

Abstract
Vascular thiol redox state has been shown to modulate vasodilator functions in large conductance Ca2+-activated K+ channels and other related channels. However, the role of vascular redox in small resistance arteries is unknown. To determine how in vivo modulation of thiol redox state impacts small resistance arteries relaxation we generated a transgenic mouse strain that overexpress thioredoxin (Trx), a small redox protein (Trx-Tg) and another strain that is Trx-deficient (dnTrx-Tg). The redox state of the mesenteric arteries (MAs) in Trx-Tg mice is found to be predominantly in reduced state; in contrast, MAs from dnTrx-Tg mice remain in oxidized state. Thus, we created an in vivo redox system of mice and isolated the 2nd-order branches of the main superior MAs from wildtype (wt), Trx-Tg or dnTrx-Tg mice to assess endothelium-dependent relaxing responsesin a wire-myograph. In MAs isolated from Trx-Tg mice we observed an enhanced IK1 channel contribution resulting in a larger endothelium-dependent hyperpolarizing (EDH) relaxation in response to indirect (ACh) and direct (NS309) opening of endothelial KCa channels. MAs derived from dnTrx-Tg mice showed both blunted NO-mediated and EDH-mediated relaxation compared to Trx-Tg mice. In a control study, diamide decreased EDH relaxations in wt mice MAs, whereas DTT improved EDH relaxations, and was able to restore the diamide-induced impairment in EDH response. Further, the basal or angiotensin II-mediated systolic blood pressure remained significantly lower in Trx-Tg mice compared to wt or dnTrx-Tg mice; thus, directly establishing redox-mediated EDHF in blood pressure control.
Keywords: thioredoxin, vascular redox, endothelium, relaxation, mesenteric artery, resistant arteries, hyperpolarization
Introduction
The endothelium plays an important role in maintaining normal vascular hemostasis by releasing vasoactive mediators, such as nitric oxide (NO), prostacyclin (PGI2), and endothelium-dependent hyperpolarization (EDH) factors in response to hemodynamic, metabolic and humeral stimuli.1 EDH is generally described as the relaxation which is independent of NO or PGI2.2 The relaxation due to EDH is dependent on the vessel type and size, being more pronounced in resistance-sized arteries (lumen diameter of < 300 μm) and arterioles, which are important regulators of vasomotor tone, local tissue perfusion and blood pressure.3, 4 Calcium-activated potassium channels (KCa) are key players in the initiation of the EDH response. In the mesenteric arterial circulation the small- and intermediate KCa channels (SK3 or KCa2.3 and IK1 or KCa3.1, respectively) are generally believed to mediate endothelial and smooth muscle hyperpolarization, since pharmacological blockade of both these channels abolishes the acetylcholine- induced EDH response.4-9
Studies with large conductance Ca2+ activated K+ channels (BKCa) have shown that chemical reductant DTT promotes channel activity; in contrast, thiol oxidizing agent diamide inhibits it in vitro.10 There are limited numbers of studies demonstrating that the BKCa channels are modulated by oxidative stress.10-12 For example, NO activates this channel leading to channel opening, whereas oxidizing agent, H2O2 inactivates this channel in isolated porcine renal arteries.13 Redox modification of sulfhydryl groups has been shown to alter BKCa channel gating, in a manner that oxidizing agents inhibited, but reducing agents like DTT and β-mercaptoethanol stimulated its activity in smooth muscle cells.10, 14 All of these studies were conducted in vitro, treating vessels with chemical oxidizers or reducers ex vivo.
Small mesenteric arteries isolated from mice are commonly used to study endothelium-dependent relaxations. We reasoned that endothelial KCa channels could be redox sensitive and thereby susceptible to redoxregulation of their activity. To determine the effect of in vivo thiol-disulfide reductase on vascular function we generated a transgenic mouse strain(Trx-Tg) that overexpress human thioredoxin (Trx), and another strain that is deficient (dnTrx-Tg) in endogenous active Trx. Because Trx knockout mice are embryonic lethal15, we generated the dnTrx-Tg transgenic mouse line, which expresses Trx that is redox inactive because the catalytic cysteines are mutated to serine (C32S, C35S).Trx is a thiol disulfide reductase with antioxidant properties and is a major regulator of cellular redox state.16 Together with Trx reductase (TrxR) it regenerates oxidatively inactivated proteins to restore their normal function and NADPH provides reducing equivalents for regeneration of Trx. Although potentially important the role of Trx in vessel redox homeostasis remains virtually unknown.
The major goal of this study was to determine whether in vivo modulation Trx redox state increases EDH response in resistance arteries. We hypothesized that redox- active Trx controls the endothelial KCa channel activity via its thiol reductase function leading to an enhanced EDH response in murine small mesenteric arteries. We also determined whether increased levels of Trx would enhance the IK1 activity, which may increase ACh-induced EDH response. We utilized our newly created Trx-Tg mice and dnTrx-Tg mice to demonstrate in vivo effects of Trx on SK3 and IK1–dependent EDH in mice mesenteric arteries. We demonstrate that whereas mesenteric arteries from Trx-Tg mice show increased EDH, the vessels from dnTrx-Tg mice had significantly lower EDH. As a result systolic blood pressure is reduced in Trx-Tg mice compared to wt or dnTrx-Tg mice
Methods
See the online-only Data Supplement for detailed methods.
Animals
To understand the effect of in vivo redox modulation on EDH response we generated transgenic mice overexpressing cytosolic Trx, and another strain that contain decreased level of active Trx in a dominant-negative manner due to mutation of active site cysteines to serine (C32S C35S) as shown in online Figure S1.
Non-Invasive Systolic Blood Pressure Measurements by Tail Cuff Method
In order to obtain reliable blood pressure measurements in conscious mice via the tail cuff method (CODA system, Kent Scientific, USA), mice were trained for 1-2 weeks using the tail cuff system. After this training period systolic blood pressures were recorded and the values obtained during three consecutive days were averaged.
Vascular Contractile Responses
After a 30 min washout period, cumulative concentration-response curves (CRC) were performed to the vasoconstrictor phenylephrine (PHE; 0.01 – 30 μmol/L), which causes α1-adrenergic-mediated contractions. The role of NO in suppressing PHE-induced contractions was assessed by incubating the same MAs with non-selective NO synthase blocker Nω-Nitro-L-arginine methyl ester(L-NAME;100 μmol/L) for 30 min followed by a CRC to PHE.
Vascular Relaxation Responses
During contraction with a single concentration of phenylephrine (PHE; 10 – 30 μmol/L), relaxing responses to the endothelium-dependent muscarinic vasodilator acetylcholine (ACh; 0.01 – 10 μmol/L) were recorded in the absence of any inhibitors (control).
EDH-mediated Relaxing Responses
EDH-mediated relaxations were recorded in the presence of L-NAME and INDO. To study the contribution of each KCa channel subtype, EDH-mediated relaxations were always studied in the combined presence of L-NAME and INDO to rule out any potential interference of NO and prostaglandins with KCa channels.171717
Direct Opening of SK3 and IK1 Channels by NS309
In a subset of MAs, the relaxing responses of the KCa channel activator 3-oxime-6,7-dichloro-1H-indole-2,3-dione (NS309; 0.01 – 3 μmol/L)18 were recorded. The same segments were then incubated 30 min with INDO (10 μmol/L) and L-NAME (100 μmol/L), and relaxing responses to NS309 were repeated.
Role of Thiol Sulfhydryl Modifying Agents on EDH Responses
In MAs derived from wt mice, the EDH-mediated relaxing responses were assessed with sulfhydryl modifying agents: diamide as a reversible oxidizing agent and DTT as a reducing agent.
Redox Assay of Trx
MAs were excised from Trx-Tg and dnTrx-Tg mice and carboxymethylation of MAs homogenates was performed as described in our previous publications.19
Thioredoxin, Thioredoxin Reductase and Peroxiredoxin Assays
Trx and Trx reductase activity assays were performed as described in our previous publications19. Peroxiredoxin (Prx) assay was performed with H2O2 as substrate using Trx-SH as electron donor in presence of mammalian TrxR and NADPH20.
Results
Vessel redox environment is predominantly oxidized in dnTrx-Tg mice, but maintained in reduced state in Trx-Tg mice
Because Trx knockout mice are embryonic lethal, we generated the dnTrx-Tg transgenic mouse line, which expresses Trx that is redox inactive because the catalytic cysteines are mutated to serine (C32S, C35S). This mutant protein competitively inhibits the reduction of endogenous Trx by TrxR, with a Ki of 1.8 μM 21. Thus, by blocking the regeneration of endogenous Trx, dnTrx-Tg functions as dominant-negative 21–23. For comparative studies, we generated a Trx-Tg transgenic mouse line that overexpresses human Trx (hTrx). To confirm the expression patterns, we conducted immunoblots with an anti-Trx antibody that recognizes both wt and mutant Trx, and found increased transgenic protein levels in MA lysates of dnTrx-Tg as well as in Trx-Tg mice (Figure 1A).We could not detect endogenous mouse Trx in the same blot although the antibody is reactive to mouse Trx.Increased expression of Trx by both dnTrx-Tg and Trx-Tg mice was detected in various organs (Figure 2S).To establish that increased expression of Trx (or dnTrx) correlates with increased (or decreased) Trx function, respectively, we determined the activity of Trx in the MAs isolated form wt, Trx-Tg or dnTrx-Tg. As expected Trx-Tg mice showed significantly higher levels of Trx activity compared to wt or dnTrx-Tg mice (Figure 1B) in the MAs. However, the activity of TrxR that regenerates Trx using NADPH remained unchanged among MAs isolated from wt, Trx-Tg or dnTrx-Tg mice (Figure 1C). To determine the effect of increased or decreased expression of Trx on vessel redox state we performed the redox state assay of Trx in MAs isolated from Trx-Tg or dnTrx-Tg mice. As shown in Figure 1D the redox state of MAs from dnTrx mice is predominantly oxidized as we did not detect any reduced Trx in pooled samples of MAs. However, significantly higher level of reduced Trx was detected in the MAs from Trx-Tg mice (Figure 1D, lane 1, lower band). These data show that the overall redox state of MAs from dnTrx-Tg mice is oxidized compared to Trx-Tg mice, which is reduced. Thus, our mice system represents an in vivo redox model, and we further studied MAs in this mice system.
Figure 1.
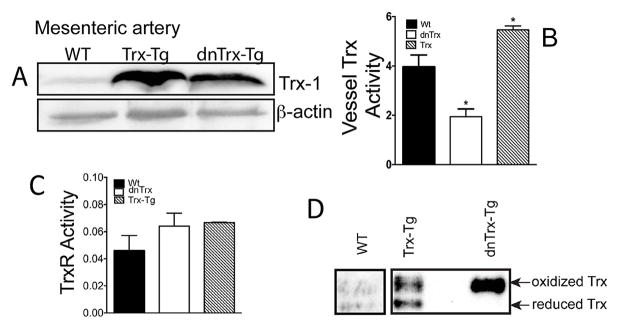
(A) Expression of Trx in mesenteric artery of wt, Trx-Tg and dnTrx-Tg mice; (B)Trx activity is increased in MAs in Trx-Tg mice, but not in dnTrx-Tg mice.(C) TrxR activity does not change in wt, Trx-Tg or dnTrx-Tg mice MAs.(D) Trx remains predominantly in the oxidized state in MAs from dnTrx mice.
Structure of wt, Trx-Tg and dnTrx-Tg mesenteric artery
The optimal diameters of segments of MAs measured in the wire-myograph were comparable for all three mice groups: 203 ± 6μm in WT, 205 ± 8 μm in Trx-Tg, and 194 ± 9μm in dnTrx-Tg mice.
Comparable contractile responses in MAs from all three mice groups
Addition of 60 mmol/L K+ KRB resulted in tensions did not differ between MAs derived from all three mice (Figure 2A) and PHE contracted MAs in a concentration-dependent manner (Figure 2B). The sensitivity (pEC50) to PHE did not differ significantly between arteries from wt, Trx-Tg, and dnTrx-Tg mice (5.61± 0.07, 5.72± 0.09 and 5.74± 0.09, respectively, Figure 2B). The maximal tension (in mN/mm) in response to 30 μM PHE was significantly increased in Trx-Tg (2.89 ± 0.12) compared to wt (2.52 ± 0.12)and dnTrx-Tg mice (2.39 ± 0.20; Figure 2B).We also studied PHE-mediated contractions in the presence of L-NAME to determine the contribution of basal NO levels in suppressing these contractions. Figure 3S shows that L-NAME caused significantly larger tension levels in response to PHE in MAs from wt and Trx-Tg mice, but not in dnTrx-Tg mice.
Figure 2.

(A) Depolarization (60 mmol/L K+ KRB)-induced, and (B) induced contraction in MAs derived from wt (white bars), Trx-Tg (blue bars) and dnTrx-Tg (red bars) mice. MAs derived from wild-type (circles), Trx-Tg (upward triangles) and dnTrx-Tg (downward triangles) mice were assessed for: (C) Endothelium-dependent relaxations to cumulative concentrations of ACh. (D) Endothelium-independent relaxations in MAs pretreated with L-NAME and indomethacin (INDO) contracted with a submaximal concentration of PHE, and assessed with cumulative concentrations of SNP. (E) NO-mediated endothelium-dependent relaxations in MAs pretreated for 30 min with INDO the IK1 channel blocker TRAM-34 and the SK3 channel blocker UCL-1684, to block EDH-mediated responses, contracted with PHE before assessing relaxing responses to cumulative concentrations of ACh. (F) Endothelium-dependent hyperpolarizing (EDH) responses in MAs treated with the NO synthase blocker L-NAME and INDO followed by cumulative concentrations of ACh. Values are means ± SEM (n=8–10 mice). * P< 0.05 Trx-Tg compared with wt, # P< 0.05 dnTrx-Tg compared with wtand Trx-Tg, ** P< 0.05 Trx-Tg compared with wtand dnTrx-Tg.
Trx-deficiency resulted in decreased NO-mediated relaxations in MAs from dnTrx- Tg mice
Since generation of NO is a major contributor to vessel relaxation, we determined the effect of in vivo redox modulation on NO-mediated relaxations in MAs. As shown in Figure 2C, endothelium-dependent ACh-mediated relaxations were significantly enhanced in MAs derived from Trx-Tg mice compared to wt mice. Sensitivity (EC50) to ACh averaged 0.16 ± 0.06μmol/L in Trx-Tg compared to 0.42 ± 0.20μmol/L in wt mice. The maximal relaxation (Emax) in response to 10 μmol/L was higher in MAs from Trx-Tg mice (96 ± 1 %) compared towt(92 ± 2 %) mice, but this did not reach statistical significance. In contrast, these ACh-mediated responses were severely blunted in MAs from dnTrx-Tg, as reflected both by a significantly larger EC50 (1.41 ± 0.65μmol/L) and lower Emax (79 ± 4 %) compared to either wt or Trx-Tg mice. This impairment in the relaxing responses of MAs from dnTrx-Tg mice was specific to the endothelium, since relaxing responses to the endothelium-independent NO donor sodium nitroprusside were comparable in MAs from all three mice groups (Figure 2D). Next, we determined NO-mediated relaxing responses in MAs by treating them with a cocktail of INDO and the endothelial KCa channel blockers TRAM-34 (1 μmol/L) and UCL-1684 (1 μmol/L),in order to block vasorelaxing prostanoid release and EDH responses, respectively. As shown in Figure 2E NO-mediated relaxing responses were significantly reduced in MAs from dnTrx-Tg mice compared to MAs from wt or Trx-Tg mice. Sensitivity (EC50) for ACh averaged 29 ± 14 μmol/L in dnTrx-Tg mice compared to 4.0 ± 2.2 μmol/L and 1.2 ± 0.3 μmol/L in wt or Trx-Tg mice, respectively (Figure 2E). Similarly, Emax values were 47 ± 7 % in dnTrx-Tg, compared to 73 ± 4 % and 77 ± 3 % in wt and Trx-Tg mice, respectively (Figure 2E).
EDH relaxations are increased in MAs from TRX-Tg mice
EDH-mediated relaxing responses were analysed by incubating MA sin the combined presence of L-NAME and INDO. EDH responses were markedly enhanced in MAs from Trx-Tg mice, compared to wt or dnTrx-Tg mice (Figure 2F). EC50 values were significantly lower in Trx-Tg (2.7 ± 1.3μmol/L) compared to wt and dnTrx-Tg mice (9.8 ± 4.7μmol/L and 16.7 ± 6.7μmol/L, respectively). Maximal relaxation averaged 74 ± 6% in Trx-Tg mice, which was significantly larger compared to wtand dnTrx-Tg mice (56 ± 8 % and 51 ± 7 %, respectively).
Effect of Trx redox on IK1 and SK3 channels in ACh-mediated EDH relaxation
The selective IK1 channel blocker TRAM-34 inhibited the EDH response to comparable levels in MAs from all three mice groups (Figure 3A). Consequently, the percentage inhibition by TRAM-34 was significantly higher in MAs derived from Trx-Tg mice compared to wt or dnTrx-Tg mice (see inserted graph Figure 3A). The selective SK3 channel blocker UCL-1684 also resulted in similar residual relaxations in MAs of all three mice groups (Figure 3B). Combined inhibition of IK1 and SK3 channels by TRAM- 34 and UCL-1684 almost completely blunted the ACh-mediated relaxations (Figure 3C).
Figure 3.
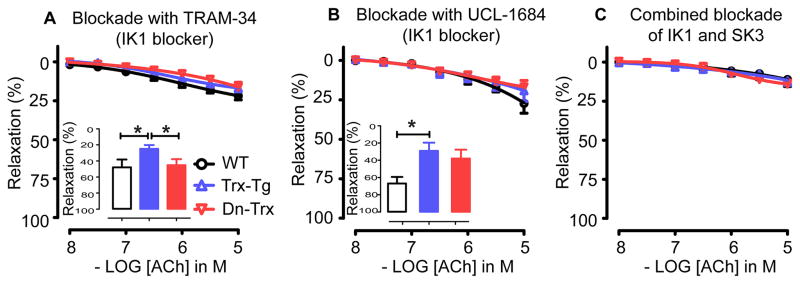
(A) EDH-mediated responses in the presence of the selective IK1 channel blocker TRAM-34 in MAs derived from wt (circles), Trx-Tg (upward triangles) and dnTrx-Tg (downward triangles) mice. Insert: Percentage inhibition of the EDH response by TRAM-34 in MAs derived from wt (white bars), Trx-Tg (blue bars) and dnTrx-Tg (red bars) mice. (B) EDH-mediated responses in the presence of the selective SK3 channel blocker UCL-1684 in MAs derived from wt (circles), Trx-Tg (upward triangles) and dnTrx-Tg (downward triangles) mice. Insert: Percentage inhibition of the EDH response by UCL-1684 in MAs derived from wt (white bars), Trx-Tg (blue bars) and dnTrx-Tg (red bars) mice. (C) EDH-mediated responses in the combined presence of TRAM-34 and UCL-1684 in MAs derived from wt (circles), Trx-Tg (upward triangles) and dnTrx-Tg (downward triangles) mice. Values are means ± SEM (n=6-8 mice). * P< 0.05 Trx-Tg compared with wt or dnTrx-Tg.
Direct opening of endothelial KCa channels by NS309 is more sensitive in MAs from Trx-Tg mice
Since ACh indirectly activates KCa channels, we investigated the effect of direct opening of IK1 and SK3 channels by NS309 on the EDH-mediated relaxation in MAs. We studied NS309-induced responses in endothelium-intact as well as endothelium-denuded segments. NS309 (0.01 – 3 μmol/L) induced potent relaxations in endothelium-intact MAs (Figure 4A). However, these relaxations were decreased in dnTrx-Tg compared to wt or Trx-Tg mice. EC50 values averaged 0.5 ± 0.2μmol/L in dnTrx-Tg compared to 0.08 ± 0.02 μmol/L and 0.07 ± 0.02 μmol/L in wt or Trx-Tg mice, respectively (Figure 4A). Endothelial denudation inhibited NS309-induced relaxations in MAs from all three mice groups indicating that the effects are endothelium-dependent (Figure 4A). Inhibition with L-NAME and INDO reduced NS309-induced relaxations in MAs from all three mice groups (Figure 4B).However, sensitivity (EC50) to NS309 was significantly higher in MAs of Trx-Tg (0.3 ± 0.1μmol/L) compared to wt(0.9 ± 0.2μmol/L) or dnTrx-Tg (0.7 ± 0.2μmol/L) mice. The pharmacological endothelial KCa channel activator SKA-31 has a 10-fold higher potency for IK1 than SK3.24 Incubation of L- NAME- and INDO-treated MAs with SKA-31 (1 μmol/L) resulted in an enhanced ACh-mediated EDH response in all mice groups, but to a greater extent in MAs from Trx-Tg mice (Figure 4C). Sensitivity was significantly larger in MAs from Trx-Tg mice (0.7 ± 0.2 μmol/L), compared to wt(4.3 ± 2.1 μmol/L) or dnTrx-Tg (4.7± 1.6 μmol/L), suggesting a greater contribution of IK1 channels in the EDH response in MAs from Trx-Tg mice.
Figure 4.
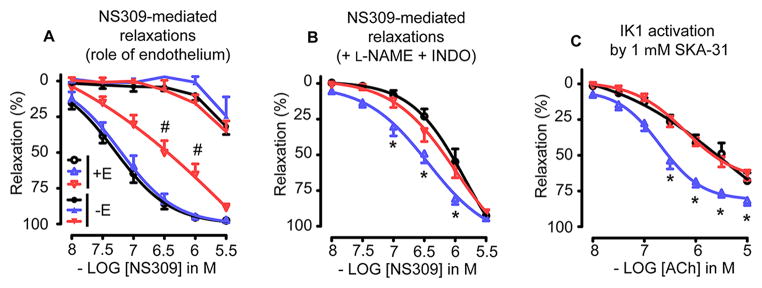
(A) NS309-mediated relaxing responses in endothelium-intact (+E, open symbols) and endothelium-denuded (-E, closed symbols) MAs derived from wt (circles), Trx-Tg (upward triangles) and dnTrx-Tg (downward triangles) mice. (B) NS309-mediated relaxing responses in MAs treated with L-NAME (100 μmol/L) and INDO (10 μmol/L) from wt (circles), Trx-Tg (upward triangles) and dnTrx-Tg (downward triangles) mice. (C) Effect of IK1 channel activator SKA-31 (1 μmol/L) in MAs treated with L-NAME and INDO followed by contraction with PHE and cumulative concentrations to ACh. MAs were isolated from wt (circles), Trx-Tg (upward triangles) and dnTrx-Tg (downward triangles) mice. Values are means ± SEM (n=8–10 mice). * P< 0.05 Trx-Tg compared with wt and dnTrx-Tg, # P< 0.05 dnTrx-Tg compared with wt and Trx-Tg.
Opening of endothelial KCa channels by NS309 is primarily mediated by IK1
Since both IK1 and SK3 are activated by NS309, we determined the effect of Trx redox on specific KCa channel opening by using NS309 in combination with a specific inhibitor of either IK1 or SK3. Inhibition of IK1 channels with TRAM-34 led to a greater rightward shiftin the response to NS309 in MAs from Trx-Tg mice compared to wtand dnTrx-Tg mice (Figure 5A). This was evident by a significantly larger shift in sensitivity (pEC50) for NS309 caused by TRAM-34 in Trx-Tg mice compared to wt or dnTrx-Tg mice (Figure 5A, insert). Blockade of SK3 channels with UCL-1684 resulted in comparable NS309-induced relaxations (Figure 5B). However, a significant leftward shift in the response to NS309 occurred in wt compared to Trx-Tg or dnTrx-Tg mice (Figure 5B, insert). Further, combined blockade by TRAM-34 and UCL-1684 led to similar relaxations as compared to TRAM-34 alone (Figure 5C).
Figure 5.
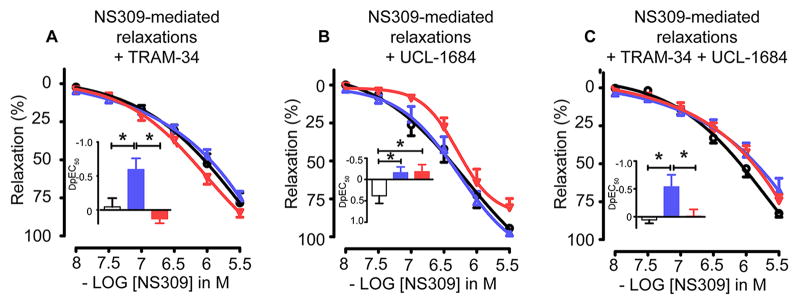
NS309-mediated relaxing responses in MAs derived from wt (circles), Trx-Tg (upward triangles) and dnTrx-Tg (downward triangles) mice. MAs were either incubated with L-NAME (100 μmol/L), INDO (10 μmol/L) and the IK1 blocker TRAM-34 (A), or the SK3 channel blocker UCL-1684 (B) or the combined presence of TRAM-34 and UCL- 1684 (C). Inserts: shift in sensitivity (ΔpEC50) for NS309 caused by IK1 inhibition by TRAM-34 for wt (white bars), Trx-Tg (blue bars), and dnTrx-Tg (red bars) mice. Values are means ± SEM (n=8–10 mice). * P< 0.05 Trx-Tg compared with wt or dnTrx-Tg.
Redox control of EDH response
Since Trx is a protein disulfide reductase, we reasoned that the enhanced EDH response in Trx-Tg mice might be mediated by the thiol reductase effect of Trx. To address this we used diamide as a reversible sulfhydryl oxidant, and DTT as a disulfide reducing agent. In MAs isolated from wt mice and contracted with PHE (3 – 10 μmol/L), diamide (0.01 – 100 μmol/L) caused a concentration-dependent inhibition of the active tension (Figure 6A). The IC50 of diamide was found to be ~2.3 μmol/L. In contrast, DTT resulted in small relaxations at lower concentrations (< 100 μmol/L) and resulted in recovery of PHE-induced active tension at higher concentrations (0.1 – 1 mmol/L; Figure 6A). Based on these characteristics we chose two concentrations for diamide and DTT, both in the micro molar range (0.1 and 1 μmol/L). To determine whether diamide-mediated “relaxing” effects were due to sulfhydryl oxidation, we examined whether DTT was able to reverse the diamide-mediated effect. As shown in Figure 6B, treatment of 1 mmol/L DTT completely reversed the diamide (20 μmol/L)-mediated reduction in PHE-induced active tension. Because of this fast-acting effect of DTT we also assessed combinations of diamide and DTT in modulating the EDH response. Figure 6C shows that 0.1 μmol/L diamide caused a significant reduction in the EDH response. Emax averaged 28 ± 3% in the presence of 0.1 μmol/L diamide and 44 ± 3 % in the absence of diamide (Figure 6C). Incubation of 1 μmol/L diamide caused a similar impairment as 0.1 μmol/L diamide (28 ± 6%; Figure 6C). Co-incubation of diamide with TRAM-34 (1 μmol/L) resulted in comparable residual relaxations (Figure 6C, insert). In contrast to diamide, DTT dose-dependently enhanced the EDH response in MAs (Figure 6D). Emax averaged 53 ± 11% in the presence of 0.1 μmol/L DTT and 68 ± 11% in the presence of 1 μmol/L DTT (Figure 6D). Again, these effects seemed to be mediated via IK1 channel activation, since co-incubation with TRAM-34 resulted in equal residual relaxations (Figure 6D, insert). The lowest concentration of DTT tested (0.1 μmol/L) was unable to reverse the diamide (0.3 μmol/L)-induced blunted EDH response, but 1 and 10 μmol/L DTT significantly restored the EDH response (Figure 6E).
Figure 6.

(A) Cumulative concentration-response curves to diamide and(DTT) in MAs derived from wt mice. (B) DTT reverses diamide-induced inhibition of PHE-induced increase in active tension. Representative tracing showing tension changes in a MAs isolated from a wt mouse contracted with PHE before, during and after application of diamide, followed by addition of DTT. (C) EDH responses (in the presence of L-NAME and INDO in MAs derived from wt mice treated without (CON, open circles), 0.1 μmol/L diamide (open diamonds) or 1 μmol/L diamide (closed diamonds). Insert: Effect of IK1 channel inhibition by TRAM-34 on the EDH responses. (D) EDH responses in MAs derived from wt mice treated without (CON, open squares), 0.1 μmol/L DTT (open squares), or 1 μmol/L DTT (closed squares). Insert: Effect of IK1 channel inhibition by TRAM-34 on the EDH responses. (E) EDH responses in MAs derived from wt mice treated with 0.3 μmol/L diamide in the additional presence of either 0.1 μmol/L DTT (circles), 1 μmol/L DTT (squares) or 10 μmol/L DTT (diamonds). (F) MAs derived from dnTrx-Tg (downward triangles) mice were treated for 30 min with either vehicle (H2O; CON) or = DTT and were assessed for endothelium-dependent relaxations to cumulative concentrations of ACh =. Values are means ± SEM (n=4–6 mice). * P< 0.05.
DTT improved ACh-induced relaxations in MAs from dnTrx-Tg mice
The dominant-negative production of catalytically inactive hTrx (C32S; C35S) in dnTrx-Tg mice competes for reduction by TrxR with oxidized Trx. Therefore, significant amount of wt mouse Trx and all of the mutant hTrx appears to remain as “oxidized Trx” in the redox assay. While DTT could reduce the oxidized wt mouse Trx, it cannot reduce the C32S and C35S mutant Trx as cysteine is substituted with serine. Therefore, we reasoned that MAs from dnTrx-Tg mice would be minimally relaxed by DTT. Incubation of MAs from dnTrx-Tg mice with 10 μmol/L DTT for 30 min, prior contraction with PHE (10 μmol/L) followed by cumulative concentrations to ACh, improved endothelium-dependent relaxations (Figure 6F), albeit with less potency.
H2O2-mediated relaxations are less sensitive in MAs from Trx-Tg mice
H2O2 is a sulfhydryl-oxidizing agent that has been shown to act as an EDHF25. Although H2O2 causes endothelium-independent relaxations in a many arteries its effect on a specific channel or its specific role in endothelial cells and/or smooth muscle cells remains far from clear26. Nevertheless, H2O2 does induce relaxations in MAs in mice, albeit at concentrations that appear toxic to vascular cells (> 3 μmol/L). We speculated that the ”relaxation” demonstrated in response to H2O2 could actually be due to its effect as an oxidizing agent similar to thiol-disulfide exchanges that we observed in diamide and DTT (Figure 6). Our data in Figure 7 shows that relaxing responses to H2O2 were significantly less sensitive in MAs from Trx-Tg mice compared to wt and dnTrx-Tg mice. These responses were significantly inhibited by catalase (Figure 7B). These results suggest that increased levels of Trx could decrease the applied concentrations of H2O2 via 2-Cys peroxiredoxins (Prx) that draw their reducing equivalents from Trx.Prx expression is known to be induced by increased levels of Trx.27 Therefore, we performed Prx activity assay of MAs isolated from wt, Trx-Tg and dnTrx-Tg mice. As shown in Figure 7C, Prx activity is significantly higher in Trx-Tg mice compared to either wt or dnTrx-Tg mice. Therefore, H2O2-mediated relaxation is decreased in Trx-Tg mice due to increased Prx activity in Trx-Tg mice, but not in wt or dnTrx-Tg mice.
Figure 7.
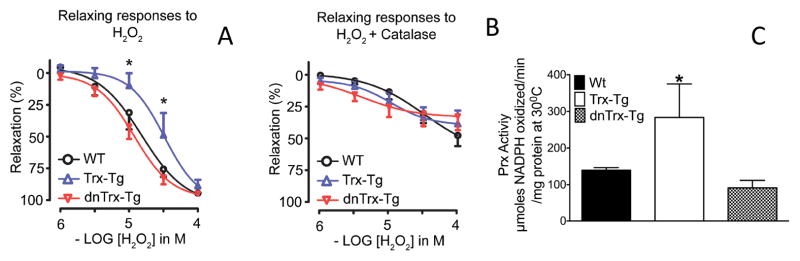
(A) In a subset of MAs, relaxing responses to hydrogen peroxide (H2O2) were analysed. Segments were contracted with PHE, followed by a CRC for H2O2. (B) After a 15 min washout period, the same segments were incubated with catalase for 30 min and CRCs to H2O2 were repeated; (C)Activity of Prx is increased in Trx-Tg MAs, but not in wt or dnTrx-Tg MAs.
Increased levels of Trx is correlated with decreased blood pressure in Trx-Tg mice, but not in wt or dnTrx-Tg mice
Our studies demonstrated that vascular relaxations are affected by redox state of the MAs. Since relaxation of MAs is directly related to blood pressure control, we determined whether modulation of vascular Trx redox would impact the basal blood pressure levels in our transgenic mice system. As shown in Figure 8, Trx-Tg mice showed significantly lower level of basal blood pressure compared either to wt mice or dnTrx-Tg mice indicating a direct effect of vascular redox homeostasis of MAs on blood pressure regulation. Further, we also determined whether higher levels of Trx could provide protection against increased blood pressure in response to angiotensin II (Ang II). As shown in Figure 8B, Ang II infusion (1000 ng/kg/min) caused an increase in blood pressure level in wt and Trx-Tg mice; however, the increase in Trx-Tg mice was significantly lower compared to wt mice, demonstrating that Ang II–mediated hypertension is attenuated by increased levels of Trx.
Figure 8.
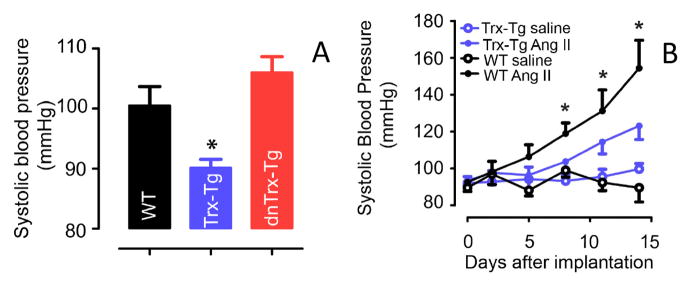
(A) The baseline blood pressure of 12 week old mice were measured as described in the methods. The blood pressure of Trx-Tg mice is significantly (P<0.05, ANOVA) lower than the wt or dnTrx-Tg mice. (B) Angiotensin II –mediated increased blood pressure is significantly decreased in Trx-Tg mice, but not in wt mice. *Significantly higher than Trx-Tg mice (P<0.05, ANOVA).
Discussion
The major finding of this study is that endothelium-dependent hyperpolarizing (EDH) relaxations in small mesenteric arteries are dependent on in vivo redox condition of vessels. This is the first report demonstrating that increased Trx expression in Trx-Tg mice results in an increased EDH response in small mesenteric arteries. In contrast, mice that are deficient in active Trx show severely blunted endothelium-dependent relaxations due to both a reduced NO- and EDH-mediating relaxation. Functionally, the reduced NO and EDH response in dnTrx-Tg was reflected by a significantly higher systolic blood pressure; in contrast, increased NO and EDH response was correlated with decreased blood pressure in Trx-Tg mice, confirming the important role of endothelial KCa channels in modulating blood pressure. Additional in vitro studies show that the EDH relaxations could be modulated by exogenous addition of sulfhydryl modifying agents such as diamide or DTT, where diamide blunted but DTT enhanced the EDH relaxation. Further, we demonstrate a pivotal role for IK1 channel activation in mediating the EDH response via redox state modulation.
It is now well accepted that endothelial SK3 and IK1 channels initiate and conduct the EDH response4–8, 28. Since combined blockade of SK3 and IK1 channels blunted ACh-mediated relaxations it is likely that these channels mediate the EDH response in these small mesenteric arteries and suggest that BKCa channels play a minor or no role as observed by others.5–7, 9, 29 Endothelial denudation drastically reduced the relaxations mediated by NS309 demonstrating that NS309 is an endothelium-dependent KCa channel opener as confirmed by others.30, 31 In the presence of L-NAME and indomethacin, NS309-induced responses were similar in MAs from wtand dnTrx-Tg mice, but both were significantly lower compared to Trx-Tg mice, suggesting an enhanced EDH response in the latter mice. NS309 has been demonstrated to initiate NO release in addition to contributing to the main EDH component in rat small mesenteric arteries.31 Our observation that NS309-induced relaxations were comparable in MAs from wt or dnTrx-Tg mice in the presence of L-NAME and INDO (whereas these responses were enhanced in the absence of these inhibitors for MA from wt compared to dnTrx-Tg mice) might be attributed to the fact that NO release was inhibited.
SKA-31 has a 10-fold higher affinity for IK1 compared to SK3 channels,24 suggesting that IK1 channels are predominantly activated. To assess this, we analyzed the role of IK1 channels in mediating NS309-induced relaxations. TRAM-34 blunted NS309-induced EDH responses to a greater extent in MAs derived from Trx-Tg mice, compared to wt or dnTrx-Tg mice (Figure 5A), suggesting a greater contribution of IK1 channels in mediating the EDH response in Trx-Tg mice. UCL-1684 did not alter NS309-induced relaxations in MAs from Trx-Tg and dnTrx-Tg, but caused a paradoxical increase in the sensitivity for NS309 in arteries from wt mice.
It is likely that in MAs from Trx-Tg mice an increased IK1 channel activity is present. We questioned whether higher levels of Trx increase IK1 channel activity. Diamide is a thiol oxidant that rapidly crosses the membrane by diffusion.32 Trx has been shown to be inactivated due to oxidation by diamide.33 Diamide has been shown to cause “relaxation” in contracted rat pulmonary arteries and bovine coronary arteries, due to closure of L-type voltage-operated calcium channels and inhibition of Ca2+ influx, hereby inhibiting the contraction.34, 35 The effects of diamide on MAs are unknown. Here we showed that diamide causes concentration-dependent relaxations in phenylephrine- contracted MAs with an EC50 value of ~ 2 – 3 μmol/L, being substantially lower than the reported EC50 value in rat pulmonary arteries (58 μmol/L).34 DTT was able to reverse the diamide-induced inhibition of contraction demonstrating the fast-acting interchangeable nature of the cellular redox state. EDH-mediated relaxations were negatively modulated by diamide and positively modulated by DTT in MAs from wt mice. Strikingly, TRAM-34 blocked EDH relaxations in the presence of diamide or DTT to comparable levels, suggesting that IK1 modulation is involved in mediating the EDH relaxation. Interestingly, DTT was able to reverse the inhibitory actions of diamide on the EDH relaxation and to improve ACh-mediated relaxations in isolated small MAs from dnTrx-Tg mice. Since in dnTrx-Tg mice human Trx is in the oxidized and mutated form, only the endogenous oxidized form could be reduced by DTT, but the mutant hTrx cannot be reduced by DTT. Hence, the beneficial effects of DTT are most likely due to reducing the endogenous murine Trx.
H2O2 causes ''relaxation" of small arteries, which has been shown to be endothelium-in dependent, and has non-specific effects on smooth muscle cells36. However, some studies have suggested that H2O2 is an EDHF; but others have shown that catalase does not inhibit EDH-mediated relaxing responses in small MAs from wt mice 26. Further, the specific effect of H2O2 on KCa channels remains largely unknown26. We speculated that H2O2 might act as an oxidizing agent, as we observed that the relaxing responses only occur in 10 μM or higher concentrations of H2O2. At this concentration H2O2 could act as a potent oxidizing agent. The decrease in relaxing responses in the MAs isolated from Trx-Tg mice could be due to the removal of H2O2 by Prxas its activity increased in MAs of Trx-Tg mice, but not in dnTrx-Tg mice. A recent study has demonstrated that MAs express lower levels of Prx compared to aorta 37, and therefore, H2O2 is a potent EDHF in MAs, but not in aorta. However, in our Trx-Tg mice MAs, the activity of Prx is higher and therefore, H2O2 could be effectively neutralized causing a dampening in relaxation, and hence may not be an EDHF. This data further supports that KCa could be effective as EDHF in MAs of Trx-Tg mice. In conclusion, our study demonstrated that in vivo Trx redox state plays an important role in regulating the endothelium-dependent arterial relaxations and arterial blood pressure. Additionally, the difference in blood pressure among three strains of mice could have resulted in vascular remodeling resulting in differential vascular reactivity in vessels of these animals. The reduced form of Trx is able to enhance EDH responses resulting in an increased IK1 channel activity and lower systolic blood pressure. In contrast, loss of active Trx(-oxidation of Trx) resulted in both a reduced NO- and EDH-mediated relaxation in response to ACh reflected by a higher systolic blood pressure. Pathological modulation of vascular Trx redox state(Figure 9) might directly result in loss of normal relaxing ability of vessels and set the stage for endothelial dysfunction and hypertension. Our observations provide new insights into the vasoprotective effects of Trx in vivo and its beneficial role in the control of hypertension.
Figure 9.
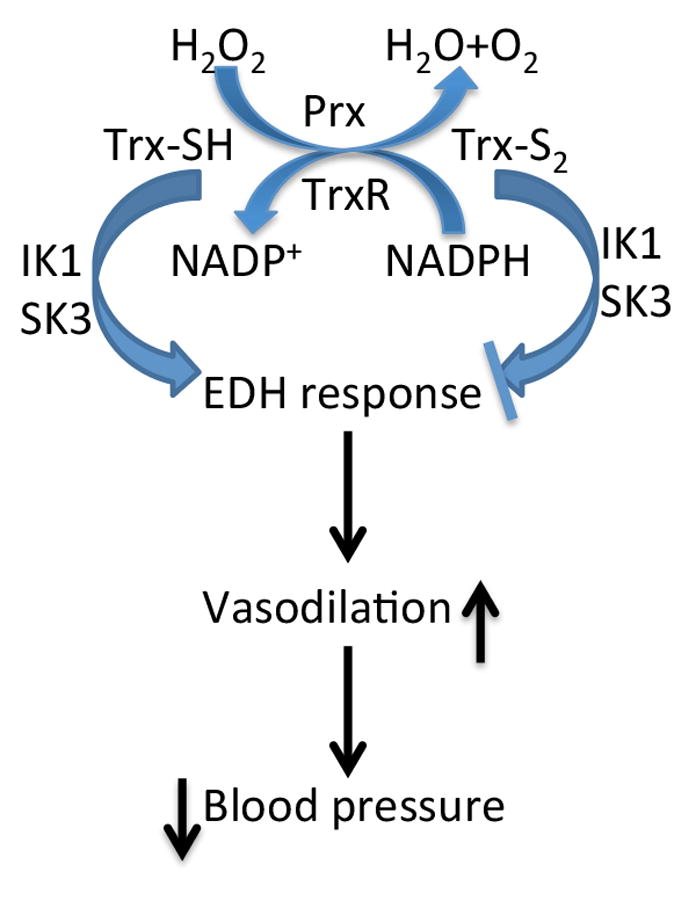
A schematic diagram presenting the role of Trx system in regulation of EDHF response and consequent blood pressure control.
Perspective
Hypertension is a major risk factor for several cardiovascular diseases including myocardial infarction and stroke. Therefore, a clear understanding of underlying mechanisms of hypertension is paramount to discovery of novel therapeutics. Our current study provides novel insight to control of vascular relaxation by modifying the redox state of vessels resulting in lower blood pressure. This new regulatory mechanism of vascular relaxation could provide important clues to develop therapeutic agent for control of hypertension.
Supplementary Material
Novelty and Significance.
What Is New?
Modulation of in vivo vessel redox state impacted relaxations in resistance-sized arteries, demonstrating that EDH and NO-mediated responses are regulated by vessel redox status, and the blood pressure is directly related to vessel redox perturbations.
What is Relevant?
The redox state of vessels is critical for relaxation responses either due to NO or EDH, and directly correlates with lower blood pressure. A shift in vessel redox to oxidized state in pathological conditions or aging could result in hypertension due to decreased relaxation by both NO and/or EDH. Therapies that target to restore the vessel redox in reduced state could be important for control or reversal of hypertension.
Summary
The study provides a mechanistic understanding of in vivo vessel redox state that influences vascular relaxations that is important in cardiovascular diseases such as hypertension and endothelial dysfunction. We demonstrated that redox perturbations modulate blood pressure by regulating the activity of endothelial KCa channels that result in NO and EDH mediated relaxations. In addition, KCa channels, specifically the IK1 channel in mesenteric arteries is modulated by redox state perturbations.
Acknowledgments
Sources of Funding: Research reported in this publication is supported by the National Heart, Lung And Blood Institute of the National Institutes of Health under Award Number R01HL107885 and R01HL109397. The content is solely the responsibility of the authors and does not necessarily represent the official view of the National Institutes of Health.
Footnotes
Disclosures: None.
References
- 1.Thomas SR, Witting PK, Drummond GR. Redox control of endothelial function and dysfunction: Molecular mechanisms and therapeutic opportunities. Antioxid Redox Signal. 2008;10:1713–1765. doi: 10.1089/ars.2008.2027. [DOI] [PubMed] [Google Scholar]
- 2.Vanhoutte PM, Shimokawa H, Tang EH, Feletou M. Endothelial dysfunction and vascular disease. Acta Physiol (Oxf) 2009;196:193–222. doi: 10.1111/j.1748-1716.2009.01964.x. [DOI] [PubMed] [Google Scholar]
- 3.Shimokawa H, Yasutake H, Fujii K, Owada MK, Nakaike R, Fukumoto Y, Takayanagi T, Nagao T, Egashira K, Fujishima M, Takeshita A. The importance of the hyperpolarizing mechanism increases as the vessel size decreases in endothelium-dependent relaxations in rat mesenteric circulation. J Cardiovasc Pharmacol. 1996;28:703–711. doi: 10.1097/00005344-199611000-00014. [DOI] [PubMed] [Google Scholar]
- 4.Hilgers RH, Todd J, Jr, Webb RC. Regional heterogeneity in acetylcholine-induced relaxation in rat vascular bed: Role of calcium-activated k+ channels. Am J Physiol Heart Circ Physiol. 2006;291:H216–222. doi: 10.1152/ajpheart.01383.2005. [DOI] [PubMed] [Google Scholar]
- 5.Waldron GJ, Garland CJ. Contribution of both nitric oxide and a change in membrane potential to acetylcholine-induced relaxation in the rat small mesenteric artery. British journal of pharmacology. 1994;112:831–836. doi: 10.1111/j.1476-5381.1994.tb13154.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eichler I, Wibawa J, Grgic I, Knorr A, Brakemeier S, Pries AR, Hoyer J, Kohler R. Selective blockade of endothelial ca2+-activated small- and intermediate-conductance k+-channels suppresses edhf-mediated vasodilation. British journal of pharmacology. 2003;138:594–601. doi: 10.1038/sj.bjp.0705075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Doughty JM, Plane F, Langton PD. Charybdotoxin and apamin block edhf in rat mesenteric artery if selectively applied to the endothelium. The American journal of physiology. 1999;276:H1107–1112. doi: 10.1152/ajpheart.1999.276.3.H1107. [DOI] [PubMed] [Google Scholar]
- 8.Hinton JM, Langton PD. Inhibition of edhf by two new combinations of k+-channel inhibitors in rat isolated mesenteric arteries. British journal of pharmacology. 2003;138:1031–1035. doi: 10.1038/sj.bjp.0705171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Edwards G, Dora KA, Gardener MJ, Garland CJ, Weston AH. K+ is an endothelium-derived hyperpolarizing factor in rat arteries. Nature. 1998;396:269–272. doi: 10.1038/24388. [DOI] [PubMed] [Google Scholar]
- 10.Wang ZW, Nara M, Wang YX, Kotlikoff MI. Redox regulation of large conductance ca(2+)-activated k+ channels in smooth muscle cells. The Journal of general physiology. 1997;110:35–44. doi: 10.1085/jgp.110.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.DiChiara TJ, Reinhart PH. Redox modulation of hslo ca2+-activated k+ channels. J Neurosci. 1997;17:4942–4955. doi: 10.1523/JNEUROSCI.17-13-04942.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Park MK, Lee SH, Lee SJ, Ho WK, Earm YE. Different modulation of ca-activated k channels by the intracellular redox potential in pulmonary and ear arterial smooth muscle cells of the rabbit. Pflugers Archiv : European journal of physiology. 1995;430:308–314. doi: 10.1007/BF00373904. [DOI] [PubMed] [Google Scholar]
- 13.Brakemeier S, Eichler I, Knorr A, Fassheber T, Kohler R, Hoyer J. Modulation of ca2+-activated k+ channel in renal artery endothelium in situ by nitric oxide and reactive oxygen species. Kidney international. 2003;64:199–207. doi: 10.1046/j.1523-1755.2003.00051.x. [DOI] [PubMed] [Google Scholar]
- 14.Ciorba MA, Heinemann SH, Weissbach H, Brot N, Hoshi T. Modulation of potassium channel function by methionine oxidation and reduction. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:9932–9937. doi: 10.1073/pnas.94.18.9932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Matsui M, Oshima M, Oshima H, Takaku K, Maruyama T, Yodoi J, Taketo MM. Early embryonic lethality caused by targeted disruption of the mouse thioredoxin gene. Dev Biol. 1996;178:179–185. doi: 10.1006/dbio.1996.0208. [DOI] [PubMed] [Google Scholar]
- 16.Das KC, Das CK. Thioredoxin, a singlet oxygen quencher and hydroxyl radical scavenger: Redox independent functions. Biochem Biophys Res Commun. 2000;277:443–447. doi: 10.1006/bbrc.2000.3689. [DOI] [PubMed] [Google Scholar]
- 17.Bolotina VM, Najibi S, Palacino JJ, Pagano PJ, Cohen RA. Nitric oxide directly activates calcium-dependent potassium channels in vascular smooth muscle. Nature. 1994;368:850–853. doi: 10.1038/368850a0. [DOI] [PubMed] [Google Scholar]
- 18.Strobaek D, Teuber L, Jorgensen TD, Ahring PK, Kjaer K, Hansen RS, Olesen SP, Christophersen P, Skaaning-Jensen B. Activation of human ik and sk ca2+-activated k+ channels by ns309 (6,7-dichloro-1h-indole-2,3-dione 3-oxime) Biochim Biophys Acta. 2004;1665:1–5. doi: 10.1016/j.bbamem.2004.07.006. [DOI] [PubMed] [Google Scholar]
- 19.Das KC, Guo XL, White CW. Induction of thioredoxin and thioredoxin reductase gene expression in lungs of newborn primates by oxygen. Am J Physiol. 1999;276:L530–539. doi: 10.1152/ajplung.1999.276.3.L530. [DOI] [PubMed] [Google Scholar]
- 20.Nelson KJ, Parsonage D. Measurement of peroxiredoxin activity. Curr Protoc Toxicol. 2011;Chapter 7(Unit 7):10. doi: 10.1002/0471140856.tx0710s49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Oblong JE, Berggren M, Gasdaska PY, Powis G. Site-directed mutagenesis of active site cysteines in human thioredoxin produces competitive inhibitors of human thioredoxin reductase and elimination of mitogenic properties of thioredoxin. J Biol Chem. 1994;269:11714–11720. [PubMed] [Google Scholar]
- 22.Das KC. C-jun nh2-terminal kinase-mediated redox-dependent degradation of ikappab: Role of thioredoxin in nf-kappab activation. J Biol Chem. 2001;276:4662–4670. doi: 10.1074/jbc.M006206200. [DOI] [PubMed] [Google Scholar]
- 23.Gallegos A, Gasdaska JR, Taylor CW, Paine-Murrieta GD, Goodman D, Gasdaska PY, Berggren M, Briehl MM, Powis G. Transfection with human thioredoxin increases cell proliferation and a dominant-negative mutant thioredoxin reverses the transformed phenotype of human breast cancer cells. Cancer Res. 1996;56:5765–5770. [PubMed] [Google Scholar]
- 24.Sankaranarayanan A, Raman G, Busch C, Schultz T, Zimin PI, Hoyer J, Kohler R, Wulff H. Naphtho[1,2-d]thiazol-2-ylamine (ska-31), a new activator of kca2 and kca3.1 potassium channels, potentiates the endothelium-derived hyperpolarizing factor response and lowers blood pressure. Molecular pharmacology. 2009;75:281–295. doi: 10.1124/mol.108.051425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shimokawa H, Matoba T. Hydrogen peroxide as an endothelium-derived hyperpolarizing factor. Pharmacological research : the official journal of the Italian Pharmacological Society. 2004;49:543–549. doi: 10.1016/j.phrs.2003.10.016. [DOI] [PubMed] [Google Scholar]
- 26.Feletou M, Vanhoutte PM. Endothelium-derived hyperpolarizing factor: Where are we now? Arterioscler Thromb Vasc Biol. 2006;26:1215–1225. doi: 10.1161/01.ATV.0000217611.81085.c5. [DOI] [PubMed] [Google Scholar]
- 27.Berggren MI, Husbeck B, Samulitis B, Baker AF, Gallegos A, Powis G. Thioredoxin peroxidase-1 (peroxiredoxin-1) is increased in thioredoxin-1 transfected cells and results in enhanced protection against apoptosis caused by hydrogen peroxide but not by other agents including dexamethasone, etoposide, and doxorubicin. Arch Biochem Biophys. 2001;392:103–109. doi: 10.1006/abbi.2001.2435. [DOI] [PubMed] [Google Scholar]
- 28.Dora KA, Sandow SL, Gallagher NT, Takano H, Rummery NM, Hill CE, Garland CJ. Myoendothelial gap junctions may provide the pathway for edhf in mouse mesenteric artery. Journal of vascular research. 2003;40:480–490. doi: 10.1159/000074549. [DOI] [PubMed] [Google Scholar]
- 29.Hilgers RH, Webb RC. Reduced expression of skca and ikca channel proteins in rat small mesenteric arteries during angiotensin ii-induced hypertension. Am J Physiol Heart Circ Physiol. 2007;292:H2275–2284. doi: 10.1152/ajpheart.00949.2006. [DOI] [PubMed] [Google Scholar]
- 30.Dalsgaard T, Kroigaard C, Misfeldt M, Bek T, Simonsen U. Openers of small conductance calcium-activated potassium channels selectively enhance no-mediated bradykinin vasodilatation in porcine retinal arterioles. British journal of pharmacology. 2010;160:1496–1508. doi: 10.1111/j.1476-5381.2010.00803.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stankevicius E, Lopez-Valverde V, Rivera L, Hughes AD, Mulvany MJ, Simonsen U. Combination of ca2+-activated k+ channel blockers inhibits acetylcholine-evoked nitric oxide release in rat superior mesenteric artery. British journal of pharmacology. 2006;149:560–572. doi: 10.1038/sj.bjp.0706886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kosower NS, Kosower EM, Wertheim B, Correa WS. Diamide, a new reagent for the intracellular oxidation of glutathione to the disulfide. Biochemical and biophysical research communications. 1969;37:593–596. doi: 10.1016/0006-291x(69)90850-x. [DOI] [PubMed] [Google Scholar]
- 33.Hashemy SI, Holmgren A. Regulation of the catalytic activity and structure of human thioredoxin 1 via oxidation and s-nitrosylation of cysteine residues. J Biol Chem. 2008;283:21890–21898. doi: 10.1074/jbc.M801047200. [DOI] [PubMed] [Google Scholar]
- 34.Schach C, Xu M, Platoshyn O, Keller SH, Yuan JX. Thiol oxidation causes pulmonary vasodilation by activating k+ channels and inhibiting store-operated ca2+ channels. American journal of physiology. Lung cellular and molecular physiology. 2007;292:L685–698. doi: 10.1152/ajplung.00276.2006. [DOI] [PubMed] [Google Scholar]
- 35.Iesaki T, Wolin MS. Thiol oxidation activates a novel redox-regulated coronary vasodilator mechanism involving inhibition of ca2+ influx. Arteriosclerosis, thrombosis, and vascular biology. 2000;20:2359–2365. doi: 10.1161/01.atv.20.11.2359. [DOI] [PubMed] [Google Scholar]
- 36.Shimokawa H, Morikawa K. Hydrogen peroxide is an endothelium-derived hyperpolarizing factor in animals and humans. J Mol Cell Cardiol. 2005;39:725–732. doi: 10.1016/j.yjmcc.2005.07.007. [DOI] [PubMed] [Google Scholar]
- 37.Burgoyne JR, Prysyazhna O, Rudyk O, Eaton P. Cgmp-dependent activation of protein kinase g precludes disulfide activation: Implications for blood pressure control. Hypertension. 2012;60:1301–1308. doi: 10.1161/HYPERTENSIONAHA.112.198754. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.