

SUMMARY
How cancer cells shift metabolism to aerobic glycolysis is largely unknown. Here we show that deficiency of α/β-hydrolase domain-containing-5 (Abhd5), an intracellular lipolytic activator that is also known as comparative gene identification-58 (CGI-58), promotes this metabolic shift and enhances malignancies of colorectal carcinomas (CRCs). Silencing of Abhd5 in normal fibroblasts induces malignant transformation. Intestine-specific knockout of Abhd5 in ApcMin/+ mice robustly increases tumorigenesis and malignant transformation of adenomatous polyps. In colon cancer cells, Abhd5 deficiency induces epithelial-mesenchymal transition by suppressing the AMPKα-p53 pathway, which is attributable to increased aerobic glycolysis. In human CRCs, Abhd5 expression falls substantially and correlates negatively with malignant features. Our study is the first to link Abhd5 to CRC pathogenesis. It suggests that cancer cells may develop aerobic glycolysis by suppressing Abhd5-mediated intracellular lipolysis.
Keywords: intracellular fat lipolysis, colorectal cancer, aerobic glycolysis, epithelial-mesenchymal transition, AMPK
INTRODUCTION
Cancer is a leading cause of death. The etiology of cancer attributes to both genetic and environmental factors. Many oncogenes and tumor suppressor genes have been identified (Vogelstein and Kinzler, 2004). Interestingly, the products of these genes are often positioned at the critical nodes of important metabolic networks, and their activities are hyper-responsive to metabolic perturbations (Jones and Thompson, 2009). For example, during metabolic stress, the AMP-activated protein kinase (AMPK) activates the tumor suppressor p53 (Feng et al., 2005), and the mammalian target of rapamycin (mTOR) seems to modulate this activation (Lee et al., 2007). Both AMPK and mTOR play critical roles in energy sensing (Hardie, 2007; Tokunaga et al., 2004) and tumorigenesis (Faubert et al., 2013; Vogt, 2001). mTOR mediates the growth signals originated from a well-known master regulator of cell metabolism, the phosphatidylinositol 3 kinase (PI3K). PI3K/Akt/mTOR pathway critically controls protein translation via activation of p70 S6 kinase (S6K) and eukaryotic initiation factor 4E (Vogt, 2001). Additionally, inborn or acquired mutations of several metabolic enzymes are associated with development and progression of several types of cancer, highlighting important roles of altered cell metabolism in cancer etiology (Das et al., 2011; DeBerardinis and Thompson, 2012; Frezza et al., 2011; Gao et al., 2012; Lyssiotis and Cantley, 2012; Mullen et al., 2012; Nomura et al., 2010; Yang et al., 2012).
In mammalian cells, glucose, fatty acids and amino acids such as glutamine are major energy sources. A hallmark of cancer cells is the glycolytic breakdown of glucose for ATP production in the presence of ample oxygen to fuel mitochondrial oxidative phosphorylation. This aerobic glycolysis is known as the “Warburg effect” (Warburg, 1956). Although the Warburg effect has been demonstrated to critically contribute to cancer pathogenesis, it is largely unknown how cancer cells shift their energy metabolism to aerobic glycolysis. Malignant tumors such as hepatocellular carcinoma and colorectal carcinoma (CRC) often show increased intracellular lipid droplet (LD) deposition (Bozza and Viola, 2010; Straub et al., 2008), implying an aberrant lipid metabolism. Cellular fat homeostasis is controlled by balanced biosynthesis and utilization. Mobilization of stored fat in adipocytes for use as energy is called lipolysis. Cytosolic fat lipolysis requires at least three different enzymes: 1) adipose triglyceride lipase (ATGL) that catalyzes the first step of lipolysis, converting triglycerides (TGs) to diacylglycerols (DAGs) (Zimmermann et al., 2004); 2) hormone sensitive lipase (HSL) mainly responsible for the conversion of DAGs to monoacylglycerols (MAGs); and 3) monoacylglycerol lipase (MAGL) that hydrolyzes MAGs to release the last fatty acyl chain from the glycerol backbone. Recently, the lipid-specific macroautophagy (lipophagy) was shown to clear some of cytosolic LDs by delivering LD-associated fat to lysosomes for degradation by acidic lipases (Singh et al., 2009). It is likely that these two intracellular pathways crosstalk with each other to ultimately determine the level of fat utilization in a cell. While increased de novo lipid biosynthesis has long been known to play an important role in cancer growth (Menendez and Lupu, 2007), the role of disrupted fat utilization in cancer development and progression has just begun to be explored. It was shown that MAGL deficiency inhibits cancer pathogenesis (Nomura et al., 2010). Recently, ATGL deficiency was shown to protect against cancer-associated cachexia (Das et al., 2011). Although the role of lipophagy in cancer remains unknown, macroautophagy is known to influence the pathogenesis of cancer (Levine and Kroemer, 2008; White and DiPaola, 2009). Nonetheless, it is currently unclear whether the defective fat utilization plays a part in shifting cancer cell’s metabolism to aerobic glycolysis.
ATGL requires a coactivator, comparative gene identification-58 (CGI-58), to achieve full TG hydrolase activity (Lass et al., 2006). CGI-58 is also known as α/β hydrolase domain-containing protein-5 (ABHD5). Mutations in human abhd5 cause Chanarin-Dorfman Syndrome (CDS) (Chanarin et al., 1975; Dorfman et al., 1974), a rare autosomal recessive genetic disease characterized by TG-rich LD accumulation in almost all tissues but fat. Mutations in human ATGL also cause a neutral lipid storage disease (Fischer et al., 2007). Despite this similarity, obvious phenotypic differences exist between ATGL and Abhd5 mutations. For example, patients with Abhd5 mutations display thickened dry skin (ichthyosis), but this is absent in patients with ATGL mutations (Fischer et al., 2007; Igal et al., 1997). Mice lacking Abhd5 die neonatally (Radner et al., 2010), but mice lacking ATGL are viable (Haemmerle et al., 2006). Liver-specific Abhd5 knockout mice develop hepatic steatohepatitis and fibrosis (Guo et al., 2013), while liver-specific ATGL knockout mice display only simple hepatic steatosis (Wu et al., 2011). These observations indicate that Abhd5 must have functions beyond activating ATGL.
We have previously shown that antisense oligonucleotide (ASO)-mediated knockdown (KD) of Abhd5 in adult mice promotes glucose disposal while inhibiting fat utilization (Brown et al., 2010). Whole-body ATGL knockout mice also show improved glucose tolerance (Haemmerle et al., 2006). These observations raised an intriguing question in cancer metabolism: Is defective fat hydrolysis a cause of aggravated glucose uptake commonly seen in cancer cells? In the Database of the Catalogue of Somatic Mutations in Cancer (COSMIC), the loss of gene copy number is frequent and the loss of heterozygosity (LOH) is present for Abhd5 gene in several human cancer types, including those of large intestine origin. These animal and human data led us to hypothesize that Abhd5 may be a potential molecular switch of cancer cell’s fuel selection. Cancer cells may choose to down-regulate Abhd5 to inhibit fat utilization, thereby enhancing their aerobic glycolysis. To test this hypothesis, in this study, we examined the role of Abhd5 in the development and progression of CRCs, the third leading cause of cancer deaths in the United States (Jemal et al., 2010). We demonstrated that Abhd5 functions as a colorectal tumor suppressor. It suppresses colorectal tumorigenesis and malignant transformation by inhibiting aerobic glycolysis and epithelial-mesenchymal transition (EMT), a major hallmark of tumor invasion and metastasis (Hanahan and Weinberg, 2011). At the cell signaling level, Abhd5 acts as an activator of AMPK and a negative regulator of PI3K/Akt/mTOR/p53 pathway to inhibit CRC growth and invasion.
RESULTS
Abhd5 KD in Colorectal Cancer Cells Promotes Invasive Capacity via Inducing Epithelial-Mesenchymal Transition
To examine whether Abhd5 has any relation to cancer biology, we first silenced Abhd5 expression in HCT116 human colon cancer cell line. Lentiviral shRNA-mediated KD of Abhd5 caused decreased apoptosis, but had little effect on proliferation (Figure S1A–C). Strikingly, Abhd5-KD cells acquired fibroblast-like morphology and were loosely scattered (Figure 1A). The highly scattered cells were significantly increased in Abhd5-KD cells. Transwell matrigel assays demonstrated that Abhd5-KD cells became more invasive than control cells as evidenced by increased cell migration (Figure 1B). These findings imply that Abhd5-KD cells may have undergone an EMT (Hanahan and Weinberg, 2011). We then measured expression levels of EMT markers. As expected, the protein level of E-cadherin, an epithelial marker, decreased while that of a mesenchymal marker Snail increased in Abhd5-KD cells (Figure 1C). To rule out the potential off-target effects of the shRNA, we used two additional shRNAs of different sequences to silence Abhd5 expression in HCT116 cells and obtained identical results (Figure S1D and S1E). In addition, we silenced Abhd5 expression in another human colon cancer cell line SW620. Similar results were observed (Figure S1F).
Figure 1. Abhd5 Suppresses EMT and Growth Advantage of CRC Cells.
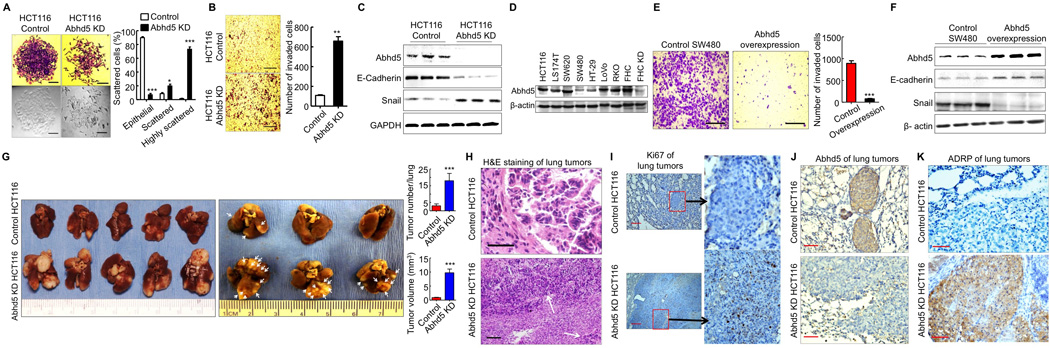
(A) Morphology (crystal violet staining and phase-contrast) of HCT116 control and Abhd5-knockdown (KD) cells. One hundred cells in each colony were counted under microscopy (200x). Ten colonies were counted for each group. Epithelial: cells attached to each other tightly; Scattered: cells scattered, but still attached to others; Highly scattered: the scattered cells that had no contact with others. *P < 0.01. ***P < 0.0001. Scale bar = 50 µm.
(B) Transwell assays. Scale bar = 200 µm.
(C) Western blots of EMT markers.
(D) Western blots of Abhd5 in colon cancer and normal colon epithelial cell lines.
(E) Transwell assays of SW480 cells overexpressing Abhd5. Scale bar = 200 µm.
(F) Western blots of EMT markers in SW480 cells overexpressing Abhd5.
(G) Lung tumor lesions induced by the tail vein injection of HCT116 cells in nude mice (left panel, nonfixed organ; right panel, formalin-fixed organ).
(H) H&E staining of the above lung tumors. Scale bar = 100 µm.
(I–K) Immunohistochemistry of Ki67, Abhd5 and ADRP in the above lung tumors. Scale bar = 100 µm.
To further confirm the role of Abhd5 in regulating EMT and migration capacity of colorectal cancer cells, we overexpressed Abhd5 in a highly aggressive SW480 human colon cancer cell line whose endogenous Abhd5 protein level is low relative to many other colon cancer cell lines (Figure 1D). Abhd5 overexpression dramatically attenuated the migration capacity of SW480 cells, suggesting reduced invasiveness (Figure 1E). It also upregulated E-cadherin and downregulated Snail expression (Figure 1F), consistent with a mesenchymal-epithelial transition (MET), a biological process opposite to EMT and indicative of reduced mesenchymal phenotypes.
Given the critical role of EMT in tumor metastasis, we examined the ectopic growth capacity of Abhd5-KD cells in vivo. Nude mice injected with Abhd5-KD HCT116 cells showed a dramatic increase of tumor lesions in the lung, the primary target organ of tail vein-injected cancer cells (Figure 1G). After fixation, increased tumor lesions became apparent (Figure 1G, middle panel). When lung surface lesions were counted and measured, the tumor number and size were significantly increased in the KD group (Figure 1G, left panel). The tumor lesions derived from Abhd5-KD but not control HCT116 cells also appeared in other tissues including subcutaneous regions, mediastinum, chest wall and adrenal gland (Figure S1G). H&E staining of the pulmonary lesions (Figure 1H) showed that the lesions from control HCT116 cells exhibited a typical glandular morphology surrounded by a fibrous layer. The majority of the pulmonary lesions originated from Abhd5-KD HCT116 cells had a necrotic core. The tumor cells at the edge failed to form glandular appearance and fibrous layer, and were often spread out to adjacent lung tissues where glandular structures were occasionally observed. This microscopic appearance indicates invasive growth of Abhd5-KD cells in the lung. Ki67-positive staining was frequently seen in the lesions derived from Abhd5-KD but not control HCT116 cells, suggesting a greater proliferation potential of KD versus control cells (Figure 1I). Immunostaining of Abhd5 demonstrated the loss of Abhd5 expression in the pulmonary tumors originated from Abhd5-KD cells (Figure 1J). The adjacent lung tissues stained positive because the antibody also recognizes the mouse Abhd5. A hallmark of Abhd5 loss is the cytosolic accumulation of LDs. As expected, the pulmonary tumors derived from Abhd5-KD cells accumulated LDs that were stained positive for ADRP (adipose differentiation-related protein), an LD coat protein (Figure 1K). These findings collectively indicate that Abhd5 KD substantially increases the growth advantage of HCT116 cells.
Abhd5 KD in Normal Cells Induces Malignant Transformation
To examine whether Abhd5 loss plays any roles in malignant transformation of normal cells, we silenced Abhd5 expression in FHC human normal colon mucosal cells and BJ human foreskin fibroblasts using lentiviral shRNAs. Transwell assays showed that Abhd5 KD in FHC cells resulted in a significant increase in cells that penetrated the matrigel between the two chambers (Figure 2A), which was associated with a reduction in E-cadherin protein (Figure 2B), indicative of EMT. Silencing Abhd5 in BJ cells (Figure 2C) did not alter cells’ in vitro growth potential (Figure 2D), but dramatically enhanced their invasive capacity across the matrigel (Figure 2E). Subcutaneous xenograft experiments with BJ cells showed that Balb/c nude mice injected with Abhd5-KD cells at the thigh developed visible tumors 2 weeks after cell injection while the mice injected with the same amount of control cells showed no tumors at the same time. Eight weeks post cell injection, the tumors were dissected out (Figure 2F and 2G). H&E staining of tumor sections revealed a sarcoma-like morphology (Figure 2H). Immunostaining identified abundant human vimentin in the tumor (Figure 2I). Collectively these data suggest that Abhd5 KD causes malignant transformation of normal colon mucosal cells and foreskin fibroblasts.
Figure 2. Abhd5 Knockdown Induces Malignant Transformation of Normal Colon Mucosal Cells and Foreskin Fibroblasts.
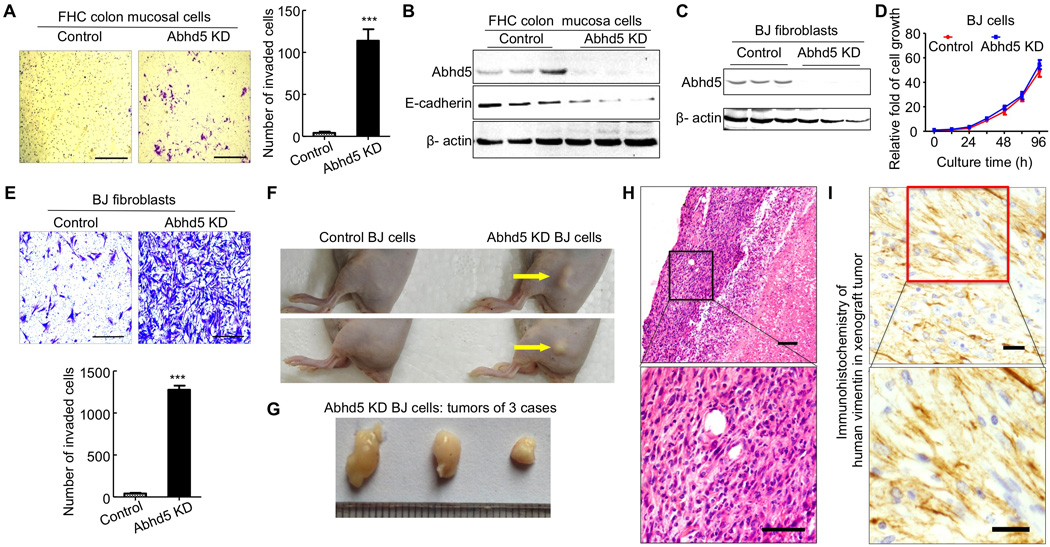
(A) Transwell assays of Abhd5-KD and control FHC cells (Passage: ~14). Scale bar = 200 µm.
(B) Western blots.
(C) Abhd5 knockdown.
(D) Growth curve of normal and Abhd5-KD BJ cells.
(E) Transwell assays. Scale bar = 200 µm.
(F) Gross appearance of subcutaneous xenografts in Balb/c nude mice 8 weeks after injection of control or Abhd5-KD BJ cells (5×104 cells per mouse).
(G) Subcutaneous xenografts dissected from the mice described in (F).
(H) H&E staining of the above subcutaneous xenografts. Scale bar = 100 µm.
(I) Immunohistochemistry of human vimentin in the above xenografts. Scale bar = 50 µm.
Intestine-specific Inactivation of Abhd5 in ApcMin/+Mice Promotes Tumorigenesis and Malignant Transformation of Intestinal Adenomatous Polyps
To establish a causal role for Abhd5 loss in CRC pathogenesis in vivo, we crossed the adenomatous polyposis coli (Apc)Min/+ allele (Su et al., 1992) into the intestine-specific Abhd5 knockout mice (Xie et al., 2014). Strikingly, selective inactivation of Abhd5 in the intestine of ApcMin/+ male mice (ApcMin/+/Abhd5f/f/Cre+) caused a significant increase of tumor number and size in both small intestine and colorectum (Figure 3A–D). Similar changes were observed in female mice (data not shown). Unlike humans carrying Apc mutations, who develop macroscopic tumors mainly in the colorectum, regular ApcMin/+ mice develop adenomatous polyps mainly in small intestine, and the colorectal polyps are rare (Su et al., 1992). Interestingly, all Abhd5-deficient ApcMin/+ mice showed extensive distension of colorectum and frequent rectal prolapse, which was unprecedented in regular ApcMin/+ mice at the same age. The average tumor number and size increased significantly in the colorectum of Abhd5-deficient ApcMin/+ mice (Figure 3C and 3D). Due to robust increases in tumor burden, these animals developed severe anemia at 100 days of age, as evidenced by a dramatic decline in red blood cells and blood hemoglobin (Figure 3E), as well as by increased spleen size and weight (Figure 3F and 3G). Very impressively, all of the aforementioned phenotypes were gene dose-dependent.
Figure 3. Intestine-Specific Knockout of Abhd5 Promotes Tumorigenesis and Aggressiveness of Tumors in ApcMin/+ Mice.
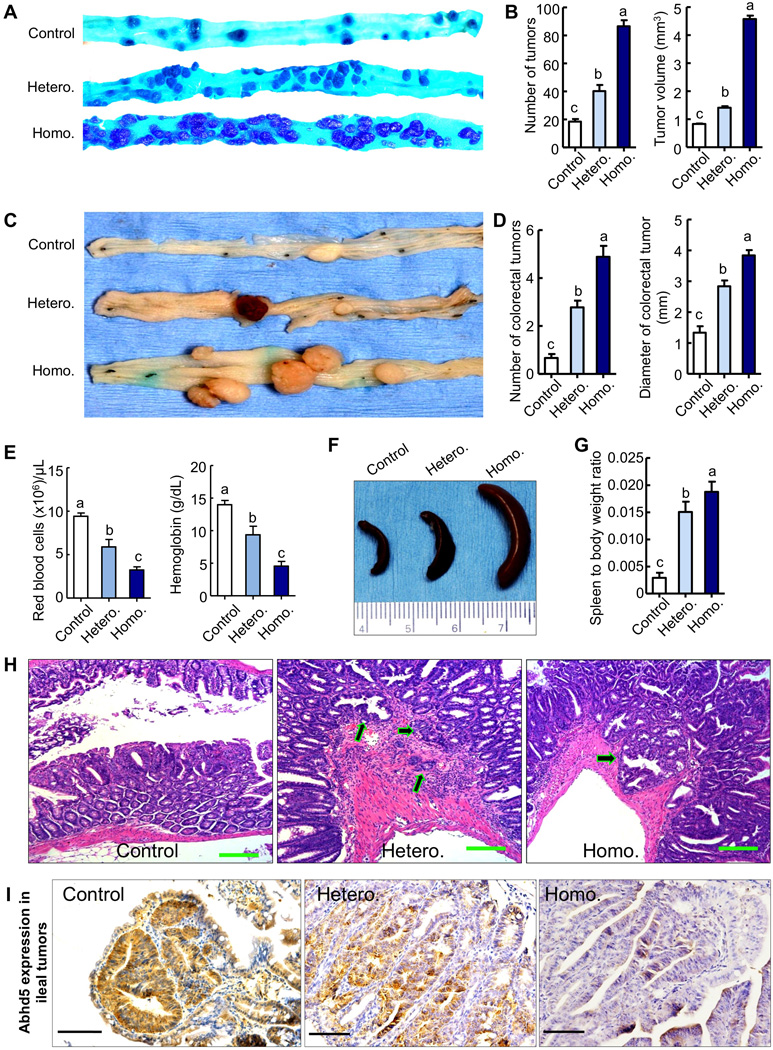
(A) Macroscopic appearance of tumors in the distal ilia of 100-day-old male mice. Control, ApcMin/+/Abhd5+/+/Cre+; Hetero, ApcMin/+/Abhd5f/+/Cre+; Homo, ApcMin/+/Abhd5f/f/Cre+.
(B) Statistical analysis of tumor number and size in the entire small intestine.
(C, D) Macroscopic appearance (C) of tumors and statistical analysis of tumor number and size (D) in the colorectum of 100-day-old mice.
(E) Red blood cell counts in 100-day-old mice.
(F, G) Macroscopic appearance (E) of spleens and statistical analysis (F) of spleen-to-body weight ratios in 100-day-old mice.
(H) Representative H&E sections of tumors in the small intestine of control, heterozygous (Hetero.) and homozygous (Homo.) intestine-specific Abhd5 knockout male mice at 100 days of age. Arrows point to invasive glands. Scale bar = 100 µm.
(I) Immunohistochemistry of Abhd5 in the tumors described in (H). Different letters associated with individual bars represent significant statistical differences among the groups (One-way ANOVA; P < 0.0001; n = 9 in all experiments). Scale bar = 100.
Besides increased tumor number and size, many tumors formed in Abhd5-deficient ApcMin/+ mice were highly dysplastic with cauliflower-like surfaces, profound architectural distortion, and cytological alterations to a level that was highly uncommon in human or murine polyps but characteristic of carcinomas, including the tightly and irregularly arranged tall, columnar cells, dark stain, atypical glandular proliferation with back-to-back phenomenon and the two glands sharing the same glandular wall, loss or disorder of cell polarity, dark stain, increased ratios of cell nucleus to cytoplasm, pleomorphic nuclei with prominent nucleoli, and activities of mitosis and pathological mitosis (Figure 3A, 3C, 3H and S2A). There was an increased stromal reaction to the cancer as evidenced by increased expression of CD31, an angiogenic marker (Figure S2B). We frequently observed adenocarcinomas in the submucosa and muscularis propria in the ilea of Abhd5-deficient ApcMin/+ mice (Figure 3H), a feature of malignancy that was not present in the control mice of the same age. Even a single Abhd5-null allele significantly enhanced the invasiveness of ApcMin/+ tumors (Figure 3H, Hetero.). Immunostaining of Abhd5 in the intestinal tumors revealed that the expression of this single Abhd5 allele was not inactivated (Figure 3I). Abhd5-deficient tumors showed accumulation of ADRP (adipose differentiation-related protein) (Figure S3A), an LD coat protein. Notably, one out of nine Abhd5-deficient ApcMin/+ male at 100 days had liver metastases (Figure S3B) from aggressive intestinal tumors that clearly penetrated the muscle layer (Figure S3C). H&E staining of the liver tumors often revealed a necrotic core and the glandular structures at the edge that penetrated into the liver tissue (Figure S3D). The intestinal origin of liver tumors was confirmed by immunostaining of CK20 (Figure S3E), a marker of intestinal epithelial cells, as well as CDX-2 (Figure S3F), an intestinal transcription factor. This evidence from Abhd5-knockout mice demonstrates that loss of Abhd5 promotes the malignant transformation of benign intestine adenomas.
Loss of Abhd5 Is a Hallmark of CRCs
In the COSMIC database, Abhd5 gene shows the loss of heterozygosity and frequent copy number variation (CNV) (mostly loss of copy number) in several human cancer types including cancer of large intestine. To further probe the relevance of our cell and animal findings to human pathophysiology, we analyzed human CRC samples and patient history. Since it has been reported that cancer tissues relative to normal tissues of their origins often accumulate more LDs (Accioly et al., 2008; Straub et al., 2010), we first stained LDs in CRC tissues and their adjacent normal colon tissues from 10 patients (8 colon and 2 rectal cancer patients) to determine if these previous observations can be recapitulated in our cohort of CRC specimens. Consistently, all CRC tissues examined displayed increased LDs (Figure 4A). Next we immunostained Abhd5 in a tissue chip containing normal colon mucosa and colon specimens of hyperplasia, colitis, adenoma and carcinoma. Abhd5 expression levels were substantially decreased in CRCs versus normal and benign colon pathological alterations, but there were no differences in Abhd5 expression among normal colon mucosa, hyperplasia and colitis tissues (Figure 4B and 4C). The gradual loss of Abhd5 expression from normal colon mucosa to paracancerous tissue to CRC region was often seen in one tissue section (Figure 4D). Since some degrees of Abhd5 loss were found in adenomas (Figure 4C), we speculated that loss of Abhd5 might be associated with the malignant transformation of adenomas, a critical step of CRC development. We compared Abhd5 expression levels in colorectal adenomas between the patients with and those without malignant transformation (Figure S4A). Consistently, the risk of malignant transformation was significantly higher in adenomas with extensive loss of Abhd5 compared to those with less or without Abhd5 loss (Figure S4B). Additionally, in human CRCs, Abhd5 expression levels were substantially lower in stages M1 and IV than stages M0 and I–III (Figure 4E and 4F). CRC patients with recurrence after surgical resection exhibited lower Abhd5 expression relative to those with no recurrence (Figure 4G). CRC patients with lower Abhd5 expression had poorer prognosis than those with higher Abhd5 expression (Figure 4H). Our findings from animal and human studies support that Abhd5 deficiency promotes CRC progression.
Figure 4. Loss of Abhd5 Expression Correlates Positively with CRC Development and Progression in Humans.
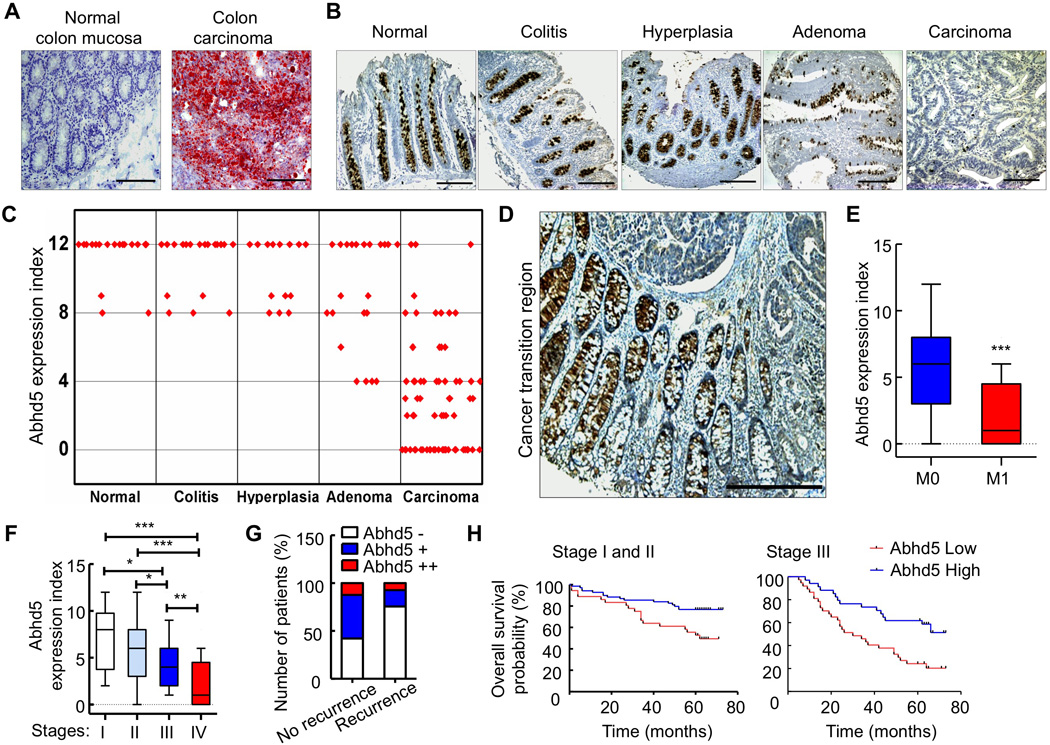
(A) Oil-red O staining of the normal colon mucosa and the colon carcinoma from the same patient. Ten patients examined and similar results obtained. Scale bar = 200 µm.
(B, C) Representative immunostaining images (B, Scale bar = 200 µm) and statistical analysis (One-way ANOVA) of expression index (C) of Abhd5 in colon tissues of different diseases. P < 0.001 between carcinoma and each of other disease stages.
(D) A representative immunostaining image of Abhd5 in a human colon carcinoma and its adjacent normal tissue, showing gradual loss of Abhd5 expression. Scale bar = 200 µm.
(E, F) Statistical analysis of Abhd5 expression levels between M0 CRCs (n = 218) and M1 CRCs (n = 34) (E) (Student t-test), and among CRCs of different stages (F) (Oneway ANOVA). I, n = 18; II, n = 117; III, n = 83; IV, n = 34. *P < 0.01. **P < 0.001. ***P < 0.0001.
(G) Fisher’s Exact Test of the correlation between Abhd5 expression levels and the risk of recurrence in Stage I and II CRC patients (n = 57 in No recurrence; n = 41 in Recurrence; P = 0.004).
(H) Statistical analysis of the correlation between Abhd5 expression levels and the Overall Survival of CRC patients (Stages I, & II, n = 105; Stage III, n = 71). P < 0.01 (Kaplan-Meier Survival Curves).
p53 is Critically Involved in Abhd5 Deficiency-induced EMT in Colorectal Cancer Cells
To explore the molecular mechanisms underlying Abhd5 deficiency-induced EMT, we employed cultured cells as a model. Transcription factor Snail plays an essential role in regulating EMT. It was shown that Snail directly binds to the promoter region of E-cadherin to inhibit transcription of E-cadherin (Cano et al., 2000). Many factors are known to suppress Snail expression, one of which is the potent tumor suppressor p53 (Hahn et al., 2013; Siemens et al., 2011). We found that total p53 protein was reduced in Abhd5-KD FHC normal colon mucosal cells (Figure S5A). Similar reduction in total p53 protein was observed in Abhd5-KD HCT116 cells (Figures 5A and S5B) and Abhd5-KD SW620 cells (Figure S5C). Interestingly, phosphorylation of p53 was increased at Serine 392 and decreased at Serine 15, 37 and 46 of p53 in Abhd5-KD HCT116 cells (Figure 5A). Cell fractionation studies showed that p53 protein was reduced in both cytosol and nucleus, with a more dramatic reduction in the nucleus (Figure 5B). Immunofluorescence staining also revealed a substantial reduction of p53 protein in the nucleus (Figure 5C). Consistently, mRNAs for several known target genes of p53 were significantly reduced (Figure 5D). The opposite was observed when Abhd5 was overexpressed in HCT116 cells (Figure 5E and 5F). Abhd5 deficiency does not seem to induce malignant properties via other classical pathways involved in carcinogenesis because Abhd5 KD had no effects on protein levels of c-myc and PTEN, and reduced levels of oncogenic Ras protein, phosphorylated ERK, and p-catenin (Figure S5D and S5E). On the other hand, forced expression of human p53 in Abhd5-KD HCT116 cells prevented Abhd5 deficiency from causing EMT (Figure 5G and 5H). Additionally, when Abhd5 was knocked down in p53-null HCT116 cells, E-cadherin protein was not decreased (Figure 5I). Reduced E-cadherin was largely responsible for driving EMT in Abhd5-KD HCT116 cells because overexpression of human E-cadherin in these cells attenuated their invasiveness (Figure S5F). In intestinal tumors from apcMin/+ mice lacking Abhd5, p53 and E-cadherin expression levels were reduced and Snail expression was increased (Figure S5G). Collectively, these data demonstrate that Abhd5 deficiency in HCT116 cells promotes EMT and cell invasion via inhibiting p53 expression and functions.
Figure 5. Abhd5 Deficiency Induces EMT via inactivating p53.
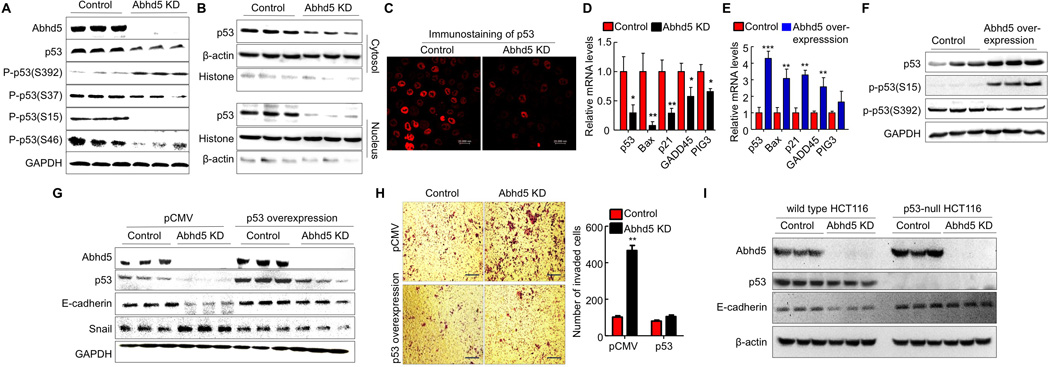
(A) Western blots of total p53 and phosphorylated (P-) p53 levels in HCT116 cells.
(B) Western blots of p53 protein in the cytosol and nucleus of HCT116 cells.
(C) Immunofluorescence staining of p53 in HCT116 cells.
(D) The mRNA levels of p53 target genes in HCT116 cells.
(E) The mRNA levels of p53 target genes in HCT116 cells overexpressing human Abhd5.
(F) Western blots of p53 protein in HCT116 cells overexpressing human Abhd5.
(G) Normalization of E-cadherin and Snail expression by forced expression of p53 in Abhd5-KD HCT116 cells.
(H) Attenuation of increased cell migration by forced expression of p53 in Abhd5-KD HCT116 cells. Scale bar = 200 µm.
(I) Western blots of Abhd5, p53, E-cadherin, and β-actin in p53-null and parental wild-type control HCT116 cells.
Abhd5 KD in HCT116 Cells Enhances Aerobic Glycolysis
To probe the metabolic and cellular basis for Abhd5 deficiency-induced EMT, we examined the fat and glucose utilization. As expected, Abhd5 KD increased cellular content of triglycerides in HCT116 cells (Figure 6A). Abhd5 deficiency is known to cause intracellular lipid accumulation by inhibiting triglyceride catabolism instead of increasing triglyceride synthesis (Brown et al, 2007; Guo et al., 2013; Lass et al., 2006; Radner et al., 2010). We found that triglyceride hydrolase activity was reduced in Abhd5-KD cell lysate (Figure 6B). While this was consistent with a role of Abhd5 in promoting ATGL’s activity (Lass et al., 2006), ATGL-KD HCT116 cells displayed reduced invasion capacity, increased p53 protein and unaltered Snail expression, though a slight reduction in E-cadherin (Figure S6A and S6B). Their growth was substantially attenuated (Figure S6C). In human CRC specimens, immunostaining did not show any changes in ATGL expression levels between normal colon mucosa and CRC tissues (Figure S6D). These data demonstrate that ATGL deficiency does not increase, or correlate with, malignant properties of CRCs. Abhd5 must have ATGL-independent functions, which, together with its ATGL coactivator function, ultimately determine the biological behaviors of cancer cells. One of such functions may be its role in activating macroautophagy, a cellular process that is critically involved in the pathogenesis of cancer (Levine and Kroemer, 2008; White and DiPaola, 2009). The microtube-associated protein 1 light chain 3β-II (LC3b-II) is a lipidated form of LC3b-I and a reliable marker for the state of autophagy (Klionsky et al., 2012). We found that Abhd5-KD cells showed decreased, but ATGL-KD cells displayed increased, LC3b-II protein levels under normal culture conditions (Figure 6C), though both cells exhibited reduced LC3B-II protein levels when cultured in the serum-free medium, a condition that is useful in examining the potential role of a protein in autophagy induction, but is absent physiologically. Clearly lots of studies are required to address potential roles of Abhd5 and ATGL in regulating autophagy and perhaps lipophagy in the future.
Figure 6. Abhd5 Deficiency-Induced EMT in CRC Cells Depends on Glucose Uptake and Aerobic Glycolysis.
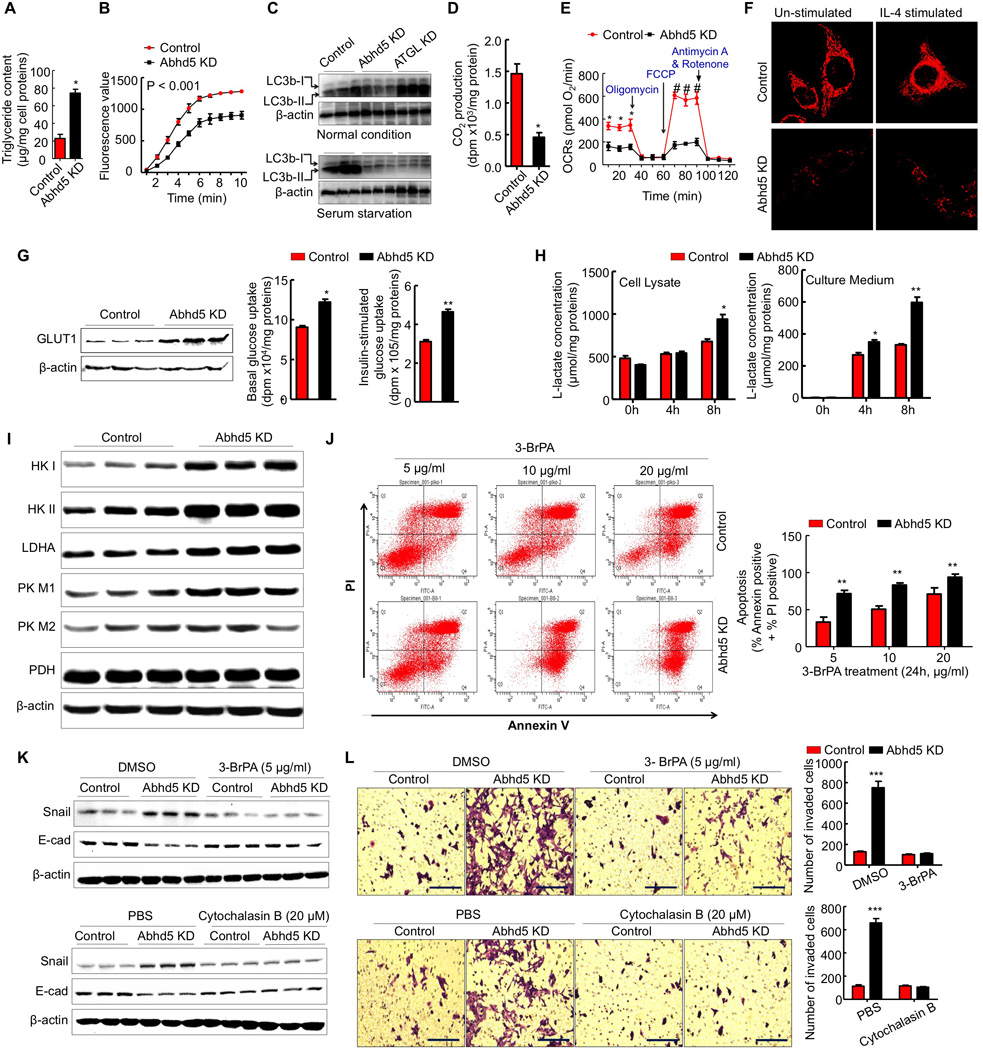
(A) Triglyceride content of control and Abhd5-KD HCT116 cells.
(B) Triglyceride hydrolase activity of HCT116 cell lysates.
(C) Western blots of LC3b protein in HCT116 cells.
(D) CO2 produced from [14C]palmitic acid in HCT116 cells (n = 4). *P < 0.01.
(E) Seahorse Assays of mitochondrial oxygen consumption rates (OCRs) in HCT116 cells (n = 3). *P < 0.01.
(F) Mitochondria stained with Mitotracker-Red FM in HCT116 cells. Scale bar = 10 µm.
(G) GLUT1 protein expression as well as basal and insulin-stimulated uptake of [3H]2-deoxy-D-glucose in HCT116 cells (n = 3). *P < 0.05. **P < 0.01.
(H) Lactate concentrations in the lysate and culture medium of HCT116 cells (n = 3). *P < 0.05. **P < 0.01.
(I) Western blots of glycolytic proteins in HCT116 cells.
(J) Flow cytometry analysis of apoptosis of HCT116 cells treated with a glycolysis inhibitor 3-bromopyruvate (BrPA).
(K) Western blots of EMT markers in Abhd5-KD HCT116 cells treated with a glycolysis inhibitor 3-BrPA (5 µg/ml, 24h) or a glucose transport inhibitor cytochalasin B (20 µM, 24h).
(L) Transwell assays of Abhd5-KD HCT116 cells treated with 3-BrPA (5 µg/ml, 48h) or cytochalasin B (20 µM, 48h). Scale bar = 100 µm.
To determine how Abhd5 deficiency affects cell metabolism, we first measured fatty acid oxidation potential of Abhd5-KD HCT116 cells because Abhd5-KD hepatoma cells have reduced fatty acid oxidation (Brown et al., 2007). Consistently CO2 production from [14C]palmitic acid was significantly decreased in Abhd5-KD HCT116 cells (Figure 6D). Given that mitochondria are the major sites of fatty acid, we analyzed mitochondrial functions in our cells by Seahorse studies. Abhd5-KD HCT116 cells exhibited a significant reduction in both basal mitochondrial oxygen consumption rates and mitochondrial oxidative capacity (Figure 6E). Mitotracker-Red staining revealed a dramatic reduction in mitochondria in Abhd5-KD cells under both basal and interleukin (IL)-4-stimulated conditions (Figure 6F), indicating impaired mitochondrial biogenesis.
Impairment of mitochondrial functions may cause the Warburg effect in cancer cells (Warburg, 1956). We have previously shown that Abhd5 KD in mice promotes glucose disposal while inhibiting fat utilization (Brown et al., 2010). Consistently, glucose transporter-1 (GLUT-1), glucose uptake under both basal and insulin-stimulated states, and lactate production were significantly increased in Abhd5-KD HCT116 cells (Figure 6G and 6H). So did protein levels of glycolytic enzymes, including hexokinase I (HK-I), hexokinase II (HK-II), lactate dehydrogenase A (LDHA), and pyruvate kinase muscle isozyme I (PK-M1) (Figure 6I). Similar changes were also seen in Abhd5-KD SW620 cells except HK II whose expression was unaltered (Figure S7A). Additionally expression levels of GLUT-1, HK-II and LDHA were increased in the intestinal tumor from apcMin/+ mice lacking intestinal Abhd5 (Figure S7B). Cell apoptosis assays showed that Abhd5-KD versus control HCT116 cells were more sensitive to 3-bromopyruvatic acid (3-BrPA), a glycolysis inhibitor (Figure 6J). Very impressively, 3-BrPA or cytochalasin B (a glucose transport inhibitor) reversed the effects of Abhd5 KD on EMT in HCT116 cells (Figure 6K and 6L). These results suggest that Abhd5 may act as a molecular switch of fuel selection, loss of which turns off mitochondrial fat oxidation and turns on aerobic glycolysis (the Warburg effect).
Reduced AMPK Phosphorylation Mediates p53 Suppression and EMT in Abhd5-KD Colorectal Cancer Cells
To determine if altered fuel metabolism influences cell’s energy balance, we measured cellular contents of AMP and ATP. The ratio of AMP/ATP was significantly reduced in Abhd5-KD HCT116 cells (Figure 7A). Treatment with cytochalasin B or 3-BrPA completely prevented this reduction (Figure 7B), indicating that enhanced glucose uptake and glycolysis were responsible for reduced AMP/ATP ratio. The AMP-activated protein kinase (AMPK) is an energy sensor (Hardie, 2007). When cellular energy is abundant (AMP decreases while ATP increases), AMPK phosphorylation is suppressed to stimulate anabolic pathways. Consistent with the decreased AMP:ATP ratio, phosphorylation of AMPKα was substantially decreased in Abhd5-KD HCT116 cells (Figure 7C) and SW620 cells (Figure S7A), but not in ATGL-KD HCT116 cells (Figure S7C). It has been reported that glucose deprivation activates AMPK activity (Yun et al., 2005). To determine if reduced AMPK phosphorylation in Abhd5-KD cells resulted from augmented glucose uptake and glycolysis, we treated our cells with cytochalasin B (a glucose uptake inhibitor) or 3-BrPA (a glycolysis inhibitor). Indeed, inhibiting glucose uptake or glycolysis almost completely rescued AMPKα phosphorylation in Abhd5-KD HCT116 cells (Figure 7C).
Figure 7. Aerobic Glycolysis Induces p53 Suppression via Inactivating AMPK in Abhd5-deficient Cells.
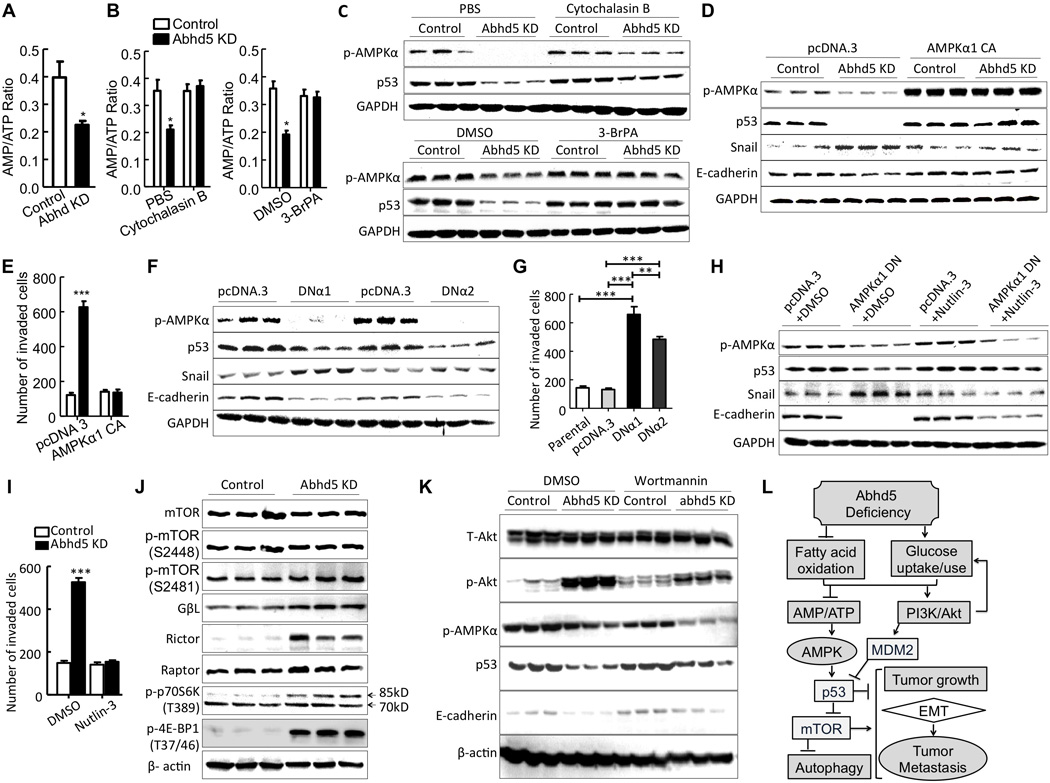
(A) The AMP/ATP ratio in HCT116 cells.
(B) The AMP/ATP ratio in HCT116 cells treated with cytochalasin B (20 µM, 12h) or 3-BrPA (5 µg/ml, 12h).
(C) Western blots of p53 and phosphorylated (p)-AMPKα proteins in HCT116 cells treated with cytochalasin B (20 µM, 24h) and 3-BrPA (5 µg/ml, 24h).
(D) Western blots of p53, Snail, E-cadherin and phosphorylated (p)-AMPKα proteins in HCT116 cells expressing constitutively active (CA) AMPKα1.
(E) Transwell assays of HCT116 cells expressing CA-AMPKα1.
(F) Western blots of HCT116 cells expressing a dominant negative (DN) AMPKα1 (DNα1) or AMPKα2 (DNα2).
(G) Transwell assays of HCT116 cells expressing DNα1 or DNα2.
(H) Western blots of HCT116 cells expressing CA-AMPKα1 in the presence of Nutlin-3 (10 µM, 24h).
(I) Transwell assays of HCT116 cells expressing CA-AMPKα1 in the presence of Nutlin-3 (10 µM, 48h).
(J) Western blots of mTOR pathway proteins in HCT116 cells.
(K) Western blots of HCT116 cells treated with or without Wortmannin (100 nmol/L for 24 h).
(L) Proposed mechanisms underlying Abhd5 deficiency-induced malignancy in CRCs. *P < 0.05, **P < 0.001, ***P < 0.0001.
It has been shown that AMPK activation induces p53 phosphorylation on serine 15, which stimulates the transcriptional activity of p53 (Jones et al., 2005). Recently AMPK was shown to function as a negative regulator of the Warburg effect to suppress tumor growth (Faubert et al., 2013). Given that Abhd5-deficient HCT116 cells shifted their fuel metabolism to aerobic glycolysis (Figure 6) and displayed loss of p53 phosphorylation on serine 15 (Figure 5A), we hypothesized that inhibition of AMPK phosphorylation may mediate suppression of p53 activity, thereby inducing EMT in HCT116 cells. In agreement, cytochalasin B or 3-BrPA treatment that rescued AMPKα phosphorylation also rescued p53 expression in these cells (Figure 7C). Importantly, constitutive activation of AMPKα in Abhd5-KD HCT116 cells normalized expression levels of p53 and EMT markers (Figure 7D), and inhibited cells’ invasive capacity (Figure 7E). The opposite effects were observed when the dominant negative form of AMPKα1 or AMPKα2 was introduced to these cells (Figure 7F and 7G). In addition, Nutlin-3, an activator of p53 activity, rescued the dominant negative AMPKα-induced changes in p53 and EMT markers (Figure 7H) as well as the invasive capacity (Figure 7I) of HCT116 cells. These findings together indicate that AMPK functions as an upstream regulator of p53 to control the energy shift-induced EMT in Abhd5-KD HCT116 cells.
Studies have linked AMPK to mTOR pathway (Lee et al., 2007). In Abhd5-KD HCT116 cells, mTOR phosphorylation (serine 2481), GβL, rictor, raptor, p70S6 kinase (85kDa) phosphorylation (threonine 389), and the eukaryotic initiation factor-4E-binding protein-1 (4E-BP1) phosphorylation (threonine 37/46) were all increased, though total mTOR and mTOR phosphorylation at serine 2448 remained unchanged (Figure 7J). This finding indicates that the mTOR pathway is activated in these cells. Since mTOR mediates growth signals originated from activation of the PI3K-Akt pathway, we examined effects of Abhd5 KD on Akt phosphorylation in HCT116 cells. Akt phosphorylation at serine 473 was highly induced in Abhd5-KD cells (Figure 7K). Inhibition of PI3K by Wortmannin partially restored expression levels of p53 and E-cadherin, but failed to rescue AMPK phosphorylation (Figure 7K), suggesting that Akt is upstream of p53, but downstream of AMPK, or in a pathway parallel to AMPK pathway. PI3K-Akt pathway and AMPK pathways may converge and jointly regulate the p53-mTOR pathway (Figure 7L) as demonstrated previously (Feng et al., 2005; Jones et al., 2005).
DISCUSSION
In this study we for the first time demonstrate that Abhd5 (CGI-58), a lipolytic activator, is a tumor suppressor in colorectal cancer development and progression. Our results from studies of cell behaviors, genetically engineered animal models and human patients strongly support the following mechanisms (Figure 7K): Abhd5 acts as a fuel switch whose deficiency inhibits mitochondrial fatty acid oxidation (fat catabolism) and promotes glucose uptake and utilization. Due to defective fat catabolism, Abhd5-deficient cells display severe mitochondrial dysfunction. Increased glucose is channeled to aerobic glycolysis (the Warburg Effect). Cells gain more energy (reduced AMP:ATP ratio) due to increased glucose influx that is likely promoted by activation of PI3K/Akt pathway. As a result, AMPK phosphorylation is suppressed. Activation of PI3K/Akt pathway and suppression of AMPK phosphorylation inhibit p53 activity. Consequently, mTOR pathway is activated and autophagy is inhibited. Thus, Abhd5 deficiency-induced metabolic reprogramming activates a potent oncogenic signaling network at least in colorectal cancer cells.
Abhd5 was reported to promote triglyceride hydrolysis by activating ATGL’s triglyceride hydrolase activity in vitro (Lass et al., 2006). However we found no correlations between ATGL expression levels and malignant features in human CRC samples examined (Figure S6). This finding may not be surprising because mutations of Abhd5 and ATGL cause several distinct phenotypes in both humans and mice (Fischer et al., 2007; Haemmerle et al., 2006; Lefevre et al., 2001; Radner et al., 2010), suggesting that Abhd5 possesses ATGL-independent functions. Perhaps, Abhd5 has other lipase targets besides ATGL. Alternatively, it may be unnecessary to reduce ATGL expression to inhibit its activity as down-regulation of its coactivator Abhd5 may achieve the same goal. Interestingly, monoacylglycerol lipase (MAGL), the enzyme that cleaves the last hydrocarbon chain on the glycerol backbone of triglyceride during fat lipolysis, was shown to be upregulated in aggressive human cancer cells and primary tumors, and promote tumor aggressiveness (Nomura et al., 2010). While this study appears to be contrary to ours and another report (Das et al., 2011) because they showed that inhibition of a lipolytic enzyme prevents tumor development and progression, one should not expect similar metabolic consequences of Abhd5 or ATGL mutations and MAGL deficiency. Their substrates are different. Abhd5 and ATGL are responsible for triglyceride breakdown. MAGL hydrolyzes monoacylglycerols. While the LDs induced by Abhd5 or ATGL mutations may sequester oncogenic lipids inside the cell, MAGL mutations reduce free fatty acids and their metabolites (Nomura et al., 2010).
Aerobic glycolysis is a hallmark of cancer cell metabolism (Warburg, 1956), but molecular mediators of this metabolic rewiring are poorly understood. Many oncogenic proteins and tumor suppressors may regulate carcinogenesis through their regulatory roles in bioenergetics/metabolic pathways (Vogelstein and Kinzler, 2004). Here we demonstrate that Abhd5 deficiency exacerbates the Warburg effect by inhibiting fat catabolism. Metabolic rewiring often shifts energy balance in a cell. For a glucose molecule, the energy (quantity of ATP) produced from aerobic glycolysis is substantially lower than that from mitochondrial oxidative phosphorylation. However, when the flux rate of glucose through the pathway is much faster, glycolysis relative to oxidative phosphorylation has the potential to produce more ATPs (Guppy et al., 1993). Consistently, Abhd5-deficient cancer cells display a positive energy balance as evidenced by decreased AMP:ATP ratio, despite their preferential use of aerobic glycolysis as an energy source. This energy surplus is sensed by AMPK. AMPK was recently shown to function as a negative regulator of the Warburg effect to suppress tumor growth (Faubert et al., 2013), which is totally consistent with our current findings.
It has been shown that glucose deprivation-induced AMPK activation enhances p53 functions by increasing total p53 protein, p53 phosphorylation at serine 15, and p53-dependent transcription (Jones et al., 2005). The activation of p53 by AMPK likely suppresses glycolysis to restore cell’s energy balance by promoting catabolism (Jones and Thompson, 2009; Kruse and Gu, 2006). Our results are in agreement with these early studies. Considering that Abhd5-deficient cells are bioenergetically forced to ferment glucose for energy production, it appears that Abhd5 deficiency creates a futile cycle to worsen the aerobic glycolysis via suppressing the AMPK-p53 pathway. This futile cycle may explain why Abhd5 deficiency alone is able and sufficient to induce malignant transformation of normal fibroblasts in culture and xenograft model (Figure 2) and to further enhance the aggressiveness of HCT116 cancer cells whose baseline aerobic glycolysis level is already high.
An important finding from our studies is the key role of Abhd5 in limiting EMT of colorectal cancer cells. EMT is critically involved in tumor metastasis (Chaffer and Weinberg, 2011). Consistently, we showed that Abhd5 deficiency in HCT116 cells enhances cell’s invasiveness in matrigel assays and growth advantage in the lung of the nude mice. Our data collectively support that EMT induction in Abhd5-deficient cells is a direct result of the suppression of the AMPK-p53 pathway. Thus we have identified Abhd5 as a novel factor that links fuel metabolism to EMT through the AMPK-p53 pathway.
ApcMin/+ mice develop adenomatous polyps due to Wnt pathway activation (Behrens et al., 1998; Rubinfeld et al., 1993). Normally these polyps are mainly located in the distal segment of small intestine and rarely seen in colorectum (Su et al., 1992). All Abhd5-deficient ApcMin/+ mice were found to bear significantly increased tumors of large size in colon and rectum, highlighting a strong tumor suppression role of Abhd5 in colorectal carcinogenesis. But this role of Abhd5 does not seem to be mediated by β-catenin, c-myc, Ras or PTEN because none of these oncogenic proteins is elevated in Abhd5-KD cells. Our in vivo and in vitro observations instead demonstrate that Abhd5 itself is a tumor suppressor and its loss directly causes malignant transformation. This tumor suppressor role of Abhd5 likely contributes to the strong negative correlation between Abhd5 expression levels and CRC malignant properties in human CRC tissues. Colorectal tumor cells may choose to down-regulate Abhd5 to promote the Warburg effect, thereby enhancing their malignancy. The downregulation of Abhd5 may partly explain the epidemic of cytosolic LD deposition seen in human colorectal and other cancers (Accioly et al., 2008; Straub et al., 2010) because a hallmark of Abhd5 deficiency is the cytosolic LD accumulation. Our findings raise an intriguing question: How is Abhd5 expression downregulated in human malignant tumors? Is it at the epigenetic, transcriptional, posttranscriptional, and/or posttranslational level? Is Abhd5 genetically mutated? Although the loss of copy number and heterozygosity of Abhd5 gene is seen in many cancers including CRCs (COSMIC database), which supports Abhd5 as a tumor suppressor, extensive genetic and epigenetic screening of Abhd5 gene sequences in a large panel of human tumors is warranted. We did not see any differences in Abhd5 expression among normal colon mucosa, colitis and colon hyperplasia, but did observe a partial loss of Abhd5 expression in adenomas and an extensive loss in CRCs, implying that Abhd5 deficiency may be implicated in adenoma development and importantly in malignant transformation of adenomas to CRCs.
In summary, genetic inactivation of Abhd5 in normal cells and APCMin/+ mice facilitates tumorigenesis and malignant transformation. Silencing of Abhd5 in colorectal cancer cells inhibits fatty acid oxidation and enhances glucose uptake and aerobic glycolysis. This metabolic reprogramming promotes carcinogenesis and EMT-driven malignancy by activating PI3K/Akt pathway and suppressing AMPK-p53 pathway. Loss of Abhd5 expression occurs frequently in human colorectal carcinomas and is strongly associated with malignant properties. These results strongly argue for Abhd5 as a colorectal tumor suppressor whose expression level may serve as a biomarker for assessing the malignant transformation risk of the tumor.
EXPERIMENTAL PROCEDURES
Creation of ApcMin/+ Mice Lacking Intestinal Abhd5
Intestine-specific Abhd5 knockout mice (Xie et al., 2014) were generated by mating Abhd5-floxed mice created in our lab (Guo et al., 2013) with B6.SJL-Tg (Vil-cre)977Gum/J mice (Jackson Laboratory, Stock #: 004586). A male ApcMin/+ mouse on the C57BL/6J background was purchased from the Jackson Laboratory (Stock #: 002020) and crossed with female intestine-specific Abhd5 knockout mice to produce ApcMin/+ mice lacking Abhd5 in the intestine and their control littermates for experiments. The Institutional Animal Care and Use Committee at the University of Maryland approved all animal procedures.
Cells
All human colorectal colon cancer cell lines, FHC human normal colon mucosal cell line and BJ human fibroblast cell line were purchased from ATCC. The p53-null HCT116 cell line and its parental HCT116 cell line were obtained from Dr. Bert Vogelstein’s lab at the Johns Hopkins University School of Medicine.
Human Samples
Tissue chips and biopsies were used. The tumors were staged by three anatomic pathologists blinded to the patient information, in the Department of Pathology, Southwest Hospital, according to the Union for International Cancer Control (UICC) classification system. The Ethics Committee of Southwest Hospital, the Third Military Medical University, approved all human experiments.
Packaging and Transfection of Lentiviruses
Human Abhd5 and ATGL shRNA constructs for lentivirus packaging were purchased from Open Biosystems, Inc. and OriGene. The following sequences were used to silence Abhd5 expression in human cells: AAGATCACTGAAACTGGAATG, TCTTTGCACCAACAGACCTGTCTATGCTT (TL306992A) and AGACGATACTGTGACAGAATACATCTACC (TL306992C). The following sequence was used to silence ATGL expression in human cells: AAGTGGGATATAATGACATTC.
Plasmids and Transfection
Construction of constitutively active (CA) and dominant-negative (DN) α1 and α2 AMPK expression vectors was described previously (Stein et al., 2000). The expression plasmid of human p53 (# SC119832) and the control empty vector pCMV6-XL5 were purchased from ORIGENE.
Antibodies, Immunohistochemistry, Immunofluorescence, Oil-red O Staining, Protein Extraction, Western Blotting, Preparation of Nuclear Extracts and Quantitative Real-Time PCR
Routine methods were used these experiments.
Flow Cytometry of Apoptosis
Cell apoptosis was assessed by flow cytometry of Annexin V/PI (Sigma) staining.
Transwell Assay
The migration ability of cells was assessed using Transwell chambers with polycarbonate membrane filters with 24-well inserts (6.5 mm diameter and 8 µm pore size) (Corning Life Sciences, Corning, NY, USA). The membrane filters were coated with 1.5 mg/ml Matrigel™ (BD Biosciences, Franklin Lakes, NJ, USA) before use.
Lipase Activity Assay
The lipase activity of cells was assessed using the fluorogenic ester substrate 4-methylumbelliferyl heptanoate (MUH) (Sigma).
Deoxyglucose Uptake and Lactate Assays
Glucose uptake of cells was assayed using [3H]-2-deoxy-D-glucose as a tracer in the absence and presence of cytochalasin B. Lactate production was measured using a commercial assay kit.
Fatty Acid Oxidation Assay
The [14C]palmitic acid was used as the substrate in the assay.
Seahorse Assays for Mitochondria Functions
Cell mitochondrial oxygen consumption rates (OCRs) were assayed in 96-well plates by using a XF Cell Mito Stress Test Kit on the XFe96 Extracellular Flux Analyzer.
Staining of Mitochondria
Cells were stained with MitoTracker-Red FM in the absence or presence of interleukin 4 (IL-4).
Intracellular Contents of AMP and ATP
Cells were extracted by perchloric acid and the lysate was analzyed by HPLC on a Waters C18 column, using a Waters 2695 Separations Module and a 2487 Dual Absorbance Detector.
Tumor Counting and Size Measurement in Mice
Fixed intestines were stained with methylene blue. The location and size of each tumor were recorded and analyzed by ImageJ Software (NIH).
Ectopic Growth Model
Four-to-six week-old Balb/c nude female mice were purchased from NCI/NIH at Fredrick, Maryland. The metastatic ability of cells was assessed by tail vein injection of cells. Animals were sacrificed 8 weeks after injection and tissues examined for cancer cell lesions.
Subcutaneous Xenograft Models
Four-to-six week-old Balb/c nude mice were purchased from the Experimental Animal Center, the Institute of Laboratory Animal Sciences (China). The mice were subcutaneously injected with BJ cells at the thighs. The mice were examined weekly and sacrificed 8 weeks after injection for xenograft collection.
Statistical Analysis
Data are expressed as Mean ± SEM (Standard Error of the Mean).
For more details on each method and the statistical analysis, please see the Extended Experimental Procedures available online.
Supplementary Material
ACKNOWLEDGMENTS
We thank technical assistance of Jianbing Zhang, Xianfeng Wang, Rongbin Zhou, and Shun Lei. This work was supported in part by Award Numbers R01DK085176 (L.Y.) and R01DK084172 (H.S.) from the National Institute of Diabetes and Digestive and Kidney Diseases, Award Number R01HL107500 (B.X.) from the National Heart, Lung and Blood Institute, and by grant numbers 81370063 (J.O.) and 81172115 (H.L.) from the National Natural Science Foundation of China. Mouse strains described are available to qualified investigators upon completion of a standard institutional material transfer agreement.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
SUPPLEMENTAL INFORMATION
Supplemental Information includes Extended Experimental Procedures, and seven figures, and can be found with this article online.
Author Contribution
L.Y., H.L., J.O., H.S., and B.X. conceived and designed the experiments. J.O., H.M., Y.M., F.G., J.D., X.W., J.Z., and G.X. performed the experiments. J.O. and J.D. analyzed the data. L.Y. and J. O. wrote and revised the paper.
REFERENCES
- Accioly MT, Pacheco P, Maya-Monteiro CM, Carrossini N, Robbs BK, Oliveira SS, Kaufmann C, Morgado-Diaz JA, Bozza PT, Viola JP. Lipid bodies are reservoirs of cyclooxygenase-2 and sites of prostaglandin-E2 synthesis in colon cancer cells. Cancer research. 2008;68:1732–1740. doi: 10.1158/0008-5472.CAN-07-1999. [DOI] [PubMed] [Google Scholar]
- Behrens J, Jerchow BA, Wurtele M, Grimm J, Asbrand C, Wirtz R, Kuhl M, Wedlich D, Birchmeier W. Functional interaction of an axin homolog, conductin, with beta-catenin, APC, and GSK3beta. Science. 1998;280:596–599. doi: 10.1126/science.280.5363.596. [DOI] [PubMed] [Google Scholar]
- Bozza PT, Viola JP. Lipid droplets in inflammation and cancer. Prostaglandins, leukotrienes, and essential fatty acids. 2010;82:243–250. doi: 10.1016/j.plefa.2010.02.005. [DOI] [PubMed] [Google Scholar]
- Brown JM, Betters JL, Lord C, Ma Y, Han X, Yang K, Alger HM, Melchior J, Sawyer J, Shah R, et al. CGI-58 knockdown in mice causes hepatic steatosis but prevents diet-induced obesity and glucose intolerance. Journal of lipid research. 2010;51:3306–3315. doi: 10.1194/jlr.M010256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JM, Chung S, Das A, Shelness GS, Rudel LL, Yu L. CGI-58 facilitates the mobilization of cytoplasmic triglyceride for lipoprotein secretion in hepatoma cells. Journal of lipid research. 2007;48:2295–2305. doi: 10.1194/jlr.M700279-JLR200. [DOI] [PubMed] [Google Scholar]
- Cano A, Perez-Moreno MA, Rodrigo I, Locascio A, Blanco MJ, del Barrio MG, Portillo F, Nieto MA. The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nature cell biology. 2000;2:76–83. doi: 10.1038/35000025. [DOI] [PubMed] [Google Scholar]
- Chaffer CL, Weinberg RA. A perspective on cancer cell metastasis. Science. 2011;331:1559–1564. doi: 10.1126/science.1203543. [DOI] [PubMed] [Google Scholar]
- Chanarin I, Patel A, Slavin G, Wills EJ, Andrews TM, Stewart G. Neutral-lipid storage disease: a new disorder of lipid metabolism. Br Med J. 1975;1:553–555. doi: 10.1136/bmj.1.5957.553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das SK, Eder S, Schauer S, Diwoky C, Temmel H, Guertl B, Gorkiewicz G, Tamilarasan KP, Kumari P, Trauner M, et al. Adipose triglyceride lipase contributes to cancer-associated cachexia. Science. 2011;333:233–238. doi: 10.1126/science.1198973. [DOI] [PubMed] [Google Scholar]
- DeBerardinis RJ, Thompson CB. Cellular metabolism and disease: what do metabolic outliers teach us? Cell. 2012;148:1132–1144. doi: 10.1016/j.cell.2012.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorfman ML, Hershko C, Eisenberg S, Sagher F. Ichthyosiform dermatosis with systemic lipidosis. Arch Dermatol. 1974;110:261–266. [PubMed] [Google Scholar]
- Faubert B, Boily G, Izreig S, Griss T, Samborska B, Dong Z, Dupuy F, Chambers C, Fuerth BJ, Viollet B, et al. AMPK Is a Negative Regulator of the Warburg Effect and Suppresses Tumor Growth In Vivo. Cell metabolism. 2013;17:113–124. doi: 10.1016/j.cmet.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Z, Zhang H, Levine AJ, Jin S. The coordinate regulation of the p53 and mTOR pathways in cells. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:8204–8209. doi: 10.1073/pnas.0502857102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer J, Lefevre C, Morava E, Mussini JM, Laforet P, Negre-Salvayre A, Lathrop M, Salvayre R. The gene encoding adipose triglyceride lipase (PNPLA2) is mutated in neutral lipid storage disease with myopathy. Nature genetics. 2007;39:28–30. doi: 10.1038/ng1951. [DOI] [PubMed] [Google Scholar]
- Frezza C, Pollard PJ, Gottlieb E. Inborn and acquired metabolic defects in cancer. J Mol Med (Berl) 2011;89:213–220. doi: 10.1007/s00109-011-0728-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X, Wang H, Yang JJ, Liu X, Liu ZR. Pyruvate kinase M2 regulates gene transcription by acting as a protein kinase. Molecular cell. 2012;45:598–609. doi: 10.1016/j.molcel.2012.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo F, Ma Y, Kadegowda AK, Xie P, Liu G, Liu X, Miao H, Ou J, Su X, Zheng Z, et al. Deficiency of liver Comparative Gene Identification-58 causes steatohepatitis and fibrosis in mice. Journal of lipid research. 2013;54:2109–2120. doi: 10.1194/jlr.M035519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guppy M, Greiner E, Brand K. The role of the Crabtree effect and an endogenous fuel in the energy metabolism of resting and proliferating thymocytes. European journal of biochemistry / FEBS. 1993;212:95–99. doi: 10.1111/j.1432-1033.1993.tb17637.x. [DOI] [PubMed] [Google Scholar]
- Haemmerle G, Lass A, Zimmermann R, Gorkiewicz G, Meyer C, Rozman J, Heldmaier G, Maier R, Theussl C, Eder S, et al. Defective lipolysis and altered energy metabolism in mice lacking adipose triglyceride lipase. Science. 2006;312:734–737. doi: 10.1126/science.1123965. [DOI] [PubMed] [Google Scholar]
- Haemmerle G, Moustafa T, Woelkart G, Buttner S, Schmidt A, van de Weijer T, Hesselink M, Jaeger D, Kienesberger PC, Zierler K, et al. ATGL-mediated fat catabolism regulates cardiac mitochondrial function via PPAR-alpha and PGC-1. Nature medicine. 2011;17:1076–1085. doi: 10.1038/nm.2439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn S, Jackstadt R, Siemens H, Hunten S, Hermeking H. SNAIL and miR-34a feed-forward regulation of ZNF281/ZBP99 promotes epithelial-mesenchymal transition. The EMBO journal. 2013;32:3079–3095. doi: 10.1038/emboj.2013.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Hardie DG. AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat Rev Mol Cell Biol. 2007;8:774–785. doi: 10.1038/nrm2249. [DOI] [PubMed] [Google Scholar]
- Igal RA, Rhoads JM, Coleman RA. Neutral lipid storage disease with fatty liver and cholestasis. J Pediatr Gastroenterol Nutr. 1997;25:541–547. doi: 10.1097/00005176-199711000-00011. [DOI] [PubMed] [Google Scholar]
- Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA: a cancer journal for clinicians. 2010;60:277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- Jones RG, Plas DR, Kubek S, Buzzai M, Mu J, Xu Y, Birnbaum MJ, Thompson CB. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Molecular cell. 2005;18:283–293. doi: 10.1016/j.molcel.2005.03.027. [DOI] [PubMed] [Google Scholar]
- Jones RG, Thompson CB. Tumor suppressors and cell metabolism: a recipe for cancer growth. Genes & development. 2009;23:537–548. doi: 10.1101/gad.1756509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2012;8:445–544. doi: 10.4161/auto.19496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruse JP, Gu W. p53 aerobics: the major tumor suppressor fuels your workout. Cell metabolism. 2006;4:1–3. doi: 10.1016/j.cmet.2006.06.004. [DOI] [PubMed] [Google Scholar]
- Lass A, Zimmermann R, Haemmerle G, Riederer M, Schoiswohl G, Schweiger M, Kienesberger P, Strauss JG, Gorkiewicz G, Zechner R. Adipose triglyceride lipase-mediated lipolysis of cellular fat stores is activated by CGI-58 and defective in Chanarin-Dorfman Syndrome. Cell metabolism. 2006;3:309–319. doi: 10.1016/j.cmet.2006.03.005. [DOI] [PubMed] [Google Scholar]
- Lee CH, Inoki K, Karbowniczek M, Petroulakis E, Sonenberg N, Henske EP, Guan KL. Constitutive mTOR activation in TSC mutants sensitizes cells to energy starvation and genomic damage via p53. The EMBO journal. 2007;26:4812–4823. doi: 10.1038/sj.emboj.7601900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefevre C, Jobard F, Caux F, Bouadjar B, Karaduman A, Heilig R, Lakhdar H, Wollenberg A, Verret JL, Weissenbach J, et al. Mutations in CGI-58, the gene encoding a new protein of the esterase/lipase/thioesterase subfamily, in Chanarin-Dorfman syndrome. American journal of human genetics. 2001;69:1002–1012. doi: 10.1086/324121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyssiotis CA, Cantley LC. SIRT6 puts cancer metabolism in the driver’s seat. Cell. 2012;151:1155–1156. doi: 10.1016/j.cell.2012.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menendez JA, Lupu R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nature reviews Cancer. 2007;7:763–777. doi: 10.1038/nrc2222. [DOI] [PubMed] [Google Scholar]
- Mullen AR, Wheaton WW, Jin ES, Chen PH, Sullivan LB, Cheng T, Yang Y, Linehan WM, Chandel NS, DeBerardinis RJ. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature. 2012;481:385–388. doi: 10.1038/nature10642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomura DK, Long JZ, Niessen S, Hoover HS, Ng SW, Cravatt BF. Monoacylglycerol lipase regulates a fatty acid network that promotes cancer pathogenesis. Cell. 2010;140:49–61. doi: 10.1016/j.cell.2009.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radner FP, Streith IE, Schoiswohl G, Schweiger M, Kumari M, Eichmann TO, Rechberger G, Koefeler HC, Eder S, Schauer S, et al. Growth retardation, impaired triacylglycerol catabolism, hepatic steatosis, and lethal skin barrier defect in mice lacking comparative gene identification-58 (CGI-58) The Journal of biological chemistry. 2010;285:7300–7311. doi: 10.1074/jbc.M109.081877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinfeld B, Souza B, Albert I, Muller O, Chamberlain SH, Masiarz FR, Munemitsu S, Polakis P. Association of the APC gene product with beta-catenin. Science. 1993;262:1731–1734. doi: 10.1126/science.8259518. [DOI] [PubMed] [Google Scholar]
- Siemens H, Jackstadt R, Hunten S, Kaller M, Menssen A, Gotz U, Hermeking H. miR-34 and SNAIL form a double-negative feedback loop to regulate epithelial-mesenchymal transitions. Cell Cycle. 2011;10:4256–4271. doi: 10.4161/cc.10.24.18552. [DOI] [PubMed] [Google Scholar]
- Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, Tanaka K, Cuervo AM, Czaja MJ. Autophagy regulates lipid metabolism. Nature. 2009;458:1131–1135. doi: 10.1038/nature07976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein SC, Woods A, Jones NA, Davison MD, Carling D. The regulation of AMP-activated protein kinase by phosphorylation. The Biochemical journal. 2000;345(Pt 3):437–443. [PMC free article] [PubMed] [Google Scholar]
- Straub BK, Herpel E, Singer S, Zimbelmann R, Breuhahn K, Macher-Goeppinger S, Warth A, Lehmann-Koch J, Longerich T, Heid H, Schirmacher P. Lipid droplet-associated PAT-proteins show frequent and differential expression in neoplastic steatogenesis. Modern pathology : an official journal of the United States and Canadian Academy of Pathology, Inc. 2010;23:480–492. doi: 10.1038/modpathol.2009.191. [DOI] [PubMed] [Google Scholar]
- Straub BK, Stoeffel P, Heid H, Zimbelmann R, Schirmacher P. Differential pattern of lipid droplet-associated proteins and de novo perilipin expression in hepatocyte steatogenesis. Hepatology. 2008;47:1936–1946. doi: 10.1002/hep.22268. [DOI] [PubMed] [Google Scholar]
- Su LK, Kinzler KW, Vogelstein B, Preisinger AC, Moser AR, Luongo C, Gould KA, Dove WF. Multiple intestinal neoplasia caused by a mutation in the murine homolog of the APC gene. Science. 1992;256:668–670. doi: 10.1126/science.1350108. [DOI] [PubMed] [Google Scholar]
- Tokunaga C, Yoshino K, Yonezawa K. mTOR integrates amino acid- and energy-sensing pathways. Biochemical and biophysical research communications. 2004;313:443–446. doi: 10.1016/j.bbrc.2003.07.019. [DOI] [PubMed] [Google Scholar]
- Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nature medicine. 2004;10:789–799. doi: 10.1038/nm1087. [DOI] [PubMed] [Google Scholar]
- Vogt PK. PI 3-kinase, mTOR, protein synthesis and cancer. Trends in molecular medicine. 2001;7:482–484. doi: 10.1016/s1471-4914(01)02161-x. [DOI] [PubMed] [Google Scholar]
- Warburg O. On the origin of cancer cells. Science. 1956;123:309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- White E, DiPaola RS. The double-edged sword of autophagy modulation in cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2009;15:5308–5316. doi: 10.1158/1078-0432.CCR-07-5023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu JW, Wang SP, Alvarez F, Casavant S, Gauthier N, Abed L, Soni KG, Yang G, Mitchell GA. Deficiency of liver adipose triglyceride lipase in mice causes progressive hepatic steatosis. Hepatology. 2011;54:122–132. doi: 10.1002/hep.24338. [DOI] [PubMed] [Google Scholar]
- Xie P, Guo F, Ma Y, Zhu H, Wang F, Xue B, Shi H, Yang J, Yu L. Intestinal cgi-58 deficiency reduces postprandial lipid absorption. PloS one. 2014;9:e91652. doi: 10.1371/journal.pone.0091652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Xia Y, Hawke D, Li X, Liang J, Xing D, Aldape K, Hunter T, Alfred Yung WK, Lu Z. PKM2 phosphorylates histone H3 and promotes gene transcription and tumorigenesis. Cell. 2012;150:685–696. doi: 10.1016/j.cell.2012.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun H, Lee M, Kim SS, Ha J. Glucose deprivation increases mRNA stability of vascular endothelial growth factor through activation of AMP-activated protein kinase in DU145 prostate carcinoma. The Journal of biological chemistry. 2005;280:9963–9972. doi: 10.1074/jbc.M412994200. [DOI] [PubMed] [Google Scholar]
- Zimmermann R, Strauss JG, Haemmerle G, Schoiswohl G, Birner-Gruenberger R, Riederer M, Lass A, Neuberger G, Eisenhaber F, Hermetter A, Zechner R. Fat mobilization in adipose tissue is promoted by adipose triglyceride lipase. Science. 2004;306:1383–1386. doi: 10.1126/science.1100747. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.