

Abstract
Purpose
The optimal end point for randomized phase II trials of anticancer therapies remains controversial. We simulated phase II trials by resampling patients from N9741, a randomized phase III trial of chemotherapy regimens for metastatic colorectal cancer, and compared the power of various end points to detect the superior therapy (FOLFOX [infusional fluorouracil, leucovorin, and oxaliplatin] had longer overall survival than both IROX [irinotecan plus oxaliplatin] and IFL [irinotecan and bolus fluorouracil plus leucovorin]).
Methods
Tumor measurements and progression-free survival (PFS) data were obtained for 1,471 patients; 1,002 had consistently measured tumors and were resampled (5,000 replicates) to simulate two-arm, randomized phase II trials with α = 0.10 (one sided) and 20 to 80 patients per arm. End points included log ratio of tumor size at 6, 12, and 18 weeks relative to baseline; time to tumor growth (TTG), estimated using a nonlinear mixed-effects model; and PFS. Arms were compared using rank sum tests for log ratio and TTG and a log-rank test for PFS.
Results
For FOLFOX versus IFL, TTG and PFS had similar power, with both exceeding the power of log ratio at 18 weeks; for FOLFOX versus IROX, TTG and log ratio at 18 weeks had similar power, with both exceeding the power of PFS. The best end points exhibited > 80% power with 60 to 80 patients per arm.
Conclusion
TTG is a powerful end point for randomized phase II trials of cytotoxic therapies in metastatic colorectal cancer; it was either comparable or superior to PFS and log ratio at 18 weeks. Additional studies will be needed to clarify the potential of TTG as a phase II end point.
INTRODUCTION
The phase III success rate in oncology, estimated at 47% for drugs that entered clinical studies between 1993 and 2004, continues to lag behind other therapeutic areas.1 These data imply that phase II trials are not informative enough to identify therapies with the greatest probability of success compared with standard of care. Randomized phase II trials are increasingly common, and there is evidence to suggest that their use will increase the success rate of phase III trials.2,3 However, with randomized trials, there are a variety of primary end points to consider and few data to indicate which are optimal to inform go/no-go decisions at the end of phase II.
Because of power limitations, overall survival (OS) is usually not a feasible end point in phase II and may be limited by the availability of therapies after disease progression that affect survival. The National Cancer Institute Investigational Drug Steering Committee consensus recommendations state that either response-based end points (when significant tumor shrinkage is expected) or progression-free survival (PFS) should generally be used in randomized phase II trials of anticancer therapies, with response and progression typically defined by RECIST.4,5 However, overall response rate (ORR) and PFS both have limitations that result from the categorization of continuous tumor size data, and both are limited by the subjective timing of radiographic assessments. Although PFS has previously been shown to be an acceptable surrogate for OS in first-line therapy for metastatic colorectal cancer,6,7 both ORR and PFS correlate poorly or only modestly with OS in other disease settings.8–10
Alternatives to conventional ORR and PFS end points have emerged in an attempt to overcome some of these limitations. After the initial proposal of Lavin,11 Karrison et al12 suggested the log ratio of tumor size at 8 weeks compared with baseline as the primary end point of a randomized phase II trial. Jaki et al13 pointed out that change in tumor size would require significantly smaller sample sizes than ORR or PFS in comparative phase II trials. Change in tumor size correlates strongly with OS in patients with advanced solid tumors treated in phase I trials,14 and nonlinear mixed-effects models describing tumor growth inhibition (TGI) have been used to demonstrate how early change in tumor size can predict OS in metastatic colorectal cancer and non–small-cell lung cancer.15,16 Claret et al17 introduced the concept of time to tumor growth (TTG), derived from individual TGI parameter estimates, and showed that TTG was better than early change in tumor size at predicting OS in two phase III studies in metastatic colorectal cancer. Because these novel end points treat tumor size as a continuous variable, we refer to them as tumor dynamic end points.
In the design of randomized phase II trials, the principal question is which end point has the greatest power to detect a true difference among therapies. Early end points are of great value because they minimize time from the start of the trial to the primary analysis that informs go/no-go decisions. One way to approach the question is to resample patients from randomized phase III trials with significant differences in meaningful end points to simulate randomized phase II trials using both tumor dynamic and conventional end points. After resampling a large number of times and comparing the arms in each replicate, the power of an end point is simply the percentage of replicates in which a significant difference is found. We previously applied this approach to a randomized phase III trial of sorafenib versus placebo in renal cell carcinoma, but the results were not generalizable to other therapies or other tumor types, and we did not consider model-based end points in that work.18
The N9741 trial, which enrolled patients between 1999 and 2001, randomly assigned patients to one of three chemotherapy regimens for previously untreated metastatic colorectal cancer.19 At the time the trial was designed, IFL (irinotecan and bolus fluorouracil plus leucovorin) was the standard of care, whereas FOLFOX (infusional fluorouracil, leucovorin, and oxaliplatin) and IROX (irinotecan plus oxaliplatin) were investigational therapies. The primary end point was time to progression (TTP), and long-term results demonstrated a statistically significant advantage for FOLFOX compared with either of the other regimens with respect to TTP and OS.20 In our study, we had access to tumor measurement, PFS, and OS data for patients in these three arms of N9741. We chose PFS rather than TTP because it is the preferred regulatory end point and is more widely used.21 We resampled these patients and simulated randomized phase II trials of FOLFOX versus IFL and FOLFOX versus IROX to compare tumor dynamic end points and PFS based on their power to detect differences between therapies.
METHODS
Individual Patient Data
After obtaining institutional review board approval, data were obtained in an electronic, deidentified format from the North Central Clinical Trials Group for 1,471 patients enrolled onto N9741 and treated with IFL, FOLFOX, or IROX. Data included bidimensional measurements of target lesions from computed tomography (CT) scans every 6 weeks, treatment assignment, PFS in days, OS in days, and whether PFS and OS events were censored. We excluded 469 patients from our analyses; 222 had no tumor measurement data (nonmeasurable disease), and 247 had missing data (Fig 1). The accrual period was shorter for the IFL and IROX arms than for the FOLFOX arm, which explains the unequal assignment of patients in this randomized trial.22
Fig 1.

Flow diagram indicating reasons for exclusion from analyses and number of patients with imputed data by treatment arm. FOLFOX, infusional fluorouracil, leucovorin, and oxaliplatin; IFL, irinotecan and bolus fluorouracil plus leucovorin; IROX, irinotecan plus oxaliplatin. (*) Early missing data indicate that patient was missing ≥ two lesion measurements in first 24 weeks but had not experienced disease progression or died at that time.
Tumor size was defined as the sum of the longest diameters of the target lesions measured consistently across the first four CT scans (baseline, 6 ± 3 weeks, 12 ± 3 weeks, and 18 ± 3 weeks). Patients who missed a CT scan at one of these time points had tumor size imputed by linear interpolation between CT scans (Fig 1). Those who experienced a PFS event before one of the first four CT scans also had tumor size imputed. For those who experienced clinical progression, tumor size was imputed to growth of 20% compared with baseline. For those who died, tumor size was imputed to the largest percentage increase in the first four scans across all patients. Imputations were performed only once, and imputed data were carried forward up to 18 weeks.
Resampling Simulations
Random sampling with replacement was carried out at the level of the individual patient. Sampling with replacement effectively treats the empiric cumulative distribution function as reflective of the population distribution. For each simulated trial, 5,000 replicates were performed. Simulated trials were classified as significant or nonsignificant according to statistical criteria, which we outline here.
Design and End Points Evaluated
Randomized, two-arm, phase II trials were simulated with randomization at a ratio of 1:1 and sample sizes of 20, 40, 60, and 80 patients per arm. These simulated trials were evaluated using the following end points: log ratio of tumor size at 6, 12, and 18 weeks relative to baseline; TTG; and PFS. To compare PFS and TTG with the log ratio end point at 18 weeks with comparable trial length, PFS and tumor size data for TTG were censored at 150 days from the time the last patient was accrued, with patients randomly assigned (before resampling) an accrual time between 0 and 180 days (total trial time, 330 days).
The TGI model was previously developed and includes two key parameters: first, the tumor growth rate constant (KL, day −1); and second, the tumor growth inhibition rate (KDE0, day −1), which decreases exponentially over time according to λ (day −1).15 These parameters can be used to estimate TTG: TTG = (log[KDE0] − log[KL]) ÷ λ.17 KL would be expected to be smaller and/or KDE0 larger for a more effective therapy, resulting in a longer TTG. Each of the parameters is treated as a random effect with an assumed lognormal distribution. Patient-specific estimates are obtained, leading to an estimated TTG for each patient. Because it is possible for TTG to exceed the observation limit for some patients, rank-based methods for comparing arms were used to assign these patients the highest rankings. The population models were estimated using the first-order conditional estimation algorithm with interaction (NONMEM, version 7.2; http://www.iconplc.com/jp/technology/products/nonmem/).
Statistical Analyses
Statistical analyses were conducted with standard software (R, version 2.15.2; http://www.r-project.org/). A one-sided α (type I error rate) of 0.10 was used in all cases. N9741 showed that FOLFOX was superior to both IROX and IFL. When comparing FOLFOX with IROX or IFL, FOLFOX was considered the investigational arm and IROX or IFL the control arm. Trials were significant if patients in the investigational arm did significantly better than patients in the control arm (one-sided P < .10). The power for each end point was the percentage of 5,000 replicates that were statistically significant in favor of the investigational arm. Type I error rates were obtained by resampling both arms from the FOLFOX arm. Treatment arms were compared using a rank sum test for log ratio and TTG and a log-rank test for PFS.
RESULTS
For patients included in the analyses from N9741 (n = 1,002), PFS and OS were comparable to those previously reported.20 PFS was longer in the FOLFOX arm compared with both IROX (P = .0024) and IFL (P < .001) arms (Fig 2A). OS was longer in the FOLFOX arm compared with both IROX (P < .001) and IFL (P < .001) arms (Fig 2B). The proportional hazards assumption was met when comparing any combination of the three arms with respect to PFS or OS. Table 1 summarizes results for early change in tumor size and TTG by treatment arm for all patients, and Appendix Figure A1 (online only) shows waterfall plots for the three treatment arms at weeks 6, 12, and 18. Average percent reduction in tumor size was 2× to 3× greater in the FOLFOX arm compared with the other two arms at all three time points, and median TTG was almost 50% longer. These serve as the true distributions from which individual patient data were resampled.
Fig 2.
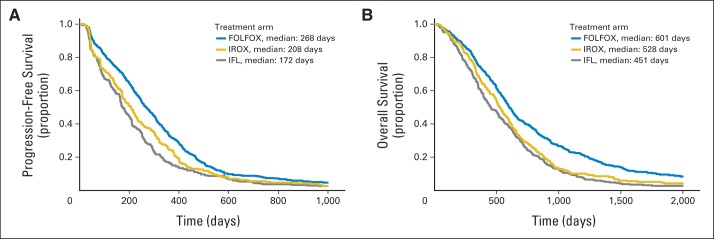
Kaplan-Meier analyses of (A) progression-free and (B) overall survival by treatment arm for patients included in analyses. FOLFOX, infusional fluorouracil, leucovorin, and oxaliplatin; IFL, irinotecan and bolus fluorouracil plus leucovorin; IROX, irinotecan plus oxaliplatin.
Table 1.
Summary Statistics for Early Change in Tumor Size and TTG by Treatment Arm
| Change (%) | Mean | Median | SD |
|---|---|---|---|
| FOLFOX (n = 444) | |||
| Week 6 | −16.2 | −15.4 | 26.4 |
| Week 12 | −22.3 | −29.4 | 41.3 |
| Week 18 | −24.2 | −33.3 | 48.8 |
| TTG, days | 237.9 | 237.1 | 189.0 |
| IROX (n = 255) | |||
| Week 6 | −8.0 | −10.0 | 27.1 |
| Week 12 | −11.0 | −16.7 | 42.1 |
| Week 18 | −9.4 | −17.1 | 50.9 |
| TTG, days | 176.3 | 165.4 | 169.9 |
| IFL (n = 303) | |||
| Week 6 | −7.6 | −10.0 | 30.8 |
| Week 12 | −10.9 | −17.7 | 41.4 |
| Week 18 | −7.3 | −17.8 | 52.1 |
| TTG, days | 165.4 | 173.2 | 154.2 |
Abbreviations: FOLFOX, infusional fluorouracil, leucovorin, and oxaliplatin; IFL, irinotecan and bolus fluorouracil plus leucovorin; IROX, irinotecan plus oxaliplatin; SD, standard deviation; TTG, time to tumor growth.
Table 2 summarizes the results of resampling simulations for FOLFOX versus IFL, FOLFOX versus IROX, and FOLFOX versus FOLFOX for various end points and sample sizes. For FOLFOX versus IFL and FOLFOX versus IROX, log ratio at 18 weeks had greater power than log ratio at 6 or 12 weeks. TTG and PFS with data censored at 150 days after the last patient was accrued were comparable to log ratio at 18 weeks in trial length. For FOLFOX versus IFL, TTG and PFS had greater power than log ratio at 18 weeks. For example, with 60 patients per arm, the powers for TTG, PFS, and log ratio at 18 weeks were 88.0%, 88.5%, and 76.0%, respectively. For FOLFOX versus IROX, TTG and log ratio at 18 weeks had greater power than PFS. For example, with 60 patients per arm, the powers for TTG, log ratio at 18 weeks, and PFS were 72.4%, 72.1%, and 58.3%, respectively. Regardless of end point or sample size, the rejection rate for FOLFOX versus FOLFOX was approximately equal to the stipulated type I error rate of 10%.
Table 2.
Resampling Simulation Results for FOLFOX Versus IFL, FOLFOX Versus IROX, and FOLFOX Versus FOLFOX for Various End Points and Sample Sizes
| End Point | Sample Size Per Arm (No.) |
|||||||
|---|---|---|---|---|---|---|---|---|
| 20 |
40 |
60 |
80 |
|||||
| Significant Trials (%)* | 95% CI | Significant Trials (%)* | 95% CI | Significant Trials (%)* | 95% CI | Significant Trials (%)* | 95% CI | |
| FOLFOX v IFL | ||||||||
| PFS | 55.1 | 53.9 to 56.2 | 77.1 | 76.2 to 78.1 | 88.5 | 87.8 to 89.3 | 94.6 | 94.1 to 95.1 |
| TTG | 54.2 | 53.0 to 55.3 | 75.7 | 74.7 to 76.7 | 88.0 | 87.2 to 88.7 | 93.3 | 92.8 to 93.9 |
| Log ratio at 6 weeks | 37.0 | 35.9 to 38.2 | 54.5 | 53.3 to 55.6 | 65.2 | 64.1 to 66.4 | 73.7 | 72.6 to 74.7 |
| Log ratio at 12 weeks | 37.9 | 36.8 to 39.1 | 57.1 | 56.0 to 58.3 | 68.3 | 67.3 to 69.4 | 76.8 | 75.8 to 77.8 |
| Log ratio at 18 weeks | 43.9 | 42.7 to 45.0 | 64.3 | 63.2 to 65.4 | 76.0 | 75.0 to 77.0 | 84.6 | 83.8 to 85.5 |
| FOLFOX v IROX | ||||||||
| PFS | 33.8 | 32.7 to 34.9 | 48.4 | 47.2 to 49.6 | 58.3 | 57.1 to 59.4 | 66.6 | 65.5 to 67.7 |
| TTG | 41.7 | 40.6 to 42.9 | 59.6 | 58.5 to 60.7 | 72.4 | 71.4 to 73.5 | 81.7 | 80.8 to 82.6 |
| Log ratio at 6 weeks | 39.5 | 38.3 to 40.6 | 57.0 | 55.9 to 58.2 | 69.1 | 68.1 to 70.2 | 78.1 | 77.2 to 79.1 |
| Log ratio at 12 weeks | 39.6 | 38.5 to 40.8 | 56.8 | 55.7 to 58.0 | 68.1 | 67.0 to 69.1 | 77.6 | 76.6 to 78.5 |
| Log ratio at 18 weeks | 41.6 | 40.5 to 42.8 | 59.3 | 58.1 to 60.4 | 72.1 | 71.1 to 73.1 | 80.3 | 79.4 to 81.2 |
| FOLFOX v FOLFOX† | ||||||||
| PFS | 9.8 | 9.1 to 10.5 | 10.2 | 9.5 to 10.9 | 10.3 | 9.6 to 11.0 | 10.8 | 10.1 to 11.5 |
| TTG | 9.9 | 9.2 to 10.6 | 10.0 | 9.3 to 10.7 | 10.3 | 9.6 to 11.0 | 9.2 | 8.5 to 9.9 |
| Log ratio at 6 weeks | 9.3 | 8.6 to 10.0 | 11.1 | 10.4 to 11.9 | 10.0 | 9.3 to 10.7 | 10.1 | 9.4 to 10.8 |
| Log ratio at 12 weeks | 10.0 | 9.3 to 10.7 | 11.0 | 10.3 to 11.8 | 10.2 | 9.5 to 10.9 | 9.7 | 9.1 to 10.4 |
| Log ratio at 18 weeks | 10.2 | 9.5 to 11.0 | 10.5 | 9.8 to 11.2 | 10.4 | 9.7 to 11.1 | 9.6 | 9.0 to 10.3 |
Abbreviations: FOLFOX, infusional fluorouracil, leucovorin, and oxaliplatin; IFL, irinotecan and bolus fluorouracil plus leucovorin; IROX, irinotecan plus oxaliplatin; PFS, progression-free survival; TTG, time to tumor growth.
Of 5,000 replicates.
Null hypothesis.
Figure 3 shows results of resampling simulations for FOLFOX versus IFL and FOLFOX versus IROX across sample sizes. The power for each end point increased with sample size, but the relative power of the end points did not change. For FOLFOX versus IFL, the power exceeded 80% at 60 patients per arm and 90% at 80 patients per arm with TTG and PFS. For FOLFOX versus IROX, the power exceeded 80% at 80 patients per arm with TTG and log ratio at 18 weeks.
Fig 3.

Summary of results from resampling simulations for randomized phase II trials of (A) infusional fluorouracil, leucovorin, and oxaliplatin (FOLFOX) versus irinotecan and bolus fluorouracil plus leucovorin (IFL) and (B) FOLFOX versus irinotecan plus oxaliplatin (IROX) with various early end points. PFS, progression-free survival; TTG, time to tumor growth.
DISCUSSION
Although randomized phase III trials may not be necessary for anticancer therapies that are highly effective,23 the majority of novel therapies will continue to be scrutinized for incremental improvements compared with standard of care. In these cases, the choice of an end point for a randomized phase II trial affects two metrics: first, efficiency of drug development; and second, success of the phase III trial (if one is undertaken). Efficiency is inversely related to sample size and follow-up time, so early end points with high power maximize efficiency. Success of the phase III trial is dependent on making the correct go/no-go decision at the end of phase II. Early end points with greater power to detect differences between therapies at the same type I error rate improve the likelihood that a go decision will be made for a truly effective therapy, while providing confidence that a no-go decision can be made if the phase II trial is negative. A more powerful end point also allows investigators to reduce the prespecified type I error (false-positive rate) with less impact on sample size.
The results of our study do not provide a clear winner regarding an optimal end point for randomized phase II trials; however, there are two interesting conclusions. First, TTG had comparable or better power than PFS and log ratio at 18 weeks in both of the comparisons (FOLFOX v IFL and FOLFOX v IROX), suggesting that it may be an attractive end point for randomized phase II trials. TTG can provide ≥ 80% power at reasonable sample sizes (60 to 80 patients per arm) for randomized phase II trials that are being conducted in multicenter consortia for relatively common cancers. Second, the superiority of PFS versus log ratio depends on the therapies being compared. In our study, the difference between the best and worst therapies (FOLFOX and IFL) was more powerfully demonstrated by PFS, whereas the difference between the best and second-best therapies (FOLFOX and IROX) was more powerfully demonstrated by log ratio. It is not necessarily surprising that tumor dynamic end points (log ratio and TTG) would be better able to detect subtle differences compared with PFS. If both therapies are delaying progression, but one is shrinking tumors more than the other, log ratio and TTG may detect differences better than PFS at early time points.
There are a number of limitations in our study. First, resampling simulations were based on data from a single phase III trial in a single disease/setting (previously untreated metastatic colorectal cancer). The results might not be generalizable to other therapies or other diseases/settings. Nonetheless, our results are consistent with the findings of Claret et al17 indicating that TTG was better than tumor size ratio for predicting OS in previously untreated metastatic colorectal cancer in a multivariable model. Our results also add to our previous work demonstrating the utility of an early log ratio end point for detecting the efficacy of sorafenib in renal cell carcinoma.18 Second, the log ratio and TTG end points required imputation of tumor size data for early progression or death to avoid bias from informative dropout. We attempted to minimize bias by including these imputations and using a rank sum test for comparisons between arms. Another approach, proposed by Wason and Seaman,24 is an augmented binary method, in which binary criteria (new lesions, death, and toxicity) are considered along with change in tumor size. Third, we excluded some patients from our analyses because they had nonmeasurable disease or lacked consistently measured tumor size data; < 35% of patients from each arm were excluded, and their exclusion did not change the results for PFS or OS. However, the exclusion of patients with early missing data may have mitigated the power of the log ratio at 6 weeks end point. Finally, we acknowledge the limitations inherent in the tumor size data from this relatively dated trial, which used WHO rather than modern criteria (RECIST version 1.1) for measuring target lesions. Recent work suggests that there is significant interoccasion and interobserver variability in tumor size measurements25 and that semiautomated volumetric measurements may capture drug effects better than unidimensional measurements with comparable variability.26,27
Our results may be interpreted as divergent from those of previous studies. Kaiser28 found that percentage change in tumor size and tumor burden modeling were not better than PFS for phase II decision making by resampling data from six phase II and III trials in three cancers (colorectal, breast, and non–small-cell lung cancers). In the five positive trials considered by Kaiser, the hazard ratios for PFS of 0.50 to 0.65 indicated a larger effect size compared with FOLFOX versus IROX (hazard ratio, 0.79) in our study, in which TTG and log ratio at 18 weeks were more powerful than PFS. The optimal selection of TTG versus log ratio versus PFS will depend on the relative magnitude of the true difference among therapies for these metrics, and this will vary from case to case. Ultimately, the question becomes which of these surrogate measures best predicts the outcome in a subsequent phase III trial. If PFS will be the preferred end point over OS in the phase III trial, PFS may be the preferred end point for the phase II trial.29 PFS may also be the preferred metric in disease settings where it has been demonstrated that PFS is an acceptable surrogate for OS. Two studies found that change in tumor size was not better than categorical metrics (complete/partial response v stable disease v progressive disease) for predicting OS using data from N9741.30,31 Although these results might seem to contradict ours given the overlap in the source data, the methodology differed in that they used landmark analyses to measure associations with OS. The resampling approach used in our study does not test how well end points associate with OS, but rather how powerful they are for making the correct go/no-go decision at the end of phase II.
In conclusion, this study supports the consideration of TTG estimated from nonlinear mixed-effects modeling of tumor measurements as a powerful end point for randomized phase II trials, based on an example in metastatic colorectal cancer. Although we do not recommend using TTG as the primary end point for future trials based on this study alone, it should be measured as a secondary end point, and additional studies should compare it with PFS and log ratio. In the end, the question is an empirical one: Which phase II end point will most often lead to the correct go/no-go decision? This question is perhaps best answered by synthesizing data across multiple trials in various diseases/settings, as is currently being done in colorectal cancer.32
Acknowledgment/Grant Support
We thank Laurent Claret, PhD, and Rene Bruno, PhD, for sharing the NONMEM code for their tumor growth inhibition model.
Appendix
Fig A1.

Waterfall plots depicting change in tumor size at 6, 12, and 18 weeks for (A) infusional fluorouracil, leucovorin, and oxaliplatin (FOLFOX), (B) irinotecan plus oxaliplatin (IROX), and (C) irinotecan and bolus fluorouracil plus leucovorin (IFL) treatment arms.
Footnotes
Supported by Award No. K12CA139160 from the National Cancer Institute (M.R.S.) and by the Biostatistics Laboratory at the University of Chicago, a core facility supported by University of Chicago Comprehensive Cancer Center Support Grant No. P30CA014599.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute or the National Institutes of Health.
Authors' disclosures of potential conflicts of interest are found in the article online at www.jco.org. Author contributions are found at the end of this article.
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Disclosures provided by the authors are available with this article at www.jco.org.
AUTHOR CONTRIBUTIONS
Conception and design: Manish R. Sharma, Daniel J. Sargent, Theodore G. Karrison
Collection and assembly of data: Manish R. Sharma, Elizabeth Gray, Daniel J. Sargent, Theodore G. Karrison
Data analysis and interpretation: All authors
Manuscript writing: All authors
Final approval of manuscript: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Resampling the N9741 Trial to Compare Tumor Dynamic Versus Conventional End Points in Randomized Phase II Trials
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. For a detailed description of the disclosure categories, or for more information about ASCO's conflict of interest policy, please refer to the Author Disclosure Declaration and the Disclosures of Potential Conflicts of Interest section in Information for Contributors.
Manish R. Sharma
No relationship to disclose
Elizabeth Gray
No relationship to disclose
Richard M. Goldberg
Honoraria: sanofi-aventis, Eli Lilly, Biothera
Research Funding: sanofi-aventis (Inst), Bayer HealthCare Pharmaceuticals (Inst), Immunomedix (Inst), Merck (Inst)
Travel, Accommodations, Expenses: sanofi-aventis, Merck
Daniel J. Sargent
Consulting or Advisory Role: Roche, Novartis, Abbvie, Bayer HealthCare Pharmaceuticals
Research Funding: Celgene (Inst), Diagnocure (Inst)
Theodore G. Karrison
No relationship to disclose
REFERENCES
- 1.DiMasi JA, Reichert JM, Feldman L, et al. Clinical approval success rates for investigational cancer drugs. Clin Pharmacol Ther. 2013;94:329–335. doi: 10.1038/clpt.2013.117. [DOI] [PubMed] [Google Scholar]
- 2.Tang H, Foster NR, Grothey A, et al. Comparison of error rates in single-arm versus randomized phase II cancer clinical trials. J Clin Oncol. 2010;28:1936–1941. doi: 10.1200/JCO.2009.25.5489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sharma MR, Stadler WM, Ratain MJ. Randomized phase II trials: A long-term investment with promising returns. J Natl Cancer Inst. 2011;103:1093–1100. doi: 10.1093/jnci/djr218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Seymour L, Ivy SP, Sargent D, et al. The design of phase II clinical trials testing cancer therapeutics: Consensus recommendations from the clinical trial design task force of the National Cancer Institute Investigational Drug Steering Committee. Clin Cancer Res. 2010;16:1764–1769. doi: 10.1158/1078-0432.CCR-09-3287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1) Eur J Cancer. 2009;45:228–247. doi: 10.1016/j.ejca.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 6.Buyse M, Burzykowski T, Carroll K, et al. Progression-free survival is a surrogate for survival in advanced colorectal cancer. J Clin Oncol. 2007;25:5218–5224. doi: 10.1200/JCO.2007.11.8836. [DOI] [PubMed] [Google Scholar]
- 7.Tang PA, Bentzen SM, Chen EX, et al. Surrogate end points for median overall survival in metastatic colorectal cancer: Literature-based analysis from 39 randomized controlled trials of first-line chemotherapy. J Clin Oncol. 2007;25:4562–4568. doi: 10.1200/JCO.2006.08.1935. [DOI] [PubMed] [Google Scholar]
- 8.Burzykowski T, Buyse M, Piccart-Gebhart MJ, et al. Evaluation of tumor response, disease control, progression-free survival, and time to progression as potential surrogate end points in metastatic breast cancer. J Clin Oncol. 2008;26:1987–1992. doi: 10.1200/JCO.2007.10.8407. [DOI] [PubMed] [Google Scholar]
- 9.Halabi S, Rini B, Escudier B, et al. Progression-free survival as a surrogate endpoint of overall survival in patients with metastatic renal cell carcinoma. Cancer. 2014;120:52–60. doi: 10.1002/cncr.28221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Laporte S, Squifflet P, Baroux N, et al. Prediction of survival benefits from progression-free survival benefits in advanced non-small-cell lung cancer: Evidence from a meta-analysis of 2334 patients from 5 randomised trials. BMJ Open. doi: 10.1136/bmjopen-2012-001802. [epub ahead of print on March 13, 2013] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lavin PT. An alternative model for the evaluation of antitumor activity. Cancer Clin Trials. 1981;4:451–457. [PubMed] [Google Scholar]
- 12.Karrison TG, Maitland ML, Stadler WM, et al. Design of phase II cancer trials using a continuous endpoint of change in tumor size: Application to a study of sorafenib and erlotinib in non small-cell lung cancer. J Natl Cancer Inst. 2007;99:1455–1461. doi: 10.1093/jnci/djm158. [DOI] [PubMed] [Google Scholar]
- 13.Jaki T, André V, Su TL, et al. Designing exploratory cancer trials using change in tumour size as primary endpoint. Stat Med. 2013;32:2544–2554. doi: 10.1002/sim.5716. [DOI] [PubMed] [Google Scholar]
- 14.Jain RK, Lee JJ, Ng C, et al. Change in tumor size by RECIST correlates linearly with overall survival in phase I oncology studies. J Clin Oncol. 2012;30:2684–2690. doi: 10.1200/JCO.2011.36.4752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Claret L, Girard P, Hoff PM, et al. Model-based prediction of phase III overall survival in colorectal cancer on the basis of phase II tumor dynamics. J Clin Oncol. 2009;27:4103–4108. doi: 10.1200/JCO.2008.21.0807. [DOI] [PubMed] [Google Scholar]
- 16.Wang Y, Sung C, Dartois C, et al. Elucidation of relationship between tumor size and survival in non-small-cell lung cancer patients can aid early decision making in clinical drug development. Clin Pharmacol Ther. 2009;86:167–174. doi: 10.1038/clpt.2009.64. [DOI] [PubMed] [Google Scholar]
- 17.Claret L, Gupta M, Han K, et al. Evaluation of tumor-size response metrics to predict overall survival in Western and Chinese patients with first-line metastatic colorectal cancer. J Clin Oncol. 2013;31:2110–2114. doi: 10.1200/JCO.2012.45.0973. [DOI] [PubMed] [Google Scholar]
- 18.Sharma MR, Karrison TG, Jin Y, et al. Resampling phase III data to assess phase II trial designs and endpoints. Clin Cancer Res. 2012;18:2309–2315. doi: 10.1158/1078-0432.CCR-11-1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goldberg RM, Sargent DJ, Morton RF, et al. A randomized controlled trial of fluorouracil plus leucovorin, irinotecan, and oxaliplatin combinations in patients with previously untreated metastatic colorectal cancer. J Clin Oncol. 2004;22:23–30. doi: 10.1200/JCO.2004.09.046. [DOI] [PubMed] [Google Scholar]
- 20.Sanoff HK, Sargent DJ, Campbell ME, et al. Five-year data and prognostic factor analysis of oxaliplatin and irinotecan combinations for advanced colorectal cancer: N9741. J Clin Oncol. 2008;26:5721–5727. doi: 10.1200/JCO.2008.17.7147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.US Department of Health and Human Services. Guidance for industry: Clinical trial endpoints for the approval of cancer drugs and biologics. http://www.fda.gov/downloads/drugsGuidanceComplianceRegulatoyInformation/Guidance/UCM071590.pdf.
- 22.Goldberg RM, Sargent DJ, Morton RF, et al. NCCTG study N9741: Leveraging learning from an NCI Cooperative Group phase III trial. Oncologist. 2009;14:970–978. doi: 10.1634/theoncologist.2009-0175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sharma MR, Schilsky RL. Role of randomized phase III trials in an era of effective targeted therapies. Nat Rev Clin Oncol. 2012;9:208–214. doi: 10.1038/nrclinonc.2011.190. [DOI] [PubMed] [Google Scholar]
- 24.Wason JM, Seaman SR. Using continuous data on tumour measurements to improve inference in phase II cancer studies. Stat Med. 2013;32:4639–4650. doi: 10.1002/sim.5867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Oxnard GR, Zhao B, Sima CS, et al. Variability of lung tumor measurements on repeat computed tomography scans taken within 15 minutes. J Clin Oncol. 2011;29:3114–3119. doi: 10.1200/JCO.2010.33.7071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhao B, Oxnard GR, Moskowitz CS, et al. A pilot study of volume measurement as a method of tumor response evaluation to aid biomarker development. Clin Cancer Res. 2010;16:4647–4653. doi: 10.1158/1078-0432.CCR-10-0125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhao B, Tan Y, Bell DJ, et al. Exploring intra- and inter-reader variability in uni-dimensional, bi-dimensional, and volumetric measurements of solid tumors on CT scans reconstructed at different slice intervals. Eur J Radiol. 2013;82:959–968. doi: 10.1016/j.ejrad.2013.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kaiser LD. Tumor burden modeling versus progression-free survival for phase II decision making. Clin Cancer Res. 2013;19:314–319. doi: 10.1158/1078-0432.CCR-12-2161. [DOI] [PubMed] [Google Scholar]
- 29.Broglio KR, Berry DA. Detecting an overall survival benefit that is derived from progression-free survival. J Natl Cancer Inst. 2009;101:1642–1649. doi: 10.1093/jnci/djp369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.An MW, Mandrekar SJ, Branda ME, et al. Comparison of continuous versus categorical tumor measurement-based metrics to predict overall survival in cancer treatment trials. Clin Cancer Res. 2011;17:6592–6599. doi: 10.1158/1078-0432.CCR-11-0822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huen JM, Grothey A, Branda ME, et al. Tumor status at 12 weeks predicts survival in advanced colorectal cancer: Findings from NCCTG N9741. Oncologist. 2011;16:859–867. doi: 10.1634/theoncologist.2011-0064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sargent DJ, Buyse M, Matheson A, et al. The ARCAD clinical trials program: An update and invitation. Oncologist. 2012;17:188–191. doi: 10.1634/theoncologist.2011-0332. [DOI] [PMC free article] [PubMed] [Google Scholar]