

Abstract
Oncogenic KRAS mutations found in 20–30% of all non-small cell lung cancers (NSCLC) are associated with chemoresistance and poor prognosis. Here we demonstrate that activation of the cell protective stress response gene NRF2 by KRAS is responsible for its ability to promote drug resistance. RNAi-mediated silencing of NRF2 was sufficient to reverse resistance to cisplatin elicited by ectopic expression of oncogenic KRAS in NSCLC cells. Mechanistically, KRAS increased NRF2 gene transcription through a TPA response element (TRE) located in the NRF2 promoter. In a mouse model of mutant KrasG12D-induced lung cancer, we found that suppressing the NRF2 pathway with the chemical inhibitor brusatol enhanced the antitumor efficacy of cisplatin. Co-treatment reduced tumor burden and improved survival. Our findings illuminate the mechanistic details of KRAS-mediated drug resistance and provide a preclinical rationale to improve the management of lung tumors harboring KRAS mutations with NRF2 pathway inhibitors.
Keywords: KRAS, NRF2, Brusatol, chemoresistance, TPA Response Element
Introduction
The RAS genes encode a family of membrane-associated 21 kDa GTP-binding proteins, including HRAS, KRAS and NRAS that control cell growth, differentiation, and apoptosis. By switching from the GTP-bound active form to the GDP-bound inactive form, RAS proteins function as a molecular switch to turn on or off their downstream effectors (1,2). Although each of the three RAS genes can be mutated in human cancers, KRAS mutations are the most common. Oncogenic KRAS mutations occur in approximately 30% of all cancer types and in 20–30% of non-small cell lung cancers (NSCLC) (3). These oncogenic mutations frequently occur as point mutations in codons 12, 13, or 61, each resulting in a protein with impaired GTPase activity, and therefore, constitutive activation of RAS signaling (3,4). A large body of literature has reported that cancers with oncogenic KRAS mutations are resistant to anti-cancer drug treatments and thus patients with these malignancies have poor prognoses (5–9). Mechanistically, chemoresistance may be explained by: (i) mutation or overexpression of a therapeutically targeted protein, (ii) inactivation of the drug, (iii) reduced drug uptake, (iv) enhanced efflux of the drug, or (v) the recovery of drug-induced DNA lesions by DNA repair enzymes (10).
NRF2 is a transcription factor that regulates the antioxidant response by inducing the expression of genes bearing an antioxidant response element (ARE) in their regulatory regions. Activation of the NRF2 pathway promotes cell survival during oxidative stress or xenobiotic insult (11–14). Importantly, many of the NRF2 target genes, including drug metabolizing enzymes, antioxidant enzymes, and drug transporters, play a crucial role in determining drug resistance (14). Examples of NRF2 target genes that may confer enhanced drug processing include: Glutamate-cysteine ligase (GCLC/GCLM), thioredoxin reductase 1 (TXNRD1), aldo-keto reductase (AKR), glutathione S-transferase (GST), multidrug resistance-associated protein 2 (MRP2), NAD(P)H quinone oxidoreductase 1 (NQO1), and heme oxygenase 1 (HMOX1).
It has been demonstrated that NRF2 has a dual role in cancer. First, NRF2 is involved in chemoprevention. Oxidative stress is implicated in the initiation and progression of cancer. Under oxidative stress, NRF2 induces the transcription of hundreds of cellular protective genes to combat potentially carcinogenic reactive intermediates. As evidence for this protective role, many chemopreventive compounds have been identified as NRF2 activators (11–15), and Nrf2-null mice are highly susceptible to chemical carcinogens and are no longer protected by chemopreventive compounds (16,17). Second, recent findings point to a “dark side” of NRF2 which promotes cancer (18). Many studies have shown that cancers can harbor somatic mutations in NRF2, KEAP1, or CUL3 that disrupt the KEAP1-mediated negative regulation of NRF2, resulting in a constitutive high level of NRF2 (19–22), which correlates with chemoresistance in cancer cells (13,18,23–26). Discovery of the cancer-promoting activity of NRF2 has prompted us to identify compounds that inhibit the NRF2 pathway (27). We previously identified a potent NRF2 pathway inhibitor, brusatol, which inhibits the NRF2-mediated protective response at sub-nanomolar concentrations. Brusatol treatment also enhances the efficacy of chemotherapeutics in an NRF2-dependent manner in both cell culture and murine A549 xenograft models.
Previous studies have demonstrated that NRF2 is primarily regulated at the protein level by the ubiquitin proteasome system (UPS). Under physiological conditions, NRF2 levels are low in all organs due to tight regulation by KEAP1, a substrate adaptor protein for a Cullin3-based E3 ubiquitin ligase (28–31). Under these basal conditions, this E3 ligase constantly targets NRF2 for ubiquitylation and subsequent proteasomal degradation. Upon activation of the pathway by oxidative or electrophilic stress, the enzymatic activity of the E3 ligase is inhibited, resulting in stabilization of NRF2 and transcriptional activation of its target genes (13). Recently, another E3 ubiquitin ligase, β-TrCP-Skp1-Cul1-Rbx1, was also found to ubiquitylate Nrf2 (32–34). Additionally, we have identified another E3 ubiquitin ligase, HRD1, that compromises the NRF2-mediated cytoprotective mechanism during the pathogenesis of liver cirrhosis (35). All these studies indicate that NRF2 is controlled at the protein level through protein stability modulation. Furthermore, many NRF2 modulators, including small molecules and endogenous proteins, upregulate NRF2 signaling by increasing the stability and thus the protein level of NRF2 without affecting its mRNA level (13). Interestingly, a recent study reported in a murine model a 1.6-fold increase in the mRNA level of Nrf2 in response to activation of oncogenic alleles of KrasG12D, B-RafV619E, and c-MycERT12 (36). However, the molecular mechanisms underlying increased Nrf2 transcription were not reported. In the current study, we provide strong evidence that oncogenic KRAS transcriptionally upregulates the mRNA levels of NRF2 through a TRE enhancer located in the proximal promoter of NRF2. More importantly, we show that inhibition of the NRF2 pathway by brusatol was able to overcome KRAS-mediated chemoresistance, and thus enhanced the efficacy of cisplatin.
Materials and Methods
Cell lines and cell culture
HBE cells were obtained from Dr. Dieter Gruenert, whose lab generated and characterized them (37); the rest were purchased from the American Type Culture Collection (ATCC), where they were tested and authenticated by STR (short tandem repeat) and were maintained at 37°C in a humidified incubator containing 5% CO2. HEK293 cells were grown in modified Eagle’s medium (MEM) supplemented with 10% fetal bovine serum (FBS), 1% L- glutamine, and 0.01% gentamicin, 0.1 mM nonessential amino acids (Cellgro) and 1mM sodium pyruvate (Gibco). HBE cells were maintained in MEM medium supplemented with 10% FBS, 1% L- glutamine, and 0.01% gentamicin. BEAS-2B cells were maintained in F-12 HAM’s medium (Thermo Scientific) supplemented with 1% L-glutamine, 2 mg/ml insulin (Sigma), 10 μg/ml epidermal growth factor (EGF) (Fisher Scientific), 2.5 mg/ml transferrin (Sigma), 0.05 mM dexamethasone (Sigma), 10 μg/ml cholera toxin (LIST biological), and endothelial cell growth supplement (ECGS) (Millipore). NCI-H292, NCI-H23, NCI-H838, NCI-H1299, NCI-H1703, A549, and Calu-1 cells were maintained in RPMI 1640 supplemented with 10% FBS, 1% L-glutamine, and 0.01% gentamicin.
Construction of recombinant DNA molecules
The KRASWT, KRASG12D and KRASDN expression vectors were constructed by cloning a PCR-generated fragment into pcDNA 3.1 (Invitrogen). Deletion fragments of the NRF2 promoter sequence were amplified by PCR from genomic DNA extracted from HBE cells and cloned into pGL4.22 (Promega). The fragment named “TRE”, located between bases +267 to +273 in NRF2’s promoter sequence, including a sequence similar to AP-1 recognition site (“TGCGTCA”), was purchased (Sigma) and inserted into pGL4.22 after end-repairing and annealing the two oligos together. All the sequences were confirmed by direct nucleotide sequencing. See Supplementary Material and Methods for details.
Transfection of siRNA and cDNA
Transfection of cDNA was performed using Lipofectamine 2000 (Invitrogen). Hiperfect (Qiagen) was used for transfection of small interfering RNA (siRNA). NRF2 siRNA#3 (SI00657937), NRF2 siRNA#5 (SI0387289) and Control siRNA (1027281) were purchased from Qiagen. Both siRNA#3 and siRNA#5 were able to specifically reduce the Nrf2 protein level without off-target effects (Fig. S1). siRNA#5 was used for all the data presented in this manuscript.
Luciferase reporter gene assay
HEK293 cells were transfected with several deletion fragments of NRF2 promoter-luciferase constructs along with thymidine kinase (TK)-Renilla luciferase (internal control). Luciferase activities were measured with the dual-luciferase reporter assay system (Promega). Experiments were performed in triplicates.
Cell viability
Cisplatin-induced toxicity was measured by functional impairment of mitochondria using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) (Sigma) as previously described (38). Approximately 1.5 × 104 NCI-H292 cells per well were seeded in a 96-well plate and transfected with either siRNA or vectors expressing KRASDN, KRASG12D or KRASWT before treatment with the indicated concentrations of cisplatin for 48 hours. Experiments were done in triplicates.
mRNA extraction and real-time quantitative reverse transcription-PCR (qRT-PCR)
Total mRNA was extracted using TRIzol (Invitrogen) according to the manufacturer’s instructions. cDNA was synthesized using equal amounts of mRNA and the Transcriptor first-strand cDNA synthesis kit (Promega). The detailed TaqMan probe and primer sequences can be seen in the Supplementary Materials and Methods. Real-time PCR (RT-PCR) was performed as previously described (38). Reactions for each sample were done in duplicate, and the experiment was repeated three times. Results are expressed as relative mRNA levels normalized to GAPDH.
Quantification of cDNA amounts for Nrf2, Keap1, Nqo1, Akr1b10, Akr1c1, Gclm, Hmox1 and β-actin from tissues was performed with KAPA SYBR FASR qPCR Kit (Kapa Biosystems). All primer sets were designed with Primer 3 free online software and synthesized by Sigma. Sequences can be found in the Supplementary Materials and Methods. The RT-PCR was performed as previously described (38). All reporter gene and RT-PCR analyses were done in duplicates and repeated in three independent experiments.
Immunoblot analysis
Protein expression from cell lines and lung tissues was assessed by SDS-PAGE electrophoresis and immunoblotting as described previously (38). For details on sample collection see Supplementary Materials and Methods.
Apoptotic cell death (TUNEL)
Briefly, lung epithelium tissue sections were pretreated with proteinase K (15 μg/ml) in10 mM Tris/HCl (PH 7.8) at 37°C for 30 min, and an in situ cell death detection kit (Roche, IN, USA) was used for detecting apoptotic cell death according to the manufacturer’s instructions. Tissue sections were then costained with Hoechst and analyzed with a fluorescence microscope (Zeiss Observer.Z1 microscope with the Slidebook software).
Materials and antibodies
Cisplatin and U0126 were purchased from Sigma-Aldrich. HRP-conjugated secondary antibodies as well as primary antibodies against NRF2, KEAP1, p-ERK, ERK, KRAS, GCLM, HMOX1, AKR1B10, AKR1C1, NQO1, Ki67, Lamin-A, and GAPDH were purchased from Santa Cruz Biotechnology. Primary antibody against γ-H2Ax was purchased from Bethyl Laboratories, Inc. Primary antibody against 8-oxo-dG was purchased from Trevigen.
Experimental animals
6 week-old C57BL6 LSL-KrasG12D mice were purchased from Jackson Laboratory. The CCSPCre mice were reported previously (39–41). CCSPCre / LSL-KrasG12D mice were generated by cross breeding a mouse harboring the LSL-KrasG12D allele with a mouse containing Cre recombinase inserted into the Clara cell secretory protein (CCSP) locus. All mice were housed in specific, pathogen-free conditions and handled in accordance with the Institutional Animal Care policies. Mice were intraperitoneally injected with cisplatin and brusatol for both short and long term treatments. Tissues were harvested at the indicated time points and the mice were monitored daily for evidence of disease or death.
Tissue collection, hematoxylin and eosin (H&E) staining and immune-histochemistry (IHC)
Lung tissues were isolated at the indicated time points (Figure 4A). After images were collected, surface tumors were weighted and counted using a dissecting microscope. One half of the lung was directly frozen in liquid nitrogen and stored at −80°C for total RNA extraction and for immunoblot analysis, and the other half was fixed in 10% buffered formalin and embedded in paraffin. 5 μm sections were cut and stained with H&E. IHC analysis was performed as previously described (38).
Figure 4.

Brusatol cotreatment reduces tumor burden and enhances survival. Two sets of experiments were performed: short-term treatment (left panel) and long-term treatment (right panel). (A) Treatment regimens: 16 week-old CCSPCre/Cre/LSL-KrasG12D/+ mice were injected i.p. with PBS (Ctrl), cisplatin (Cis), brusatol (Bru), or cisplatin plus brusatol (Bru+Cis) at the indicated time point (black arrows). (B) Kaplan Meier survival curves of CCSPCre/Cre/LSL-KrasG12D/+ mice treated with Ctrl, Cis, Bru, or Bru+Cis. (C) The representative lung image of CCSPCre/Cre/LSL-KrasG12D/+ mice from each treatment group (n=10) (D) Wet lung weights. (E) The total number of surface tumors. (F) The number of surface tumors > 2 mm. (G) The number of surface tumors > 5 mm (*p< 0.05, Ctrl vs. treatment groups; #p< 0.05, Cis vs. Bru+Cis groups).
Oxidative DNA damage
A monoclonal antibody against 8-Oxop7, 8-dihydro-2′-deoxyguanosine (8-oxo-dG) (Trevigen, Gaithersburg, MD) was used for the detection of oxidative DNA damage. The staining was performed as previously described (42).
Detection of mRNA level in the fresh tumor lung tissue slides by double-stranded locked nucleic acid (LNA) probes
Four LNA probes were designed as previously described (43) to detect the relative gene expression levels of Nrf2, Hmox1, Nqo1 and Akr1c1 in both tumor and normal lung tissues. A β-actin probe and a random probe were designed as the positive and negative controls. The gold nanorod (GNR)-LNA complex solution including 0.1 μM LNA probe and 2.5 × 1011 GNR/ml was added to the lung slices in 24 well plates. After incubation for 8 h at 37°C, the slides were washed with PBS 3 times and imaged with an inverted fluorescence microscope with an HQ2 CCD camera. Data collection and imaging analysis were performed in Image J.
Statistical analysis
Results are presented as the mean ± SEM of at least three independent experiments performed in duplicate or triplicate. Statistical tests were performed using SPSS 10.0. Un-paired Student’s t-tests were used to compare the means of two groups. One-way analysis of variance was applied to compare the means of three or more groups. p < 0.05 was deemed significant.
Results
Expression of oncogenic KRAS enhances cisplatin resistance by upregulating the NRF2-mediated protective response
To show that chemoresistance observed in tumors expressing oncogenic KRAS is associated with activation of NRF2 signaling, we first compared the NRF2 protein level to that of the phosphorylated form of the extracellular signal-regulated kinase (p-ERK; a readout of KRAS activation) in several lung cell lines (Fig. S2). Their response to cisplatin-mediated toxicity was measured and LD50s are listed (Table S1). The coding regions of NRF2 and KRAS were sequenced and the status of each gene is listed; the mutation information of KEAP1 and TP53 was obtained from the literature (Table S1). By combining the results in Fig. S2 with the information in Table S1, we selected a human lung epithelial carcinoma cell line (NCI-H292) and an immortalized but not transformed human bronchial epithelial cell line (HBE) for further studies. Both NCI-H292 and HBE have no mutations in KEAP1, NRF2, TP53, or KRAS and have basal levels of p-ERK (Fig. S2). Overexpression of KRASG12D or, to a lesser extent, wild type KRAS (KRASwt) enhanced cell viability in response to cisplatin treatment, whereas overexpression of a dominant negative KRAS mutant (KRASDN) reduced cell viability in both cell lines (Fig. 1A and 1C, upper panels). The effect of KRAS overexpression in cisplatin resistance was shown to be NRF2-dependent, since it was lost when NRF2 expression was silenced by siRNA (Fig. 1A and 1C, lower panels). We noticed that the LD50 for HBE cells shifted from 9.6 μM (Table S1) to 3 μM (Fig. 1C) after transfection with control siRNA, whereas transfection of control siRNA had no effect on the LD50 of NCI-H292 (LD50=2.3, Table S1 and Fig. 1A). This might be due to the fact that HBE, a primary cell line, is more sensitive to the transfection reagent, compared to the cancer cell line NCI-H292.
Figure 1.

Expression of oncogenic KRAS enhances chemoresistance by upregulating the NRF2-mediated protective response. (A and C) Overexpression of the oncogenic form of KRAS protected NCI-H292 and HBE cells against cisplatin-mediated cell toxicity in an NRF2-dependent manner. NCI-H292 and HBE cells were transfected with control siRNA (upper panel) or NRF2 siRNA (lower panel) for 24 h, followed by transfection of an empty vector or the indicated KRAS mutant. At 24 h post-transfection of cDNA, the indicated dose of cisplatin was added and cell viability was measured 48 h after cisplatin treatment. Data are expressed as mean ± SEM (*p< 0.05, KRAS vs. control group). (B and D) A positive relationship between activation of the KRAS pathway and the NRF2 pathway. An aliquot of cells, transfected and treated as described in (A and C), was harvested, and cell lysates were subjected to immunoblot analysis.
As expected, overexpression of KRASG12D and KRASWT activated the ERK pathway as indicated by enhanced p-ERK, while total ERK remained the same (Fig. 1B and 1D). Interestingly, overexpression of KRASG12D and KRASWT also increased the levels of NRF2 and its target genes, GCLM and HMOX1 (Fig. 1B and 1D), indicating activation of the NRF2 pathway. Conversely, ectopic expression of KRASDN slightly reduced p-ERK, NRF2, GCLM, and HMOX1 levels (Fig. 1B and 1D). Immunoblot analyses confirmed NRF2 silencing by NRF2-siRNA since expression of NRF2, GCLM and HMOX1 were dramatically reduced (Fig. 1B and 1D). Taken together, these results demonstrate that the KRAS-ERK pathway positively regulates the NRF2 pathway, indicating that KRAS-mediated cisplatin resistance may be due to activation of the NRF2 pathway.
KrasG12D-induced lung tumor tissues have higher levels of Nrf2 and its target genes
To confirm that oncogenic KRAS upregulates the NRF2 pathway a murine lung cancer model (LSL-KrasG12D/+) was used. Cre-virus intratracheal infection of LSL KrasG12D/+ mice for 8 weeks resulted in multiple lung adenomas. Tumor tissues from each mouse were pooled and Kras activation and the expression of Nrf2 and its target genes in tumor tissues were compared with those in normal lung tissues. As expected, Kras was activated in tumors, where marked elevation of p-Erk was only observed in tumor but not normal tissues. Comparatively, Erk was expressed equally in tumor vs. normal tissues (Fig. 2A). Remarkably, the expression of Nrf2 and its target genes Akr1b10, Akr1c1, Gclm, and Nqo1 was dramatically increased in tumor tissues as compared to the corresponding normal tissues. Notably, the activation of the Nrf2 pathway by oncogenic Kras was more substantial in mice than in cultured cells (compare Fig. 2A with Fig. 1B). Immunohistochemistry (IHC) analyses demonstrated that Nrf2 and Nqo1 were highly expressed in tumors compared to adjacent normal tissues or normal control lungs (Fig. 2B and S4). Additionally, the mRNA levels of Nrf2, Hmox1, Nqo1, Gclm, Akr1b10, and Akr1c1 were significantly higher in tumor than in normal tissues, whereas Keap1 mRNA was expressed at a similar level in both tissues (Fig. 2C). To further confirm the real-time RT-PCR data and to visualize the mRNA expression in tumor vs. normal tissues, we utilized our newly developed method, a gold nanorod-locked nucleic acid complex for single cell gene expression detection in living cells and tissues (44). The fluorescence intensities of Nrf2, Nqo1, Hmox1 and Akr1c1 were higher in tumor tissues compared to the adjacent normal tissues, whereas the signal for a random probe or β-actin was similar between both (Fig. 2D). Taken together, these results demonstrate that KrasG12D upregulates the Nrf2 pathway by increasing the level of Nrf2 mRNA.
Figure 2.

KrasG12D-induced lung tumor tissues have higher levels of Nrf2 and its target genes. (A) Activation of the Kras pathway in lung tumors correlated with higher levels of Nrf2, Gclm and Nqo1. Tissue lysates from Kras-induced tumors and normal lungs from age-matched mice were subjected to immunoblot analysis with the indicated antibodies. Each lane contains a pooled lung tumor sample or a piece of normal lung tissue from individual mice. (B) Nrf2 and Nqo1 were expressed at higher levels in lung tumor tissues than normal lung tissues. H&E staining (HE) and immunohistochemical analyses, with anti-Nrf2 or anti-Nqo1 antibodies, of lung tissue sections were performed (n=3 in each group; one representative image from each group is shown; scale bar: 100 μm). (C) The mRNA levels of Nrf2 and its target genes were elevated in the Kras-induced lung tumor tissues compared to normal lung tissues. Total RNAs were extracted from tumor and normal lung tissues. The relative mRNA level of the indicated genes was determined by quantitative real-time RT-PCR. Data are expressed as mean ± SEM (*p< 0.05 tumor vs. normal tissue, n=3 mice per group, the sample was run in duplicate). The experiment was repeated twice and similar results obtained. (D) Elevated mRNA levels of Nrf2 and its target genes were observed in real-time in Kras-induced tumor tissues, compared to the adjacent normal lung tissues. mRNA expression in fresh tissue slides was detected by gold nanorod-locked nucleic acid complexes. Representative bright field and fluorescence images of lung slice with random, β-actin, Nrf2, Nqo11, Hmox1 and Akr1c1 probes are shown (n=3 mice per group). The yellow dashed line in the bright field delineates tumors (scale bar: 100 μm; upper panel). Relative fluorescence (florescence intensity/area) was plotted (lower panel). The data are expressed as mean ± SEM (*p< 0.05 Kras-induced tumor vs. normal tissues).
KRAS transcriptionally activates NRF2 through the TRE
To understand how activation of the KRAS-ERK pathway upregulates the NRF2 mRNA levels, we made a series of reporter constructs with truncated NRF2 proximal promoters cloned upstream of a luciferase gene. KRASG12D and KRASwt, but not KRASDN, significantly enhanced the luciferase activity in all constructs except for construct F5-R1, indicating that potential enhancer sequence(s) are flanked by primers F4 and F5 (+227 to +403) (Fig. 3A, left panel). Similarly, another set of reporter gene analyses localized enhancer sequence(s) to the R3-R4 region (+234 to +343) (Fig. S3). A computational search for enhancers identified a TRE sequence between +267 and +273. Therefore, the TRE sequence TGCGTAC flanked by 15 nucleotides on both sides was inserted into the luciferase reporter gene vector (TRE, Fig. 3A, right panel). TRE was upregulated by KRASG12D and KRASWT, not KRASDN (Fig. 3A, right panel). Next, the TRE core sequence TGCGTCA was mutated to AACGTCA in both F3-R1 and F4-R1 constructs (F3-R1 Mu and F4-R1 Mu). KRASG12D and KRASWT were no longer able to enhance luciferase activities of F3-R1 Mu and F4-R1 Mu (Fig. 3A, right panel). These results demonstrate that KRAS upregulates NRF2 mRNA through the TRE. To confirm that this KRAS-mediated transcriptional upregulation of NRF2 is through MEK-ERK signaling, a MEK inhibitor U0126 was used. U0126 suppressed p-ERK and inhibited both basal and KRAS-induced NRF2, GCLM, and HMOX1 protein (Fig. 3B, left panel) and mRNA levels (Fig. 3B, right panel).
Figure 3.

KRAS transcriptionally activates NRF2 through the TRE. (A) Identification of a TRE (267 nt TGCGTCA 273 nt) in the promoter of NRF2. The different human NRF2 promoter constructs were cloned upstream of a luciferase reporter gene (the sites where primer pairs bind are illustrated). These constructs were co-transfected into HEK293 cells alone with a control, KRASDN, KRASG12D or KRASWT expression vector for 48h. Dual luciferase activities were measured. The experiment was repeated three times, each with triplicate samples. Data are expressed as mean ± SEM (*p< 0.05 Ctrl group vs. KRAS) (left panel). TRE refers to a construct where the TRE sequence TGCGTAC flanked by 15 nt on both sides was inserted into the cloning site of the luciferase reporter gene vector (left panel). F3-R1 Mu or F4-R1 Mu is a construct where the TRE sequence (TGCGTCA) in the F3-R1 or F4-R1 construct was mutated to AACGTCA. Dual luciferase activities with these constructs were measured as described (right panel). (B) KRAS upregulated NRF2 and its target genes through activation of ERK. HEK293 cells were either transfected with empty vector, KRASDN, KRASG12D or KRASWT for 24 h. Cells were treated with 10 μM U0126 for 4 h after overnight starvation. mRNAs were extracted and the relative mRNA level of NRF2, GCLM, HMOX1 was then determined by quantitative real-time RT-PCR. The experiment was repeated three times, each with triplicate samples. The data are expressed as mean ± SEM (*p< 0.05 Ctrl group vs. KRAS) (upper panel). Cell lysates from another set of the same experiment were subjected to immunoblot analysis (lower panel).
Brusatol cotreatment reduces tumor burden and enhances survival
To test if brusatol cotreatment is able to overcome KRAS tumor resistance to cisplatin, an LSL-KrasG12D/+ murine lung cancer model was chosen. In order to generate consistent tumor numbers in the lung, CCSPCre/Cre mice were crossed with LSL-KrasG12D/+ to generate CCSPCre/Cre/LSL-KrasG12D/+ mice. These mice developed multiple lesions such as atypical adenomatous hyperplasia, adenoma, and adenocarcinoma by 16 weeks of age (39–41). Two sets of experiments were carried out: a short-term treatment experiment, which consisted of one treatment regimen (Fig 4A, left panel) to test if the combination of brusatol with cisplatin could sensitize cancer cells to overcome intrinsic resistance; and a long-term treatment experiment, which consisted of two treatment regimens (Fig 4A, right panel) to test the possible role of Nrf2 in long-term cisplatin-induced resistance (acquired resistance), as was previously observed in this model (10). Mice in the untreated control group had a median survival of 9 months and died by ten months (Fig 4B). In the cisplatin single agent treated group, all mice died at 13 months in the short-term (left panel) and 90% at 18 months in the long-term studies (right panel); brusatol single agent treatment showed similar effects. The survival of the cotreatment group significantly increased in both short-term and long-term experiments. Cotreatment increased the median survival from 11.5 months (single agent) to 15 months in the short term experiment, and from 12.5 and 13.5 (single agent) to 15.5 months in the long-term experiment. At the end of our experiments, 20% of the short-term and 40% of the long-term mice in the cotreatment group survived beyond 20 months. Mice were harvested 40 days post-injection in the short-term group or 62 days post-injection in the long-term group for evaluation of the lungs. Morphological differences were readily apparent in the lung from the treated vs. untreated groups. The lungs in the cotreated groups showed the most normal tissue texture (Fig. 4C), had the lowest wet-lung weights (Fig. 4D), the lowest number of grossly visible surface tumors (Fig. 4E), and the smallest size of tumors (Figs. 4F and G) in both the short- and the long-term studies. In the short-term studies, only the untreated group had one tumor with a diameter > 5 mm, while in the long-term study there were no tumors > 5 mm in the cotreatment group, but 5 mice in the control, 2 mice in the cisplatin group, and 3 mice in the brusatol group had them (Fig. 4G). Next, we classified the histopathology of lesions observed in different groups (AAH and adenomas vs. adenocarcinomas). The percentage of mice with adenocarcinomas in the cotreatment group was relatively low (Table S2). The number of lesions was lowest in the cotreatment group (Fig. S7).
Brusatol cotreatment enhances the efficacy of cisplatin through Nrf2 inhibition in KrasG12D tumors
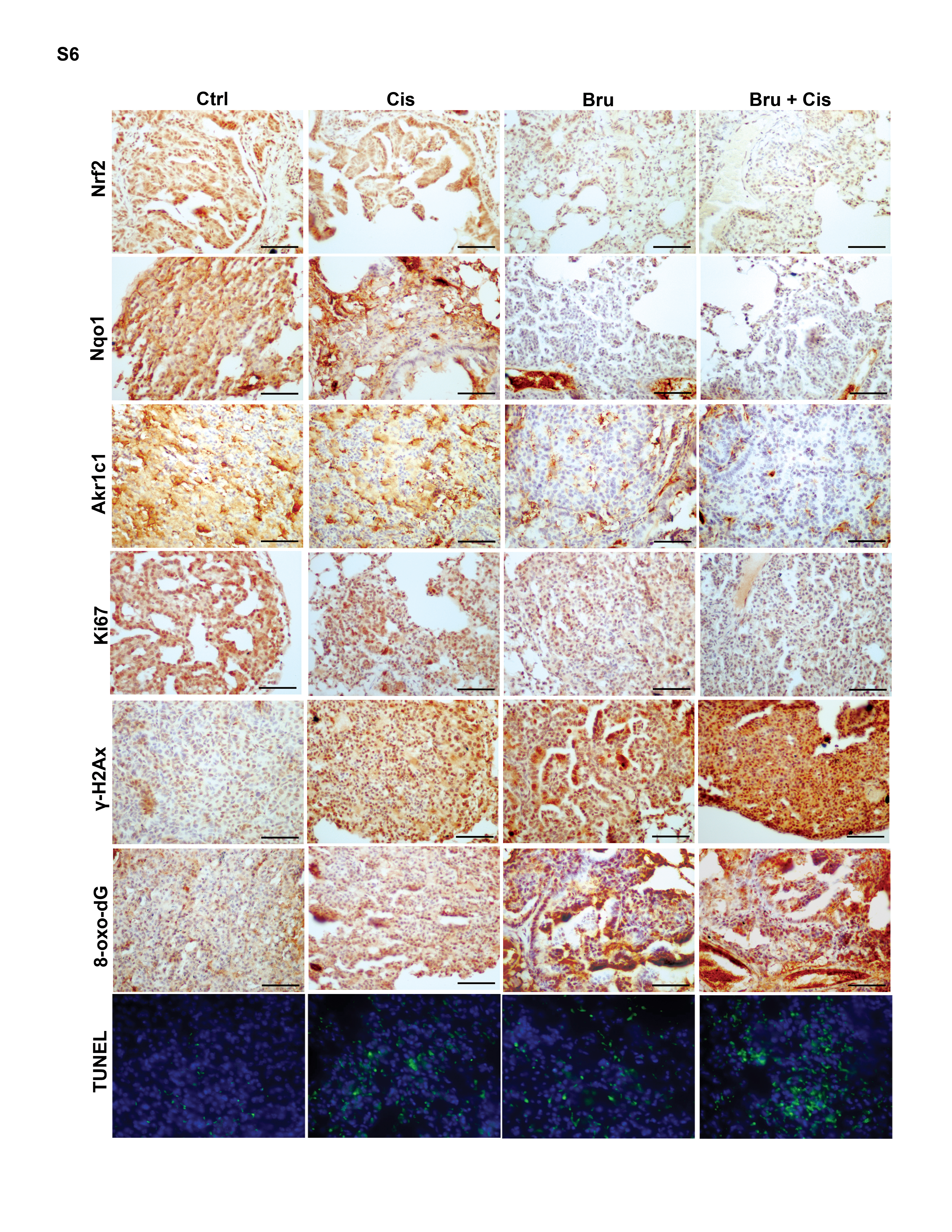
As expected, brusatol treatment markedly suppressed the protein levels of Nrf2, Akr1b10, Akr1c1, Nqo1, and Gclm, without affecting proteins in the Kras pathway, as indicated by p-Erk (Fig. 5A). Cisplatin treatment resulted in a slight increase in p-Erk (Fig. 5A) consistent with previous findings (45). Brusatol also decreased the mRNA levels of Nqo1, Akr1b10, Akr1c1, Hmox1, and Gclm without affecting Nrf2 and Keap1 mRNA levels (Fig. 5B). Brusatol and cisplatin cotreatment significantly reduced tumor volume, as measured by relative tumor area vs. total area of H&E stained lung tissue sections (Fig. 5C). A reduction of the Nrf2, Nqo1, and Akr1c1 protein levels was also confirmed by IHC analyses (Fig. 5D; Fig. S5 and S6). Cisplatin treatment decreased tumor cell proliferation and this was further reduced by cisplatin and brusatol cotreatment. γ-H2ax staining showed that the greatest DNA damage occurred in the tumors of mice cotreated with brusatol and cisplatin. Similarly, oxidative damage was the highest in the tumors of cotreated mice as measured by IHC staining of 8-oxo-dG. Measurement of apoptotic cell death using TUNEL analysis indicated that the largest degree of cell death occurred in the cotreatment group. Collectively, these results demonstrate that brusatol-mediated inhibition of the Nrf2 pathway enhanced the efficacy of cisplatin treatment through reduced cell proliferation, enhanced DNA damage, and increased apoptotic cell death.
Figure 5.

Brusatol cotreatment enhances the efficacy of cisplatin through Nrf2 inhibition in KrasG12D tumors. Results from Short-term treatment (left panel) and long-term treatment (right panel) are shown. (A) Brusatol treatment significantly inhibited the Nrf2 signaling pathway. Lung tissue lysates from each group were subjected to immunoblot analysis with the indicated antibodies. Each lane contains a lung tissue sample from individual mice (the upper panel). (B) Brusatol inhibited the mRNA level of Nrf2 target genes. An aliquot of the same lung tissue sample was used for quantitative real-time RT-PCR to measure the relative mRNA level of Nrf2, Keap1, Nqo1, Akr1b10, Akr1c1, Hxom1 and Gclm. Data are expressed as mean ± SEM. (n=3, *p < 0.05 Ctrl vs. treated group). (C) The lowest tumor volume was observed in the cotreatment group (Bru+Cis). A representative H&E staining of lung tissues from each group (n=10) is shown. The ratio of tumor area/total lung area was quantified (*p< 0.05 control vs. treatment groups, #p< 0.05 Cis vs. Bru+Cis groups). (D) IHC staining with Nrf2, NQO1, AKR1C1, Ki67, γ-H2Ax and 8-oxo-dG antibodies of lung tissue sections from CCSPCre/Cre/LSL-KrasG12D/+ mice treated with PBS, Cis, Bru, or Bru+Cis (scale bar: 100 μm). Lung sections were stained with TUNEL as a measurement of cell death and hoechst was included to label the nucleus. A representative image from each group (n=3) is shown.
Discussion
In the present study, we found that oncogenic mutation of KRAS or KRAS over expression enhanced resistance of cells to cisplatin in an NRF2-dependent manner (Fig. 1). We then investigated the molecular mechanism of KRAS-mediated chemoresistance and found a novel mode of NRF2 activation by KRAS. Distinct from the previously defined UPS-mediated NRF2 regulatory mechanisms, we demonstrate that KRAS is able to transcriptionally activate NRF2 though the KRAS-ERK pathway. A TRE enhancer sequence located between +267 to +273 of the NRF2 promoter was identified, and its upregulation by oncogenic KRAS was confirmed (Figs. 2 & 3). Using our newly established GNR-LNA technique for detecting single cell mRNA expression in living tissues, we observed a marked increase in the mRNA level of Nrf2 in lung tumor tissues compared to the adjacent normal tissues (Fig. 2D, Nrf2 panel), further confirming that activation of the Nrf2 pathway by KRAS is through enhanced Nrf2 mRNA expression. Notably, Kras-mediated upregulation of Nrf2 and its target genes was more robust in the in vivo murine system when tumor tissues were compared with normal tissues, than in the in vitro cell-based system when different forms of KRAS were ectopically expressed and their effects compared (Fig. 1–3). Additionally, we presented a potential means of mitigating KRAS-induced drug resistance through cotreatment with chemotherapeutics (cisplatin) and an NRF2 inhibitor (brusatol) (Figs. 4–5).
Cancer is the second leading cause of death in developed countries; it is responsible for about 600,000 deaths in America annually, and the incidence and mortality have been steadily increasing. Lung cancer is one of the most commonly diagnosed cancers, comprising 15–30% of total cancer cases. In NSCLC, the prevalence of oncogenic KRAS mutations is approximately 20–30% (3). Lung cancer is the leading cause of cancer-related death worldwide, with a five-year survival rate of less than 15%. The death rate for lung cancer has increased dramatically over the past several decades, even though death rates for other cancer types either remain the same or have decreased. Currently, radiation and platinum-based drugs are the standard treatments (46,47). However, the toxicity profiles and high rate of relapse with platinum compounds limit both their usefulness and effectiveness. Therefore, there is an urgent need to develop new adjuvants to enhance the efficacy of platinum-based treatments and circumvent chemoresistance.
Recent studies have clearly demonstrated the association between high NRF2 activity and chemoresistance in cancers. For example, somatic gain-of-function mutations of NRF2, or somatic loss-of-function mutations of either KEAP1 or CULLIN3 (CUL3) are frequently found in lung cancer. KEAP1 mutations were identified at a frequency of 50% (6/12) or 19% (10/54) in NSCLC cancer cell lines or tumor samples, respectively. In addition, loss of heterozygosity at 19p13.2, where KEAP1 is located, was observed at a frequency of 61% or 41% in NSCLC cell lines (72 samples in total) or in tumor tissues (39 samples in total) (20). In a similar study, somatic mutations were found in 5/65 (8%) patients who had adenocarcinoma (ADC), squamous cell carcinoma (SCC), or large cell carcinoma (LCC) (21). Another study that looked at the NRF2 and KEAP1 protein levels in 304 NSCLC tissues reported that 26% of the studied cohort had high nuclear NRF2 levels, while 56% had low KEAP1 levels (22). Similarly, mutations in NRF2 that disrupt KEAP1-mediated repression can also result in high NRF2 activity. For example, NRF2 mutations were found in 10/125 (8.0%) lung cancer cases (48). In this study, we extended the previous findings that link high NRF2 expression in NSCLC with cisplatin resistance in KRAS positive lung cancers. As demonstrated, KRAS upregulated NRF2 mRNA, which activated NRF2-mediated protective mechanisms, conferring cisplatin resistance.
Inhibiting the NRF2-mediated protective mechanism to enhance the efficacy of cancer therapeutics represents an innovative approach to cancer treatment. As reported previously, we isolated brusatol from Brucea javanica (L) Merr., a plant native to Southeast Asia and Australia, which inhibits NRF2 (27). We previously demonstrated that brusatol was able to sensitize a broad spectrum of cancer cell lines and A549-derived xenografts to many chemotherapeutic drugs in an NRF2-dependent manner (27). Here, we further explored the idea that brusatol can be developed as an adjuvant to enhance the efficacy of chemotherapeutic drugs using a pre-clinical lung adenocarcinoma model in CCSPCre/LSL-KrasG12D mice where tumors were induced by oncogenic Kras. Compared to cisplatin or brusatol treatment alone, cotreatment of brusatol and cisplatin significantly reduced the number and the size of the tumors, and improved survival (Fig. 4). In addition to intrinsic resistance, we also demonstrated that brusatol is more effective in long-term cisplatin-induced resistance (acquired resistance) (Fig. 4). Brusatol cotreatment inhibited the Nrf2 protective mechanism and led to decreased cell proliferation, enhanced oxidative DNA damage, and apoptotic cell death (Fig. 5). In the current study, we did not observe any adverse effects with the regimens used in the C57BL6 strain. However, higher doses of brusatol were observed to cause a decrease in the body weight of nude mice. Therefore, local delivery of brusatol, such as aerosol administration into the lung, may be superior over the systemic i.p. injection, which warrants further investigation. In summary, our results demonstrate that oncogenic activation of KRAS and KRAS amplification can activate the NRF2-mediated protective mechanism resulting in chemoresistance. Therefore, brusatol, an NRF2 inhibitor, can be used as an adjuvant to sensitize tumors with KRAS activation, in addition to those tumors resulting from KEAP1 or NRF2 mutations. This work provides a framework for the development of NRF2 inhibitors into therapeutic drugs to combat chemoresistance. Future studies investigating the contribution of KRAS-mediated transcriptional upregulation of NRF2 in chemoresistance using human lung tumor tissues will define the penetrance of this mechanism of resistance.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
Grant Support
This research article was funded by CA154377 and ES015010 (D.D.Z.), DP2OD007161 (P.K. Wong) and ES006694 (a center grant).
Special thanks to Montserrat Rojo de la Vega for proofreading this paper.
Footnotes
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed
References
- 1.Shields JM, Pruitt K, McFall A, Shaub A, Der CJ. Understanding Ras: ‘it ain’t over ‘til it’s over’. Trends Cell Biol. 2000;10(4):147–54. doi: 10.1016/s0962-8924(00)01740-2. [DOI] [PubMed] [Google Scholar]
- 2.Vojtek AB, Der CJ. Increasing complexity of the Ras signaling pathway. The Journal of biological chemistry. 1998;273(32):19925–8. doi: 10.1074/jbc.273.32.19925. [DOI] [PubMed] [Google Scholar]
- 3.Califano R, Landi L, Cappuzzo F. Prognostic and predictive value of K-RAS mutations in non-small cell lung cancer. Drugs. 2012;72 (Suppl 1):28–36. doi: 10.2165/1163012-S0-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 4.Bar J, Damianovich M, Hout Siloni G, Dar E, Cohen Y, Perelman M, et al. Genetic mutation screen in early non--small-cell lung cancer (NSCLC) specimens. Clinical lung cancer. 2014;15(2):159–65. doi: 10.1016/j.cllc.2013.11.005. [DOI] [PubMed] [Google Scholar]
- 5.Rejiba S, Wack S, Aprahamian M, Hajri A. K-ras oncogene silencing strategy reduces tumor growth and enhances gemcitabine chemotherapy efficacy for pancreatic cancer treatment. Cancer science. 2007;98(7):1128–36. doi: 10.1111/j.1349-7006.2007.00506.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Halaschek-Wiener J, Wacheck V, Schlagbauer-Wadl H, Wolff K, Kloog Y, Jansen B. A novel Ras antagonist regulates both oncogenic Ras and the tumor suppressor p53 in colon cancer cells. Mol Med. 2000;6(8):693–704. [PMC free article] [PubMed] [Google Scholar]
- 7.Wei F, Liu Y, Bellail AC, Olson JJ, Sun SY, Lu G, et al. K-Ras mutation-mediated IGF-1-induced feedback ERK activation contributes to the rapalog resistance in pancreatic ductal adenocarcinomas. Cancer letters. 2012;322(1):58–69. doi: 10.1016/j.canlet.2012.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim WY, Prudkin L, Feng L, Kim ES, Hennessy B, Lee JS, et al. Epidermal growth factor receptor and K-Ras mutations and resistance of lung cancer to insulin-like growth factor 1 receptor tyrosine kinase inhibitors. Cancer. 2012;118(16):3993–4003. doi: 10.1002/cncr.26656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Saki M, Toulany M, Rodemann HP. Acquired resistance to cetuximab is associated with the overexpression of Ras family members and the loss of radiosensitization in head and neck cancer cells. Radiother Oncol. 2013;108(3):473–8. doi: 10.1016/j.radonc.2013.06.023. [DOI] [PubMed] [Google Scholar]
- 10.Oliver TG, Mercer KL, Sayles LC, Burke JR, Mendus D, Lovejoy KS, et al. Chronic cisplatin treatment promotes enhanced damage repair and tumor progression in a mouse model of lung cancer. Genes Dev. 2010;24(8):837–52. doi: 10.1101/gad.1897010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kensler TW, Wakabayashi N, Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu Rev Pharmacol Toxicol. 2007;47:89–116. doi: 10.1146/annurev.pharmtox.46.120604.141046. [DOI] [PubMed] [Google Scholar]
- 12.Jeong WS, Jun M, Kong AN. Nrf2: a potential molecular target for cancer chemoprevention by natural compounds. Antioxid Redox Signal. 2006;8(1–2):99–106. doi: 10.1089/ars.2006.8.99. [DOI] [PubMed] [Google Scholar]
- 13.Jaramillo MC, Zhang DD. The emerging role of the Nrf2-Keap1 signaling pathway in cancer. Genes Dev. 2013;27(20):2179–91. doi: 10.1101/gad.225680.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hayes JD, McMahon M, Chowdhry S, Dinkova-Kostova AT. Cancer chemoprevention mechanisms mediated through the Keap1-Nrf2 pathway. Antioxid Redox Signal. 2010;13(11):1713–48. doi: 10.1089/ars.2010.3221. [DOI] [PubMed] [Google Scholar]
- 15.Magesh S, Chen Y, Hu L. Small molecule modulators of Keap1-Nrf2-ARE pathway as potential preventive and therapeutic agents. Med Res Rev. 2012;32(4):687–726. doi: 10.1002/med.21257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ramos-Gomez M, Kwak MK, Dolan PM, Itoh K, Yamamoto M, Talalay P, et al. Sensitivity to carcinogenesis is increased and chemoprotective efficacy of enzyme inducers is lost in nrf2 transcription factor-deficient mice. Proc Natl Acad Sci U S A. 2001;98(6):3410–5. doi: 10.1073/pnas.051618798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Khor TO, Huang MT, Prawan A, Liu Y, Hao X, Yu S, et al. Increased susceptibility of Nrf2 knockout mice to colitis-associated colorectal cancer. Cancer Prev Res (Phila) 2008;1(3):187–91. doi: 10.1158/1940-6207.CAPR-08-0028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lau A, Villeneuve NF, Sun Z, Wong PK, Zhang DD. Dual roles of Nrf2 in cancer. Pharmacol Res. 2008;58(5–6):262–70. doi: 10.1016/j.phrs.2008.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ooi A, Wong JC, Petillo D, Roossien D, Perrier-Trudova V, Whitten D, et al. An antioxidant response phenotype shared between hereditary and sporadic type 2 papillary renal cell carcinoma. Cancer Cell. 2011;20(4):511–23. doi: 10.1016/j.ccr.2011.08.024. [DOI] [PubMed] [Google Scholar]
- 20.Singh A, Misra V, Thimmulappa RK, Lee H, Ames S, Hoque MO, et al. Dysfunctional KEAP1-NRF2 interaction in non-small-cell lung cancer. PLoS Med. 2006;3(10):e420. doi: 10.1371/journal.pmed.0030420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ohta T, Iijima K, Miyamoto M, Nakahara I, Tanaka H, Ohtsuji M, et al. Loss of Keap1 function activates Nrf2 and provides advantages for lung cancer cell growth. Cancer research. 2008;68(5):1303–9. doi: 10.1158/0008-5472.CAN-07-5003. [DOI] [PubMed] [Google Scholar]
- 22.Solis LM, Behrens C, Dong W, Suraokar M, Ozburn NC, Moran CA, et al. Nrf2 and Keap1 abnormalities in non-small cell lung carcinoma and association with clinicopathologic features. Clinical cancer research : an official journal of the American Association for Cancer Research. 2010;16(14):3743–53. doi: 10.1158/1078-0432.CCR-09-3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jiang T, Chen N, Zhao F, Wang XJ, Kong B, Zheng W, et al. High levels of Nrf2 determine chemoresistance in type II endometrial cancer. Cancer research. 2010;70(13):5486–96. doi: 10.1158/0008-5472.CAN-10-0713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang XJ, Sun Z, Villeneuve NF, Zhang S, Zhao F, Li Y, et al. Nrf2 enhances resistance of cancer cells to chemotherapeutic drugs, the dark side of Nrf2. Carcinogenesis. 2008;29(6):1235–43. doi: 10.1093/carcin/bgn095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stacy DR, Ely K, Massion PP, Yarbrough WG, Hallahan DE, Sekhar KR, et al. Increased expression of nuclear factor E2 p45-related factor 2 (NRF2) in head and neck squamous cell carcinomas. Head Neck. 2006;28(9):813–8. doi: 10.1002/hed.20430. [DOI] [PubMed] [Google Scholar]
- 26.Shibata T, Kokubu A, Gotoh M, Ojima H, Ohta T, Yamamoto M, et al. Genetic alteration of Keap1 confers constitutive Nrf2 activation and resistance to chemotherapy in gallbladder cancer. Gastroenterology. 2008;135(4):1358–68. 68 e1–4. doi: 10.1053/j.gastro.2008.06.082. [DOI] [PubMed] [Google Scholar]
- 27.Ren D, Villeneuve NF, Jiang T, Wu T, Lau A, Toppin HA, et al. Brusatol enhances the efficacy of chemotherapy by inhibiting the Nrf2-mediated defense mechanism. Proc Natl Acad Sci U S A. 2011;108(4):1433–8. doi: 10.1073/pnas.1014275108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang DD, Lo SC, Cross JV, Templeton DJ, Hannink M. Keap1 is a redox-regulated substrate adaptor protein for a Cul3-dependent ubiquitin ligase complex. Mol Cell Biol. 2004;24(24):10941–53. doi: 10.1128/MCB.24.24.10941-10953.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cullinan SB, Gordan JD, Jin J, Harper JW, Diehl JA. The Keap1-BTB protein is an adaptor that bridges Nrf2 to a Cul3-based E3 ligase: oxidative stress sensing by a Cul3-Keap1 ligase. Mol Cell Biol. 2004;24(19):8477–86. doi: 10.1128/MCB.24.19.8477-8486.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Furukawa M, Xiong Y. BTB protein Keap1 targets antioxidant transcription factor Nrf2 for ubiquitination by the Cullin 3-Roc1 ligase. Mol Cell Biol. 2005;25(1):162–71. doi: 10.1128/MCB.25.1.162-171.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kobayashi A, Kang MI, Okawa H, Ohtsuji M, Zenke Y, Chiba T, et al. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol Cell Biol. 2004;24(16):7130–9. doi: 10.1128/MCB.24.16.7130-7139.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rada P, Rojo AI, Chowdhry S, McMahon M, Hayes JD, Cuadrado A. SCF/{beta}-TrCP promotes glycogen synthase kinase 3-dependent degradation of the Nrf2 transcription factor in a Keap1-independent manner. Mol Cell Biol. 2011;31(6):1121–33. doi: 10.1128/MCB.01204-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rada P, Rojo AI, Evrard-Todeschi N, Innamorato NG, Cotte A, Jaworski T, et al. Structural and functional characterization of Nrf2 degradation by the glycogen synthase kinase 3/beta-TrCP axis. Mol Cell Biol. 2012;32(17):3486–99. doi: 10.1128/MCB.00180-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chowdhry S, Zhang Y, McMahon M, Sutherland C, Cuadrado A, Hayes JD. Nrf2 is controlled by two distinct beta-TrCP recognition motifs in its Neh6 domain, one of which can be modulated by GSK-3 activity. Oncogene. 2013;32(32):3765–81. doi: 10.1038/onc.2012.388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wu T, Zhao F, Gao B, Tan C, Yagishita N, Nakajima T, et al. Hrd1 suppresses Nrf2-mediated cellular protection during liver cirrhosis. Genes Dev. 2014;28(7):708–22. doi: 10.1101/gad.238246.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.DeNicola GM, Karreth FA, Humpton TJ, Gopinathan A, Wei C, Frese K, et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature. 2011;475(7354):106–9. doi: 10.1038/nature10189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cozens AL, Yezzi MJ, Kunzelmann K, Ohrui T, Chin L, Eng K, et al. CFTR expression and chloride secretion in polarized immortal human bronchial epithelial cells. Am J Respir Cell Mol Biol. 1994;10(1):38–47. doi: 10.1165/ajrcmb.10.1.7507342. [DOI] [PubMed] [Google Scholar]
- 38.Tao S, Zheng Y, Lau A, Jaramillo MC, Chau BT, Lantz RC, et al. Tanshinone I activates the Nrf2-dependent antioxidant response and protects against As(III)-induced lung inflammation in vitro and in vivo. Antioxid Redox Signal. 2013;19(14):1647–61. doi: 10.1089/ars.2012.5117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Moghaddam SJ, Li H, Cho SN, Dishop MK, Wistuba II, Ji L, et al. Promotion of lung carcinogenesis by chronic obstructive pulmonary disease-like airway inflammation in a K-ras-induced mouse model. Am J Respir Cell Mol Biol. 2009;40(4):443–53. doi: 10.1165/rcmb.2008-0198OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Iwanaga K, Yang Y, Raso MG, Ma L, Hanna AE, Thilaganathan N, et al. Pten inactivation accelerates oncogenic K-ras-initiated tumorigenesis in a mouse model of lung cancer. Cancer research. 2008;68(4):1119–27. doi: 10.1158/0008-5472.CAN-07-3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sutherland KD, Song JY, Kwon MC, Proost N, Zevenhoven J, Berns A. Multiple cells-of-origin of mutant K-Ras-induced mouse lung adenocarcinoma. Proc Natl Acad Sci U S A. 2014;111(13):4952–7. doi: 10.1073/pnas.1319963111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zheng Y, Tao S, Lian F, Chau BT, Chen J, Sun G, et al. Sulforaphane prevents pulmonary damage in response to inhaled arsenic by activating the Nrf2-defense response. Toxicol Appl Pharmacol. 2012;265(3):292–9. doi: 10.1016/j.taap.2012.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Riahi R, Dean Z, Wu TH, Teitell MA, Chiou PY, Zhang DD, et al. Detection of mRNA in living cells by double-stranded locked nucleic acid probes. Analyst. 2013;138(17):4777–85. doi: 10.1039/c3an00722g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Riahi R, Wang S, Long M, Li N, Chiou PY, Zhang DD, et al. Mapping photothermally induced gene expression in living cells and tissues by nanorod-locked nucleic acid complexes. ACS Nano. 2014;8(4):3597–605. doi: 10.1021/nn500107g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Arany I, Megyesi JK, Kaneto H, Price PM, Safirstein RL. Cisplatin-induced cell death is EGFR/src/ERK signaling dependent in mouse proximal tubule cells. Am J Physiol Renal Physiol. 2004;287(3):F543–9. doi: 10.1152/ajprenal.00112.2004. [DOI] [PubMed] [Google Scholar]
- 46.Belinsky SA, Stefanski SA, Anderson MW. The A/J mouse lung as a model for developing new chemointervention strategies. Cancer research. 1993;53(2):410–6. [PubMed] [Google Scholar]
- 47.Schiller JH. Current standards of care in small-cell and non-small-cell lung cancer. Oncology. 2001;61 (Suppl 1):3–13. doi: 10.1159/000055386. [DOI] [PubMed] [Google Scholar]
- 48.Kim YR, Oh JE, Kim MS, Kang MR, Park SW, Han JY, et al. Oncogenic NRF2 mutations in squamous cell carcinomas of oesophagus and skin. J Pathol. 2010;220(4):446–51. doi: 10.1002/path.2653. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.