

Abstract
The ability to repair DNA is important for the conservation of genetic information of living organisms. Cells have a number of ways to restore a damaged DNA, such as direct DNA repair, base excision repair, and nucleotide excision repair. One of the proteins that can perform direct repair of DNA bases is Escherichia coli AlkB. In humans, there are 9 identified AlkB homologs, including AlkB homolog 1 (ALKBH1). Many of these proteins catalyze the direct oxidative dealkylation of DNA and RNA bases and, as such, have an important role in repairing DNA from damage induced by alkylating agents. In addition to the dealkylase activity, ALKBH1 can also function as an apyrimidinic/apurinic lyase and was proposed to have a distinct lyase active site. To our knowledge, no crystal structure or complete homology model of ALKBH1 protein is available. In this study, we have used homology modeling to predict the structure of ALKBH1 based on AlkB and Duffy-binding-like domain crystal structures as templates. Molecular dynamics simulations were subsequently performed on the predicted structure of ALKBH1. The positions of two disulfide bonds or a zinc-finger motif and a disulfide bond were predicted and the importance of these features was tested by mutagenesis. Possible locations for the lyase active site are proposed based on the analysis of our predicted structures and previous experimental results.
1. Introduction
Living cells depend on the conservation of genetic information stored in DNA. However, DNA is subject to various chemical reactions that can modify its structure and result in loss of its function. To ensure that genetic information is preserved and correctly propagated, cells have various pathways to protect the genetic code. For this purpose, cells can directly repair modified DNA bases, respond by regulating DNA transcription, stop their growth cycle, or undergo apoptosis [1].
Alkylation of DNA bases is an example of modifications that can result in DNA damage and mutations. There are various endogenous and exogenous factors that can induce alkylation [2], [3] and [4]. Eukaryotic cells can repair alkylation damage by a number of ways: direct de-alkylation of DNA bases, base excision repair, and nucleotide excision repair [5]. Direct dealkylation can be performed by such Escherichia coli proteins as Ada and AlkB [6], [7] and [8].
AlkB repairs DNA bases by catalyzing an oxidative dealkylation reaction. In humans, there are 9 identified AlkB homologs: ALKBH1 through ALKBH8 (alternatively named ABH1-ABH8) and FTO. The crystal structures of AlkB, ALKBH2, and ALKBH3 revealed an interesting mechanism: these enzymes flip out a nucleotide into the active site, which contains the Fe(II) cation coordinated by two histidines, an aspartate, and α-ketoglutarate [9], [10] and [11]. It has been shown that enzymes in this family vary in the base eversion mechanism [12]. The reaction also requires molecular oxygen. In the course of the reaction the damaged DNA base is restored, α-ketoglutarate is transformed into succinate plus carbon dioxide, and formaldehyde is released [9]. AlkB and its homologs share a characteristic double-stranded beta-helix or “jelly-roll” fold [9], [10] and [11]. The residues in the active site of known structures of AlkB and its homologs are strictly conserved [10], [11] and [13]. Analysis of a crystal structure of AlkB suggested that the molecular oxygen necessary for the reaction diffuses to the active site of the enzyme through a putative oxygen tunnel [13]. QM/MM calculations and energy decomposition analysis performed for AlkB revealed a total of 9 residues around its active site that are important for the catalytic activity of the enzyme [14]. AlkB is a dynamic protein and when it has no bound cofactors it is mobile and can have different conformations [15].
AlkB and its homologs differ in their affinity to DNA and RNA substrates and act on different nucleotides [16]. AlkB can repair 1-methyladenine and 3-methylcytosine and can act on both single- and double-stranded DNA [7] and [17]. ALKBH3 was shown to have a preference for single-stranded DNA, and ALKBH2 for double-stranded DNA [18] and [19]. AlkB and ALKBH3 can also repair 1-methyladenine and 3-methylcytosine in RNA [20]. AlkB, ALKBH2, and ALKBH3 repair 3-methylthymine and 1-ethyladenine in DNA [21], [22] and [23]. 3-Ethylcytidine in DNA and 1-methylguanine in RNA also can be substrates for AlkB [22] and [24]. ALKBH1 catalyzes demethylation of single-stranded DNA and RNA and repairs 3-methylcytosine, but it does not act on 1-methyladenine [25].
In addition to having the DNA repair function, AlkB and its homologs could be involved in the regulation of DNA methylation and DNA expression [26]. Advances in understanding the cellular functions of dealkylases could result in the development of new medications. Alkylating agents have been used as chemotherapeutic drugs to treat cancer. Interfering with the ability of cancer cells to remove alkyl groups from DNA bases could increase the potency of the drugs used in chemotherapy [27] and [28].
ALKBH1 has unique structural and functional characteristics. Phylogenetic analysis revealed that evolutionarily ALKBH1 is closer to AlkB than to other AlkB homologs [29]. However, ALKBH1 is almost two times larger than AlkB. In addition to the dealkylase function, ALKBH1 also has an apyrimidinic/apurinic (AP) lyase activity, and the lyase active site has been proposed to be distinct from the dealkylase active site [30], [31] and [32]. Upon performing the lyase catalysis, ALKBH1 covalently binds to the 5′ DNA product [31]. In addition, it has been proposed that ALKBH1 is implicated in the regulation of methylation patterns of histone H2A and is necessary for cell differentiation and neural development [33]. According to experimental studies by Müller et al., ALKBH1 is not involved in the base excision repair pathway [34].
A complete structure of ALKBH1 could provide insights into the mechanism and functions of the enzyme. To our knowledge, there is currently no complete folded model of ALKBH1. Vidyarthy et al. used a homology model approach to predict the structure of a region of ALKBH1 that has high sequence identity with AlkB [35]. Because ALKBH1 is substantially larger than AlkB, almost half of its sequence is not covered by the AlkB template. In this study we searched for proteins with known structures that could be used as additional templates along with AlkB. The complete tertiary structure of ALKBH1 was then predicted using the Rosetta suite of programs and molecular dynamics simulations were performed with the AMBER software. Specific residues were predicted to participate in disulfide or zinc-finger domains, and site-directed mutagenesis was carried out to assess the importance of these features. In addition, the structure was analysed and a search for a possible lyase active site was performed.
2. Methods
2.1. Template Search
With 389 residues, the ALKBH1 protein is too large for ab initio structure prediction, and thus a homology modeling approach was applied to predict its structure. AlkB and its homologs with known structures can serve as templates for the structure prediction of ALKBH1. Phylogenetic analysis revealed a close evolutionary relationship between AlkB and ALKBH1[29]. Based on these findings, AlkB (PDB ID: 3I3Q) was selected as a template. A search for additional templates was performed since ALKBH1 is larger than AlkB and has sections in its sequence that are not covered by the AlkB template. A BLAST search for sequences similar to ALKBH1 did not provide peptides with known structures that could be used as templates. A further search was performed in MODBASE, a database of annotated protein structures [36]. The search resulted in 11 structures with various degrees of sequence identity to ALKBH1. These structures were used as leads for further sequence alignment analysis. In particular, the structure of a Duffy-binding-like domain (DBL) from Plasmodium knowlesi (PDB ID: 2C6J) was found to have 29% sequence identity with ALKBH1. Sequence alignments were performed with the T-Coffee server [37]. As shown in Figure 1, DBL aligns to areas in the ALKBH1 sequence that are different from those that align to AlkB. There are also regions in ALKBH1 that align to both the DBL and the AlkB templates. DBL and AlkB together cover 88% of the sequence of ALKBH1. These two proteins were selected to serve as templates for the ALKBH1 structure prediction.
Figure 1.

Sequence alignments of AlkB and DBL with ALKBH1. Alignments with average and high scores as per T-coffee reports are color coded: orange for AlkB and cyan for DBL.
2.2. Homology Modeling
Comparative modeling application of the Rosseta 3.4 suite of programs was used to predict the structure of ALKBH1 [38]. This program was chosen as it has shown successful results in protein structure prediction [39]. Based on the model of ALKBH1 developed by Vidyarthy et al. [35] and our sequence alignment analysis, the sequence of ALKBH1 was divided into 5 consecutive sections. The 1st (residues 1–74), 3rd (residues 187–293) and 5th (residues 369–389) sections align with the AlkB sequence, while the 2nd (residues 75–186) and 4th (residues 294–368) sections align with the sequence of DBL. Two structures were modeled: one based on the AlkB template (AlkB part) and the other based on the DBL template (DBL part).
To run Rosetta, the DBL and AlkB sections of the ALKBH1 sequence were aligned to the sequences of the respective templates (See supplementary Figure S1). Three residues were introduced into the sequence of the AlkB part of ALKBH1 to compensate for the excluded DBL sections: threonine and asparagine in place of the 2nd sections and alanine in place of the 4th section. These residues were selected to match those in the aligned regions of the AlkB template. Also, there was a region in the ALKBH1 sequence that aligned to both the AlkB and DBL templates and thus was included in both modeled structural units. Introduced residues and overlapping regions were subsequently removed from the modeled structures with preference given to the AlkB model as it had overall higher alignment scores.
Ten thousand structures were generated for each of the two parts. The generated models were grouped in clusters according to structure deviations and ranked by energy score. For each of the two parts of ALKBH1, models from the largest cluster with the lowest energy were selected to ensure that the predicted structures are more energetically favorable and are consistent with the entropic effect of protein folding. For the AlkB part, there was only one cluster, indicating high target template sequence identity. For the DBL part, the largest cluster contained 78% of the generated structures and included the lowest energy structure. The selected structures of the DBL and AlkB parts of ALKBH1 were spliced together and oriented to form a tertiary ALKBH1 structure by forming 4 connections in Chimera (Figure 2)[40].
Figure 2.
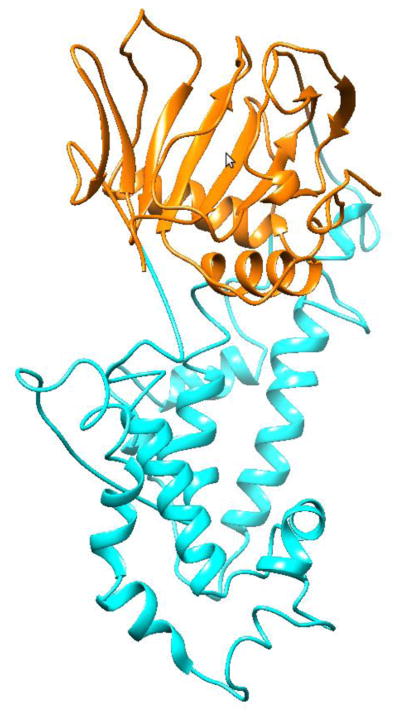
Reconstituted ALKBH1 structure. The part colored in orange corresponds to the AlkB template, in cyan – to the DBL template.
2.3. Energy Minimization and Molecular Dynamics in AMBER
The loops that link the two modeled parts of ALKBH1 were minimized in vacuum. 1000 cycles of the minimization were run with the sander program in AMBER12 [41] with the first 700 cycles using the steepest decent method. The ibelly option was set to allow only atoms in the linking loops to move. Subsequently, gas minimization for the whole structure was run for 1000 cycles. The resulting structure was solvated using TIP3P water [42], and neutralized by adding 28,644 water molecules and one sodium ion with the tleap tool resulting in a total of 92,039 atoms in the final system. Molecular dynamics (MD) simulation was first run with the ALKBH1 structure restrained under constant pressure conditions until the density of water reached 1g cm−3. Subsequently, the MD simulation was run under constant volume conditions with the restraints on ALKBH1 being gradually released. The unrestrained simulation was run for 100 ns using the ff99SB force field at 300 K temperature with the Langevin thermostat, a cut-off for non-bonded interactions of 8 Å and the sPME method for long-range electrostatics, and a time step of 1 fs. MD simulations were performed with the pmemd program in AMBER12 [41]. Two simulations were run: one with two pairs of cysteines specified as disulfide bridge forming residues, and another without such specification. Disulfide bonds were predicted based on our analysis of ALKBH1 structure and reports obtained from the DIpro server developed by Cheng et al. [43].
2.4. Site-Directed Mutagenesis
Mutants were created according to the QuickChange site directed mutagenesis protocol (Qiagen) using the respective plasmids and appropriate primers (Supplementary table S1). Plasmids were isolated with the Qiagen miniprep kit according to the manufacturer’s instructions and the introduction of the mutations was confirmed by sequencing of the DNA.
2.5. Cell Growth and Protein Purification
Plasmids were transformed into E. coli CodonPlus RIPL cells and the wild-type and variant proteins were expressed and purified as previously described [30]. Fractions were examined by sodium dodecyl sulfate polyacrylamide gel electrophoresis for their protein content and purity. The desired samples were pooled and subjected to buffer exchange into 20 mM Tris, pH 8, containing 1 mM EDTA (TE) by using a disposable G25 column (GE Healthcare) according to the manufacturer’s instructions.
2.6. Apurinic/Apyrimidinic (AP)-Site Lyase Activity Assays
AP lyase activity assays were essentially performed as described previously [31]. Briefly, single-stranded oligonucleotide 5′-AACTTCGTGCAGGCATGGTAG(dU)TTGTCTACT-3 (10 μM) was incubated with uracil DNA glycosylase (UDG) in UDG buffer for 30 min at 37°C to create the AP site. For double-stranded DNA substrates, the complementary oligonucleotide 5′-AGTAGACAAG(dU)GACCATGCCTGCACGAAGTT-3′ was annealed prior to the UDG treatment by placing the sample in a boiling water bath and cooling it to room temperature.
Standard activity assays were performed with 5 μM protein and 1 μM DNA in TE buffer. The reactions were incubated for 1 h at 37°C. When appropriate, methoxyamine was then added to the sample to a final concentration of 10 mM to reduce unspecific cleavage of the AP site and the samples were subjected to proteinase K treatment (2 U/sample) for 30 min at 65°C. DNA products were analyzed by native or denaturing PAGE and the oligonucleotides were visualized either by staining with ethidium bromide or by using a 5′- or 3′-FAM labeled substrate and detecting the fluorescence using a phosphorimager.
3. Results and Discussion
3.1. Overall Predicted Structure
The obtained structures of each of the two parts of ALKBH1 were analyzed in regard to differences from and similarities to their respective templates. Superposition of the AlkB template (PDB ID: 3I3Q) with the predicted structure of the respective part of ALKBH1 revealed that the alkylase active site residues, two histidines and an aspartate (H231, D233, and H287), are structurally conserved (Figure 3a). When the predicted AlkB part of ALKBH1 was superposed onto AlkB with cofactors (PDB ID: 3BI3), ALKBH1 residues Y222, L228, S245, F254, and N220 located in proximity to the active site were found to be also structurally conserved.
Figure 3.
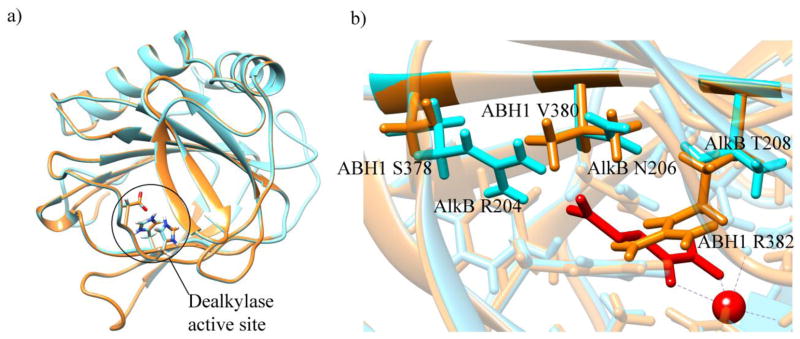
Superposed structures of ALKBH1 (orange) and the AlkB template (cyan). (a) Dealkylase active site residues. (b) Non-conserved residues in proximity to the active site. Metal ion and α-ketoglutarate of the AlkB structure are shown in red.
Residues that were not conserved were also analyzed. In the place of AlkB residues T208, N206, and R204, the predicted ALKBH1 structure had R382, V380, and S378, respectively. The arginine residue in the ALKBH1 structure in the place of T208 of AlkB overlaps with the α-ketoglutarate cofactor of AlkB when the two structures are superposed (Figure 3b). A mirror difference in the location of AlkB residue R204, with the ALKBH1 structure having a serine there, does bring a guanidine group close to the active site in AlkB, but from a different side. Such structural variation could result in a different orientation of cofactors in AlkB and ALKBH1 proteins and could modulate substrate selectivity.
Superposition of the DBL template (PDB ID: 2C6J) with the respective predicted part of ALKBH1 and analysis of the residues that were shown to be necessary for recognition of cell surface receptors by DBL as described by Singh et al. [44], revealed that only one residue was conserved: K75 of DBL superposed with K87 of ALKBH1.
3.2. Analysis of Cysteine Residues
The ALKBH1 sequence has 7 cysteines, which could have structural and functional roles. Using DIpro, a disulfide bond predictor server [43], three possible disulfide bonds were identified from the sequence of ALKBH1: C118–C129, C330–C371, and C304–C322, listed in the order of decreasing probability. Analysis of the predicted structures of ALKBH1 revealed that the disulfide bond C118–C129 is possible without significant modifications of the tertiary structure of ALKBH1. However, C330–C371 and C304–C322 disulfide bridges would require substantial changes to the structure and thus are less feasible. The DBL protein, which was used as a template to predict the structure of a respective part of ALKBH1, has a total of 12 cysteines that form 6 disulfide bonds. Superposition of the DBL template and the DBL part of ALKBH1 revealed that the ALKBH1 residues C304 and C371 correspond to two disulfide bridge-forming cysteines in the DBL structure. Formation of a disulfide bridge between these residues requires a small change to the predicted tertiary structure of ALKBH1. Thus, we propose that ALKBH1 has two disulfide bridges: C118–C129 and C304–C371 (Figure 4).
Figure 4.
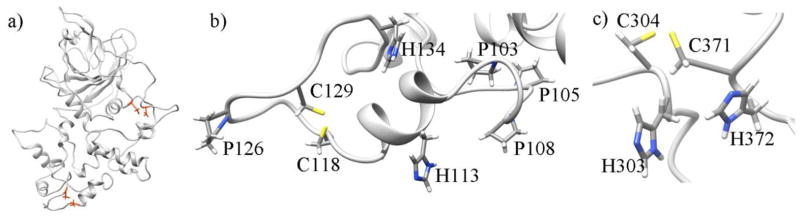
(a) Location of the two proposed disulfide bridges in the structure of ALKBH1; cysteines are shown in orange. (b) C118–C129 disulfide bridge with nearby histidines and prolines (c) C304–C371 disulfide bridge with contiguous histidines.
3.3. MD simulations
Two MD simulations were performed: with and without the two proposed disulfide bridges, each for 100 ns. With the disulfide bonds specified, the RMSD of the ALKBH1 structure stabilizes after 60 ns and overall is lower than the RMSD of the simulation without the disulfide bonds (Figure 5a). All RMSD plots were calculated for the backbone atoms with the first structures after gas minimization as reference points. RMSD analysis of each of the two parts of ALKBH1 showed that the AlkB part has a stable RMSD of around 5 Å for both systems. It can be seen in Figure 5b that the presence of the disulfide bridges has a strong stabilizing effect on the DBL part. Higher stability of the structure with the disulfide bonds is the result of lower degrees of freedom in comparison to the structure that did not have such constraint and could indicate that the ALKBH1 protein requires the formation of disulfide bonds in order to fold correctly and maintain its structural stability and function.
Figure 5.

RMSD plots for (a) ALKBH1 with and without disulfide bridges (DB); (b) AlkB and DBL parts of ALKBH1 with and without DB.
The high RMSD observed can be partly explained by a hinge movement of the DBL part around the AlkB part that can be seen in the superposition of the starting structure onto snapshots throughout the simulation (Figure 6). Loop reorganization and partial unfolding close to the flexible loops, particularly in the DBL part, also contributes to the high RMSD level. Here, we point out that the predicted AlkB and DBL parts to form the full ALKBH1 homology model were obtained separately and were spliced together without any structural information (as explained in subsection 2.2). Thus, the relaxation of the overall structure observed as a rigid body rotation of the DBL part with respect to the AlkB part during the MD simulation is not unexpected. Indeed, it is observed that both systems (with and without disulfide bonds) show a rapid decrease in potential energy in the first 5 ns (see Figure S2). This shows that the initial spliced structure is in a high energy conformation and the hinge motion results in a more stable structure.
Figure 6.

Superposition of ALKBH1 at the beginning (cyan) of the unrestrained simulation and the final structure (orange) after 100 ns of MD simulation.
3.4. Site-directed mutagenesis studies
Residues in proximity to the proposed disulfide bonds were further analyzed. For the C118–C129 bond, it was noticed that there is a histidine located 4 residues away from each of the two cysteines (H113 and H134, respectively) (Figure 4b). It was also found that there are four prolines (P126, P103, P105, P108), three of which are between these two cysteines. The disulfide bond forms a closed loop containing residues 118 through 129. Given the location of C118, C129, H113, and H134, as well as the flexibility of adjacent regions, these four residues could alternatively form a 2Cys2His zinc finger domain that could potentially play a role in DNA sequence recognition. Interestingly, cysteines that may form the second proposed disulfide bridge, C371 and C304, have contiguous histidine residues, H372 and H303 (Figure 4c).
Site-directed mutagenesis studies were carried out to test the importance of the two predicted disulfide bonds and the proposed zinc-finger motif. Thus, eight variant proteins (H113A, C118A, C129A, H134A, H303A, C304A, C371A, and H372A forms of ALKBH1) were produced in recombinant E. coli cells and purified. The AP lyase activity of these proteins was indistinguishable from the wild-type protein, thus ruling out the need for an intact disulfide or zinc finger for DNA binding or cleavage at the AP site. These studies do not, however, rule out the existence of the predicted disulfides or the presence of a zinc-finger domain in the native protein. In addition to the single-site variants, seven double-site substitutions (H113A/C118A, H113A/C129A, C118A/C129A, C129A/H134A, H303A/C304A, C304A/C371A and C371A/H372A) were examined. Most of these variants were unaffected in their AP lyase activity with the exception of the H113A/C118A and C129A/H134A proteins. The former showed 83±12.6%, the latter 65±9.5% of wild-type ALKBH1 activity using our standard assay conditions. A quadruple variant (H113A/C118A/C129A/H134A) was also created and analyzed, but showed no further reduction in its DNA cleaving activity than the double variant C129A/H134A (69±8% of wild-type ALKBH1). Taken together, these data suggest that the residues in this region of the protein are important for AP lyase activity, which is in agreement with the previous findings that K133 is the most critical active site lysine (see below).
3.5. Analysis of Lysine Residues
There are 21 lysines in the sequence of ALKBH1. Experimental studies have shown that ALKBH1 can function as an AP lyase, with lysine residues likely to act as nucleophiles in catalysis [31]. Also, it has been shown that residues in the N-terminus of ALKBH1, possibly lysines, can covalently bind to a cleaved DNA product [31]. Mutagenesis and activity assays studies have revealed that the lyase function of ALKBH1 is lowest in the K133A and K25A variants and is further decreased in a double mutant K133A/K154A [31]. It was proposed that K133 could function as a primary lyase nucleophile, with other lysines, such as K154, being able to substitute for it in the K133A variant [31]. The K182A variant also has reduced activity, though to a lesser extent than does K133A ALKBH1 [31].
Our analysis of the snapshot of the predicted structure of ALKBH1with disulfide bridges at the 100th ns of the MD simulation revealed that K133, K154, and K182 are located within 5 Å of serine residues S136, S147, and S181, respectively (see Figure 7). The distances between the residues were identified as the distances between the oxygen atoms of the hydroxyl groups of serines and the nitrogen atoms of the amino groups of lysines as these atoms could have electrostatic interactions. K133 and K182 adopt conformations where they could be activated by proximate serines for a significant amount of time during the simulation. Conversely, K154 exhibits a relatively long distance to S147 for most of the simulation time, and only gets closer at the end of the simulation. Serine residues having higher pKas could act as nucleophiles or increase the nucleophilicity of nearby lysines enabling them to perform the lyase catalytic function. In addition, it has been shown that serines can form a covalent bond with DNA cleavage products [45]. Serines could also facilitate cleavage by forming a hydrogen bond with the phosphate group of DNA, thus directing lysines to the right location for the nucleophilic attack [46].
Figure 7.

Serines in proximity to active lysine residues. Snapshots at the 100th ns of the simulation are shown for the three pairs: (a) K133 and S136 (b) K154 and S147 (c) K182 and S181. See supplementary Figure S3 for the dynamics of the distances throughout the simulation.
Proposed compensation of the lyase activity in the K133A variant by K154 could be explained by the proximity of the two lysines to each other (see Figure 8) [31]. Substituting K133 could result in a change of the orientation of K154 and S147, which could result in an increase in their catalytic ability. Alternatively, K133 and K154 could play a role in DNA recognition and non-covalent binding prior to the cleavage of the DNA molecule. When the positively charged lysines are mutated to alanines, the affinity of ALKBH1 to the negatively charged DNA backbone could be lowered as a result of decreased electrostatic interactions between the two molecules. If the C118, C129, H113, and H134 residues form a zinc finger domain that has a DNA binding function, other residues in this region of the ALKBH1 structure, such as K133 and K154, could also play a role in binding to DNA.
Figure 8.
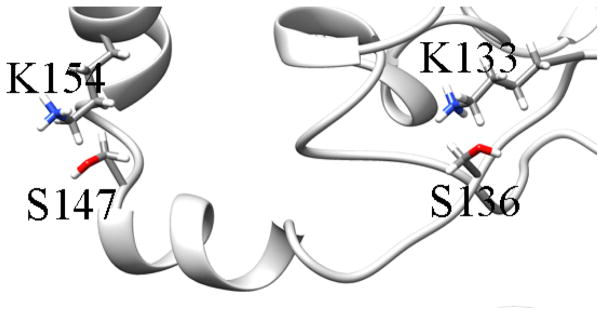
Region between residues K154 and K133.
The amino group of K25 is close to a carboxylate oxygen of D21 throughout the course of the simulation (Figure 9) and the resulting structure revealed that K25 is located in a hydrophobic region formed by F23, Y189, F192, and H188. Such an environment could significantly increase the reactivity of K25 as a nucleophile, thus making K25 a possible candidate to form and maintain a covalent bond with DNA. The K25 and D21 pair resembles the active site of Endonuclease III, which has active lysine and aspartate residues located by a hydrophobic pocket [47]. Analysis of distances from the nitrogen of the amino group of K25 to the nearby residues revealed that the distance to F192 was on average the shortest (6.6 Å), and was more stable than distances to the other residues in the region (Figure 9).
Figure 9.

Residues in proximity to K25. Shown is a snapshot at the 100th ns of the simulation with disulfide bridges. See supplementary Figure 4 for the dynamics of distances.
4. Conclusions
We have developed a homology model of ALKBH1 based on two templates with known structures: AlkB (PDB ID: 3I3Q) and DBL (PDB ID: 2C6J). Two structural units of ALKBH1, each corresponding to one of the templates, were calculated separately and subsequently combined to form the tertiary structure of ALKBH1. The Rosetta suite of programs was used to predict possible structures and to select the most feasible of them. Subsequently, MD simulations were run on the full structure using the AMBER software. Based on our final model, we propose two disulfide bridges exist in the structure of ALKBH1: C118–C129, and C304–C371. Alternatively, residues C118 and C129 together with H113 and H134 could form a zinc finger domain that could play a role in DNA recognition and binding. While site-directed mutagenesis studies could not distinguish between these interpretations, the importance of these residues was confirmed by the reduced AP lyase activity of the double and quadruple variants. The environments of lysine residues that were experimentally shown by Müller et al. [31] to be implicated in the lyase activity of ALKBH1 were analysed. Residues K133, K154, K182 are located in proximity to serines S136, S147, S181, respectively. These serines could act as nucleophiles or enhance the nucleophilicity of the nearby lysines, they could form covalent or hydrogen bonds with DNA, they could play a role of directing lysines for a nucleophilic attack. K25 is located in proximity to D21 in a hydrophobic pocket formed by F192, F23, and Y189. We propose K25 as a possible nucleophile that forms a covalent bond with the DNA product.
Supplementary Material
Highlights.
Homology modeling is used to predict the full structure of ABH1 protein.
Disulfide bonds and a zinc finger domain in the structure of ABH1 are proposed.
Mutagenesis is performed on residues predicted to form disulfide bonds/zinc finger.
Environment of residues important for lyase function is analyzed.
Acknowledgments
This work was supported in part by the National Institutes of Health (GM063584 to R.P.H. and GM108583-A1 to G.A.C.). Computing time from Wayne State’s C&IT is gratefully acknowledged. P.S. thanks Wayne State University for a graduate research fellowship during this work.
Footnotes
Primers and plasmids used to create the ABH1 variants.
Sequence alignments of ALKBH1 parts to the respective templates.
Potential energy plots of the MD simulations.
Plots of the dynamics of the distances between selected serine and lysine residues.
Plots of the dynamics of the distances between K25 and proximal residues.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sancar A, Lindsey-Boltz LA, Ünsal-Kaçmaz K, Linn S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annual Review Biochemistry. 2004;73:39–85. doi: 10.1146/annurev.biochem.73.011303.073723. [DOI] [PubMed] [Google Scholar]
- 2.Rydberg B, Lindahl T. Nonenzymatic methylation of DNA by the intracellular methyl group donor S-adenosyl-l-methionine is a potentially mutagenic reaction. EMBO J. 1982;1:211–216. doi: 10.1002/j.1460-2075.1982.tb01149.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Taverna P, Sedgwick B. Generation of an endogenous DNA-methylating agent by nitrosation in Escherichia coli. J Bacteriol. 1996;178:5105–5111. doi: 10.1128/jb.178.17.5105-5111.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hecht SS. DNA adduct formation from tobacco-specific N-nitrosamines. Mutat Res. 1999;424:127–142. doi: 10.1016/s0027-5107(99)00014-7. [DOI] [PubMed] [Google Scholar]
- 5.Drabløs F, Feyzi E, Aas PA, Vaagbø CB, Kavli B, Bratlie S, Pena-Diaz J, Otterlei M, Slupphaug G, Krokan HE. Alkylation damage in DNA and RNA - repair mechanisms and medical significance. DNA Repair. 2004;3:1389–1407. doi: 10.1016/j.dnarep.2004.05.004. [DOI] [PubMed] [Google Scholar]
- 6.Sedgwick B. Repairing DNA-methylation damage. Nature Reviews Molecular Cell Biology. 2004;5:148–157. doi: 10.1038/nrm1312. [DOI] [PubMed] [Google Scholar]
- 7.Trewick SC, Henshaw TF, Hausinger RP, Lindahl T, Sedgwick B. Oxidative demethylation by Escherichia coli AlkB directly reverts DNA base damage. Nature. 2002;419:174–178. doi: 10.1038/nature00908. [DOI] [PubMed] [Google Scholar]
- 8.Mishina Y, He C. Oxidative dealkylation DNA repair mediated by the mononuclear non-heme iron AlkB proteins. Journal of Inorganic Biochemistry. 2006;100:670–678. doi: 10.1016/j.jinorgbio.2005.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yi C, Jia G, Hou G, Dai Q, Zhang W, Zheng G, Jian X, Yang CG, Cui Q, He C. Iron-catalysed oxidation intermediates captured in a DNA repair dioxygenase. Nature. 2010;468:330–333. doi: 10.1038/nature09497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang CG, Yi C, Duguid EM, Sullivan CT, Jian X, Rice PA, He C. Crystal structures of DNA/RNA repair enzymes AlkB and ABH2 bound to dsDNA. Nature. 2008;452:961–965. doi: 10.1038/nature06889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sundheim O, Vagbo CB, Bjoras M, Desousa MML, Talstad V, Aas PA, Drablos F, Krokan HE, Tainer JA. Slupphaug Human ABH3 structure and key residues for oxidative demethylation to reverse DNA/RNA damage. EMBO J. 2006;25:3389–3397. doi: 10.1038/sj.emboj.7601219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sundheim O, Talstad VA, Vågbø CB, Slupphaug G, Krokan HE. AlkB demethylases flip out in different ways. DNA Repair. 2008;7:1916–1923. doi: 10.1016/j.dnarep.2008.07.015. [DOI] [PubMed] [Google Scholar]
- 13.Yu B, Edstrom WC, Benach J, Hamuro Y, Weber PC, Gibney BR, Hunt JF. Crystal structures of catalytic complexes of the oxidative DNA/RNA repair enzyme AlkB. Nature. 2006;439:879–884. doi: 10.1038/nature04561. [DOI] [PubMed] [Google Scholar]
- 14.Fang D, Lord RL, Cisneros GA. Ab initio QM/MM calculations show an intersystem crossing in the hydrogen abstraction step in dealkylation catalyzed by AlkB. J Phys Chem B. 2013;117 (21):6410–6420. doi: 10.1021/jp403116e. [DOI] [PubMed] [Google Scholar]
- 15.Bleijlevens B, Shivarattan T, van den Boom KS, de Haan A, van der Zwan G, Simpson PJ, Matthews SJ. Changes in protein dynamics of the DNA repair dioxygenase AlkB upon binding of Fe2+ and 2-oxoglutarate. Biochemistry. 2012;51:3334–3341. doi: 10.1021/bi201699e. [DOI] [PubMed] [Google Scholar]
- 16.Sedgwick B, Batesb PA, Paikc J, Jacobsa SC, Lindahla T. Repair of alkylated DNA: Recent advances. DNA repair. 2007;6:429–442. doi: 10.1016/j.dnarep.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 17.Falnes PO, Johansen RF, Seeberg E. AlkB-mediated oxidative demethylation reverses DNA damage in Escherichia coli. Nature. 2002;419:178–182. doi: 10.1038/nature01048. [DOI] [PubMed] [Google Scholar]
- 18.Falnes PO, Bjoras M, Aas PA, Sundheim O, Seeberg E. Substrate specificities of bacterial and human AlkB proteins. Nucleic Acids Res. 2004;32:3456–3461. doi: 10.1093/nar/gkh655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mishina Y, Lee CJ, He C. Interaction of human and bacterial AlkB proteins with DNA as probed through chemical cross-linking studies. Nucleic Acids Research. 2004;32:1548–1554. doi: 10.1093/nar/gkh319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aas A, Otterlei M, Falnes PO, Vågbø CB, Skorpen F, Akebari M, Sundheim O, Bjørås M, Slupphaug G, Seeberg E, Krokan HE. Human and bacterial oxidative demethylases repair alkylation damage in both RNA and DNA. Nature. 2003;421:859–863. doi: 10.1038/nature01363. [DOI] [PubMed] [Google Scholar]
- 21.Koivisto P, Robins P, Lindahl T, Sedgwick B. Demethylation of 3-methylthymine in DNA by bacterial and human DNA dioxygenases. J Biol Chem. 2004;279:40470–40474. doi: 10.1074/jbc.M407960200. [DOI] [PubMed] [Google Scholar]
- 22.Falnes PØ. Repair of 3-methylthymine and 1-methylguanine lesions by bacterial and human AlkB proteins. Nucleic Acids Research. 2004;32:6260–6267. doi: 10.1093/nar/gkh964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Duncan T, Trewick SC, Koivisto P, Bates PA, Lindahl T, Sedgwick B. Reversal of DNA alkylation damage by two human dioxygenases. Proc Natl Acad Sci U S A. 2002;99:16660–16665. doi: 10.1073/pnas.262589799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Delaney JC, Essigmann JM. Mutagenesis, genotoxicity, and repair of 1-methyladenine, 3-alkylcytosines, 1-methylguanine, and 3-methylthymine in AlkB Escherichia coli. Proc Natl Acad Sci U S A. 2004;101:14051–14056. doi: 10.1073/pnas.0403489101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Westbye MP, Feyzi E, Aas PA, Vagbo CB, Talstad VA, Kavli B, Hagen L, Sundheim O, Akbari M, Liabakk NB, Slupphaug G, Otterlei M, Krokan HE. Human AlkB homolog 1 is a mitochondrial protein that demethylates 3-methylcytosine in DNA and RNA. J Biol Chem. 2008;283:25046–25056. doi: 10.1074/jbc.M803776200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Monfort A, Wutz A. Breathing-in epigenetic change with vitamin C. EMBO reports. 2013;14:337–346. doi: 10.1038/embor.2013.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fu D, Calvo JA, Samson LD. Balancing repair and tolerance of DNA damage caused by alkylating agents. Nat Rev Cancer. 2012;12:104–120. doi: 10.1038/nrc3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hurley LH. DNA and its associated processes as targets for cancer therapy. Nat Rev Cancer. 2002;2:188–200. doi: 10.1038/nrc749. [DOI] [PubMed] [Google Scholar]
- 29.Kurowski MA, Bhagwat AS, Papaj G, Bujnicki JM. Phylogenomic identification of five new human homologs of the DNA repair enzyme AlkB. BMC Genomics. 2003;4:1471–2164. doi: 10.1186/1471-2164-4-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Müller TA, Meek K, Hausinger RP. Human AlkB homologue 1 (ABH1) exhibits DNA lyase activity at abasic sites. DNA Repair. 2010;9:58–65. doi: 10.1016/j.dnarep.2009.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Müller TA, Andrzejak MM, Hausinger RP. A covalent protein – DNA 5′-product adduct is generated following AP lyase activity of human ALKBH1 (AlkB homologue 1) Biochem J. 2013;452:509–518. doi: 10.1042/BJ20121908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Korvald H, Falnes PO, Laerdahl JK, Bjoras M, Alseth I. The Schizosaccharomyces pombe AlkB homolog Abh1 exhibits AP lyase activity but no demethylase activity. DNA Repair. 2012;11:453–462. doi: 10.1016/j.dnarep.2012.01.014. [DOI] [PubMed] [Google Scholar]
- 33.Ougland R, Lando D, Jonson I, Dahl JA, Moen MN, Nordstrand LM, Torbjorn R, Rognes T, Lee JT, Klungland A, Kouzarides T, Larsen E. ALKBH1 is a histone H2A dioxygenase involved in neural differentiation. Stem Cells. 2012;30(12):2672–2682. doi: 10.1002/stem.1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Müller TA, Yu K, Hausinger RP, Meek K. ALKBH1 is dispensable for abasic site cleavage during base excision repair and class switch recombination. PLoS ONE. 2013;8(6):e67403. doi: 10.1371/journal.pone.0067403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vidyarthy AS, Das SS. Homology modeling and function prediction of hABH1, involving in repair of alkylation damaged DNA. Interdiscip Sci Comput Life Sci. 2011;3:175–181. doi: 10.1007/s12539-011-0087-4. [DOI] [PubMed] [Google Scholar]
- 36.Pieper U, Webb U, Barkan DT, Schneidman-Duhovny D, Schlessinger A, Braberg H, Yang Z, Meng EC, Pettersen EF, Huang CC, Datta RS, Sampathkumar P, Madhusudhan MS, Sjolander K, Ferrin TE, Burley SK, Sali A. MODBASE, a database of annotated comparative protein structure models and associated resources. Nucleic Acids Research. 2001;39:465–474. doi: 10.1093/nar/gkq1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Notredame C, Higgins DG, Heringa J. T-Coffee: A novel method for fast and accurate multiple sequence alignment. J Mol Biol. 2000;302(1):205–17. doi: 10.1006/jmbi.2000.4042. [DOI] [PubMed] [Google Scholar]
- 38.Chivian D, Baker D. Homology modeling using parametric alignment ensemble generation with consensus and energy-based model selection. Nucleic Acids Research. 2006;34(17):e112. doi: 10.1093/nar/gkl480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kaufmann KW, Lemmon GH, DeLuca SL, Sheehan JH, Meiler J. Practically useful: what the ROSETTA protein modeling suite can do for you. Biochemistry. 2010;49:2987–2998. doi: 10.1021/bi902153g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera-a visualization system for exploratory research and analysis. J Comput Chem. 2004;25(13):1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 41.Case DA, Darden TA, Cheatham TE, III, Simmerling CL, Wang J, Duke RE, Luo R, Walker RC, Zhang W, Merz KM, Roberts B, Hayik S, Roitberg A, Seabra G, Swails J, Goetz AW, Kolossváry I, Wong KF, Paesani F, Vanicek J, Wolf RM, Liu J, Wu X, Brozell SR, Steinbrecher T, Gohlke H, Cai Q, Ye X, Wang J, Hsieh M-J, Cui G, Roe DR, Mathews DH, Seetin MG, Salomon-Ferrer R, Sagui C, Babin V, Luchko T, Gusarov S, Kovalenko A, Kollman PA. AMBER 12. University of California; San Francisco: 2012. [Google Scholar]
- 42.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79:926–935. [Google Scholar]
- 43.Cheng J, Saigo H, Baldi P. Large-scale prediction of disulphide bridges using kernel methods, two-dimensional recursive neural networks, and weighted graph matching. Proteins. 2006;62(3):617–629. doi: 10.1002/prot.20787. [DOI] [PubMed] [Google Scholar]
- 44.Singh SK, Hora R, Belrhali H, Chitnis CE, Sharma A. Structural basis for Duffy recognition by the malaria parasite Duffy-binding-like domain. Nature. 2006;439:741–744. doi: 10.1038/nature04443. [DOI] [PubMed] [Google Scholar]
- 45.Katzman M, Mack JP, Skalka AM, Lies J. A covalent complex between retroviral integrase and nicked substrate DNA. Proc Natl Acad Sci U S A. 1991;88(11):4695–4699. doi: 10.1073/pnas.88.11.4695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Strahs D, Zhu C, Cheng B, Chen J, Tse-Dinh Y. Experimental and computational investigations of Ser10 and Lys13 in the binding and cleavage of DNA substrates by Escherichia coli DNA topoisomerase I. Nucleic Acids Research. 2006;34(6):1785–1797. doi: 10.1093/nar/gkl109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fromme JC, Verdine GL. Structure of a trapped endonuclease III-DNA covalent intermediate. The EMBO Journal. 2003;22(13):3461–3471. doi: 10.1093/emboj/cdg311. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.