

Abstract
A new study identifies homozygous missense mutations in SGOL1, which encodes a component of the cohesin complex, in a newly described disorder termed Chronic Atrial and Intestinal Dysrhythmia (CAID) syndrome. These findings implicate cohesin in the regulation of intrinsic cardiac and intestinal rhythm and further expand the growing group of disorders termed the cohesinopathies.
Ever since the early work of German biologist and cytogeneticist Walther Flemming (1843–1905) describing chromatin and mitotic chromosomes1, a critical area of investigation has been the identification of those factors that hold sister chromosomes together during the mitotic process and allow for precisely timed and accurate chromosome segregation to daughter cells at the end of mitosis. The first identification of such components—termed ‘stability of mini chromosomes’ and, subsequently, ‘structural maintenance of chromosomes’, or SMC, proteins—in yeast2,3 led to the rapid delineation of a complex of proteins termed the cohesin complex and its regulators that serve the canonical role of sister chromatid cohesion, both in meiosis and mitosis4,5. The cohesin complex effectively forms a ringlike structure that ‘embraces’ the sister chromatids throughout replication. Significantly knocking down or eliminating key members of the cohesin complex is in general lethal to the organism, resulting in severe defects in chromatid segregation and aneuploidy. It was therefore surprising that mutations in components of the cohesin complex result in very specific developmental disorders in humans, collectively termed cohesinopathies, suggesting that cohesin might have roles in biological processes beyond sister chromatid segregation4–6.
The growing collection of cohesinopathies include Cornelia de Lange syndrome (CdLS), which is caused by dominant mutations in the genes encoding cohesin regulators (NIPBL and HDAC8) and cohesin structural components (SMC1A, SMC3 and RAD21); Roberts syndrome (RBS), which is caused by recessive mutations in the ESCO2 gene encoding a cohesin regulator; Warsaw breakage syndrome (WBS), which is caused by recessive mutations in the DNA helicase gene DDX11; and X-linked α-thalassemia mental retardation syndrome, caused by dominant mutations in the ATRX gene. Now, Gregor Andelfinger and colleagues report a new cohesinopathy called CAID syndrome that is caused by recessive mutations in the SGOL1 gene (encoding shugoshin-like 1)7.
Components of the cohesin pathway
The cohesin ring is composed of two SMC proteins, SMC1A and SMC3, a kleisin protein RAD21 and a kleisin-binding protein (STAG1 or STAG2) (Fig. 1). The association of cohesin rings with chromatin is a dynamic process and is tightly controlled by multiple regulatory proteins at various cell cycle stages, including the NIPBL-MAU2 heterodimer loading complex4,5,8, DNA replication factor C and the DNA helicase DDX11, which establish cohesion9, the ESCO1 and ESCO2 acetyltransferases that stabilize bound cohesin rings, the cohesinreleasing complex (WAPAL and PDS5A), and the polo-like kinase (PLK1) and aurora B kinase (AURKB)4,5,8. Pericentromeric and some chromosome- arm cohesins, however, are spared from this process through protection by the shugoshin (SGOL) and protein phosphatase 2A complex, which binds and dephosphorylates centromeric cohesin and PDS5-bound CDCA5 (ref. 9). Most of the cohesin rings released from chromatin during mitosis are recycled through SMC3 deacetylation by the cohesin deacetylase HDAC8 (refs. 4,10).
Figure 1.
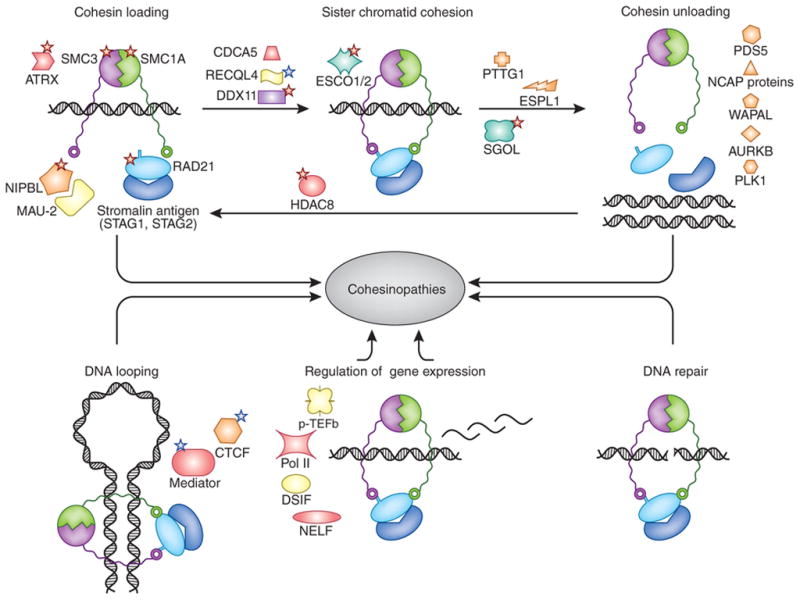
Cohesin structural and regulatory components and functions. Shown are the canonical function of cohesin in sister chromatid cohesion (top) and its non-canonical roles in genome regulation and DNA looping, regulation of gene expression and DNA repair (bottom). Key cohesin structural components and regulators are depicted. Red stars indicate proteins whose mutated forms are implicated in known cohesinopathies (NIPBL, SMC1A, SMC3, RAD21 and HDAC8 associated with CdLS, ATRX associated with X-linked α-thalassemia mental retardation syndrome, DDX11 associated with WBS, ESCO2 associated with RBS, SGOL1 associated with CAID syndrome), and blue stars indicate proteins implicated in other developmental disorders not classified as cohesinopathies (RECQL4 associated with Rothmund-Thomson, Baller-Gerold and RAPADILINO syndromes, MED12 associated with Opitz-Kaveggia, Ohdo and Lujan-Fryns syndromes, CTCF associated with autosomal dominant mental retardation 21).
Whereas the cohesin complex is well known for its canonical role in sister chromatid cohesion during cell division, cohesin and its regulators have been implicated more recently as a key regulator of multiple basic cellular processes, including DNA damage repair, regulation of gene expression, organization of chromosome architecture and centriole engagement8. The critical role of cohesin in human development was first evidenced by the discoveries that heterozygous mutations in NIPBL and homozygous or compound heterozygous mutations in ESCO2 were responsible for CdLS and RBS, respectively4–6.
Molecular mechanisms of cohesinopathies
The underlying molecular mechanism for most cohesinopathies is an active field of research. Whereas disturbances in cell cycle progression and DNA damage repair are observed for some individuals with CdLS, RBS and WBS, their contribution to disease pathogenesis is unclear6. In CdLS probands heterozygous for NIPBL mutations, cells maintain about 70% of the normal levels of NIPBL transcript and sister chromatid cohesion defects are absent11. Mounting evidence suggests that widespread alterations in gene expression downstream of mutations in the cohesin pathway likely account for the multisystem developmental phenotypes seen in CdLS and other cohesinopathies. The Drosophila melanogaster homolog of NIPBL, Nipped-B, is required to mediate the long-range activation of two essential genes, cut and Ultrabithorax, suggesting a role for cohesin in the regulation of gene expression12. The Drosophila homologs of SMC1A, STAG1, STAG2 and RAD21 are required for the development of postmitotic neurons and salivary gland cells, providing direct proof of cohesion-independent roles for cohesin in gene expression13. Together, these findings strongly argue that disruptions of the functions of cohesin and its regulators in gene regulation likely underlie pathogenic contributors to the clinical manifestations seen in the cohesinopathies.
A large body of data shows that cohesin localization on chromosomes is distinct in different species and different cell types. A unifying theme, however, is that cohesin tends to bind to actively transcribed genes4. Drosophila cohesin and Nipped-B colocalize throughout the genome and preferentially bind to the promoter and coding regions of actively transcribed genes. Their binding sites overlap with those of RNA polymerase II but are generally excluded from silenced regions marked by trimethylation of histone H3 at lysine 27 (ref. 4). In mammalian cells, cohesin binds to a large number of sites throughout the genome and colocalizes with CTCF14. In mouse embryonic stem cells, cohesin colocalizes with the mediator complex around enhancer and core promoter regions and moderates cell type–specific expression by bridging DNA looping15. Altogether, cohesin likely regulates expression via multiple modes, including DNA loop formation and divergent interaction with tissue-specific transcriptional regulators.
In the study reported in this issue, Chetaille et al.7 report homozygous mutations in the SGOL1 gene in 17 subjects from 14 families with a combination of sick sinus syndrome and chronic intestinal pseudo-obstruction representing a new disorder they have termed CAID syndrome. The authors show a role for SGOL1 in cohesin and cell cycle regulation and heterochromatin repulsion at the centromeres, suggesting that CAID syndrome constitutes a cohesinopathy. The clinical manifestations of the syndrome, limited to the autonomic rhythmic functioning of cardiac and intestinal cells, represent a novel phenotype among cohesinopathies and raises important pathophysiological questions. For example, are these differences in phenotype the result of a disruption of the cell type– and developmental stage–specific gene regulatory functions of cohesin or of an alteration in cell cycling, senescence or another SGOL1-specific function beyond cohesin regulation? The functional consequences of cohesin disruption in the cohesinopathies, with their highly specific yet diverse phenotypes, still require much work in understanding the manifold roles that the cohesin complex has in regulating developmental processes.
Footnotes
COMPETING FINANCIAL INTERESTS
The author declares no competing financial interests.
References
- 1.Paweletz N. Nat Rev Mol Cell Biol. 2001;2:72–75. doi: 10.1038/35048077. [DOI] [PubMed] [Google Scholar]
- 2.Larionov VL, et al. Curr Genet. 1985;10:15–20. doi: 10.1007/BF00418488. [DOI] [PubMed] [Google Scholar]
- 3.Strunnikov AV, Larionov VL, Koshland D. J Cell Biol. 1993;123:635–648. doi: 10.1083/jcb.123.6.1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dorsett D, Merkenschlager M. Curr Opin Cell Biol. 2013;25:327–333. doi: 10.1016/j.ceb.2013.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Losada A. Nat Rev Cancer. 2014;14:389–393. doi: 10.1038/nrc3743. [DOI] [PubMed] [Google Scholar]
- 6.Brooker AS, Berkowitz KM. Methods Mol Biol. 2014;1170:229–266. doi: 10.1007/978-1-4939-0888-2_11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chetaille P, et al. Nat Genet. 2014;46:1245–1249. doi: 10.1038/ng.3113. [DOI] [PubMed] [Google Scholar]
- 8.Nasmyth K. Nat Cell Biol. 2011;13:1170–1177. doi: 10.1038/ncb2349. [DOI] [PubMed] [Google Scholar]
- 9.Liu H, Rankin S, Yu H. Nat Cell Biol. 2013;15:40–49. doi: 10.1038/ncb2637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Deardorff MA, et al. Nature. 2012;489:313–317. doi: 10.1038/nature11316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kawauchi S, et al. PLoS Genet. 2009;5:e1000650. doi: 10.1371/journal.pgen.1000650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rollins RA, Morcillo P, Dorsett D. Genetics. 1999;152:577–593. doi: 10.1093/genetics/152.2.577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pauli A, et al. Curr Biol. 2010;20:1787–1798. doi: 10.1016/j.cub.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wendt KS, et al. Nature. 2008;451:796–801. doi: 10.1038/nature06634. [DOI] [PubMed] [Google Scholar]
- 15.Kagey MH, et al. Nature. 2010;467:430–435. doi: 10.1038/nature09380. [DOI] [PMC free article] [PubMed] [Google Scholar]