

Abstract
As mediators of the first enzymatic step in glucose metabolism, hexokinases (HKs) orchestrate a variety of catabolic and anabolic uses of glucose, regulate antioxidant power by generating NADPH for glutathione reduction, and modulate cell death processes by directly interacting with the voltage-dependent anion channel (VDAC), a regulatory component of the mitochondrial permeability transition pore (mPTP). Here we summarize the current state-of-knowledge about HKs and their role in protecting the heart from ischemia/reperfusion (I/R) injury, reviewing: 1) the properties of different HK isoforms and how their function is regulated by their subcellular localization; 2) how HKs modulate glucose metabolism and energy production during I/R; 3) the molecular mechanisms by which HKs influence mPTP opening and cellular injury during I/R; 4) how different metabolic and HK profiles correlate with susceptibility to I/R injury and cardioprotective efficacy in cancer cells, neonatal hearts, and normal, hypertrophied and failing adult hearts, and how these difference may guide novel therapeutic strategies to limit I/R injury in the heart.
Introduction
In the heart, ischemic preconditioning (IPC) is a process whereby repeated brief episodes of ischemia/reperfusion (I/R) protect the heart from injury during a subsequent prolonged I/R episode [1]. Although much research has been devoted to this phenomenon and its other variants, including pharmacologic preconditioning (PPC) and ischemic post-conditioning (IPoC), the underlying mechanisms of cardioprotection remain elusive. Common to all of these cardioprotective strategies is activation of a signaling cascade called the Reperfusion Injury Salvage Kinase (RISK) pathway involving phosphatidylinositol-3-kinase (PI3K), Akt, glycogen synthase kinase 3 beta (GSK3β) and other enzymes [2] (Fig. 1). In addition, cardioprotective signaling can also be transmitted via the Survivor Activating Factor Enhancement (SAFE) pathway involving the activation of tumor necrosis factor alpha (TNFα) and signal transducer and activator of transcription-3 (STAT-3) [3-6], which may crosstalk with the RISK pathway. Although the details are still fuzzy, these signals appear to converge ultimately on the mitochondrion [7], and avert cell death by inhibiting mitochondrial permeability transition pore (mPTP) opening in the inner mitochondrial membrane. Of note, activation of the RISK pathway depends on a modest burst of reactive oxygen species (ROS) production during the preconditioning ischemia, because if ROS scavengers are present during this period, cardioprotection is abolished [8]. When the RISK pathway is activated by pre- or post-conditioning, the massive ROS burst that typically occurs after prolonged I/R in the absence of pre- or post-conditioning is markedly attenuated [9]. This may be a major factor in averting the mPTP opening and cell death, since oxidative stress is one of the major factors promoting mPTP opening [10].
Fig. 1.
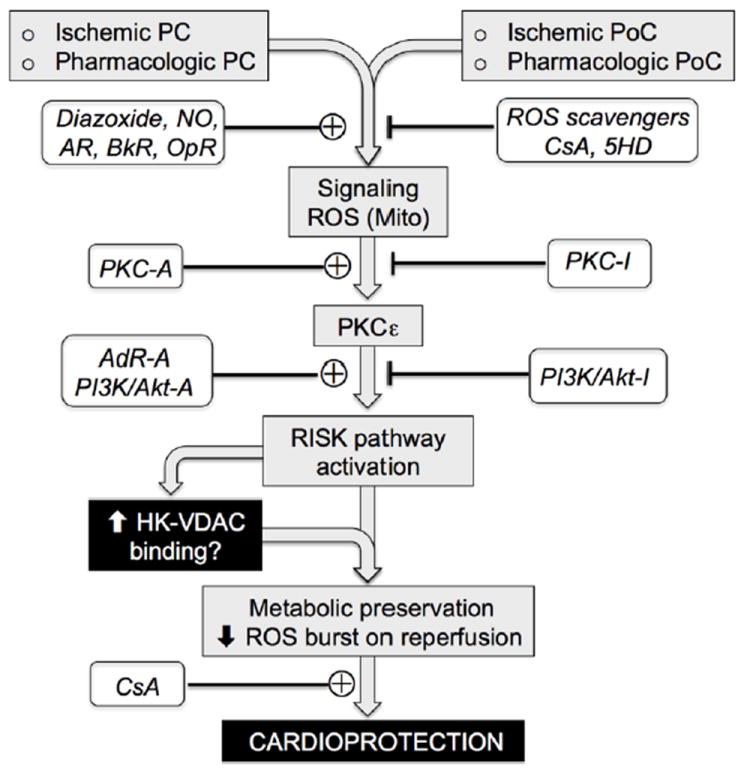
Overview of cardioprotective signaling by ischemic and pharmacologic pre-conditioning (PC) and post-conditioning (PoC). ROS-dependent PKCε activation (often triggered by ROS originating from mitochondria due to mitoKATP channel or transient low conductance mPTP opening) or other pathways such as adenosine receptor (AdR) stimulation lead to activation of the RISK pathway, initiating a signaling cascade that suppresses the massive ROS burst during reperfusion and prevents long-lasting mPTP opening. The Akt component of the RISK pathway enhances HK2 binding to mitochondria, contributing to an improved metabolic profile during prolonged ischemia and reduced ROS burst upon reperfusion characteristic of the cardioprotected state. Various activators (-A) and inhibitors (-I) of these steps are indicated. AR, adrenergic receptors; BkR, bradykinin receptors; OpR, opiate receptors; CsA, cyclosporine A; 5HD, 5-hydroxydecanoate; PI3K, phosphoinositol-3-kinase.
The finding that post-conditioning confers almost equivalent cardioprotection as pre-conditioning suggests that RISK pathway activation at any time point up and during early reperfusion is the main requirement for effective cardioprotection [11]. Once activated, the cardioprotected state has two phases. The early phase, attributed to acute post-translational modification of target proteins by the RISK pathway, lasts about 1-2 hours. Cardioprotection then dissipates, but returns again within 12-24 hours, and lasts for another 48-72 hours. This late phase is related to gene reprogramming and new protein synthesis triggered by RISK pathway activation [10].
How do hexokinases (HKs) fit into this picture? During low-flow ischemia or anoxia, glucose metabolism becomes the major source for ATP production via anaerobic glycolysis, and HKs mediates the first step in this process, namely the conversion of glucose to glucose-6-phosphate (G6P). G6P is a hub metabolite that can be directed to a number of catabolic or anabolic fates (Fig. 2). The main catabolic fate is glycolysis, which first generates ATP anaerobically via conversion to pyruvate and lactate, and then aerobically by mitochondrial oxidation of pyruvate and lactate. The main anabolic fates of G6P are two-fold: glycogen synthesis to store energy for deferred use, and the pentose phosphate shunt to generate ribose-5-phosphate (R5P). The conversion of G6P to R5P is a major source of cytoplasmic NADPH generation which is critical for maintaining antioxidant function by regenerating reduced glutathione (GSH) from oxidized glutathione (GSSG). R5P is also a precursor for synthesis of nucleotides, amino acids and fatty acids. Small amounts of G6P are also used by the hexosamine pathway for O-GlyNAtion of proteins, and some of the G6P directed to pyruvate is used in the TCA cycle for amino acid and fatty acid synthesis via anaplerosis.
Fig. 2.
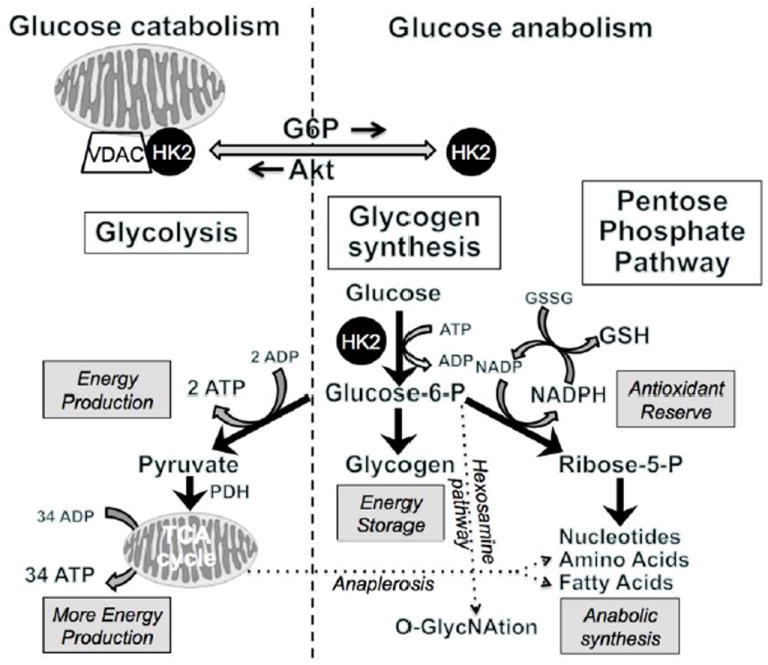
Summary of catabolic, anabolic and antioxidant pathways in glucose metabolism. HK2 binding to mitochondria favors glycolysis, whereas cytoplasmic HK2 favors glycogen synthesis and the pentose phosphate shunt, which provides NADPH to reduce oxidized glutathione (GSSG) to reduced glutathione (GSH) and thereby bolsters antioxidant reserve.
In addition to acting as the gatekeeper for the metabolic and antioxidant roles of glucose, HKs have also been found to regulate mPTP opening directly. First described in the cancer field, the high-affinity HK isoforms HK1 and HK2 are strongly anti-apoptotic when bound to the outer mitochondrial membrane, putatively to the voltage-dependent anion channel (VDAC), an important regulator of the mPTP [12, 13].
Thus, HKs are multifunctional proteins that orchestrate metabolic, antioxidant and direct anti-cell death effects. These functions are also strongly influenced by the subcellular distribution of HKs, with mitochondrially-bound HK promoting glucose catabolism and anti-cell death effects, while cytoplasmic HK promotes anabolic usages and antioxidant effects (Fig. 2). The predominant HK isoform in adult heart, HK2, dynamically shuttles between the mitochondria and cytoplasm in response to changes in intracellular G6P, pH and the cardioprotective signaling pathway Akt [14-22]. These factors set the stage for HKs to have a critical influence on the susceptibility of the heart to I/R injury.
Here we summarize the current state-of-knowledge about HKs and their role in protecting the heart from I/R injury. We begin by discussing the properties of different HK isoforms and how their function is regulated by their subcellular localization. Next, we discuss how HK modulates glucose metabolism and energy production during I/R, and then review the molecular mechanisms by which HKs influence mPTP opening and cellular injury during I/R. Finally, we describe the differences in susceptibility to oxidative or I/R injury in cancer cells, neonatal hearts, and normal, hypertrophied and failing adult hearts in relation to their HK and metabolic profiles, and discuss potential therapeutic implications.
Properties of HK isoforms
HKs are comprised of a family of four isoforms. HKI and HK2 are the most abundant, with HKI (“the brain HK”) ubiquitous in most tissues, especially brain and red blood cells [23, 24] where glycolysis plays a critical role in energy production. In contrast, HK2 (“the muscle HK”) is found primarily in insulin-sensitive tissues such as adipocytes, adult skeletal and cardiac muscle [25, 26]. Few data are available for human heart, but a recent report indicates that in non-dilated human atrial tissue, HKI is the most abundant isoform [27]. In mouse and human skeletal muscle [25, 28], however, HKII accounts for >80% of total HK activity. Importantly, both HKI and HK2 contain a hydrophobic amino terminal mitochondrial binding motif, which is not present in the HK3 or HK4 (glucokinase) isoforms.
The idea that subcellular locations of HKI and HK2 are important in regulating glucose metabolism was first postulated by Wilson, who stated that “the Type I isozyme bound to actively phosphorylating mitochondria facilitates introduction of glucose into glycolysis, with the final stages of glucose metabolism occurring in the mitochondria. In contrast, Type II and to some extent Type III isozymes serve primarily anabolic function to provide G6P for glycogen synthesis or lipid synthesis via the pentose phosphate pathway” (see review [29]). Subsequent work by multiple investigators [14-22, 30, 31] has demonstrated that unlike other HK isoforms, the interaction of HK2 with mitochondria is not static, but is regulated by factors such as glucose, G6P and kinases such as Akt. Thus, a picture has emerged of HK2 as a multifunctional orchestrator of glucose metabolism: channeling G6P into glycogen and the pentose phosphate pathways when localized in the cytoplasm, and preferentially shuttling G6P to glycolysis and oxidative phosphorylation when bound to mitochondria [31, 32]. In contrast, HKI, due to its strong mitochondrial binding, primarily facilitates glycolysis [30, 31], although under some non-physiological conditions may also contribute to glycogen synthesis [33]. HK3 and HK4 are cytoplasmic, since they lack a mitochondrial binding motif, and serve primarily anabolic functions. HK4 (glucokinase), however, also has the ability to shuttle to the nucleus, perhaps playing a role in gene transcription/new protein synthesis.
HKI and HK2 are inhibited allosterically by their product, G6P, and their sensitivity to G6P inhibition decreases when HKs are bound to mitochondria [34, 35]. Physiological levels of orthophosphate (Pi) counter the G6P inhibition of HKI [24, 36, 37], but not HK2. In fact, Pi may cause further HK2 inhibition. Based on these observations, Wilson [29] proposed that “reciprocal changes in intracellular levels of G6P and Pi are closely associated with cellular energy status, and that the response of HK1 to these effectors adapts it for catalytic function by adjusting glucose flow into glycolytic metabolism. In contrast, HK2 serves primarily anabolic functions.”
G6P also plays a critical role in regulating HK2 binding to mitochondria. Elevated levels of intracellular G6P weaken HK2 binding to mitochondria, causing the enzyme to translocate to the cytoplasm and facilitate anabolic glucose metabolism. Acidosis similarly induces HK2 translocation from mitochondria to cytoplasm [38].
Regulation of glucose metabolism by HK during I/R
During acute myocardial ischemia, anaerobic glycolysis and glycogenolysis assume the central role for energy production when oxidative phosphorylation cannot occur because of a lack of oxygen. The shift to anaerobic metabolism entails rapid increases in glucose uptake, glycogenolysis, and glycolytic flux [39]. During severe ischemia, however, the accumulation of protons and glycolytic intermediates eventually inhibits glycolytic flux and anaerobic ATP production ceases after 20-30 min [40, 41]. The relative contribution of the glycolytic/glycogenolytic pathway to energy production is highly dependent on the severity of ischemia, with virtually no change up to moderate ischemia (reduction of coronary flow by <75%) to virtually 100% during total global ischemia. In the moderate ischemia case, glucose uptake remains unchanged, but glucose metabolism is directed from oxidation to lactate production [42]. Increased glucose uptake during ischemia is further stimulated by insulin [43]. Promoting glucose metabolism with glucose and insulin has been used successfully to protect hearts from I/R injury [44, 45], although the clinical utility has been limited [46].
In the adult heart, the subcellular localization of HK2 shifts during ischemia [47] due to the dissociation of HK2, but not HKI, from the mitochondria to cytoplasm [30, 48, 49] in response to intracellular acidification and G6P accumulation [38]. Upon reperfusion after a period of ischemia, the activity of HK is increased in both the cytosolic and mitochondrial compartments [50], probably promoted by activation of Akt signaling via the RISK pathway. Under these conditions of Akt-enhanced HK2 binding to mitochondria, the heart is in a cardioprotected state. After IPC, HK2 dissociates more slowly during a subsequent prolonged ischemic episode, and this correlates with a slower rate of ATP depletion than during ischemia in unprotected hearts [51]. The maintained fraction of HK bound to mitochondria may also serve to facilitate glycolysis during reperfusion, which plays a critical role in recovery [52].
Given the multiple roles of HK in orchestrating metabolic, antioxidant and direct cell death effects from different subcellular locations, it is natural to ask whether the parallels between the cardioprotective status and HK binding to mitochondria are mere associations or are causally-linked. A growing body of evidence now supports a causal relationship. HK binding to mitochondria in cardiomyocytes has been demonstrated to confer profound protection against cell death by preventing mPTP opening [18, 53, 54]. Following prolonged ischemia, cytochrome c release, ROS production and infarct size at the reperfusion have been shown to parallel the amount of HK dissociated from mitochondria [38]. Similar to IPC, several cardioprotective interventions acting through the RISK pathway also increase mitochondrial HK activity and reduce I/R injury in isolated rat hearts [55]. The cardioprotective actions of the volatile anesthetics isoflurane and sevoflurane, whose effects are dependent on the PI3K/Akt signaling [56], have been shown to increase mitochondrial HK activity in in vivo adult rat hearts [57]. Akt has been shown to directly phosphorylate HK2, which inhibits G6P-mediated dissociation from mitochondria and increases cell viability after stress [18, 58]. Finally, disrupting HK2 binding to mitochondria with targeted peptides has been found to increase I/R injury and attenuate cardioprotection by IPC, and genetic reduction of HK2 levels in heterozygous HK2 knock out mice increased susceptibility to I/R injury [59, 60].
Although HK localizes mainly to either the cytosol or mitochondria [29-31] and cardioprotection is associated with increased mitochondrial HK binding, some studies have identified a component of HK located in intracellular vesicles within the cardiomyocyte [48, 61], supporting older data that HK may also be associated with membrane structures other than mitochondria [62]. Although no changes in HK activity seems to occur in this microsomal compartment with ischemia or IPC, HK protein levels in this compartment during ischemia are affected by IPC [48]. It is conceivable that the HK in these microsomes is somehow involved in IPC, but further research will be necessary to elucidate the role, if any.
In the next section, we discuss the possible underlying molecular mechanisms by which HK binding to mitochondria may protect the heart from I/R injury.
Molecular actions of HK that impact mPTP opening during I/R
The mPTP protein complex
The mPTP is a multi-protein complex, whose pore-forming component is now believed to consist of F0-F1 ATP synthase dimers in the inner mitochondrial membrane [63]. The mPTP forms at contact sites between the inner and outer mitochondrial membranes. The other main proteins in the complex, which regulate pore function, include VDAC in the outer mitochondrial membrane, the adenine nucleotide translocator (ANT) in the inner mitochondrial membrane, cyclophilin D, and perhaps the mitochondria phosphate carrier (PiC). Other proteins with regulatory roles include the benzodiazepine receptor, creatine kinase and HK.
Once formed, mPTP can open in either a transient low conductance mode that may be protective [64-66], or a long-lasting high conductance mode that eventually irreversibly damages the mitochondria and promotes cell death signaling. The transient low conductance openings may be important for allowing mitochondrial to release accumulated matrix Ca by transiently depolarizing, thereby avoiding matrix Ca overload leading to long-lasting mPTP. Transient mPTP openings are also associated with a burst of superoxide production [67] that may contribute to the ROS-dependent triggering of the RISK pathway during IPC (Fig. 1), since blocking transient mPTP opening during IPC with CsA has been shown to abolish cardioprotection [68], similar to the effect of ROS scavengers administered during IPC [8].
Long-lasting mPTP opening, on the other hand, leads to: 1) an abrupt complete dissipation of the mitochondrial membrane potential, which converts mitochondria from ATP generators into ATP consumers as F0-F1 ATP synthase hydrolyzes ATP to pump protons out of the matrix in a futile effort to restore membrane potential; 2) loss of solutes with molecular masses <1.5 kDa, including key metabolic co-factors such as pyridine nucleotides essential for oxidative phosphorylation; 3) influx of water due to the high oncotic pressure of the matrix that swells the mitochondrial inner membrane and can eventually rupture the outer mitochondrial membrane rupture, releasing pro-apoptotic proteins such a cytochrome c [69, 70].
It has been shown that both HKI and HK2 interact with VDAC in the outer mitochondrial membrane [20, 71-73], and that this requires the presence of their N-terminal hydrophobic domain [74, 75]. The interaction of HK with mitochondria is known to protect against cell death in many cell types [76-78] and mPTP opening is inhibited by the interaction with HK2 [69]. Other proteins in the mPTP complex influence HK binding to VDAC. Inhibition of ANT with bonkregic acid, which locks the ANT in a closed state and is an mPTP inhibitor, reduced HKI and HK2 binding to isolated mitochondria, while treatment with atractyloside, which locks the ANT in an open conformation and is an mPTP inducer, increased HK binding [79]. Inhibition of CypD with the mPTP blocker cyclosporine A (CsA) also decreased HK2 binding to mitochondria, as did siRNA knockdown of CypD [80].
Since VDAC is both a component of the mPTP multiprotein complex and a key regulator of mitochondrial bioenergetics, its interaction with HK raises the following question: are the protective effects of mitochondrial HK binding against mPTP opening a direct effect on mPTP function, an indirect effect mediated by the metabolic effects of HK when bound to VDAC, or both? The evidence to date is limited but suggests that both direct (nonmetabolic) and indirect (metabolic) factors are important. For example, studies have shown that glucose must be present in order for mitochondrially-bound HK to offer protection against cell death [16, 53, 81], implying that enzymatic HK activity to generate G6P is critical. On the other hand, experiments overexpressing HK2 mutants lacking enzymatic activity, but with intact binding to mitochondria, conferred partial protection against H2O2-induced injury in cultured cardiomyocytes [18, 48, 53]. Conversely, HK2 mutants with intact enzymatic activity, but no mitochondrial binding, also exhibited partial protection, suggesting that cytoplasmic HK2 may confer some benefit as well. Whether this is also true in the setting of I/R injury, however, remains to be established.
Cytoprotective effects of HK during I/R
There are at least five mechanisms by which HK binding to VDAC may exert cytoprotective effects during I/R that avert long-lasting mPTP opening (Fig. 3 and see [26] for review). The first mechanism is a direct (nonmetabolic) action and the remaining four are indirect (metabolic) actions. They are:
Fig. 3.
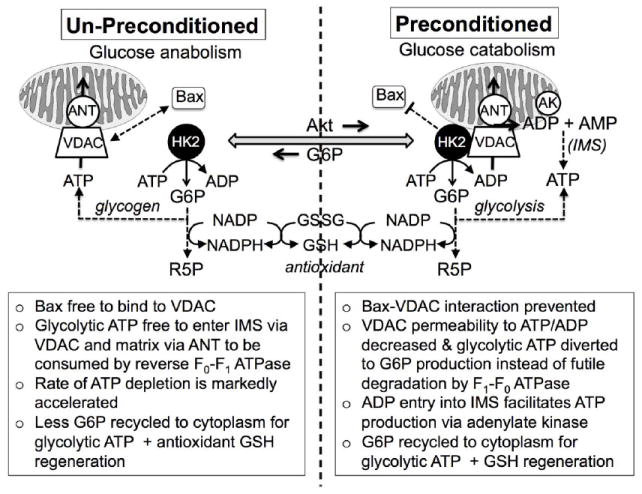
Summary of possible mechanisms by which enhanced HK2 binding to VDAC in the preconditioned heart improves the metabolic profile and averts mPTP opening during I/R. IMS, intermembrane space; ANT, adenine nucleotide transporter; AK, adenylate kinase
Interference with translocation of pro-apoptotic proteins (Bax, Bad) to VDAC. In cellular studies employing oxidant stress, HK binding to mitochondria has been shown to be cytoprotective by structurally interfering with the ability of pro-apoptotic proteins such as Bax and Bad to translocate from the cytoplasm and bind to VDAC [82]. However, the relevance to I/R injury is unclear, since studies performed in isolated hearts [38, 59] or skeletal muscle [83] have not been able to show a correlation between HK-mediated protection and Bax/Bak levels at the mitochondria in the setting of I/R. Bax binding to VDAC is implicated in the formation of a megachannel in the outer mitochondrial membrane through which large molecules, including cytochrome c, can pass [84, 85]. Release of cytochrome c into the cytoplasm is a major trigger of the apoptotic signaling cascade, which feeds back to induce long-lasted mPTP opening [86]. In addition, cytochrome c plays an important antioxidant role [82], and its loss from the intermembrane space leaves mitochondria more susceptible to oxidative stress-induced mPTP opening during reperfusion.
Reduction in futile ATP consumption by reverse F0-F1 ATPase. When bound to VDAC under aerobic conditions, HK utilizes ATP to generate G6P in close proximity to the outer mitochondrial membrane, facilitating its glycolytic conversion to pyruvate as a substrate for oxidative phosphorylation. During ischemia, however, the lack of oxygen inhibits electron transport and the matrix rapidly depolarizes. Pyruvate can no longer be oxidized and is converted to lactate. G6P as well as other glycolytic intermediates accumulate intracellularly. Under these conditions, matrix F0-F1 ATPase operates in reverse, consuming ATP to pump protons in a futile attempt to restore membrane potential. Studies using F0-F1 ATPase inhibitors such as oligomycin show that this futile ATP consumption accounts for as much as 50-70% cellular ATP depletion during early ischemia [87, 88]. In this setting, VDAC acts as the critical gatekeeper for glycolytically-generated ATP to enter the IMS, where it is then transported by ANT into matrix to be consumed in this futile cycle. ATP and ADP can readily permeate the outer mitochondrial membrane when VDAC is in its open state, but are virtually impermeant when VDAC is closed [89]. It has been shown that HK binding promotes the closed state of VDAC, reducing the permeability to adenine nucleotides [90]. Under de-energized conditions, slowing of adenine nucleotide transport in and out of the mitochondria by inhibiting VDAC opening significantly attenuates myocardial ischemia-reperfusion injury [91]. Thus, one cytoprotective effect of HK binding to VDAC may be to reduce VDAC’s permeability to adenine nucleotides and thereby restrict the entry of glycolytic ATP into the intermembrane space and subsequently into the matrix via ANT, resulting in less futile ATP degradation by reverse F0-F1 ATPase. In addition, even when the VDAD pore is open, HK depletes local [ATP] in its immediate vicinity by dephosphorylating ATP to ADP, thereby favoring the entry of ADP, rather than ATP, into the IMS when the VDAC pore is open. This further reduces futile ATP consumption by reverse F0-F1 ATPase.
Facilitation of ATP production by adenylate kinase in the IMS. By hydrolyzing glycolytic ATP to ADP near the VDAC pore, HK facilitates entry of ADP, instead of ATP, into the IMS. The preferential transport of ADP into the IMS stimulates ATP generation by adenylate kinase, which catalyzes the reaction reaction 2ADP ↔ AMP + ATP. The regenerated ATP can then exit to IMS via VDAC to support cytoplasmic bioenergetic functions. Thus glycolytic ATP that would have been futilely consumed by reverse F0-F1 ATPase in the matrix is diverted to ATP regeneration by adenylate kinase.
Facilitation of glycolytic ATP generation. A fourth cytoprotective effect relates to the fact that in the process of dephosphorylating ATP to ADP before it can enter the IMS through VDAC, HK generates additional G6P, which is recycled to support additional glycolytic ATP generation.
Facilitation of antioxidant power by the pentose phosphate shunt. Alternatively, the G6P generated by mitochondrially-bound HK can be remain in the cytoplasm and be utilized by the pentose phosphate shunt to produce NADPH for regenerating GSH from GSSG, thereby enhancing antioxidant reserve [92].
The combination of these five factors may explain why IPC, by activating Akt and enhancing HK binding to VDAC, slows the rate of ATP decline during the subsequent prolonged ischemic episode [93], and also attenuates the massive burst of ROS that occurs upon reperfusion after prolonged ischemia by bolstering cytochrome c- and GSH-related antioxidant power. The combination of reduced Bax/Bad interaction with VDAC, less futile ATP consumption by reverse F0-F1 ATPase, enhanced adenylate kinase-mediated ATP generation in the IMS, and increased G6P production to facilitate both glycolytic ATP generation and antioxidant power via the pentose phosphate shunt may allow the heart to preserve a better metabolic profile during ischemia, and be better prepared to limit oxidative injury following reperfusion. In combination, these actions may avert long-lasted mPTP opening and limit cell death in pre-conditioned hearts (Fig. 3).
HK and metabolic profiles in cancer, developing hearts, and normal and diseased adult hearts
Differences in susceptibility to I/R injury and efficacy of IPC in various cells types in relation to their metabolic profiles may provide additional clues about the role of HK in cardioprotection. Specifically, cancer cells exhibit Warburg-like metabolic profiles characterized by enhanced anaerobic glycolysis, reduced glucose oxidation, upregulated glycolytic enzymes (including HK) and down-regulated oxidative phosphorylation enzymes, elements of which are shared by neonatal hearts, and hypertrophied or failing adult hearts (Table 1). Neonatal hearts, like cancer cells, are intrinsically less susceptible to I/R injury, and are not further protected by IPC like normal adult hearts [94]. Hypertrophied and failing adult hearts, on the other hand, show some elements of the Warburg metabolic profile, but are not necessarily less susceptible to I/R injury or as effectively cardioprotected by IPC compared to normal adult hearts [95-98]. Here we review the relationship between susceptibility to I/R injury, efficacy of IPC and the metabolic profiles in these different cell types.
Table 1.
Summary of HK isoforms, metabolic profiles, susceptibility to I/R injury and ability to protect by IPC in different settings.
| Tissue | Predominant HK isoform | Metabolic Profile | Susceptibility to I/R injury | Protection by IPC | ||
|---|---|---|---|---|---|---|
| Glycolysis | Glu Ox | FA Ox | ||||
| Adult heart | HK2 | Low | Low | High | High | High |
| Cancer cells | HK2 | High | Low | Low | Low | ? |
| Fetal/Neonatal heart | HK1 | High | High | Low | Low | Low |
| Hypertrophied adult heart | HK2 | High | Low | Low | High | High |
| Failing adult heart | HK2 | Low | Low | Low | Low? | Low? |
| Diabetic adult heart | HK1 | Low | Low | High | Low (short term diabetes) | Low |
| High (long term diabetes) | ||||||
FA= fatty acid; Glu=glucose; Ox=oxidation
Cancer
In 1956, Otto Warburg observed that the rate of anaerobic glycolysis was abnormally high in cancer cells, despite a smaller fraction of this glucose being oxidized by mitochondria due to a preferential channeling of pyruvate to lactate. This ‘Warburg effect’ indicates that cancer cells prefer anaerobic glycolysis for energy production, rather than mitochondrial oxidative phosphorylation of either glucose or fatty acids [99, 100]. The preference for anaerobic energy production by glycolysis may be important for allowing tumor cells to proliferate when O2 supply is limited by the absence of a well-developed vascular supply,and also confers increased resistance to apoptosis.
HK plays a pivotal role in this metabolic shift and increased resistance to cell death, with the activity of HK2 and to some extent HKI being markedly elevated in rapidly growing tumors which exhibit high rates of glycolysis [71, 101-103]. Two factors contribute to this enhanced activity: increased expression of HK’s by as much as 100-fold, and a propensity for the HKs to bind to VDAC in the outer mitochondrial membrane in tumor cells [12, 103, 104]. Mitochondrial binding of the tumor HKs has been shown to provide the HK with preferential access to mitochondrially-generated ATP to phosphorylate glucose, [71] and to reduce its sensitivity to product inhibition by G6P [105], an important regulator of HK activity in normal cells [20, 105]. Secondly, HK binding to VDAC is integral to the protection against apoptosis in cancer cells by inhibiting cytochrome c release and mPTP opening [13], as described earlier.
The Warburg-like pattern of increased HK activity and redistribution to mitochondria is not solely the hallmark of rapidly growing cancer cells, but is also observed in other tissues including brain, skeletal muscle and heart at various developmental stages in health and disease.
Fetal and neonatal heart
An increase in HKI expression/activity signals a switch from pyruvate to glucose metabolism in the preimplantation embryo [106, 107], and HKI activity increases significantly in the morula and blastocyst stages [108-110]. From this point through the early perinatal period, cardiac energy metabolism relies heavily on glucose metabolism, facilitated by the continuous availability of glucose from the mother to the fetus through the placental circulation. Mitochondrial oxidative metabolism is poorly developed, with glycolysis as the primary energy source [111-114]. This high glycolytic profile seems to facilitate the proliferative state of developing fetal cardiomyocytes [111, 113, 114]. Even after birth, as mitochondrial metabolism matures, glycolysis is still the preferred energetic pathway for several weeks, driven in part by expression of insulin-independent isoforms of the glucose transporter (GLUT1) and HKI, with its strong binding to mitochondria. Thus, HKI is the predominant isoform in embryonic, fetal and neonatal hearts [115] facilitating their high glycolytic rate [30]. After birth, the transition from a continuous supply of placental glucose to an intermittent mixed carbohydrate-fat oral diet during nursing, induces a switch to the insulin-dependent isoforms GLUT4 and HK2 [30, 116], along with down-regulation of other glycolytic enzymes and upregulation of oxidative phosphorylation enzymes [117]. These changes confer a preference for fatty acids, lactate and ketone bodies over glucose in the adult heart.
The Warburg-like metabolic profile in the developing heart is associated with an increased resistance to ischemic/hypoxic injury as compared to adult myocardium [118-120]. This greater resistance to cell death may involve both the increased capacity of anaerobic glycolysis to maintain ATP and the stronger binding of HKI than HK2 to mitochondria, which suppresses mPTP opening [121]. Indeed, whereas in adult hearts, strengthening of HK2 binding to mitochondria is essential for the protective effects of IPC [59], this is not the case for neonatal hearts. In neonatal hearts, the inherently stronger binding of HKI to mitochondria is not further increased by IPC, and the intrinsically high resistance of the newborn heart to I/R injury also cannot be further increased by IPC or adaptation to chronic hypoxia [122]. Neonatal cardiac mitochondria also have been shown to be less sensitive to mPTP opening [123], further strengthening the idea that the high affinity binding of HKI to mitochondria in neonatal heart may contribute to the increased resistance to I/R injury.
Cardiac hypertrophy
Under normal aerobic conditions, the adult heart prefers fatty acid oxidation for energy production. Anaerobic glycolysis accounts for only about 5% of total ATP generation [124], and pyruvate and lactate oxidation less than 40%. Even when no exogenous fatty acids are provided to an isolated working heart, fatty acids derived from breakdown of endogenous lipids still account for substantial ATP production [117]. Cardiac hypertrophy induces a switch to increased anaerobic glucose utilization, with reduced or unchanged glucose oxidation and reduced fatty acid oxidation, i.e. a reappearance of the fetal Warburg-like metabolic pattern [98, 125, 126]. This metabolic change is attributed to both transcriptional down-regulation of oxidative phosphorylation genes (by master regulatory genes such as PGC-1α) and up-regulation of glucose metabolism genes (by master regulatory genes such as HIF-1 α), and by post-translational activation of AMP-activated protein kinase (AMPK), which increases glucose uptake and stimulates glycolysis. Glucose oxidation does not increase commensurately, however, since AMPK activation also reduces pyruvate oxidation by phosphorylating pyruvate dehydrogenase kinase (PDK), which inhibits pyruvate dehydrogenase (PDH).
In addition to glycolysis, other glucose metabolic pathways including the pentose phosphate pathway, the hexosamine biosynthesis pathway and anaplerosis are also altered in hypertrophied and failing adult hearts [127] [128].
As a key enzyme in glucose metabolism, HK plays an important role in the metabolic switch during hypertrophy [129]. Increased levels and activity of HK have been measured in hypertrophied hearts. [130] Moreover, it was recently shown in transgenic mouse models that overexpressing HK2 attenuated the response to a hypertrophic stimulus, [92] and, conversely, knocking down HK2 increased susceptibility [131]. HK’s role in promoting antioxidant function via the pentose phosphate pathway (Fig. 2) was proposed as the mechanism [92]. Despite the conversion to a metabolic profile resembling the neonatal pattern, however, hypertrophied adult hearts do not in general exhibit reduced susceptibility to I/R injury, and in most, but not all studies, can still be protected by IPC [95-97].
Heart failure
In the failing heart, the metabolic changes are more variable [128], influenced by both the pathogenesis and stage of heart failure at which the metabolic profile has been characterized. Both glycolytic and oxidative phosphorylation genes are usually down-regulated in severe heart failure, and glucose oxidation is reduced. However, fatty acid oxidation can be either increased or decreased (for review, see [98]). Thus, unlike the Warburg pattern characteristic of cancer, neonatal and hypertrophied adult hearts, failing adult hearts exhibit a spectrum of changes in which both energy production and energy transfer are impaired, described as an “energy-starved” profile [132]. Pharmacologic interventions that increase glucose oxidation (even when concomitantly inhibiting fatty acid oxidation) have been shown to be beneficial in failing hearts [133-141].
HK levels in heart failure are variable, and, instead of increased, may be unchanged [132] or reduced [142]. Nevertheless, some studies have reported that failing hearts are intrinsically less susceptible to I/R injury and less protected by IPC, resembling maximally cardioprotected neonatal hearts [143]. Whether HK overexpression is beneficial to the failing adult hearts is unknown at the present time, but is an important issue, since therapies to prevent I/R injury are highly clinically relevant to the human heart failure population.
Diabetic heart
Development of diabetes is associated with a shift in cardiac metabolism away from glucose metabolism towards fatty acid oxidation. This metabolic remodeling is accompanied by a significant decrease in cardiac HKII, but not HKI, protein content [62]. In addition, the diabetic heart displays an altered response to I/R and IPC, with short-term diabetes frequently offering protection against I/R but an attenuated response to IPC, whereas long-term diabetes results in both worsened outcome after I/R and loss of IPC protective effects [144]. Changes in HK may play a role in these altered responses of diabetic hearts to I/R and IPC. Indeed, altered cardiac HK cellular trafficking has been shown in the short-term type I diabetic heart [145], with the major difference in HK cellular trafficking between healthy and diabetic hearts occurring at early reperfusion after an IPC episode. Further research will be necessary to elucidate the role of HKs in diabetic cardiomyopathy.
Summary, conclusions and therapeutic implications
HKs are powerful orchestrators of glucose metabolism, determining whether glucose is directed catabolically to glycolysis, or anabolically to glycogen synthesis, the pentose phosphate shunt, where it can be utilized to increase antioxidant power and synthesize nucleotides, amino acids or fatty acids, or the hexosamine pathway mediating O-GlycNAtion signaling. The HK2 isoform which predominates in adult heart actively translocates between mitochondria and the cytoplasm to facilitate these catabolic or anabolic functions, respectively. When bound to VDAC in the outer mitochondrial membrane, HK1 and HK2 are highly protective against I/R injury by suppressing long-lasted mPTP openings. This protective effect of HK binding to VDAC can be explained by a combination of direct (nonmetabolic) actions which inhibit mPTP pore opening, and indirect (metabolic) effects which limit futile ATP consumption by mitochondria, enhance glycolytic ATP production by glycolysis and adenylate kinase, and increase antioxidant power. In the heart, IPC strengthens HK2 binding to mitochondria possibly via Akt signaling, a component of the RISK pathway, thereby increasing resistance to mPTP opening and cell death upon reperfusion. Disrupting HK binding to mitochondria with peptides has been shown to increase I/R injury and attenuate cardioprotection by IPC, and heterozygous HK2 knock out mice show increased susceptibility to I/R injury.
Based on the scope of direct anti-cell death and favorable metabolic actions of HK’s during I/R, it is intriguing to speculate that enhanced HK binding to VDAC may be one of the long sought after end-targets of cardioprotective signaling. If so, then strategies to strengthen the HK-VDAC interaction may have therapeutic promise. Overexpression of HK2 has been shown to protect cultured neonatal myocytes from oxidative injury [53], but similar experiments have yet to be carried out in adult hearts. HK1 overexpression may be particularly promising, since HK1 remains bound to mitochondria and does not normally translocate to the cytoplasm like HK2. HK1 is also the predominant isoform in neonatal hearts, which are highly resistant to I/R injury, probably because they are already maximally cardioprotected. Cancer cells predominantly upregulate HK2 in massive amounts which confers cytoprotective effects. In hypertrophied adult hearts, however, the more modest upregulation of HK2 in concert with other glycolytic enzymes does not appear to confer increased resistance to I/R injury as in cancer cells. The difference may be that in cancer cells, basal Akt activity is typically high, thereby reinforcing HK2 binding to mitochondria. This is consistent with the finding that hypertrophied hearts can still be cardioprotected by IPC, suggesting that HK2 binding to mitochondria is not maximized. It should now be possible to explore these issues and the therapeutic potential of HK-based gene delivery techniques in adult hearts, by comparing the ability of HK1 and HK2, and corresponding HK mutants which lack either enzymatic activity or the mitochondrial binding motif, to protect against I/R injury.
Highlights.
Hexokinases orchestrate glucose metabolism, antioxidant function and cell death
These functions are dynamically regulated by hexokinase’s subcellular localization
Hexokinase expression is modified in a variety of disease states
Hexokinase may be a key target of cardioprotective signaling in heart
Acknowledgments
Funding sources
This work was supported by the NIH/NHLBI grants R01 HL117385, HL101228, American Heart Association Western States Affiliate Post-doctoral Research Fellowship 11POST6110007, American Heart Association Western States Affliliate Grant in Aid 13GRNT14650074 and the Laubisch and Kawata endowments.
Footnotes
Disclosures
Guillaume Calmettes: None. Scott John: None. Paavo Korge: None. Peipei Ping: None. Bernard Ribalet: None; James N. Weiss, None.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Bibliography
- 1.Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation. 1986;74:1124–36. doi: 10.1161/01.cir.74.5.1124. [DOI] [PubMed] [Google Scholar]
- 2.Murphy E, Steenbergen C. Preconditioning: the mitochondrial connection. Annu Rev Physiol. 2007;69:51–67. doi: 10.1146/annurev.physiol.69.031905.163645. [DOI] [PubMed] [Google Scholar]
- 3.Lacerda L, Somers S, Opie LH, Lecour S. Ischaemic postconditioning protects against reperfusion injury via the SAFE pathway. Cardiovasc Res. 2009;84:201–8. doi: 10.1093/cvr/cvp274. [DOI] [PubMed] [Google Scholar]
- 4.Lecour S. Activation of the protective Survivor Activating Factor Enhancement (SAFE) pathway against reperfusion injury: Does it go beyond the RISK pathway? J Mol Cell Cardiol. 2009;47:32–40. doi: 10.1016/j.yjmcc.2009.03.019. [DOI] [PubMed] [Google Scholar]
- 5.Lecour S. Multiple protective pathways against reperfusion injury: a SAFE path without Aktion? J Mol Cell Cardiol. 2009;46:607–9. doi: 10.1016/j.yjmcc.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 6.Tamareille S, Mateus V, Ghaboura N, Jeanneteau J, Croue A, Henrion D, et al. RISK and SAFE signaling pathway interactions in remote limb ischemic perconditioning in combination with local ischemic postconditioning. Basic Res Cardiol. 2011;106:1329–39. doi: 10.1007/s00395-011-0210-z. [DOI] [PubMed] [Google Scholar]
- 7.Murphy E. Primary and secondary signaling pathways in early preconditioning that converge on the mitochondria to produce cardioprotection. Circ Res. 2004;94:7–16. doi: 10.1161/01.RES.0000108082.76667.F4. [DOI] [PubMed] [Google Scholar]
- 8.Penna C, Mancardi D, Rastaldo R, Pagliaro P. Cardioprotection: a radical view Free radicals in pre and postconditioning. Biochim Biophys Acta. 2009;1787:781–93. doi: 10.1016/j.bbabio.2009.02.008. [DOI] [PubMed] [Google Scholar]
- 9.Vanden Hoek T, Becker LB, Shao ZH, Li CQ, Schumacker PT. Preconditioning in cardiomyocytes protects by attenuating oxidant stress at reperfusion. Circ Res. 2000;86:541–8. doi: 10.1161/01.res.86.5.541. [DOI] [PubMed] [Google Scholar]
- 10.Weiss JN, Korge P, Honda HM, Ping P. Role of the mitochondrial permeability transition in myocardial disease. Circ Res. 2003;93:292–301. doi: 10.1161/01.RES.0000087542.26971.D4. [DOI] [PubMed] [Google Scholar]
- 11.Hausenloy DJ, Tsang A, Mocanu MM, Yellon DM. Ischemic preconditioning protects by activating prosurvival kinases at reperfusion. Am J Physiol Heart Circ Physiol. 2005;288:H971–6. doi: 10.1152/ajpheart.00374.2004. [DOI] [PubMed] [Google Scholar]
- 12.Mathupala SP, Ko YH, Pedersen PL. Hexokinase II: cancer’s double-edged sword acting as both facilitator and gatekeeper of malignancy when bound to mitochondria. Oncogene. 2006;25:4777–86. doi: 10.1038/sj.onc.1209603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shoshan-Barmatz V, Zakar M, Rosenthal K, Abu-Hamad S. Key regions of VDAC1 functioning in apoptosis induction and regulation by hexokinase. Biochim Biophys Acta. 2009;1787:421–30. doi: 10.1016/j.bbabio.2008.11.009. [DOI] [PubMed] [Google Scholar]
- 14.Galluzzi L, Kepp O, Tajeddine N, Kroemer G. Disruption of the hexokinase-VDAC complex for tumor therapy. Oncogene. 2008;27:4633–5. doi: 10.1038/onc.2008.114. [DOI] [PubMed] [Google Scholar]
- 15.Gimenez-Cassina A, Lim F, Cerrato T, Palomo GM, Diaz-Nido J. Mitochondrial hexokinase II promotes neuronal survival and acts downstream of glycogen synthase kinase-3. J Biol Chem. 2009;284:3001–11. doi: 10.1074/jbc.M808698200. [DOI] [PubMed] [Google Scholar]
- 16.Gottlob K, Majewski N, Kennedy S, Kandel E, Robey RB, Hay N. Inhibition of early apoptotic events by Akt/PKB is dependent on the first committed step of glycolysis and mitochondrial hexokinase. Genes Dev. 2001;15:1406–18. doi: 10.1101/gad.889901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Majewski N, Nogueira V, Robey RB, Hay N. Akt inhibits apoptosis downstream of BID cleavage via a glucose-dependent mechanism involving mitochondrial hexokinases. Mol Cell Biol. 2004;24:730–40. doi: 10.1128/MCB.24.2.730-740.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Miyamoto S, Murphy AN, Brown JH. Akt mediates mitochondrial protection in cardiomyocytes through phosphorylation of mitochondrial hexokinase-II. Cell Death Differ. 2008;15:521–9. doi: 10.1038/sj.cdd.4402285. [DOI] [PubMed] [Google Scholar]
- 19.Pastorino JG, Hoek JB, Shulga N. Activation of glycogen synthase kinase 3beta disrupts the binding of hexokinase II to mitochondria by phosphorylating voltage-dependent anion channel and potentiates chemotherapy-induced cytotoxicity. Cancer Res. 2005;65:10545–54. doi: 10.1158/0008-5472.CAN-05-1925. [DOI] [PubMed] [Google Scholar]
- 20.Rose IA, Warms JV. Mitochondrial hexokinase. Release, rebinding, and location. J Biol Chem. 1967;242:1635–45. [PubMed] [Google Scholar]
- 21.Skaff DA, Kim CS, Tsai HJ, Honzatko RB, Fromm HJ. Glucose 6-phosphate release of wild-type and mutant human brain hexokinases from mitochondria. J Biol Chem. 2005;280:38403–9. doi: 10.1074/jbc.M506943200. [DOI] [PubMed] [Google Scholar]
- 22.Sui D, Wilson JE. Structural determinants for the intracellular localization of the isozymes of mammalian hexokinase: intracellular localization of fusion constructs incorporating structural elements from the hexokinase isozymes and the green fluorescent protein. Arch Biochem Biophys. 1997;345:111–25. doi: 10.1006/abbi.1997.0241. [DOI] [PubMed] [Google Scholar]
- 23.Lowry OH, Passonneau JV. The Relationships between Substrates and Enzymes of Glycolysis in Brain. J Biol Chem. 1964;239:31–42. [PubMed] [Google Scholar]
- 24.Purich DL, Fromm HJ. The kinetics and regulation of rat brain hexokinase. J Biol Chem. 1971;246:3456–63. [PubMed] [Google Scholar]
- 25.Mandarino LJ, Printz RL, Cusi KA, Kinchington P, O’Doherty RM, Osawa H, et al. Regulation of hexokinase II and glycogen synthase mRNA, protein, and activity in human muscle. Am J Physiol. 1995;269:E701–8. doi: 10.1152/ajpendo.1995.269.4.E701. [DOI] [PubMed] [Google Scholar]
- 26.Nederlof R, Eerbeek O, Hollmann MW, Southworth R, Zuurbier CJ. Targeting hexokinase II to mitochondria to modulate energy metabolism and reduce ischaemia-reperfusion injury in heart. Br J Pharmacol. 2014;171:2067–79. doi: 10.1111/bph.12363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Roosimaa M, Podramagi T, Kadaja L, Ruusalepp A, Paju K, Puhke R, et al. Dilation of human atria: increased diffusion restrictions for ADP, overexpression of hexokinase 2 and its coupling to oxidative phosphorylation in cardiomyocytes. Mitochondrion. 2013;13:399–409. doi: 10.1016/j.mito.2012.12.005. [DOI] [PubMed] [Google Scholar]
- 28.Smeele KM, ter Horst LH, Koeman A, Heikkinen S, Laakso M, Weber NC, et al. The effect of standard chow and reduced hexokinase II on growth, cardiac and skeletal muscle hexokinase and low-flow cardiac ischaemia-reperfusion injury. Lab Anim. 2011;45:160–6. doi: 10.1258/la.2011.010096. [DOI] [PubMed] [Google Scholar]
- 29.Wilson JE. Isozymes of mammalian hexokinase: structure, subcellular localization and metabolic function. J Exp Biol. 2003;206:2049–57. doi: 10.1242/jeb.00241. [DOI] [PubMed] [Google Scholar]
- 30.Calmettes G, John SA, Weiss JN, Ribalet B. Hexokinase-mitochondrial interactions regulate glucose metabolism differentially in adult and neonatal cardiac myocytes. J Gen Physiol. 2013;142:425–36. doi: 10.1085/jgp.201310968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.John S, Weiss JN, Ribalet B. Subcellular localization of hexokinases I and II directs the metabolic fate of glucose. PLoS One. 2011;6:e17674. doi: 10.1371/journal.pone.0017674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jurczak MJ, Danos AM, Rehrmann VR, Brady MJ. The role of protein translocation in the regulation of glycogen metabolism. J Cell Biochem. 2008;104:435–43. doi: 10.1002/jcb.21634. [DOI] [PubMed] [Google Scholar]
- 33.Cifuentes D, Martinez-Pons C, Garcia-Rocha M, Galina A, Ribas de Pouplana L, Guinovart JJ. Hepatic glycogen synthesis in the absence of glucokinase: the case of embryonic liver. J Biol Chem. 2008;283:5642–9. doi: 10.1074/jbc.M706334200. [DOI] [PubMed] [Google Scholar]
- 34.Azoulay-Zohar H, Israelson A, Abu-Hamad S, Shoshan-Barmatz V. In self-defence: hexokinase promotes voltage-dependent anion channel closure and prevents mitochondria-mediated apoptotic cell death. Biochem J. 2004;377:347–55. doi: 10.1042/BJ20031465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.de Cerqueira Cesar M, Wilson JE. Functional characteristics of hexokinase bound to the type a and type B sites of bovine brain mitochondria. Arch Biochem Biophys. 2002;397:106–12. doi: 10.1006/abbi.2001.2639. [DOI] [PubMed] [Google Scholar]
- 36.Ellison WR, Lueck JD, Fromm HJ. Studies on the mechanism of orthophosphate regulation of bovine brain hexokinase. J Biol Chem. 1975;250:1864–71. [PubMed] [Google Scholar]
- 37.Fang TY, Alechina O, Aleshin AE, Fromm HJ, Honzatko RB. Identification of a phosphate regulatory site and a low affinity binding site for glucose 6-phosphate in the N-terminal half of human brain hexokinase. J Biol Chem. 1998;273:19548–53. doi: 10.1074/jbc.273.31.19548. [DOI] [PubMed] [Google Scholar]
- 38.Pasdois P, Parker JE, Halestrap AP. Extent of mitochondrial hexokinase II dissociation during ischemia correlates with mitochondrial cytochrome c release, reactive oxygen species production, and infarct size on reperfusion. J Am Heart Assoc. 2013;2:e005645. doi: 10.1161/JAHA.112.005645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Morgan HE, Henderson MJ, Regen DM, Park CR. Regulation of glucose uptake in muscle. I. The effects of insulin and anoxia on glucose transport and phosphorylation in the isolated, perfused heart of normal rats. J Biol Chem. 1961;236:253–61. [PubMed] [Google Scholar]
- 40.Neely JR, Whitmer JT, Rovetto MJ. Effect of coronary blood flow on glycolytic flux and intracellular pH in isolated rat hearts. Circ Res. 1975;37:733–41. doi: 10.1161/01.res.37.6.733. [DOI] [PubMed] [Google Scholar]
- 41.Rovetto MJ, Lamberton WF, Neely JR. Mechanisms of glycolytic inhibition in ischemic rat hearts. Circ Res. 1975;37:742–51. doi: 10.1161/01.res.37.6.742. [DOI] [PubMed] [Google Scholar]
- 42.Bolukoglu H, Goodwin GW, Guthrie PH, Carmical SG, Chen TM, Taegtmeyer H. Metabolic fate of glucose in reversible low-flow ischemia of the isolated working rat heart. Am J Physiol. 1996;270:H817–26. doi: 10.1152/ajpheart.1996.270.3.H817. [DOI] [PubMed] [Google Scholar]
- 43.Chen TM, Goodwin GW, Guthrie PH, Taegtmeyer H. Effects of insulin on glucose uptake by rat hearts during and after coronary flow reduction. Am J Physiol. 1997;273:H2170–7. doi: 10.1152/ajpheart.1997.273.5.H2170. [DOI] [PubMed] [Google Scholar]
- 44.Oldfield GS, Commerford PJ, Opie LH. Effects of preoperative glucose-insulin-potassium on myocardial glycogen levels and on complications of mitral valve replacement. J Thorac Cardiovasc Surg. 1986;91:874–8. [PubMed] [Google Scholar]
- 45.Opie LH. Glucose and the metabolism of ischaemic myocardium. Lancet. 1995;345:1520–1. doi: 10.1016/s0140-6736(95)91080-8. [DOI] [PubMed] [Google Scholar]
- 46.Kloner RA. Current state of clinical translation of cardioprotective agents for acute myocardial infarction. Circ Res. 2013;113:451–63. doi: 10.1161/CIRCRESAHA.112.300627. [DOI] [PubMed] [Google Scholar]
- 47.Correa F, Garcia N, Gallardo-Perez J, Carreno-Fuentes L, Rodriguez-Enriquez S, Marin-Hernandez A, et al. Post-conditioning preserves glycolytic ATP during early reperfusion: a survival mechanism for the reperfused heart. Cell Physiol Biochem. 2008;22:635–44. doi: 10.1159/000185547. [DOI] [PubMed] [Google Scholar]
- 48.Gurel E, Smeele KM, Eerbeek O, Koeman A, Demirci C, Hollmann MW, et al. Ischemic preconditioning affects hexokinase activity and HKII in different subcellular compartments throughout cardiac ischemia-reperfusion. J Appl Physiol (1985) 2009;106:1909–16. doi: 10.1152/japplphysiol.90537.2008. [DOI] [PubMed] [Google Scholar]
- 49.Pasdois P, Parker JE, Griffiths EJ, Halestrap AP. The role of oxidized cytochrome c in regulating mitochondrial reactive oxygen species production and its perturbation in ischaemia. Biochem J. 2011;436:493–505. doi: 10.1042/BJ20101957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yeih DF, Yeh HI, Lin LY, Tsay YG, Chiang FT, Tseng CD, et al. Enhanced activity and subcellular redistribution of myocardial hexokinase after acute myocardial infarction. Int J Cardiol. 2011;149:74–9. doi: 10.1016/j.ijcard.2009.12.002. [DOI] [PubMed] [Google Scholar]
- 51.Murry CE, Richard VJ, Reimer KA, Jennings RB. Ischemic preconditioning slows energy metabolism and delays ultrastructural damage during a sustained ischemic episode. Circ Res. 1990;66:913–31. doi: 10.1161/01.res.66.4.913. [DOI] [PubMed] [Google Scholar]
- 52.Jeremy RW, Ambrosio G, Pike MM, Jacobus WE, Becker LC. The functional recovery of post-ischemic myocardium requires glycolysis during early reperfusion. JMolCell Cardiol. 1993;25:261–76. doi: 10.1006/jmcc.1993.1033. [DOI] [PubMed] [Google Scholar]
- 53.Sun L, Shukair S, Naik TJ, Moazed F, Ardehali H. Glucose phosphorylation and mitochondrial binding are required for the protective effects of hexokinases I and II. Mol Cell Biol. 2008;28:1007–17. doi: 10.1128/MCB.00224-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chiara F, Castellaro D, Marin O, Petronilli V, Brusilow WS, Juhaszova M, et al. Hexokinase II detachment from mitochondria triggers apoptosis through the permeability transition pore independent of voltage-dependent anion channels. PLoS One. 2008;3:e1852. doi: 10.1371/journal.pone.0001852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zuurbier CJ, Eerbeek O, Meijer AJ. Ischemic preconditioning, insulin, and morphine all cause hexokinase redistribution. Am J Physiol Heart Circ Physiol. 2005;289:H496–9. doi: 10.1152/ajpheart.01182.2004. [DOI] [PubMed] [Google Scholar]
- 56.Raphael J, Rivo J, Gozal Y. Isoflurane-induced myocardial preconditioning is dependent on phosphatidylinositol-3-kinase/Akt signalling. Br J Anaesth. 2005;95:756–63. doi: 10.1093/bja/aei264. [DOI] [PubMed] [Google Scholar]
- 57.Zuurbier CJ, Keijzers PJ, Koeman A, Van Wezel HB, Hollmann MW. Anesthesia’s effects on plasma glucose and insulin and cardiac hexokinase at similar hemodynamics and without major surgical stress in fed rats. Anesth Analg. 2008;106:135–42. doi: 10.1213/01.ane.0000297299.91527.74. table of contents. [DOI] [PubMed] [Google Scholar]
- 58.Roberts DJ, Tan-Sah VP, Smith JM, Miyamoto S. Akt phosphorylates HK-II at Thr-473 and increases mitochondrial HK-II association to protect cardiomyocytes. J Biol Chem. 2013;288:23798–806. doi: 10.1074/jbc.M113.482026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Smeele KM, Southworth R, Wu R, Xie C, Nederlof R, Warley A, et al. Disruption of hexokinase II-mitochondrial binding blocks ischemic preconditioning and causes rapid cardiac necrosis. Circ Res. 2011;108:1165–9. doi: 10.1161/CIRCRESAHA.111.244962. [DOI] [PubMed] [Google Scholar]
- 60.Wu R, Smeele KM, Wyatt E, Ichikawa Y, Eerbeek O, Sun L, et al. Reduction in hexokinase II levels results in decreased cardiac function and altered remodeling after ischemia/reperfusion injury. Circ Res. 2011;108:60–9. doi: 10.1161/CIRCRESAHA.110.223115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Southworth R, Davey KA, Warley A, Garlick PB. A reevaluation of the roles of hexokinase I and II in the heart. Am J Physiol Heart Circ Physiol. 2007;292:H378–86. doi: 10.1152/ajpheart.00664.2006. [DOI] [PubMed] [Google Scholar]
- 62.Katzen HM, Soderman DD, Wiley CE. Multiple forms of hexokinase. Activities associated with subcellular particulate and soluble fractions of normal and streptozotocin diabetic rat tissues. J Biol Chem. 1970;245:4081–96. [PubMed] [Google Scholar]
- 63.Giorgio V, von Stockum S, Antoniel M, Fabbro A, Fogolari F, Forte M, et al. Dimers of mitochondrial ATP synthase form the permeability transition pore. Proc Natl Acad Sci U S A. 2013;110:5887–92. doi: 10.1073/pnas.1217823110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ichas F, Mazat J-P. From calcium signaling to cell death: two conformations for the mitochondrial permeability transition pore. Switching from low- to high-conductance state. Biochimica et Biophysica Acta (BBA) - Bioenergetics. 1998;1366:33–50. doi: 10.1016/s0005-2728(98)00119-4. [DOI] [PubMed] [Google Scholar]
- 65.Ichas F, Jouaville LS, Mazat JP. Mitochondria are excitable organelles capable of generating and conveying electrical and calcium signals. Cell. 1997;89:1145–53. doi: 10.1016/s0092-8674(00)80301-3. [DOI] [PubMed] [Google Scholar]
- 66.Korge P, Yang L, Yang JH, Wang Y, Qu Z, Weiss JN. Protective role of transient pore openings in calcium handling by cardiac mitochondria. J Biol Chem. 2011;286:34851–7. doi: 10.1074/jbc.M111.239921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wang W, Fang H, Groom L, Cheng A, Zhang W, Liu J, et al. Superoxide flashes in single mitochondria. Cell. 2008;134:279–90. doi: 10.1016/j.cell.2008.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hausenloy DJ, Ong SB, Yellon DM. The mitochondrial permeability transition pore as a target for preconditioning and postconditioning. Basic Res Cardiol. 2009;104:189–202. doi: 10.1007/s00395-009-0010-x. [DOI] [PubMed] [Google Scholar]
- 69.Halestrap AP. What is the mitochondrial permeability transition pore? J Mol Cell Cardiol. 2009;46:821–31. doi: 10.1016/j.yjmcc.2009.02.021. [DOI] [PubMed] [Google Scholar]
- 70.Bernardi P. Mitochondrial transport of cations: channels, exchangers, and permeability transition. Physiol Rev. 1999;79:1127–55. doi: 10.1152/physrev.1999.79.4.1127. [DOI] [PubMed] [Google Scholar]
- 71.Arora KK, Pedersen PL. Functional significance of mitochondrial bound hexokinase in tumor cell metabolism. Evidence for preferential phosphorylation of glucose by intramitochondrially generated ATP. J Biol Chem. 1988;263:17422–8. [PubMed] [Google Scholar]
- 72.Majewski N, Nogueira V, Bhaskar P, Coy PE, Skeen JE, Gottlob K, et al. Hexokinase-mitochondria interaction mediated by Akt is required to inhibit apoptosis in the presence or absence of Bax and Bak. Mol Cell. 2004;16:819–30. doi: 10.1016/j.molcel.2004.11.014. [DOI] [PubMed] [Google Scholar]
- 73.Pastorino JG, Hoek JB. Regulation of hexokinase binding to VDAC. J Bioenerg Biomembr. 2008;40:171–82. doi: 10.1007/s10863-008-9148-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Azoulay-Zohar H, Aflalo C. Binding of rat brain hexokinase to recombinant yeast mitochondria: identification of necessary molecular determinants. J Bioenerg Biomembr. 1999;31:569–79. doi: 10.1023/a:1005469028274. [DOI] [PubMed] [Google Scholar]
- 75.Gelb BD, Adams V, Jones SN, Griffin LD, MacGregor GR, McCabe ER. Targeting of hexokinase 1 to liver and hepatoma mitochondria. Proc Natl Acad Sci U S A. 1992;89:202–6. doi: 10.1073/pnas.89.1.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Robey RB, Hay N. Mitochondrial hexokinases: guardians of the mitochondria. Cell Cycle. 2005;4:654–8. doi: 10.4161/cc.4.5.1678. [DOI] [PubMed] [Google Scholar]
- 77.Zuurbier CJ, Smeele KM, Eerbeek O. Mitochondrial hexokinase and cardioprotection of the intact heart. J Bioenerg Biomembr. 2009;41:181–5. doi: 10.1007/s10863-009-9209-7. [DOI] [PubMed] [Google Scholar]
- 78.Southworth R. Hexokinase-mitochondrial interaction in cardiac tissue: implications for cardiac glucose uptake, the 18FDG lumped constant and cardiac protection. J Bioenerg Biomembr. 2009;41:187–93. doi: 10.1007/s10863-009-9207-9. [DOI] [PubMed] [Google Scholar]
- 79.Vyssokikh M, Zorova L, Zorov D, Heimlich G, Jurgensmeier J, Schreiner D, et al. The intra-mitochondrial cytochrome c distribution varies correlated to the formation of a complex between VDAC and the adenine nucleotide translocase: this affects Bax-dependent cytochrome c release. Biochim Biophys Acta. 2004;1644:27–36. doi: 10.1016/j.bbamcr.2003.10.007. [DOI] [PubMed] [Google Scholar]
- 80.Machida K, Ohta Y, Osada H. Suppression of apoptosis by cyclophilin D via stabilization of hexokinase II mitochondrial binding in cancer cells. J Biol Chem. 2006;281:14314–20. doi: 10.1074/jbc.M513297200. [DOI] [PubMed] [Google Scholar]
- 81.Mergenthaler P, Kahl A, Kamitz A, van Laak V, Stohlmann K, Thomsen S, et al. Mitochondrial hexokinase II (HKII) and phosphoprotein enriched in astrocytes (PEA15) form a molecular switch governing cellular fate depending on the metabolic state. Proc Natl Acad Sci U S A. 2012;109:1518–23. doi: 10.1073/pnas.1108225109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Pastorino JG, Shulga N, Hoek JB. Mitochondrial binding of hexokinase II inhibits Bax-induced cytochrome c release and apoptosis. J Biol Chem. 2002;277:7610–8. doi: 10.1074/jbc.M109950200. [DOI] [PubMed] [Google Scholar]
- 83.Smeele KM, Eerbeek O, Schaart G, Koeman A, Bezemer R, Nelson JK, et al. Reduced hexokinase II impairs muscle function 2 wk after ischemia-reperfusion through increased cell necrosis and fibrosis. J Appl Physiol (1985) 2012113:608–18. doi: 10.1152/japplphysiol.01494.2011. [DOI] [PubMed] [Google Scholar]
- 84.Shimizu S, Narita M, Tsujimoto Y. Bcl-2 family proteins regulate the release of apoptogenic cytochrome c by the mitochondrial channel VDAC. Nature. 1999;399:483–7. doi: 10.1038/20959. [DOI] [PubMed] [Google Scholar]
- 85.Eskes R, Antonsson B, Osen-Sand A, Montessuit S, Richter C, Sadoul R, et al. Bax-induced cytochrome C release from mitochondria is independent of the permeability transition pore but highly dependent on Mg2+ ions. J Cell Biol. 1998;143:217–24. doi: 10.1083/jcb.143.1.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Jiang X, Wang X. Cytochrome C-mediated apoptosis. Annu Rev Biochem. 2004;73:87–106. doi: 10.1146/annurev.biochem.73.011303.073706. [DOI] [PubMed] [Google Scholar]
- 87.St-Pierre J, Brand MD, Boutilier RG. Mitochondria as ATP consumers: cellular treason in anoxia. Proc Natl Acad Sci U S A. 2000;97:8670–4. doi: 10.1073/pnas.140093597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Grover GJ, Atwal KS, Sleph PG, Wang FL, Monshizadegan H, Monticello T, et al. Excessive ATP hydrolysis in ischemic myocardium by mitochondrial F1F0-ATPase: effect of selective pharmacological inhibition of mitochondrial ATPase hydrolase activity. Am J Physiol Heart Circ Physiol. 2004;287:H1747–55. doi: 10.1152/ajpheart.01019.2003. [DOI] [PubMed] [Google Scholar]
- 89.Steenbergen C, Das S, Su J, Wong R, Murphy E. Cardioprotection and altered mitochondrial adenine nucleotide transport. Basic Res Cardiol. 2009;104:149–56. doi: 10.1007/s00395-009-0002-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Perevoshchikova IV, Zorov SD, Kotova EA, Zorov DB, Antonenko YN. Hexokinase inhibits flux of fluorescently labeled ATP through mitochondrial outer membrane porin. FEBS Lett. 2010;584:2397–402. doi: 10.1016/j.febslet.2010.04.033. [DOI] [PubMed] [Google Scholar]
- 91.Imahashi K, Schneider MD, Steenbergen C, Murphy E. Transgenic expression of Bcl-2 modulates energy metabolism, prevents cytosolic acidification during ischemia, and reduces ischemia/reperfusion injury. Circ Res. 2004;95:734–41. doi: 10.1161/01.RES.0000143898.67182.4c. [DOI] [PubMed] [Google Scholar]
- 92.McCommis KS, Douglas DL, Krenz M, Baines CP. Cardiac-specific hexokinase 2 overexpression attenuates hypertrophy by increasing pentose phosphate pathway flux. J Am Heart Assoc. 2013;2:e000355. doi: 10.1161/JAHA.113.000355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Murry CE, Richard VJ, Reimer KA, Jennings RB. Ischemic preconditioning slows energy metabolism and delays ultrastructual damage during a sustained ischemic episode. Circulation Research. 1990;66:913–31. doi: 10.1161/01.res.66.4.913. [DOI] [PubMed] [Google Scholar]
- 94.Ostadalova I, Ostadal B, Kolar F, Parratt JR, Wilson S. Tolerance to ischaemia and ischaemic preconditioning in neonatal rat heart. J Mol Cell Cardiol. 1998;30:857–65. doi: 10.1006/jmcc.1998.0653. [DOI] [PubMed] [Google Scholar]
- 95.Pantos CI, Davos CH, Carageorgiou HC, Varonos DV, Cokkinos DV. Ischaemic preconditioning protects against myocardial dysfunction caused by ischaemia in isolated hypertrophied rat hearts. Basic Res Cardiol. 1996;91:444–9. doi: 10.1007/BF00788725. [DOI] [PubMed] [Google Scholar]
- 96.Andersen A, Povlsen JA, Botker HE, Nielsen-Kudsk JE. Right ventricular hypertrophy and failure abolish cardioprotection by ischaemic pre-conditioning. Eur J Heart Fail. 2013;15:1208–14. doi: 10.1093/eurjhf/hft105. [DOI] [PubMed] [Google Scholar]
- 97.Ghosh S, Standen NB, Galinianes M. Failure to precondition pathological human myocardium. J Am Coll Cardiol. 2001;37:711–8. doi: 10.1016/s0735-1097(00)01161-x. [DOI] [PubMed] [Google Scholar]
- 98.van Bilsen M, van Nieuwenhoven FA, van der Vusse GJ. Metabolic remodelling of the failing heart: beneficial or detrimental? Cardiovasc Res. 2009;81:420–8. doi: 10.1093/cvr/cvn282. [DOI] [PubMed] [Google Scholar]
- 99.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–33. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Warburg O. On respiratory impairment in cancer cells. Science. 1956;124:269–70. [PubMed] [Google Scholar]
- 101.Pedersen PL. Tumor mitochondria and the bioenergetics of cancer cells. Prog Exp Tumor Res. 1978;22:190–274. doi: 10.1159/000401202. [DOI] [PubMed] [Google Scholar]
- 102.Bustamante E, Morris HP, Pedersen PL. Energy metabolism of tumor cells. Requirement for a form of hexokinase with a propensity for mitochondrial binding. J Biol Chem. 1981;256:8699–704. [PubMed] [Google Scholar]
- 103.Parry DM, Pedersen PL. Intracellular localization and properties of particulate hexokinase in the Novikoff ascites tumor. Evidence for an outer mitochondrial membrane location. J Biol Chem. 1983;258:10904–12. [PubMed] [Google Scholar]
- 104.Pedersen PL. Warburg, me and Hexokinase 2: Multiple discoveries of key molecular events underlying one of cancers’ most common phenotypes, the “Warburg Effect”, i.e., elevated glycolysis in the presence of oxygen. J Bioenerg Biomembr. 2007;39:211–22. doi: 10.1007/s10863-007-9094-x. [DOI] [PubMed] [Google Scholar]
- 105.Bustamante E, Pedersen PL. High aerobic glycolysis of rat hepatoma cells in culture: role of mitochondrial hexokinase. Proc Natl Acad Sci U S A. 1977;74:3735–9. doi: 10.1073/pnas.74.9.3735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Brinster RL. Hexokinase activity in the preimplantation mouse embryo. Enzymologia. 1968;34:304–8. [PubMed] [Google Scholar]
- 107.Gardner DK, Leese HJ. The role of glucose and pyruvate transport in regulating nutrient utilization by preimplantation mouse embryos. Development. 1988;104:423–9. doi: 10.1242/dev.104.3.423. [DOI] [PubMed] [Google Scholar]
- 108.Ayabe T, Tsutsumi O, Taketani Y. Hexokinase activity in mouse embryos developed in vivo and in vitro. Hum Reprod. 1994;9:347–51. doi: 10.1093/oxfordjournals.humrep.a138506. [DOI] [PubMed] [Google Scholar]
- 109.Johnson MD, Batey DW, Behr B. Quantification of hexokinase mRNA in mouse blastocysts by competitive reverse transcriptase polymerase chain reaction. Mol Hum Reprod. 1997;3:359–65. doi: 10.1093/molehr/3.4.359. [DOI] [PubMed] [Google Scholar]
- 110.Saito T, Hiroi M, Kato T. Development of glucose utilization studied in single oocytes and preimplantation embryos from mice. Biol Reprod. 1994;50:266–70. doi: 10.1095/biolreprod50.2.266. [DOI] [PubMed] [Google Scholar]
- 111.Cho YM, Kwon S, Pak YK, Seol HW, Choi YM, Park do J, et al. Dynamic changes in mitochondrial biogenesis and antioxidant enzymes during the spontaneous differentiation of human embryonic stem cells. Biochem Biophys Res Commun. 2006;348:1472–8. doi: 10.1016/j.bbrc.2006.08.020. [DOI] [PubMed] [Google Scholar]
- 112.Chung S, Arrell DK, Faustino RS, Terzic A, Dzeja PP. Glycolytic network restructuring integral to the energetics of embryonic stem cell cardiac differentiation. J Mol Cell Cardiol. 2010;48:725–34. doi: 10.1016/j.yjmcc.2009.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Chung S, Dzeja PP, Faustino RS, Perez-Terzic C, Behfar A, Terzic A. Mitochondrial oxidative metabolism is required for the cardiac differentiation of stem cells. Nat Clin Pract Cardiovasc Med. 2007;4(Suppl 1):S60–7. doi: 10.1038/ncpcardio0766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.St John JC, Ramalho-Santos J, Gray HL, Petrosko P, Rawe VY, Navara CS, et al. The expression of mitochondrial DNA transcription factors during early cardiomyocyte in vitro differentiation from human embryonic stem cells. Cloning Stem Cells. 2005;7:141–53. doi: 10.1089/clo.2005.7.141. [DOI] [PubMed] [Google Scholar]
- 115.Fritz HL, Smoak IW, Branch S. Hexokinase I expression and activity in embryonic mouse heart during early and late organogenesis. Histochem Cell Biol. 1999;112:359–65. doi: 10.1007/s004180050417. [DOI] [PubMed] [Google Scholar]
- 116.Depre C, Vanoverschelde JL, Taegtmeyer H. Glucose for the heart. Circulation. 1999;99:578–88. doi: 10.1161/01.cir.99.4.578. [DOI] [PubMed] [Google Scholar]
- 117.Lopaschuk GD, Jaswal JS. Energy metabolic phenotype of the cardiomyocyte during development, differentiation, and postnatal maturation. J Cardiovasc Pharmacol. 2010;56:130–40. doi: 10.1097/FJC.0b013e3181e74a14. [DOI] [PubMed] [Google Scholar]
- 118.Coles JG, Boscarino C, Takahashi M, Grant D, Chang A, Ritter J, et al. Cardioprotective stress response in the human fetal heart. J Thorac Cardiovasc Surg. 2005;129:1128–36. doi: 10.1016/j.jtcvs.2004.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Grice WN, Konishi T, Apstein CS. Resistance of neonatal myocardium to injury during normothermic and hypothermic ischemic arrest and reperfusion. Circulation. 1987;76:V150–5. [PubMed] [Google Scholar]
- 120.Ostadal B, Ostadalova I, Dhalla NS. Development of cardiac sensitivity to oxygen deficiency: comparative and ontogenetic aspects. Physiol Rev. 1999;79:635–59. doi: 10.1152/physrev.1999.79.3.635. [DOI] [PubMed] [Google Scholar]
- 121.Miura T, Tanno M. The mPTP and its regulatory proteins: final common targets of signalling pathways for protection against necrosis. Cardiovasc Res. 2012;94:181–9. doi: 10.1093/cvr/cvr302. [DOI] [PubMed] [Google Scholar]
- 122.Ostadalova I, Ostadal B, Jarkovska D, Kolar F. Ischemic preconditioning in chronically hypoxic neonatal rat heart. Pediatr Res. 2002;52:561–7. doi: 10.1203/00006450-200210000-00016. [DOI] [PubMed] [Google Scholar]
- 123.Milerova M, Charvatova Z, Skarka L, Ostadalova I, Drahota Z, Fialova M, et al. Neonatal cardiac mitochondria and ischemia/reperfusion injury. Mol Cell Biochem. 2010;335:147–53. doi: 10.1007/s11010-009-0251-x. [DOI] [PubMed] [Google Scholar]
- 124.Neely JR, Rovetto MJ, Oram JF. Myocardial utilization of carbohydrate and lipids. Prog Cardiovasc Dis. 1972;15:289–329. doi: 10.1016/0033-0620(72)90029-1. [DOI] [PubMed] [Google Scholar]
- 125.Kolwicz SC, Jr, Tian R. Glucose metabolism and cardiac hypertrophy. Cardiovasc Res. 2011;90:194–201. doi: 10.1093/cvr/cvr071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Bishop SP, Altschuld RA. Increased glycolytic metabolism in cardiac hypertrophy and congestive failure. Am J Physiol. 1970;218:153–9. doi: 10.1152/ajplegacy.1970.218.1.153. [DOI] [PubMed] [Google Scholar]
- 127.Stanley WC, Recchia FA, Lopaschuk GD. Myocardial substrate metabolism in the normal and failing heart. Physiol Rev. 2005;85:1093–129. doi: 10.1152/physrev.00006.2004. [DOI] [PubMed] [Google Scholar]
- 128.Doenst T, Nguyen TD, Abel ED. Cardiac metabolism in heart failure: implications beyond ATP production. Circ Res. 2013;113:709–24. doi: 10.1161/CIRCRESAHA.113.300376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Liang Q, Donthi RV, Kralik PM, Epstein PN. Elevated hexokinase increases cardiac glycolysis in transgenic mice. Cardiovasc Res. 2002;53:423–30. doi: 10.1016/s0008-6363(01)00495-3. [DOI] [PubMed] [Google Scholar]
- 130.Koehler U, Medugorac I. Left ventricular enzyme activities of the energy-supplying metabolism in Goldblatt-II rats. Res Exp Med (Berl) 1985;185:299–307. doi: 10.1007/BF01851955. [DOI] [PubMed] [Google Scholar]
- 131.Wu R, Wyatt E, Chawla K, Tran M, Ghanefar M, Laakso M, et al. Hexokinase II knockdown results in exaggerated cardiac hypertrophy via increased ROS production. EMBO Mol Med. 2012;4:633–46. doi: 10.1002/emmm.201200240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Dzeja PP, Pucar D, Redfield MM, Burnett JC, Terzic A. Reduced activity of enzymes coupling ATP-generating with ATP-consuming processes in the failing myocardium. Mol Cell Biochem. 1999;201:33–40. doi: 10.1023/a:1007016703229. [DOI] [PubMed] [Google Scholar]
- 133.Broderick TL, Quinney HA, Barker CC, Lopaschuk GD. Beneficial effect of carnitine on mechanical recovery of rat hearts reperfused after a transient period of global ischemia is accompanied by a stimulation of glucose oxidation. Circulation. 1993;87:972–81. doi: 10.1161/01.cir.87.3.972. [DOI] [PubMed] [Google Scholar]
- 134.Kantor PF, Lucien A, Kozak R, Lopaschuk GD. The antianginal drug trimetazidine shifts cardiac energy metabolism from fatty acid oxidation to glucose oxidation by inhibiting mitochondrial long-chain 3-ketoacyl coenzyme A thiolase. Circ Res. 2000;86:580–8. doi: 10.1161/01.res.86.5.580. [DOI] [PubMed] [Google Scholar]
- 135.Lee L, Campbell R, Scheuermann-Freestone M, Taylor R, Gunaruwan P, Williams L, et al. Metabolic modulation with perhexiline in chronic heart failure: a randomized, controlled trial of short-term use of a novel treatment. Circulation. 2005;112:3280–8. doi: 10.1161/CIRCULATIONAHA.105.551457. [DOI] [PubMed] [Google Scholar]
- 136.Liu B, Clanachan AS, Schulz R, Lopaschuk GD. Cardiac efficiency is improved after ischemia by altering both the source and fate of protons. Circ Res. 1996;79:940–8. doi: 10.1161/01.res.79.5.940. [DOI] [PubMed] [Google Scholar]
- 137.Liu Q, Docherty JC, Rendell JC, Clanachan AS, Lopaschuk GD. High levels of fatty acids delay the recovery of intracellular pH and cardiac efficiency in post-ischemic hearts by inhibiting glucose oxidation. J Am Coll Cardiol. 2002;39:718–25. doi: 10.1016/s0735-1097(01)01803-4. [DOI] [PubMed] [Google Scholar]
- 138.Lopaschuk GD, Barr R, Thomas PD, Dyck JR. Beneficial effects of trimetazidine in ex vivo working ischemic hearts are due to a stimulation of glucose oxidation secondary to inhibition of long-chain 3-ketoacyl coenzyme a thiolase. Circ Res. 2003;93:e33–7. doi: 10.1161/01.RES.0000086964.07404.A5. [DOI] [PubMed] [Google Scholar]
- 139.Lopaschuk GD, Wall SR, Olley PM, Davies NJ. Etomoxir, a carnitine palmitoyltransferase I inhibitor, protects hearts from fatty acid-induced ischemic injury independent of changes in long chain acylcarnitine. Circ Res. 1988;63:1036–43. doi: 10.1161/01.res.63.6.1036. [DOI] [PubMed] [Google Scholar]
- 140.McVeigh JJ, Lopaschuk GD. Dichloroacetate stimulation of glucose oxidation improves recovery of ischemic rat hearts. Am J Physiol. 1990;259:H1079–85. doi: 10.1152/ajpheart.1990.259.4.H1079. [DOI] [PubMed] [Google Scholar]
- 141.Schonekess BO, Allard MF, Lopaschuk GD. Propionyl L-carnitine improvement of hypertrophied heart function is accompanied by an increase in carbohydrate oxidation. Circ Res. 1995;77:726–34. doi: 10.1161/01.res.77.4.726. [DOI] [PubMed] [Google Scholar]
- 142.Lionetti V, Aquaro GD, Simioniuc A, Di Cristofano C, Forini F, Cecchetti F, et al. Severe mechanical dyssynchrony causes regional hibernation-like changes in pigs with nonischemic heart failure. J Card Fail. 2009;15:920–8. doi: 10.1016/j.cardfail.2009.06.436. [DOI] [PubMed] [Google Scholar]
- 143.Adb-Efattah AS, Guo J-H, Gao S-H. Failing hearts are preconditioned against myocardial infarction and exhausted their ischemic preconditioning reserve. FASEB Journal. 2011;25:1033.23. [Google Scholar]
- 144.Miki T, Itoh T, Sunaga D, Miura T. Effects of diabetes on myocardial infarct size and cardioprotection by preconditioning and postconditioning. Cardiovasc Diabetol. 2012;11:67. doi: 10.1186/1475-2840-11-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Gurel E, Ustunova S, Kapucu A, Yilmazer N, Eerbeek O, Nederlof R, et al. Hexokinase cellular trafficking in ischemia-reperfusion and ischemic preconditioning is altered in type I diabetic heart. Mol Biol Rep. 2013;40:4153–60. doi: 10.1007/s11033-013-2495-5. [DOI] [PubMed] [Google Scholar]