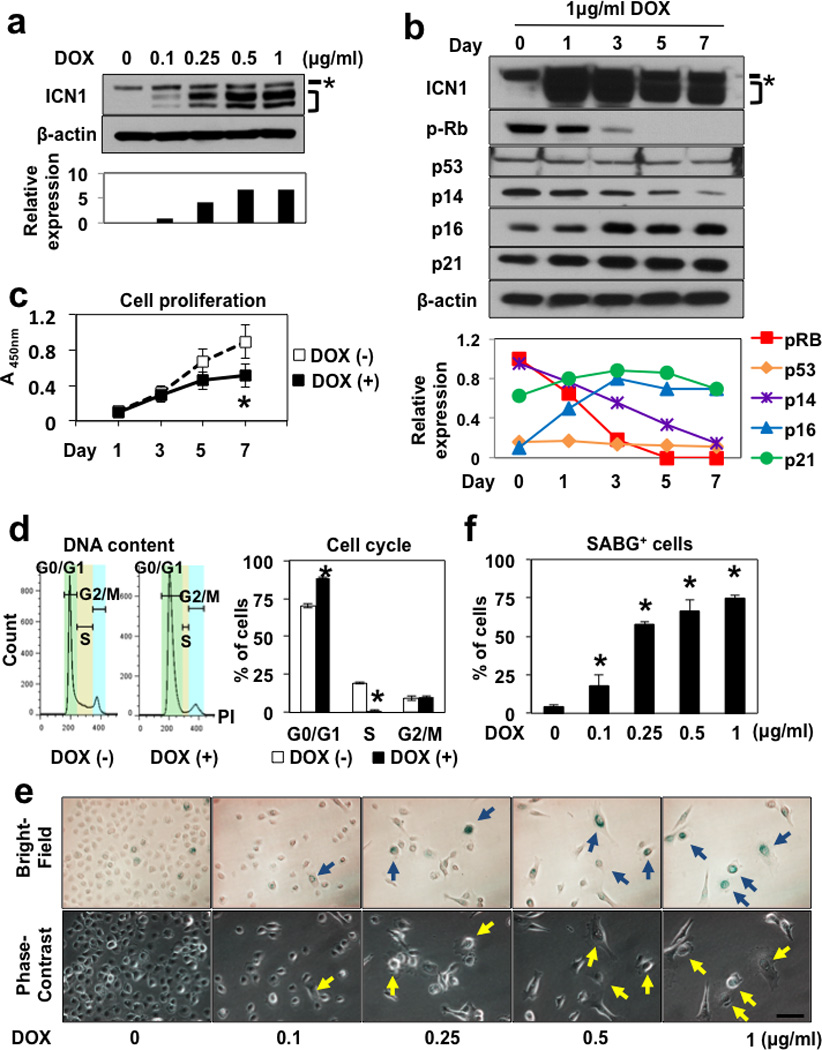
EPC2-hTERT carrying ICN1Tet-On was treated with indicated concentrations of doxycycline (DOX); or 0 µg/ml [DOX (−)] or 1 µg/ml [DOX (+)] of DOX to induce ICN1. In (a), (d) and (e)–(f), cells were exposed to DOX for 7 days. In (b) and (c), cells were exposed to DOX for indicated time period. Following DOX treatment, cells were analyzed by Western blotting for ICN1, phospho-RbS780 (p-Rb), p53 and cell-cycle regulators at indicated time points with densitometry in (a) and (b); WST1 assays for cell proliferation in (c); flow cytometry for cell-cycle in (d); and SABG assays in (e) and (f). In (a) and (b), β-actin served as a loading control. * denotes transmembrane/intracellular region of endogenous Notch1 which was suppressed by RNAi directed against Notch1 (data not shown). Bracket indicates lentivirally expressed ICN1 induced by DOX. A doublet appears consistently and may represent a posttranslational modification. Or note, anit-Notch1 (5B5) antibody was used to detect lentivirally expressed ICN1 which lacks the epitope recognized by anti-ICN1Val1744 antibody. In (c), *, P < 0.05 vs. DOX (−) at day 7 (n=6). In (d), representative histogram plots are shown. Proportions of cells in G0/G1, S and G2/M cell-cycle phases were determined. *, P < 0.05 vs. DOX (-)(n=3). In (f), representative bright-field and phase contrast images of SABG-positive cells with flat and enlarged cell morphology (arrows) as scored in (e); *, P <0.05 vs. 0 µg/ml DOX (n=6).
We next conducted RNA interference (RNAi) experiments to determine the role of canonical CSL. CSL knockdown prevented ICN1 from activating CSL-dependent transcription, allowing continued cell proliferation with antagonized Rb dephosphorylation and decreased SABG activation in EPC2-hTERT and EPC1-hTERT cells (Figure 3 and Supplementary Figures S3), suggesting that CSL-dependent transcription may mediate ICN1-induced senescence since.
Figure 3. CSL knockdown inhibits ICN1-mediated senescence in EPC2-hTERT cells.
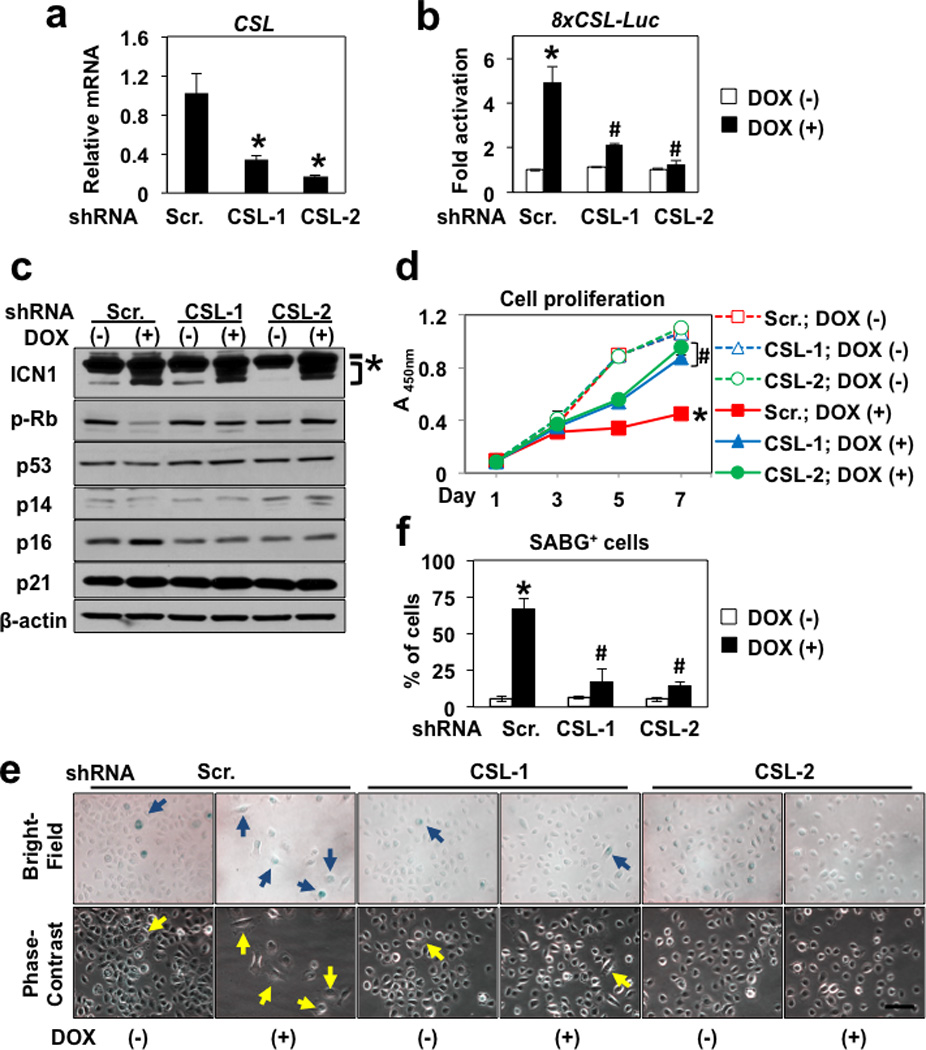
EPC2-hTERT carrying ICN1Tet-On was stably transduced with lentivirus expressing two independent shRNA sequences directed against CSL (CSL-1 and CSL-2) or a non-silencing control scramble shRNA (Scr.) sequence. In (b), cells were transiently transfected with 8xCSL-luc 24 h before DOX treatment. Cells were treated with DOX at a concentration of 0 µg/ml [DOX (−)] or 1 µg/ml [DOX (+)] to induce ICN1 for 48 h in (b) and 7 d in (c)–(f). Cells were harvested at indicated time points in (d). Following DOX treatment, cells were subjected to quantitative RT-PCR for CSL mRNA in (a); luciferase assays for 8xCSL-luc reporter activity in (b); Western blotting for ICN1, phospho-RbS780 (p-Rb), p53 and cell-cycle regulators in (c); WST1 assays for cell proliferation in (d); and SABG assays in (e) and (f). In (a), β-actin served as an internal control. *, P < 0.05 vs. Scr. (n=3). In (b), *, P < 0.05 vs. Scr. and DOX (−); #, P < 0.05 vs. Scr. and DOX (+); (n=3). In (c), β-actin served as a loading control. * denotes transmembrane/intracellular region of endogenous Notch1. Bracket indicates lentivirally expressed ICN1 induced by DOX. In (d), *, P < 0.05 vs. Scr. and DOX (−) at day 7; #, P < 0.05 vs. Scr. and DOX (+) at day 7; (n=6). In (e), representative bright-field and phase contrast images of SABG-positive cells with flat and enlarged cell morphology (arrows) as scored in (e); *, P < 0.05 vs. Scr. and DOX (−); #, P < 0.05 vs. Scr. and DOX (+); (n=6).
ICN1-induced senescence may be impaired in transformed human esophageal cells
Malignant transformation may involve inactivation of the cellular senescence check point functions, serving as a fail-safe mechanism against oncogene activation. We asked if ectopically expressed ICN1 induces senescence in transformed human esophageal cells EPC2-T, EN60 and TE11. EPC2-T is a derivative of EPC2-hTERT carrying EGFR, cyclin D1 and p53R175H transgenes and that has been further modified to express either DNMAML1, a genetic pan-Notch inhibitor, or GFP as a control 36. The p14ARF-p53 and p16INK4A-Rb pathways are compromised in EN60 cells carrying HPV E6 and E74, which target p53 and Rb for degradation or sequestration, respectively. TE11 cells show biallelic p53 inactivation37 and INK4A deletion38.
ICN1 activated CSL-dependent transcription in EPC2-T that is inhibited by DNMAML1 (Supplementary Figures S1c and S4b). Likewise, CSL knockdown prevented ICN1 from activating CSL-dependent transcription and SABG induction in EPC2-T cells (data not shown). In the absence of DNMAML1, ICN1 induced p16INK4A and Rb dephosphorylation to inhibit cell proliferation (Supplementary Figure S4b – e). Interestingly, the extent of ICN1-mediated SABG induction was limited in EPC2-T cells (40–50%) without DNMAML1 (Supplementary Figure S4d and e) as compared to parental EPC2-hTERT cells (60–80%)(Figure 2).
When tested in EN60 and TE11, ICN1 activated CSL-dependent transcription and induced Notch target genes; however, ICN1 affected little, if any, Rb phosphorylation, cell proliferation or SABG activity (Supplementary Figures S5), suggesting that oncogenic genetic alterations that limit cellular senescence check point functions may suppress ICN1-induced senescence without affecting CSL-dependent transcriptional activity.
The p16INK4A-Rb pathway may have a regulatory role in ICN1-induced senescence
We next asked how the p14ARF-p53 and p16INK4A-Rb pathways may influence ICN1-induced senescence. Notch can either activate or inhibit p53 in a contextdependent manner 39. p53 and p14ARF proteins, the latter a p53 stabilizing tumor suppressor, were unaffected or rather downregulated in EPC2-hTERT, EPC1 and EPC1-hTERT with ectopically expressed ICN1 (Figure 2b; Supplementary Figure S2a). We also examined p21 and BAX, two genes induced by oncogenic RasG12V in EPC2-hTERT in a p53-dependent manner5. ICN1 induced p21, but not BAX, mRNA in EPC1-hTERT and EPC2-hTERT cells (Supplementary Figure S6a and b). Since ICN1 induces p21 in a CSL-dependent manner 12, the failure of BAX induction may suggest the lack of p53 activation in response to ICN1. Moreover, ICN1 induced neither p21 nor BAX mRNA in EPC2-T cells expressing p53R175H (Supplementary Figure S6c). Finally, p53R175H did not prevent ICN1-induced senescence in EPC2-hTERT and EPC2-T (Supplementary Figures S7 and S4). Thus, p53 inactivation may be insufficient to negate ICN1-induced senescence; however, the above findings do not exclude the requirement of p14ARF at the onset of ICN1-induced senescence. p14ARF may also inhibit cell proliferation in a p53-independent manner 40, prompting us to perform RNAi experiments to explore cell-cycle regulators including p14ARF.
Amongst the cyclin-dependent kinase inhibitors (CDKIs), ICN1 induced p15INK4B and p16INK4A CSL-dependently in EPC2-T as antagonized by DNMAML1 (Supplementary Figures S4a and S6c). We screened their involvement in ICN1-induced senescence with siRNA sequences directed against p15INK4B and the exon 3 of INK4A (Figure 4a), the latter shared by both p14ARF and p16INK4A. RNAi directed against both p14ARF and p16INK4A, but not p15INK4B, significantly inhibited ICN1-mediated SABG activation in EPC2-T (Supplementary Figure S8), implicating the INK4A locus in ICN1-induced senescence.
Figure 4. p16INK4A may contribute to ICN1-induced senescence in EPC2-T cells.
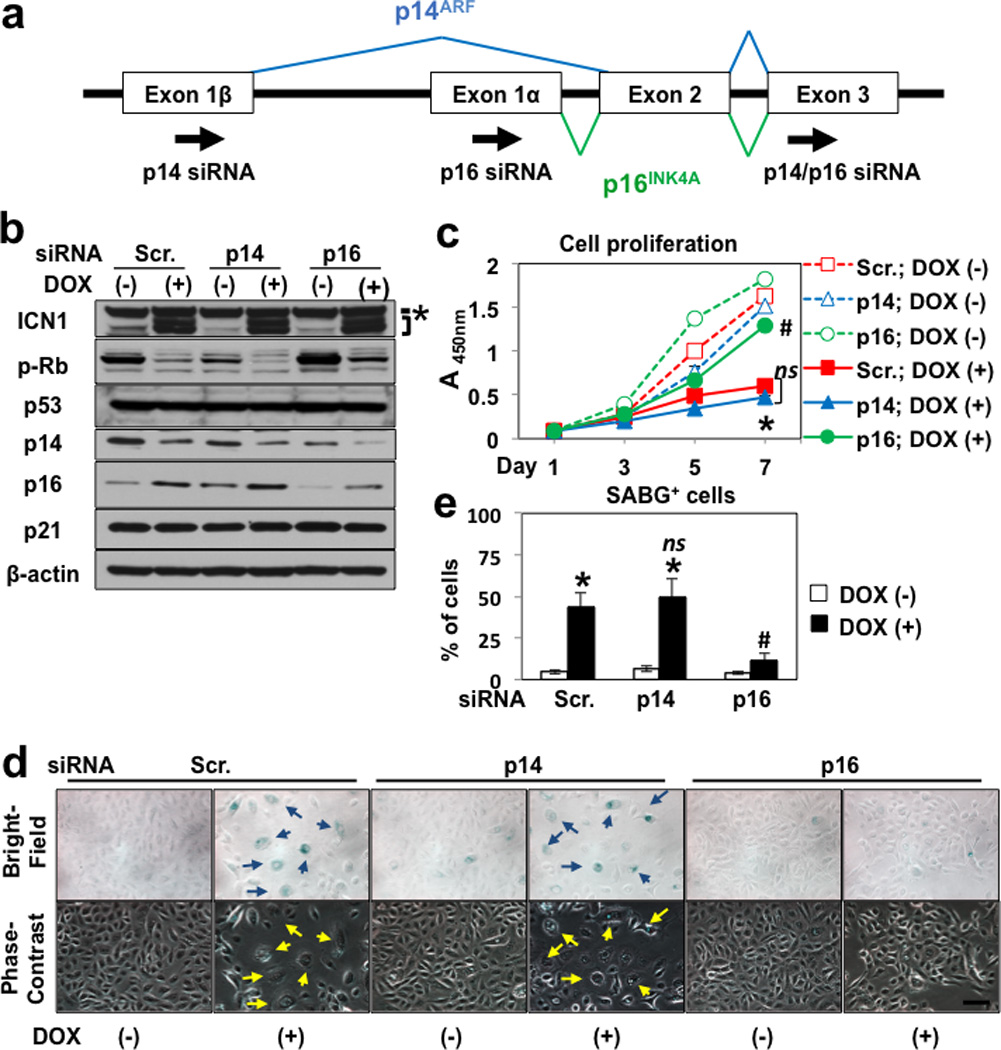
In (a), siRNA was designed to target p14ARF (p14 siRNA) and p16iNK4A (p16 siRNA) on the INK4A locus. p14/p16 siRNA was used to knockdown p14ARF and p16iNK4A concurrently as shown in Supplementary Figure S8. In (b)–(e), EPC2-T carrying ICN1Tet-On was treated with 0 µg/ml [DOX (−)] or 1 µg/ml [DOX (+)] of DOX to induce ICN1 following transfection with p14 siRNA, p16 siRNA or a non-silencing control scramble short interfering RNA (Scr.). Starting 24 h after transfection, cells were treated with DOX for 7 days in (b), (d) and (e); and indicated time points in (c); and subjected to Western blotting for ICN1, phospho-RbS780 (p-Rb), p53 and cell-cycle regulators in (b); WST1 assays for cell proliferation in (c); and SABG assays in (d) and (e). In (b), β-actin served as a loading control. * denotes transmembrane/intracellular region of endogenous Notch1. Bracket indicates lentivirally expressed ICN1 induced by DOX. In (c), *, P < 0.05 vs. Scr. and DOX (−) at day 7; #, P < 0.05 vs. Scr. and DOX (+) at day 7; ns, not significant vs. Scr. and DOX (+) at day 7 (n=6). In (d), representative bright-field and phase contrast images of SABG-positive cells with flat and enlarged cell morphology (arrows) as scored in (e); *, P < 0.05 vs. Scr. and DOX (−); #, P < 0.05 vs. Scr. and DOX (+); ns, not significant vs. Scr. and DOX (+); (n=6). Note that densitometry from (b) was summarized along with cell proliferation and SABG data in (c)–(e) in Supplementary Table S1.
To dissect the roles of p14ARF and p16INK4A in ICN1-induced senescence more specifically, we designed siRNA targeting non-overlapped sequences in p16INK4A (exon 1α) and p14ARF (exon 1β)(Figure 4a). When tested in EPC2-hTERT, EPC2-T and EPC1 cells that express both p14ARF and p16INK4A, RNAi directed against p16INK4A, but not p14ARF, prevented ICN1 from inducing senescence as corroborated by decreased Rb dephosphorylation, continuous cell proliferation and reduced SABG activity in all cell lines (Figure 4, b–e for EPC2-T; Supplementary Figures S9 and S10 for EPC2-hTERT and EPC1, respectively), suggesting that p16INK4A may have a predominant role in ICN1-induced senescence. Nevertheless, RNAi directed against p14ARF or p16INK4A revealed context-dependent functional interplays between p14ARF and p16INK4A, influencing basal cell proliferation and expression of other cell-cycle regulators as summarized in Supplementary Table S1.
Since DNMAML1 prevented ICN1 from inducing p16INK4A mRNA (Supplementary Figure S6c), we asked whether ICN1 may transcriptionally activate p16INK4A. In transfection assays using a pGL3-p16 reporter construct, ICN1 did not activate the 2.3-kb 5’-regulatory region of p16INK4A in EPC2-hTERT and EPC2-T (data not shown). Of note, the ECR browser did not detect conserved CSL-binding cis-elements within this region in silico 41 .Thus, ICN1 may not regulate p16INK4A transcriptionally through its proximal 5’-regulatory region.
HPV E6/E7 may repress TGF-β signaling to prevent Notch1-mediated senescence in transformed human esophageal keratinocytes
The HPV E6 and E7 proteins inactivate p53 and Rb, respectively. To determine if E6 and E7 may influence endogenous Notch activity, we performed RNAi experiments in EN60 cells carrying both E6 and E7 as a single fusion gene. siRNA sequences directed against either E6 or E7 suppressed both E6 and E7 transcripts (Figure 5a; and data not shown), resulting in induction of ICN1Val1744 and CDKIs with p53 stabilization, Rb dephosphorylation, reduced cell proliferation, G0/G1 cell-cycle arrest and SABG expression (Figure 5b–f; Supplementary Figures S11). Importantly, SABG activity was antagonized by concurrent knockdown of either Notch1 or p16INK4A (Figure 5e and f; Supplementary Figures S11), suggesting that endogenous Notch1 and p16INK4A may cooperate to mediate senescence when p16INK4A becomes accessible to Rb as a consequence of E7 knockdown.
Figure 5. HPV E6/E7 knockdown activates endogenous Notch1 and TGF-β signaling to induce senescence in EN60 cells.
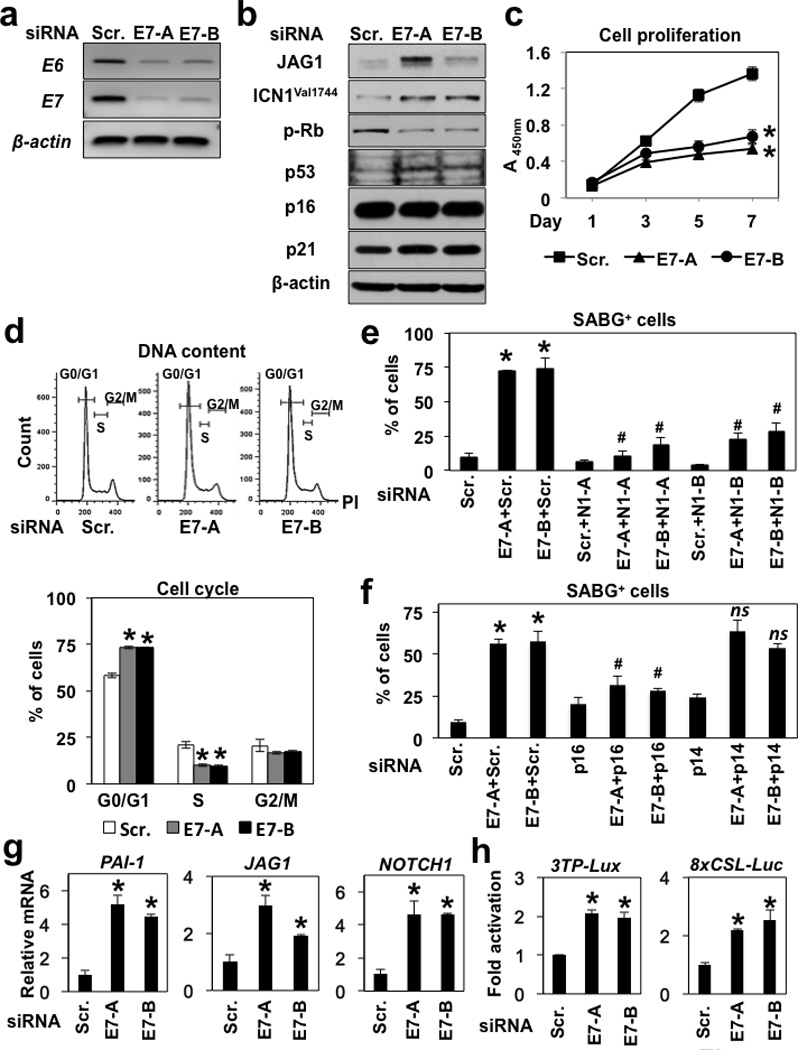
EN60 cells expressing HPV E6/E7 were transiently transfected with two independent siRNA sequences directed against either HPV E7 (E7-A and E7-B) or a non-silencing control scramble short interfering RNA (Scr.) along with or without siRNA directed against Notch1 (N1-A and N1-B), p16iNK4A (p16) or p14ARF (p14). In (h), cells were concurrently transfected with indicated reporter constructs. Cells were analyzed 7 days after transfection by RT-PCR for HPV E6 and E7 transcripts in (a); Western blotting for indicated molecules in (b); WST1 assays for cell proliferation in (c); flow cytometry for cell-cycle in (d); and SABG assays in (e) and (f); quantitative RT-PCR for indicated mRNA in (g); and luciferase assays for activation of the TGF-β (3TP-Lux) and Notch (8xCSL-luc) reporters in (h). In (a), β-actin served as an internal control. In (b), β-actin served as a loading control. In (d), representative histogram plots are shown. Proportions of cells in G0/G1, S and G2/M cell-cycle phases were determined. *, P < 0.05 vs. Scr.; (n=3). In (e) and (f), SABG-positive cells were scored (see Supplementary Figure S11 for representative photomicrographs). *, P < 0.05 vs. Scr. only; #, P < 0.05 vs. Scr. + either E7-A or E7-B; ns, not significant vs. Scr. + either E7-A or E7-B; (n=6) in (e) and (f). In (g), β-actin served as an internal control. *, P < 0.05 vs. Scr.; (n=3). In (h), *, P < 0.05 vs. Scr.; (n=4).
E7 suppresses TGF-β signaling by blocking Smad3 binding to target sequences on DNA42. Since TGF-β induces the Notch ligand JAG1 in keratinocytes43, we suspected that RNAi directed against HPV gene products may reactivate TGF-β signaling to allow Notch activation via JAG1. In agreement, TGF-β target genes PAI1 and JAG1 were found to be elevated in the presence of E7 siRNA (Figure 5b and g). Moreover, E7 knockdown resulted in the activation of both TGF-β and Notch reporter constructs in transfection assays (Figure 5h). Finally, E7 knockdown led to upregulation of Notch1 mRNA (Figure 5g). These data agree with Notch1 suppression in HPV-transformed cells19. Therefore, HPV E6/E7 may suppress TGF-β signaling to inhibit the Notch1-mediated senescence program.
Endogenous Notch1 mediates senescence in non-transformed keratinocytes in response to TGF-β stimulation
Next, we asked whether and how TGF-β may induce senescence via Notch1 in cells without HPV oncogene products. Since the majority of transformed human esophageal cells resist senescence in response to TGF-β stimulation7, we used non-transformed EPC2-hTERT cells. TGF-β induced JAG1 to enhance endogenous ICN1Val1744 expression and activate CSL-dependent transcription, leading to Rb dephosphorylation, inhibition of cell proliferation, G0/G1 cell-cycle arrest and induction of flat and enlarged cell morphology as well as SABG activity (Figure 6, a–f; Supplementary Figure S12a). Importantly, GSI inhibited the ability of TGF-β to trigger Notch1 activation, growth inhibition and SABG induction (Figure 6b, c–f; Supplementary Figure S12a), indicating that Notch activation mediates TGF-β-induced senescence. Moreover, RNAi directed against Notch1 decreased significantly TGF-β-induced Notch1 and SABG activation (Figure 6g and Supplementary Figure S12b and c). Nevertheless, GSI or Notch1 knockdown reversed G0/G1 cell-cycle arrest to a partial extent (Figure 6e; and data not shown) as corroborated by the limited antagonistic effect upon Rb dephosphorylation and cell proliferation in the presence of TGF-β (Figure 6b and d; Supplementary Figure S12b). TGF-β induced p16INK4A and p21 prior to full induction of ICN1Val1744 (Figure 6a) although Notch1 knockdown delayed Rb dephosphorylation (Supplementary Figure S12b). Thus, TGF-β may not necessarily depend upon Notch to induce cell-cycle arrest, but instead may depend largely upon Notch1 for SABG activation in this context. These data suggest that endogenous Notch1 may mediate TGF-β-induced senescence.
Figure 6. TGF-β stimulates endogenous Notch1 to mediate senescence in EPC2-hTERT cells.
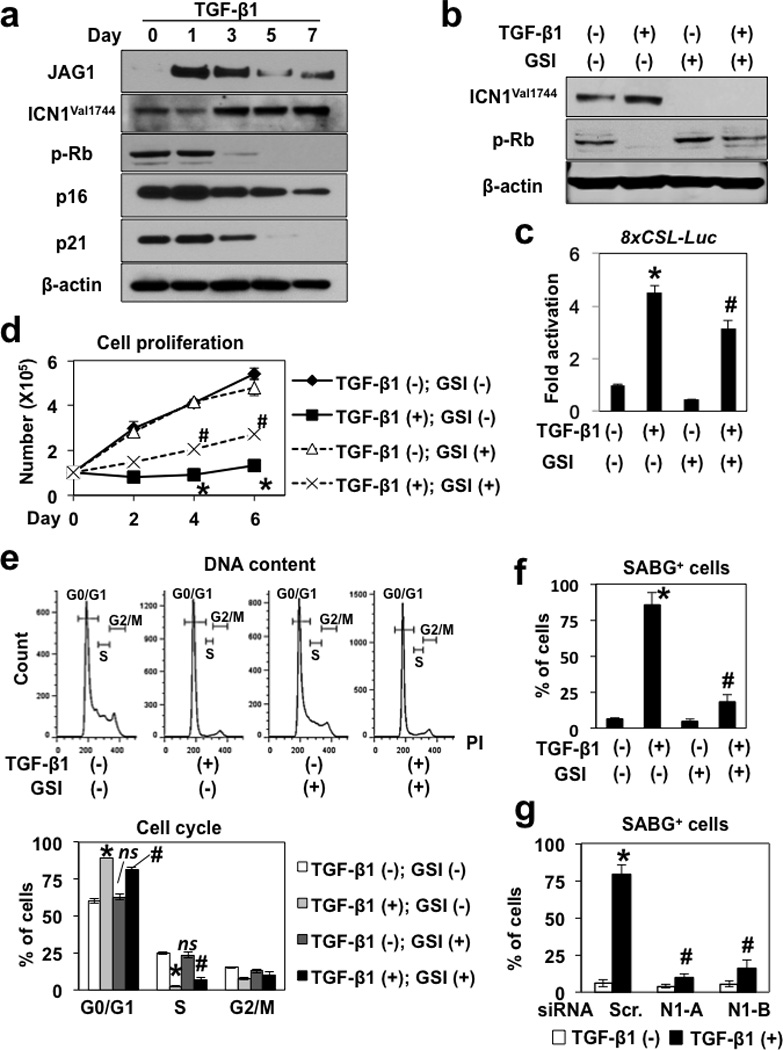
EPC2-hTERT cells were stimulated with 5 ng/ml TGF-β1 alone, or along with either GSI (1µM compound E) or DMSO (vehicle) in (a)–(f). Cells were transiently transfected with a 8xCSL-luc Notch reporter construct 24 h prior to TGF-β stimulation in (c) and siRNA directed against Notch1 (N1-A and N1-B) 24 h prior to TGF-β stimulation in (g). Cells were analyzed at indicated time points in (a) and (d); and 7 d after TGF-β stimulation in (b), (c), (e)–(g). Western blotting determined indicated molecules with β-actin serving as a loading control in (a) and (b). In (c), luciferase assays determined activation of the 8xCSL-luc Notch reporter construct. In (d), cell number was counted to determine cell proliferation. In (e), flow cytometry was done to determine cell-cycle. In (f) and (g), SABG assays were carried out and scored (see Supplementary Figure S12a and c for representative photomicrographs). In (c), *, P <0.05 vs. TGF-β1 (−) and GSI (−); #, P <0.05 vs. TGF-β1 (+) and GSI (−); (n=3). In (d), *, P <0.05 vs. TGF-β1 (−) and GSI (−); #, P <0.05 vs. TGF-β1 (+) and GSI (−); (n=3). In (e), representative histogram plots are shown. Histograms show proportions of cells in G0/G1, S and G2/M cell-cycle phases. *, P < 0.05 vs. TGF-β (−) and GSI (−); nsnot significant vs. TGF-β (−) and GSI (−); #, P < 0.05 vs. TGF-β (+) and GSI (−); (n=3). In (f), *, P <0.05 vs. TGF-β1 (−) and GSI (−); #, P <0.05 vs. TGF-β1 (+) and GSI (−); (n=6). In (g), *, P <0.05 vs. TGF-β1 (−) and Scr. (−); #, P <0.05 vs. TGF-β1 (+) and Scr.; (n=6).
Ectopically expressed ICN1 facilitates anchorage-independent cell growth and tumor formation in transformed human esophageal keratinocytes
We explored finally how ICN1 may affect tumorigenicity of transformed cells that are able to negate ICN1-induced senescence. To this end, we first conducted soft agar colony formation assays using EN60 and TE11 cells with tetracycline-inducible ICN1. ICN1 enhanced colony formation in both cell lines and stimulated colony growth in TE11, but not EN60 (Figure 7a). Next, we performed xenograft transplantation experiments. In nude mice, ICN1 greatly enhanced tumor growth by EN60 and TE11 cells (Figure 7b). Interestingly, histopathological analysis of xenograft tumors revealed a significantly increased number of less differentiated, smaller and proliferative ESCC cells upon ICN1 expression (Figure 7c and Supplementary Fig. S13). These results suggest that in response to Notch1 activation ESCC cells not only negate ICN1-induced senescence, but also gain more malignant characteristics, revealing an oncogene-like attribute of ICN1.
Figure 7. ICN1 increases malignant potentials in esophageal cells negating ICN1-mediated senescence.
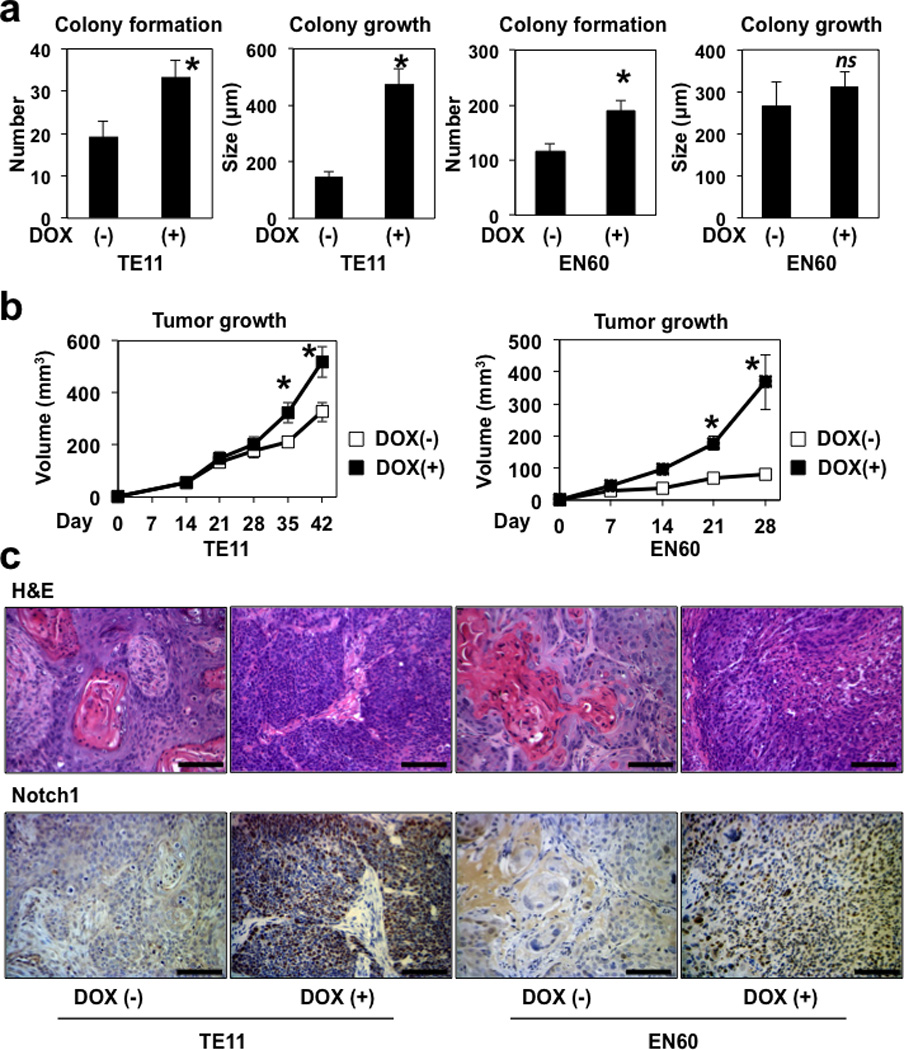
TE11 and EN60 cells carrying ICN1Tet-On were subjected to soft agar colony formation assays in (a) and xenograft transplantation experiments in (b) and (c). In (a), cells were grown for 2 weeks in soft agar in the presence [DOX (+)] or absence [DOX (−)] of 1 µg/ml DOX and photomicrographed. Colony number and size were determined per low-power field under light microscopy. *, P< 0.01 vs. DOX (−); n=6. In (b) and (c), immunodeficient mice underwent xenograft transplantation and fed with DOX-containing pellets (20 mg/kg) to induce ICN1. Tumor growth was monitored for indicated time periods in (b). Representative images for H&E and immunohistochemistry for Notch1 in resulting xenograft tumors are shown in (c). Note tumors in mice treated with DOX display less-differentiated SCC featuring smaller ESCC cells. Tumors grown in DOX-untreated control mice display well-differentiated SCC with keratin pearl formation. Scale bar, 100 µm.
Discussion
Notch1 has been implicated in replicative senescence in endothelial cells31, 32; however, this is the first study demonstrating in epithelial cells that Notch1 activates cellular senescence defined by cell-cycle arrest, morphological changes, SABG induction and molecular changes including Rb dephosphorylation. Importantly, our study sheds light on the role of cellular senescence checkpoint functions in influencing dichotomous Notch activities in the neoplastic context. Our data indicate that activated Notch1 may induce senescence in concert with intact cell-cycle checkpoint functions (Figures 1–4). When they are fully impaired, cells may negate senescence (Supplementary Figure S5), but gain more malignant characteristics in response to Notch1 activation (Figure 7). In this context, Notch1 exhibits features of an oncogene (Figure 8a). However, endogenous Notch1 mediates senescence as a downstream effector for TGF-β signaling (Figure 6). Such a function of Notch1 may be targeted for inactivation by the HPV oncogenes E6 and E7 (Figure 5), implying a feature of Notch1 as a tumor suppressor gene (Figure 8b). Like TGF-β acting as a tumor suppressor in the early stage of skin carcinogenesis while promoting tumor progression in later states44, Notch1 may have differential roles during cancer development and progression.
Figure 8. Model: Notch1 displays both oncogene and tumor suppressor attributes in a context-dependent manner.
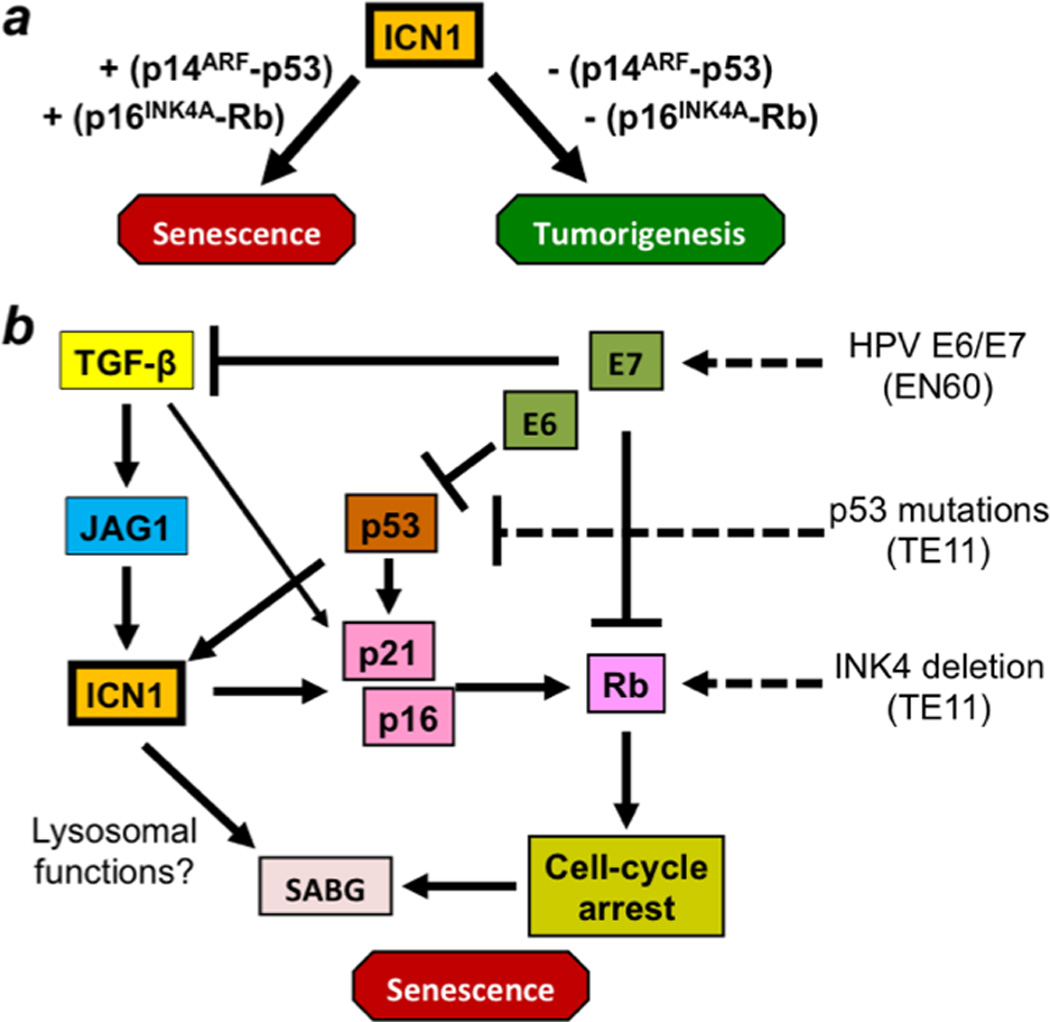
a. Like classic oncogenes (e.g. Ras and Raf), ICN1 induces senescence via intact cell-cycle checkpoint functions. In normal human esophageal keratinocytes, the p16INK4A-Rb pathway may have a predominant role in ICN1-induced senescence; however, the p14ARF, p53 and others may constitute alternative pathways to mediate senescence when the p16INK4A-Rb pathway is impaired. When cell-cycle checkpoint functions are fully impaired (e.g. concurrent Rb and p53 inactivation), cells fail to undergo senescence in response to ICN1, resulting in malignant transformation.
b. Endogenous Notch1 may serve as a tumor suppressor by mediating TGF-β-induced senescence. TGF-β has been implicated in replicative senescence51 as well as oncogene-induced senescence52, 53. TGF-β requires p53 to transactivate p2165. Notch1 may be targeted for inactivation by HPV oncogenes E6 and E7 during malignant transformation. E6 may suppress Notch1 by degrading p53 50. E7 also targets Rb for degradation or sequestration. We find that HPV E6 and E7 inhibit TGF-β signaling to prevent the activation of endogenous Notch1 and induction of p16INK4A and p21. While TGF-β may induce CDKIs independent of Notch1, Notch1 may regulate the SABG activity independent of cell-cycle regulation. Since oncogeneinduced senescence may involve autophagy and lysosomal functions66, Notch signaling may regulate the activity of SABG, a lysosomal enzyme 67, 68.
Notch activity can be influenced by the intensity or duration of ligand stimulation, differential Notch receptor paralogs, ligands and co-existing factors. Hypoxia and TGF-β are essential components in the tumor microenvironment to facilitate invasive growth of ESCC45–47. ICN1 interacts with transcription factors such as SMAD3 and HIF-1α43, 48. Notch1 activates Notch3 to induce squamous-cell differentiation markers including IVL and KRT1313. Since KRT13 expression peaked at a lower DOX concentration (supplementary Fig. S1a), a higher Notch activation may be required for senescence. Unlike squamous-cell differentiation, our data suggest that Notch1 may induce senescence independent of Notch3 (Supplementary Figures S14). We also confirm a recent report49 that ectopically expressed ICN3 induces senescence by suppressing Notch1 (Supplementary Figures S15), revealing a redundant role of Notch1 and Notch3 in senescence in epithelial cells.
How does Notch regulate senescence? Cell-cycle arrest can be mediated by p16INK4A and p21. We do not exclude p21, as postulated in Notch3-mediated senescence49, although ICN3 did not induce p21 in EPC1-hTERT (Supplementary Figure S15). Our data suggest that the p16INK4A-Rb pathway may have a predominant role in ICN1-induced senescence in normal esophageal keratinocytes expressing p16INK4A (Figure 4, Supplementary Figures S9 and S10); however, complex interplays may exist between multiple cell-cycle regulators (Figure 4, Supplementary Figures S9 and S10; Supplementary Table S1). Given limited RNAi efficiency especially in EPC2-T, p14ARF may not be unequivocally dismissed; however, ICN1 suppressed p14ARF during senescence (Figure 2b) as observed in oncogenic RasG12V-induced senescence in EPC2-hTERT5. The inability of p53R175H to prevent ICN1-induced senescence in EPC2-hTERT (Supplementary Figures S7) also diminished the role of p53. Nevertheless, p53 was upregulated in EPC2 undergoing replicative senescence with concurrent Notch1 activation (Figure 1c). HPV E6 may suppress Notch1 by degrading p53 39, 50. Finally, ICN1 induced senescence in EPC1-hTERT without detectable p16INK4A expression (Supplementary Figure S2). Thus, redundant pathways may allow Notch-induced senescence in cell and contextdependent manners.
We show for the first time that endogenous Notch1 mediates TGF-β-induced senescence (Figure 6), corroborating a tumor suppressor function of Notch1 activated in response to TGF-β stimulation. TGF-β signaling is implicated in replicative senescence51 as well as Ras-induced senescence52, 53. Concurrent expression of oncogenic Ras and DNMAML1 in human primary keratinocytes resulted in aggressive SCC 24, suggesting that DNMAML1 may inhibit Notch-mediated senescence activated by Ras during malignant transformation. Besides loss-of-function Notch1 mutations20–22, tumor suppressor activities of Notch have been suggested by Notch downregulation via p53 dysfunction24, 50 and EGFR overexpression54. Since senescence can be triggered by either oncogene activation or loss of tumor suppressor functions55, Notch suppression may allow these genetic lesions to promote carcinogenesis.
TGF-β also facilitates tumor progression. TGF-β induces JAG1 to activate Notch1 during epithelial-mesenchymal transition (EMT)43. Thus, Notch activation may contribute to tumor progression stimulated by TGF-β. Besides cancer cell invasion, metastasis and cancer stem cell regulation56, EMT may circumvent oncogene-induced senescence57. In this context, TGF-β and Notch seem to cooperate to activate EMT, but not senescence. What is a molecular mechanism facilitating the conversion of TGF-β and Notch from tumor suppressors to promoters? Transformed human esophageal keratinocytes undergo EMT in response to TGF-β stimulation, negating senescence through transcriptional repression of p16INK4A by ZEB1/2, EMT regulators7. Thus, a pre-existing dysfunctional p16INK4A/Rb-mediated cell-cycle regulatory machinery may nullify Notch-mediated senescence in transformed cells. Although our data show an accelerated growth and altered differentiation in tumors expressing ICN1 (Figure 7), ICN1 suppressed tumor growth by oral SCC cell lines carrying loss-of-function Notch1 mutations 58 where ICN1 induced SABG, but not morphological features of senescent cells in vitro. ICN3 inhibited tumor growth in cancer cell lines 49. By contrast, ICN3 induced aggressive inflammatory breast cancer cells when expressed in the mammary stem/progenitor cells in mice 59. Thus, it is possible that ICN1 may have differential roles in different subsets of intratumoral cells and/or premalignant cells. It also remains unclear how Notch promotes EMT in tumors. Such investigation is currently underway.
Materials and methods
Cell lines and treatment
EPC1 and EPC2, normal human esophageal keratinocytes and their derivatives (EPC1-hTERT, EPC2-hTERT, EPC2-T, EPC2-T-GFP and EPC2-T-DNMAML1) as well as EN60 and TE11 cells were described previously4, 7, 36, 47. Cells were counted with Countess™ Automated Cell Counter (Invitrogen) with 0.2% Trypan Blue dye to exclude dead cells. Population doubling time was determined as described 34. Cells were treated with 1 µM Compound E, a GSI or 5 ng/ml TGF-β1 as described 7, 13. Phase contrast images were acquired using a Nikon Eclipse E600 microscope.
Generation of pTRIPZ-ICN1 and pTRIPZ-ICN3
Platinum® Pfx DNA polymerase (Invitrogen) was used to amplify cDNAs by PCR for ICN1 (Arg-1761 to Lys-2555 of full-length human Notch1) with primers AgeI-ICN1 (5’-AGCAGCACCGGTGCCACCATGCGGCGGCAGCATGGCCAGCT-3’) and MluI-ICN1 (5’-AGCAGCACGCGTTTACTTGAAGGCCTCCGGAATGCGGG-3’); using MigRI-ICNX 60 as a template, ICN3 (Met-1663 to Ala-2331 of full length human Notch3) with primers AgeI-ICN3 (5’-AGCAGCACCGGTGCCACCATGGTGGCCCGGCGCAA-3’) and MluI-ICN3 (5’-AGCAGCACGCGTTCAGGCCAACACTTGCCTCTTG-3’); using pcDNA3.1-ICN3 (gift of Dr. Tao Wang) as a template. Following initial incubation at 94°C for 5 min, PCR was carried out for 35 cycles at 94°C for 15 sec for denaturing, 56°C for 30 sec for annealing, 68°C for 3 min for extension, with extended incubation at 68°C for 5 min after the final extension. The ICN1 and ICN3 PCR products were ligated into the pTRIPZ (Open Biosystems) at AgeI and MluI sites, replacing the parental sequence flanked by these restriction sites with either cDNA under the tetracyclineinducible promoter, resulting in creation of pTRIPZ-ICN1 and pTRIPZ-ICN3. All constructs were verified by DNA sequencing.
Retrovirus and lentivirus-mediated gene transfer
MigRI-ICNX and MigRI (control vector) were used as described13. The lentiviral pTRIPZ-ICN1, pTRIPZ-ICN3 and pGIPZ expressing short hairpin RNA (shRNA) directed against human CSL designated CSL-1 and CSL-2 (clone ID # V2LHS_114863 and V2LHS_263385) or a nonsilencing scramble sequence(RHS4346)(Open Biosystems) were transfected into HEK-293T cells with Lipofectamine LTX reagent (Invitrogen) to produce replication-incompetent viruses. Stable cell lines were established by drug selection for 7 days with 1 µg/ml of Puromycin (Invitrogen) for pTRIPZ and selected by FACS for pGIPZ-transduced GFP expressing cells by FACSVantage SE (Becton Dickinson).
Transient transfection for RNAi and dual-luciferase assays
Small interfering RNA (siRNA) directed against Notch1 (N1-A, HSS181550 and N1-B, HSS107249), HPV16 E7 (E7-A, s445412 and E7-B, s445413), p14/p16 (p14/p16-A, s216 and p14/p16-B, s218), p14 (5’-GATGCTACTGAGGAGCCAGCG-3’) and p16 (5’-AACGCACCGAAT AGTTACGGT-3’)(Figure 4a), p15 (p15-A,s2843 and p15-B, s2844) or a non-targeting scramble control sequence (4390843, Invitrogen) was transfected using the Lipofectamine RNAiMAX reagent (Invitrogen), following the manufacturer’s instructions.
Transient transfection of reporter plasmids and luciferase assays were performed as described previously 7, 13. Briefly, 400 ng of 8xCBF1-luc (designated as 8xCSL-luc)61, a Notchinducible reporter, or p3TP-Lux 62, a TGF-β-inducible reporter or pGL3-p16 63 containing a 2.3-kilobase p16INK4A promoter was transfected. Cells were incubated in the presence or absence of 1 µg/ml DOX to induce ICN1 in cells expressing ICN1TetOn for 48 hours before cell lysis. Alternatively, 5 ng/ml TGF-β1 was added at 24 hours after transfection and incubated for an additional 48 hours before cell lysis. The mean of firefly luciferase activity was normalized with the co-transfected Renilla luciferase activity. Transfection was carried out at least three times, and variation between experiments was not greater than 15%.
WST-1 cell proliferation assays
The WST-1 reagent (Roche) was used for colorimetric cell proliferation assays following the manufacturer’s instructions. All experiments were performed in sextuplicate.
Senescence-Associated β-galactosidase (SABG) assays
The Senescence β-Galactosidase Staining Kit (Cell Signaling, Danvers, MA) was used to stain senescent cells, which were scored by counting at least 100 cells high-power field (n=3–6) under light microscopy.
Cell-cycle analysis
Cellular DNA content was determined by flow cytometry. In brief, cells were fixed with 70% ethanol at −20 °C, washed twice with PBS and incubated with 50µg/ml propidium iodide and 200 µg/ml RNase A for 30 min at room temperature. At least 10,000 events were recorded and analyzed by FACSCalibur (BD Biosciences) with FlowJo software (Tree Star, Ashland, OR).
RNA isolation, cDNA synthesis and real-time RT-PCR
RNA extraction and cDNA synthesis were done as described 13, 36. Real-time RT-PCR was done with SYBR® Green and TaqMan® Gene Expression Assays (Applied Biosystems) for NOTCH1 (Hs01062014_m1), NOTCH3 (Hs00166432_m1), IVL (Hs00846307_s1), CK13 (Hs00999762_m1), CSL (Hs01068138_m1), JAG1 (Hs00164982_m1), HES5 (Hs01387463_g1), HES1(Hs00172878_m1), HEY1 (Hs00232618_m1), HEY2 (Hs00232622_m1), CDKN2A/p16INK4a (Hs99999189_m1), CDKN2B/p15INK4b (Hs00394703_m1), CDKN1A/p21 (Hs00355782_m1), CDKN1B/p27 (Hs00153277_m1), CDKN1C/p57 (Hs00175938_m1), PAI-1 (Hs01126606_m1) using the StepOnePlus™ Real-Time PCR System (Applied Biosystems). SYBR green reagent (Applied Biosystems) was used to quantitate mRNA for p14ARF, p16INK4A and β-actin as described 13, 36, 64. The relative level of each mRNA was normalized to β-actin as an internal control. The following primer sequences were used for RT-PCR to determine HPV E6 (E6 forward; 5’-TCAGGACCCACAGGAGCGACC-3’; E6 reverse; 5’-TCGACCGGTCCACCGACCC-3’) and E7 (E7 forward; 5’-ATGCATGGAGATACACCTACATTGC −3’; E7 reverse; 5’-CATTAACAGGTCTTCCAAAGTACGAATG-3’).
Western blot analysis
Whole cell lysates were prepared as described 13, 36. 20 µg of denatured protein was fractionated on a NuPAGE Bis-Tris 4–12% gel (Invitrogen). Following electrotransfer, Immobilon-P membranes (Millipore) were incubated with primary antibodies for Notch1 (1:1000 rat monoclonal 5B5; Cell Signaling, Danvers, MA), ICN1Val1744 (1:1000 rabbit monoclonal anticleaved NOTCH1 Val1744 D3B8; Cell Signaling), ICN3 (1:1000 rat monoclonal anti-NOTCH3 8G5; Cell Signaling), JAG1 (1:1000 rabbit monoclonal anti-Jagged1 28H8; Cell Signaling), pRb (1:1000 rabbit polyclonal anti-Phospho-Rb Ser780; Cell Signaling), p16 (1:1000 mouse monoclonal anti-Human p16 G175–1239; BD Biosciences), p15 (1:200 mouse monoclonal anti-p15 15P06; Santa Cruz), p21 (1:1000 mouse monoclonal anti-Human Cip1; BD Biosciences), p53 (1:1000 Rabbit polyclonal anti-p53; Cell Signaling) IVL (1:1000 mouse monoclonal anti-Involucrin clone SY5; Sigma-Aldrich, St Louis, MO), β-actin (1:30,000 mouse monoclonal anti-β-actin AC-74; Sigma Aldrich), Cat# A5316, and then with the appropriate HRP-conjugated secondary antibody (GE Healthcare, Piscataway, NJ). β-actin served as a loading control.
Soft agar colony formation assays
Soft agar colony formation assays were done as described 36. In brief, 1×103 cells were suspended in 0.67% agarose containing media and overlaid on top of a 1% agarose per well (24 well plate). 200 µl of medium with or without 1µg/ml DOX was added twice a week in each well and grown for 3 weeks. The colonies over 100 µm were counted following Giemsa staining.
Xenograft transplantation experiments and histopathological analysis
Xenograft transplantation experiments were done as described 36. In brief, 5 × 106 cells were suspended in 50% Matrigel and implanted subcutaneously into the dorsal skin of athymic nu/nu mice (4–6 weeks old)(Charles River Breeding Laboratories). Tumor growth was monitored and histopathological analysis was done as described13. Immunohistochemistry was done using primary antibodies for Notch1 (polyclonal anti-NOTCH1 ab27526; 1:500 at 4°C overnight with sections microwaved at pH3.0)(Abcam, Cambridge, MA), Ki67 (Rabbit monoclonal anti-Ki67 ab16667; 1:200 at 4°C overnight with sections microwaved at pH6.0 Abcam), Caspase3 (Rabbit monoclonal anti Cleaved Caspase-3 Asp175 5A1E 9664; 1:800 at 4°C overnight with sections microwaved at pH6.0 Cell Signaling). Signals were developed using the diaminobenzidine (DAB) substrate kit (Vector Laboratories, Burlingame, CA) following incubation with secondary anti-mouse IgG (Vector)(1:100 at 37°C for 30 min) or anti-rabbit IgG (Vector)(1:200 at 37°C for 30 min), and counterstained with Hematoxylin (Fisher Scientific CS401-1D). Stained objects were examined with a Nikon Microphot microscope and imaged with a digital camera. The immunohistochemical staining was assessed independently (SN and AKS) and the intensity was expressed as negative (-), weakly positive (+) or moderately positive (++). All experiments were done under approved protocols from the University of Pennsylvania.
Statistical Analysis
Data are presented as mean ± SE or mean ± SD and were analyzed by two-tailed Student’s t test. P <0.05 was considered significant.
Supplementary Material
Acknowledgments
We thank Dr. Hiroshi Shirasawa (Chiba University, Chiba, Japan) for a gift of EN60 cells. We are grateful to the Molecular Pathology & Imaging, Molecular Biology/Gene Expression and Cell Culture Core Facilities of the NIH/NIDDK Center for Molecular Studies in Digestive and Liver Diseases (P30-DK050306) and of the NIH P01CA098101. This study was supported in part by NIH Grants P01CA098101 (to SK, MN, KAW, DB, HK, SN, SO, PAG, AJK, AB, KW, JAD, HN and AKR), U01CA143056 (AKR), R01DK077005 (HN), K26 RR032714 (HN), Pennsylvania CURE Program Grant (HN), F32-CA174176 (KAW), K08DE022842 (DB), K07CA137140 (AME), University of Pennsylvania University Research Foundation Award (HN), University of Pennsylvania, Abramson Cancer Center Pilot Project Grant (HN), and the American Cancer Society RP-10-033-01-CCE (AKR).
Abbreviations
- DNMAML1
dominant negative MAML1
- DOX
doxycycline
- EMT
epithelial-mesenchymal transition
- ESCC
esophageal squamous cell carcinoma
- FACS
fluorescence activated cell sorting
- FBS
fetal bovine serum
- GSI
γ-secretase inhibitor
- GFP
green fluorescent protein
- HPV
human papilloma virus
- ICN
intracellular domain of Notch
- RNAi
RNA interference
- RT-PCR
reverse-transcription polymerase chain reaction
- SABG
senescence-associated β-galactosidase activity
- SCC
squamous cell carcinomas
- shRNA
short hairpin RNA
- Tet-On
tetracycline inducible
- TGF-β
transforming growth factor-β
Footnotes
Conflict of Interest The authors declare no conflict of interest.
References
- 1.Enzinger PC, Mayer RJ. Esophageal cancer. N Engl J Med. 2003;349:2241–2252. doi: 10.1056/NEJMra035010. [DOI] [PubMed] [Google Scholar]
- 2.Nakagawa H, Katzka D, Rustgi AK. Biology of esophageal cancer. In: AK Rustgi., editor. Gastrointestinal Cancers. London: Elsevier; 2003. pp. 241–251. [Google Scholar]
- 3.Harada H, Nakagawa H, Oyama K, Takaoka M, Andl CD, Jacobmeier B, et al. Telomerase induces immortalization of human esophageal keratinocytes without p16INK4a inactivation. Mol Cancer Res. 2003;1:729–738. [PubMed] [Google Scholar]
- 4.Sashiyama H, Shino Y, Kawamata Y, Tomita Y, Ogawa N, Shimada H, et al. Immortalization of human esophageal keratinocytes by E6 and E7 of human papillomavirus type 16. International journal of oncology. 2001;19:97–103. doi: 10.3892/ijo.19.1.97. [DOI] [PubMed] [Google Scholar]
- 5.Takaoka M, Harada H, Deramaudt TB, Oyama K, Andl CD, Johnstone CN, et al. Ha-Ras(G12V) induces senescence in primary and immortalized human esophageal keratinocytes with p53 dysfunction. Oncogene. 2004;23:6760–6768. doi: 10.1038/sj.onc.1207923. [DOI] [PubMed] [Google Scholar]
- 6.Oyama K, Okawa T, Nakagawa H, Takaoka M, Andl CD, Kim SH, et al. AKT induces senescence in primary esophageal epithelial cells but is permissive for differentiation as revealed in organotypic culture. Oncogene. 2007;26:2353–2364. doi: 10.1038/sj.onc.1210025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ohashi S, Natsuizaka M, Wong GS, Michaylira CZ, Grugan KD, Stairs DB, et al. Epidermal growth factor receptor and mutant p53 expand an esophageal cellular subpopulation capable of epithelial-to-mesenchymal transition through ZEB transcription factors. Cancer Res. 2010;70:4174–4184. doi: 10.1158/0008-5472.CAN-09-4614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Okawa T, Michaylira CZ, Kalabis J, Stairs DB, Nakagawa H, Andl CD, et al. The functional interplay between EGFR overexpression, hTERT activation, and p53 mutation in esophageal epithelial cells with activation of stromal fibroblasts induces tumor development, invasion, and differentiation. Genes Dev. 2007;21:2788–2803. doi: 10.1101/gad.1544507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim SH, Nakagawa H, Navaraj A, Naomoto Y, Klein-Szanto AJ, Rustgi AK, et al. Tumorigenic conversion of primary human esophageal epithelial cells using oncogene combinations in the absence of exogenous Ras. Cancer Res. 2006;66:10415–10424. doi: 10.1158/0008-5472.CAN-06-2104. [DOI] [PubMed] [Google Scholar]
- 10.McElhinny AS, Li JL, Wu L. Mastermind-like transcriptional co-activators: emerging roles in regulating cross talk among multiple signaling pathways. Oncogene. 2008;27:5138–5147. doi: 10.1038/onc.2008.228. [DOI] [PubMed] [Google Scholar]
- 11.Blanpain C, Lowry WE, Pasolli HA, Fuchs E. Canonical notch signaling functions as a commitment switch in the epidermal lineage. Genes Dev. 2006;20:3022–3035. doi: 10.1101/gad.1477606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rangarajan A, Talora C, Okuyama R, Nicolas M, Mammucari C, Oh H, et al. Notch signaling is a direct determinant of keratinocyte growth arrest and entry into differentiation. Embo J. 2001;20:3427–3436. doi: 10.1093/emboj/20.13.3427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ohashi S, Natsuizaka M, Yashiro-Ohtani Y, Kalman RA, Nakagawa M, Wu L, et al. NOTCH1 and NOTCH3 coordinate esophageal squamous differentiation through a CSL-dependent transcriptional network. Gastroenterology. 2010;139:2113–2123. doi: 10.1053/j.gastro.2010.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ranganathan P, Weaver KL, Capobianco AJ. Notch signalling in solid tumours: a little bit of everything but not all the time. Nat Rev Cancer. 2011;11:338–351. doi: 10.1038/nrc3035. [DOI] [PubMed] [Google Scholar]
- 15.Hijioka H, Setoguchi T, Miyawaki A, Gao H, Ishida T, Komiya S, et al. Upregulation of Notch pathway molecules in oral squamous cell carcinoma. Int J Oncol. 2010;36:817–822. doi: 10.3892/ijo_00000558. [DOI] [PubMed] [Google Scholar]
- 16.Zagouras P, Stifani S, Blaumueller CM, Carcangiu ML, Artavanis-Tsakonas S. Alterations in Notch signaling in neoplastic lesions of the human cervix. Proc Natl Acad Sci U S A. 1995;92:6414–6418. doi: 10.1073/pnas.92.14.6414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rangarajan A, Syal R, Selvarajah S, Chakrabarti O, Sarin A, Krishna S. Activated Notch1 signaling cooperates with papillomavirus oncogenes in transformation and generates resistance to apoptosis on matrix withdrawal through PKB/Akt. Virology. 2001;286:23–30. doi: 10.1006/viro.2001.0867. [DOI] [PubMed] [Google Scholar]
- 18.Weijzen S, Zlobin A, Braid M, Miele L, Kast WM. HPV16 E6 and E7 oncoproteins regulate Notch-1 expression and cooperate to induce transformation. Journal of cellular physiology. 2003;194:356–362. doi: 10.1002/jcp.10217. [DOI] [PubMed] [Google Scholar]
- 19.Talora C, Sgroi DC, Crum CP, Dotto GP. Specific down-modulation of Notch1 signaling in cervical cancer cells is required for sustained HPV-E6/E7 expression and late steps of malignant transformation. Genes & development. 2002;16:2252–2263. doi: 10.1101/gad.988902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Agrawal N, Frederick MJ, Pickering CR, Bettegowda C, Chang K, Li RJ, et al. Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science. 2011;333:1154–1157. doi: 10.1126/science.1206923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stransky N, Egloff AM, Tward AD, Kostic AD, Cibulskis K, Sivachenko A, et al. The mutational landscape of head and neck squamous cell carcinoma. Science. 2011;333:1157–1160. doi: 10.1126/science.1208130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang NJ, Sanborn Z, Arnett KL, Bayston LJ, Liao W, Proby CM, et al. Loss-of-function mutations in Notch receptors in cutaneous and lung squamous cell carcinoma. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:17761–17766. doi: 10.1073/pnas.1114669108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Agrawal N, Jiao Y, Bettegowda C, Hutfless SM, Wang Y, David S, et al. Comparative genomic analysis of esophageal adenocarcinoma and squamous cell carcinoma. Cancer Discov. 2012;2:899–905. doi: 10.1158/2159-8290.CD-12-0189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lefort K, Mandinova A, Ostano P, Kolev V, Calpini V, Kolfschoten I, et al. Notch1 is a p53 target gene involved in human keratinocyte tumor suppression through negative regulation of ROCK1/2 and MRCKalpha kinases. Genes Dev. 2007;21:562–577. doi: 10.1101/gad.1484707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li T, Wen H, Brayton C, Laird FM, Ma G, Peng S, et al. Moderate reduction of gamma-secretase attenuates amyloid burden and limits mechanism-based liabilities. J Neurosci. 2007;27:10849–10859. doi: 10.1523/JNEUROSCI.2152-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nicolas M, Wolfer A, Raj K, Kummer JA, Mill P, van Noort M, et al. Notch1 functions as a tumor suppressor in mouse skin. Nat Genet. 2003;33:416–421. doi: 10.1038/ng1099. [DOI] [PubMed] [Google Scholar]
- 27.Pan Y, Lin MH, Tian X, Cheng HT, Gridley T, Shen J, et al. gamma-secretase functions through Notch signaling to maintain skin appendages but is not required for their patterning or initial morphogenesis. Dev Cell. 2004;7:731–743. doi: 10.1016/j.devcel.2004.09.014. [DOI] [PubMed] [Google Scholar]
- 28.Proweller A, Tu L, Lepore JJ, Cheng L, Lu MM, Seykora J, et al. Impaired notch signaling promotes de novo squamous cell carcinoma formation. Cancer Res. 2006;66:7438–7444. doi: 10.1158/0008-5472.CAN-06-0793. [DOI] [PubMed] [Google Scholar]
- 29.Zhang YW, Wang R, Liu Q, Zhang H, Liao FF, Xu H. Presenilin/gamma-secretase-dependent processing of beta-amyloid precursor protein regulates EGF receptor expression. Proc Natl Acad Sci U S A. 2007;104:10613–10618. doi: 10.1073/pnas.0703903104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Demehri S, Turkoz A, Kopan R. Epidermal Notch1 loss promotes skin tumorigenesis by impacting the stromal microenvironment. Cancer Cell. 2009;16:55–66. doi: 10.1016/j.ccr.2009.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Venkatesh D, Fredette N, Rostama B, Tang Y, Vary CP, Liaw L, et al. RhoA-mediated signaling in Notch-induced senescence-like growth arrest and endothelial barrier dysfunction. Arterioscler Thromb Vasc Biol. 2011;31:876–882. doi: 10.1161/ATVBAHA.110.221945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu ZJ, Tan Y, Beecham GW, Seo DM, Tian R, Li Y, et al. Notch activation induces endothelial cell senescence and pro-inflammatory response: implication of Notch signaling in atherosclerosis. Atherosclerosis. 2012;225:296–303. doi: 10.1016/j.atherosclerosis.2012.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lowell S, Jones P, Le Roux I, Dunne J, Watt FM. Stimulation of human epidermal differentiation by delta-notch signalling at the boundaries of stem-cell clusters. Curr Biol. 2000;10:491–500. doi: 10.1016/s0960-9822(00)00451-6. [DOI] [PubMed] [Google Scholar]
- 34.Harada H, Nakagawa H, Oyama K, Takaoka M, Andl CD, Jacobmeier B, et al. Telomerase induces immortalization of human esophageal keratinocytes without p16INK4a inactivation. Mol Cancer Res. 2003;1:729–738. [PubMed] [Google Scholar]
- 35.Dickson MA, Hahn WC, Ino Y, Ronfard V, Wu JY, Weinberg RA, et al. Human keratinocytes that express hTERT and also bypass a p16(INK4a)-enforced mechanism that limits life span become immortal yet retain normal growth and differentiation characteristics. Mol Cell Biol. 2000;20:1436–1447. doi: 10.1128/mcb.20.4.1436-1447.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ohashi S, Natsuizaka M, Naganuma S, Kagawa S, Kimura S, Itoh H, et al. A NOTCH3-mediated squamous cell differentiation program limits expansion of EMT-competent cells that express the ZEB transcription factors. Cancer Res. 2011;71:6836–6847. doi: 10.1158/0008-5472.CAN-11-0846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jia LQ, Osada M, Ishioka C, Gamo M, Ikawa S, Suzuki T, et al. Screening the p53 status of human cell lines using a yeast functional assay. Mol Carcinog. 1997;19:243–253. doi: 10.1002/(sici)1098-2744(199708)19:4<243::aid-mc5>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 38.Liu Q, Yan YX, McClure M, Nakagawa H, Fujimura F, Rustgi AK. MTS-1 (CDKN2) tumor suppressor gene deletions are a frequent event in esophagus squamous cancer and pancreatic adenocarcinoma cell lines. Oncogene. 1995;10:619–622. [PubMed] [Google Scholar]
- 39.Dotto GP. Crosstalk of Notch with p53 and p63 in cancer growth control. Nat Rev Cancer. 2009;9:587–595. doi: 10.1038/nrc2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bertwistle D, Sugimoto M, Sherr CJ. Physical and functional interactions of the Arf tumor suppressor protein with nucleophosmin/B23. Mol Cell Biol. 2004;24:985–996. doi: 10.1128/MCB.24.3.985-996.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ovcharenko I, Nobrega MA, Loots GG, Stubbs L. ECR Browser: a tool for visualizing and accessing data from comparisons of multiple vertebrate genomes. Nucleic Acids Res. 2004;32:W280–W286. doi: 10.1093/nar/gkh355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee DK, Kim BC, Kim IY, Cho EA, Satterwhite DJ, Kim SJ. The human papilloma virus E7 oncoprotein inhibits transforming growth factor-beta signaling by blocking binding of the Smad complex to its target sequence. The Journal of biological chemistry. 2002;277:38557–38564. doi: 10.1074/jbc.M206786200. [DOI] [PubMed] [Google Scholar]
- 43.Zavadil J, Cermak L, Soto-Nieves N, Bottinger EP. Integration of TGF-beta/Smad and Jagged1/Notch signalling in epithelial-to-mesenchymal transition. Embo J. 2004;23:1155–1165. doi: 10.1038/sj.emboj.7600069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bierie B, Moses HL. Tumour microenvironment: TGFbeta: the molecular Jekyll and Hyde of cancer. Nat Rev Cancer. 2006;6:506–520. doi: 10.1038/nrc1926. [DOI] [PubMed] [Google Scholar]
- 45.Natsugoe S, Xiangming C, Matsumoto M, Okumura H, Nakashima S, Sakita H, et al. Smad4 and transforming growth factor beta1 expression in patients with squamous cell carcinoma of the esophagus. Clin Cancer Res. 2002;8:1838–1842. [PubMed] [Google Scholar]
- 46.Natsuizaka M, Ohashi S, Wong GS, Ahmadi A, Kalman RA, Budo D, et al. Insulin-like growth factor-binding protein-3 promotes transforming growth factor-{beta}1-mediated epithelial-to-mesenchymal transition and motility in transformed human esophageal cells. Carcinogenesis. 2010;31:1344–1353. doi: 10.1093/carcin/bgq108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Natsuizaka M, Naganuma S, Kagawa S, Ohashi S, Ahmadi A, Subramanian H, et al. Hypoxia induces IGFBP3 in esophageal squamous cancer cells through HIF-1alpha-mediated mRNA transcription and continuous protein synthesis. Faseb J. 2012;26:2620–2630. doi: 10.1096/fj.11-198598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gustafsson MV, Zheng X, Pereira T, Gradin K, Jin S, Lundkvist J, et al. Hypoxia requires notch signaling to maintain the undifferentiated cell state. Developmental cell. 2005;9:617–628. doi: 10.1016/j.devcel.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 49.Cui H, Kong Y, Xu M, Zhang H. Notch3 functions as a tumor suppressor by controlling cellular senescence. Cancer Research. 2013;73:3451–3459. doi: 10.1158/0008-5472.CAN-12-3902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yugawa T, Handa K, Narisawa-Saito M, Ohno S, Fujita M, Kiyono T. Regulation of Notch1 gene expression by p53 in epithelial cells. Mol Cell Biol. 2007;27:3732–3742. doi: 10.1128/MCB.02119-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pascal T, Debacq-Chainiaux F, Chretien A, Bastin C, Dabee AF, Bertholet V, et al. Comparison of replicative senescence and stress-induced premature senescence combining differential display and low-density DNA arrays. FEBS Lett. 2005;579:3651–3659. doi: 10.1016/j.febslet.2005.05.056. [DOI] [PubMed] [Google Scholar]
- 52.Tremain R, Marko M, Kinnimulki V, Ueno H, Bottinger E, Glick A. Defects in TGF-beta signaling overcome senescence of mouse keratinocytes expressing v-Ha-ras. Oncogene. 2000;19:1698–1709. doi: 10.1038/sj.onc.1203471. [DOI] [PubMed] [Google Scholar]
- 53.Lin S, Yang J, Elkahloun AG, Bandyopadhyay A, Wang L, Cornell JE, et al. Attenuation of TGF-beta signaling suppresses premature senescence in a p21-dependent manner and promotes oncogenic Ras-mediated metastatic transformation in human mammary epithelial cells. Molecular biology of the cell. 2012;23:1569–1581. doi: 10.1091/mbc.E11-10-0849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kolev V, Mandinova A, Guinea-Viniegra J, Hu B, Lefort K, Lambertini C, et al. EGFR signalling as a negative regulator of Notch1 gene transcription and function in proliferating keratinocytes and cancer. Nat Cell Biol. 2008;10:902–911. doi: 10.1038/ncb1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kuilman T, Michaloglou C, Mooi WJ, Peeper DS. The essence of senescence. Genes & development. 2010;24:2463–2479. doi: 10.1101/gad.1971610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871–890. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 57.Ansieau S, Bastid J, Doreau A, Morel AP, Bouchet BP, Thomas C, et al. Induction of EMT by twist proteins as a collateral effect of tumor-promoting inactivation of premature senescence. Cancer Cell. 2008;14:79–89. doi: 10.1016/j.ccr.2008.06.005. [DOI] [PubMed] [Google Scholar]
- 58.Pickering CR, Zhang J, Yoo SY, Bengtsson L, Moorthy S, Neskey DM, et al. Integrative genomic characterization of oral squamous cell carcinoma identifies frequent somatic drivers. Cancer Discov. 2013;3:770–781. doi: 10.1158/2159-8290.CD-12-0537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ling H, Sylvestre JR, Jolicoeur P. Cyclin D1-Dependent Induction of Luminal Inflammatory Breast Tumors by Activated Notch3. Cancer Research. 2013 doi: 10.1158/0008-5472.CAN-13-0409. [DOI] [PubMed] [Google Scholar]
- 60.Aster JC, Xu L, Karnell FG, Patriub V, Pui JC, Pear WS. Essential roles for ankyrin repeat and transactivation domains in induction of T-cell leukemia by notch1. Mol Cell Biol. 2000;20:7505–7515. doi: 10.1128/mcb.20.20.7505-7515.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jeffries S, Capobianco AJ. Neoplastic transformation by Notch requires nuclear localization. Mol Cell Biol. 2000;20:3928–3941. doi: 10.1128/mcb.20.11.3928-3941.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wrana JL, Attisano L, Carcamo J, Zentella A, Doody J, Laiho M, et al. TGF beta signals through a heteromeric protein kinase receptor complex. Cell. 1992;71:1003–1014. doi: 10.1016/0092-8674(92)90395-s. [DOI] [PubMed] [Google Scholar]
- 63.Mroz EA, Baird AH, Michaud WA, Rocco JW. COOH-terminal binding protein regulates expression of the p16INK4A tumor suppressor and senescence in primary human cells. Cancer Res. 2008;68:6049–6053. doi: 10.1158/0008-5472.CAN-08-1279. [DOI] [PubMed] [Google Scholar]
- 64.Maruo S, Zhao B, Johannsen E, Kieff E, Zou J, Takada K. Epstein-Barr virus nuclear antigens 3C and 3A maintain lymphoblastoid cell growth by repressing p16INK4A and p14ARF expression. Proc Natl Acad Sci U S A. 2011;108:1919–1924. doi: 10.1073/pnas.1019599108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cordenonsi M, Dupont S, Maretto S, Insinga A, Imbriano C, Piccolo S. Links between tumor suppressors: p53 is required for TGF-beta gene responses by cooperating with Smads. Cell. 2003;113:301–314. doi: 10.1016/s0092-8674(03)00308-8. [DOI] [PubMed] [Google Scholar]
- 66.Young AR, Narita M, Ferreira M, Kirschner K, Sadaie M, Darot JF, et al. Autophagy mediates the mitotic senescence transition. Genes & development. 2009;23:798–803. doi: 10.1101/gad.519709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lee BY, Han JA, Im JS, Morrone A, Johung K, Goodwin EC, et al. Senescence-associated beta-galactosidase is lysosomal beta-galactosidase. Aging Cell. 2006;5:187–195. doi: 10.1111/j.1474-9726.2006.00199.x. [DOI] [PubMed] [Google Scholar]
- 68.Kurz DJ, Decary S, Hong Y, Erusalimsky JD. Senescence-associated (beta)-galactosidase reflects an increase in lysosomal mass during replicative ageing of human endothelial cells. Journal of cell science. 2000;113(Pt 20):3613–3622. doi: 10.1242/jcs.113.20.3613. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.