

Abstract
In current orthopaedic practice, there is a need to increase the ability to reconstruct large segments of bone lost due to trauma, resection of tumors and skeletal deformities, or when normal regenerative processes have failed such as in non-unions and avascular necrosis. Bone marrow stromal cells (BMSCs, also known as bone marrow-derived mesenchymal stem cells), when used in conjunction with appropriate carriers, represent a means by which to achieve bone regeneration in such cases. While much has been done at the bench and in pre-clinical studies, moving towards clinical application requires the generation of clinical grade cells. What is described herein is an FDA-approved cell manufacturing procedure for the ex vivo expansion of high quality, biologically active human BMSCs.
Keywords: Bone marrow stromal cells, Good manufacturing practices, Cell Factories, Bioreactor
Background
The need for improved clinical procedures to enhance bone regeneration continues to grow as the average age of the population increases, resulting in a dramatic increase in fracture rate that may ultimately result in non-union, or when regenerative processes fail such as in avascular necrosis. Furthermore, healing of large bone defects caused by trauma or surgical resection of tumors often cannot be achieved due to an inadequate supply of autologous bone graft, the current gold standard. While numerous “bone fillers” are on the market, the extent to which they actually promote new bone formation is not known in many cases. Consequently, there is a real demand to develop therapies that will improve upon current clinical practice to restore form and function, and thus, the quality of life to patients suffering from skeletal defects.
Tissue engineering is currently thought of as the use of cells, scaffolds and factors, either singly or in various combinations. Although small bone defects may heal on their own with casting or other orthopaedic procedures, or by treatment with various different factors (e.g., platelet rich plasma), it is apparent that a combination of cells with an appropriate carrier is needed to successfully tackle large bone defects. While a long list of cell types have been proposed as being useful for bone regeneration, bone marrow stromal cells (also known as bone marrow-derived mesenchymal stem cells) are currently at the top of the list, due to their unique biological properties and inherent osteogenicity [1].
Based on the pioneering studies of Friedenstein and coworkers [2] and others (reviewed in [3]), it is now well established that bone marrow contains a type of non-hematopoietic stem cell that is a component of the bone marrow stromal cell (BMSC) population. These cells rapidly adhere to plastic and proliferate extensively in vitro. When populations of ex vivo-expanded BMSCs are transplanted in vivo with an appropriate carrier, a bone/marrow organ is formed, composed of bone with identifiable osteocytes, rimmed with active osteoblasts, hematopoiesis-supportive stroma and marrow adipocytes, all of donor origin, and hematopoietic cells of recipient origin [4, 5]. These multipotent cells arise from rare clonogenic BMSCs that are found on the subluminal surfaces of bone marrow sinusoids, otherwise known as pericytes, and are able to self-renew as was established via serial transplantation assays of clonogenic cells in vivo [6]. With the documentation of a bona fide stem cell (a skeletal stem cell, SSC) within the population, BMSCs are an attractive cell source for bone regeneration due to their ability to support bone turnover, as is required throughout life. SSCs/BMSCs generate osteogenic progenitors, and in addition, they also support hematopoiesis (one of their defining characteristics) and osteoclast formation, and lastly, the BMSC population contains the self-renewing SSC necessary for bone turnover.
SSCs/BMSCs and cells with similar characteristics derived from other connective tissues (collectively known as “mesenchymal stem cells”) are currently being used in clinical trials not only for bone regeneration, but for the treatment of non-skeletal diseases and disorders (see clinicaltrials.gov). However, the vast majority of these trials are not related to bone regeneration by the cells themselves, but rather to the so-called paracrine, immunomodulatory and immunosuppressive effects that these cells purportedly exert. These later effects have not been pinpointed to the subset of SSCs within the BMSC population, but to the population as a whole [7]. On the contrary, regeneration of a bone/marrow organ is dependent on SSCs. While more mature osteogenic cells may be used to generate bone, the ability for bone turnover to occur is greatly diminished in the absence of SSCs [1]. Due to the rarity of SSCs/BMSCs in bone marrow, insufficient numbers of cells can be isolated through the use of a variety of cell sorting strategies for direct use in bone regeneration. Ex vivo expansion is required. Thus, maintenance of the subset of the SSCs within the BMSC population is of high importance during the process of ex vivo expansion [1, 7]
We, along with others around the world, have established a facility for the generation of GMP-compliant SSC/BMSC populations (The NIH BMSC Transplantation Center). The mission of the Center is to develop clinical grade BMSCs that maintain their biological activities, and to provide clinical investigators with support to generate required regulatory documents [IRB approved clinical protocols; FDA approved Investigational New Drug applications, INDs)]. In doing so, we developed a Drug Master File and several INDs in compliance with the Code of Federal Regulation for Food and Drugs (21 CFR 600 – Biologics, http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?CFRPart=600) that address the issues of identity, purity, potency, efficacy, stability, sterility and safety. Below is a general description of process by which we have generated autologous cells that maintain their essential biological activities. Autologous cells have been generated for treatment of cardiovascular disease, but have not yet been utilized for bone regeneration. The Center has also generated allogeneic cells for use in clinical protocols for treatment of acute Graft vs. Host Disease and inflammatory bowel disease. Very specific details on the manufacturing procedure can be found in Sabatino, et al. [8], and Stroncek, et al. [9]. There is no difference in the processing of autologous versus allogeneic cells. The major difference is related to donor eligibility for generation of allogeneic cells.
Methods
Donors
Due to the fact that upon differentiation, SSCs/BMSCs express histocompatibility antigens, it is clear that cells used for persistent bone regeneration must be autologous. The required screening and inclusion criteria of autologous donors are far less onerous than what is required for related or third party allogeneic donors. Autologous donors are consented prior to screening. In our donor evaluation and marrow acquisition protocol (NIH #10-CC-0053, NCT01071577), autologous donors must be over the age of 18 yrs and able to give informed consent. They must have generally normal clinical parameters, and females must be proven to not be pregnant. Autologous donors are excluded if their medical history shows blood disorders or abnormalities, prolonged bleeding, and if testing is positive for active tuberculosis infection, HIV1/2, HBV, HCV, HTVL 1/2, T. Cruzi, HBsAg, syphilis, and West Nile virus (this testing is as required by the Standards for Blood Banks and Transfusion Services, American Association of Blood Banks). If at the time of marrow collection, they are pregnant, febrile, and display abnormal cardiac parameters, they may also be excluded or deferred.
Bone marrow collection
Procurement of starting material for ex vivo expansion in our Center is by bone marrow aspiration (unilateral or bilateral, 12 mls maximum per side collected in heparinized tubes). The quality of the aspirate is evaluated histologically to establish that normal tri-lineage hematopoiesis is present and to determine the presence or absence of bone spicules. After dilution of the aspirate by ~3.5x with BMSC growth medium (αMEM containing Glutamax, 20% lot-selected, non-heat-inactivated FBS, 10 μg/ml gentamicin sulfate) and creation of a single cell suspension, the total number of nucleated cells is determined, and if it is below 20 × 106 cells, the sample is discarded (an in process control). Additionally, a 1 ml aliquot is evaluated by FACS for CD3 and CD34 content and viability, and for performing the colony forming efficiency assay (the closest approximation of the number of SSCs present in the aspirate [10, 14]). These data are used retrospectively to correlate with the outcomes of the ex vivo expansion [8].
Method for ex vivo expansion of BMSCs
Preliminary studies
In establishing our process, a number of decisions regarding culture conditions and vessels were made based on preliminary studies. A number of studies have demonstrated successful expansion of BMSCs by substituting human platelet lysate (hPL) for 20% FBS [11], however, our preliminary experiments indicated a great deal of variability that depended on the platelet lysate donor. This variability may be reduced by the use of commercially available products (e.g., a platelet-derived media supplement made by GwoWei Technology Co. Ltd, Taipei, Taiwan), but more testing is required to ensure that cells grown with this supplement retain their functionality. In addition, the use of human-derived media supplements requires far more testing for the presence of potential pathogens than does FBS. Serum-free supplements are also commercially available, however, the high cost excluded this possibility. Consequently, we elected to use FBS, although it has been reported that some recipients of cell products grown in FBS have developed hypersensitivity to it [12]. For this reason, we have included extensive washing of the cells at the end of our process. Lastly, there are a number of reports indicating that low oxygen tension increases the proliferation rate of BMSCs [13], however, it is not yet clear if cells generated in this fashion maintain their biological activity upon in vivo transplantation, and standard tissue culture chambers with atmospheric oxygen and 5% CO2 levels were selected.
With regards to the type of culture vessels, three choices were available: flasks, cell factories or a bioreactor. The use of flasks is extremely labor intensive in terms of feeding, and also runs the risk of contamination. However, the cell surface area of even a single layer cell factory precludes plating of the bone marrow aspirate at an appropriate density. For this reason we opted for the use of T-75 flasks for the initial plating (P0), whereby virtually all of the aspirate could be used, followed by expansion in 2-layer cell factories, and subsequent expansion in 10-layer cell factories (Fig. 1). In addition, the factories were outfitted with sterile tubing connected to bags of medium and a peristaltic pump for cell seeding and medium exchange (Fluid Transfer Tube Set, Baxa Corporation, Englewood, CO), making this a semi-closed system and less prone to contamination (Fig. 2). In our validation runs, it was determined that cells grown in the closed system displayed virtually identical cell surface markers as cells grown in flasks (Table 2). Importantly, like cells grown in flasks (Fig. 3a), cells grown in the cell factories were able to form a complete bone/marrow organ upon in vivo transplantation into immunocompromised mice (Fig. 3b). While the use of this system is more advantageous than the use of flasks, a totally closed system with automated features for media exchange would be less labor intensive and occupy less space. We had the opportunity to beta-test the Quantum Cell Expansion System (Terumo BCT, Lakewood, CO), which has recently become commercially available. Our preliminary experiments indicated that the Quantum bioreactor performed very well (Table 2, Fig 3c); however, the cost of the equipment and supplies needed was several fold higher than that of the semi-closed system, and precluded its use in our Center. The important point is that our system generates cells that faithfully recapitulate their important biological properties as demonstrated by the in vivo transplantation assay.
Fig. 1.

Scheme of for the ex vivo expansion of bone marrow stromal cells in a GMP-compliant semi-closed system. The current Drug Master file accommodates harvest for final cell product at either P2 or P3.
Fig. 2.

Modifications of the semi-closed system using commercially available sterile connectors, tubing and a peristaltic pump to minimize potential contamination of the cultures.
Table 2.
Comparison of cell surface markers present on BMSCs generated by ex vivo expansion in flasks, in a semi-closed system, and a bioreactor
| Marker | Flasks | Cell Factories | Bioreactor |
|---|---|---|---|
| CD29 | 97% | 100% | 100% |
| CD73 | 100% | 100% | 100% |
| CD90 | 100% | 100% | 100% |
| CD105 | 100% | 100% | 100% |
| CD146 | 84% | 80% | 95% |
| Hematopoietic markers | <5% | <6% | <6% |
Fig. 3.
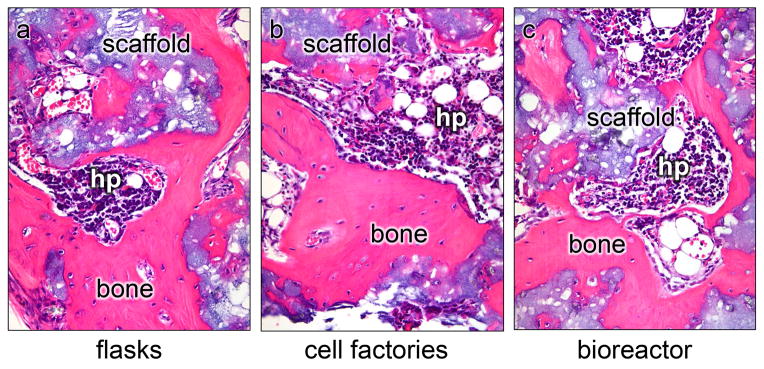
The in vivo transplantation assay for assessment of the differentiation capacity of BMSCs. Cells grown under in three types of vessels (flasks, cell factories, bioreactor). Cells in flasks and the cell factories were plated and passaged identically. Due to the surface area of the fibers in the bioreactor and the set up of the system, the ability to control plating density between passages was not as precise as with the flasks and the semi-closed system. Cells were harvested at P3, and 2 × 106 cells were attached to hydroxyapatite/tricalcium scaffold (a part of Collagraft, Zimmer, Warsaw, IN) as previously described. Cells grown in flasks (a), the semi-closed factories (b), or the bioreactor (c), were assessed for their ability to form a bone/marrow organ, whereby bone matrix, osteocytes, osteoblasts, hematopoiesis-supportive stroma and marrow adipocytes are of donor origin, while hematopoiesis (hp) is of recipient origin. Cells from all three types of vessels were comparable.
Processing (Fig. 1)
All reagents used in the cell processing procedure used in our preliminary experiments and in our FDA-approved manufacturing procedure (Drug Master File) were obtained from companies that could provide certificates of analyses (CoA) verifying their suitability for use in the generation of cell products to be used in humans.
Primary Culture (P0)
In primary culture, 2–3 × 105 cells/cm2 are seeded into T-75 flasks, and the medium is replaced 24 hours later to remove non-adherent cells, and every three days thereafter. Flasks are evaluated for colony formation and if no colonies appear by 7 days, the cultures are discarded. At day 10, if the flasks are >70% confluent, they are harvested, if <70%, they are re-fed. If >70% confluency is not achieved by day 13, the flasks are also discarded. Cells are released with recombinant trypsin (TryPLE Express, Invitrogen, CA), which is inactivated by the addition of BMSC growth medium (contains FBS). Cells are counted and assessed for viability, sterility and cell surface marker expression. CD73, CD90, CD105 and CD146 were selected as positive markers (Table 1). While we recognize that many other markers could be used, it is apparent that these markers (and others) are not specific for SSCs/BMSCs [14]. However, if these markers are not expressed by the vast majority of the cells, it is a major indication that the population is compromised. Additionally, CD34, CD45, CD14 and CD11b are used as markers to detect the contamination of the cultures by hematopoietic cells (Table 1). Cultures are discarded if sterility testing is positive.
Table 1.
BMSC lot release criteria
| Test | Method | Criteria |
|---|---|---|
| CD73 | FACS | >80% positive |
| CD90 | FACS | >80% positive |
| CD105 | FACS | >80% positive |
| CD146 | FACS | >60% positive |
| CD11b | FACS | <5% positive |
| CD14 | FACS | <5% positive |
| CD34 | FACS | <5% positive |
| CD45 | FACS | <5% positive |
| Purity (Bovine Transferrin) | ELISA | <10 ng/ml |
| Viability | Trypan Blue | >70% |
| Sterility | Bactec Plus, aerobic and anaerobic | No growth after 14 days |
| Mycoplasma | PCR | Negative |
| Endotoxin | Limulus Amebocyte Lysate | <5.0 EU/ml |
P1
Enzymatically released cells are seeded into 2-layer cell factories (Cell Factory, Easy Fill 2-Trays, Nunc A/S, Roskilde, Denmark) at 2,500 – 4000 cells/cm2 along with BMSC growth medium, facilitated by the use of the peristaltic pump, and up to four 2-layer factories could be accommodated. The medium is exchanged at day 3, and cells are passaged with TryPLE at day 5 or 6, depending on meeting the criterion of >70% confluency as described above. If <70% confluent, cultures are terminated. Depending on the number of 2-layer factories used, if the volume of released cells is less than 1200 mls, the cells are centrifuged at 406 × g for 10 minutes. If greater than 1200 mls, a cell washer is used (Cobe 2991 Cell processor, Caridian BCT, Lakewood, CO). The resulting cell pellets are re-suspended in BMSC growth medium, and an aliquot is taken for determination of cell number and viability (in process testing).
P2
The same procedure as described for P1 is applied for the generation of P2 cultures with the exception that the Cobe device is used exclusively for cell concentration. For P2, up to four 10-layer cell factories can be accommodated. Our current Drug Master File allows for another passage (P3) following the same procedure described above, using up to eight 10-layer cell factories.
At the end of P2 or P3, the process described in our Drug Master File ends. What happens to the cells next depends on the clinical application, and specifications of the clinical protocols and INDs that have been approved by the FDA. Our current FDA-approved IND for autologous cells calls for harvesting the cells at P2. The cells are released with TryPLE, which is then inactivated by the use of 5% heat-inactivated AB plasma in HBSS to reduce the level of FBS in the cell suspension. P2 cells are washed extensively (5X) with saline using the Cobe device and resuspended in saline at a concentration of cells according to the approved clinical protocol and IND. This wash procedure would most likely be modified for the use of the cells in bone regeneration in combination with an appropriate scaffold. Given experience at the bench, washing with serum-free BMSC growth medium would most likely be efficacious. In addition, in order to generate sufficient cells for repair of large bone defects, P3 cells may also be used with the modified wash procedure. If an excess number of cells are generated at either P2 or P3, it is envisioned that they could be frozen for future use if needed, according to an SOP (standard operating procedure) used in two of our FDA-approved INDs, as detailed in Sabatino et al [8], or destroyed, based on a choice provided to the patient during the consenting process.
Release criteria
Each cell lot is released only after the identity, purity, potency, efficacy, stability, sterility and safety are documented, reviewed, and judged as passing defined lot release criteria, as specified in the clinical protocol submitted with each IND, but generally follow items listed in Table 1. Aliquots of P2 cells from each cell lot are analyzed for cell surface markers (identity) and purity (level of bovine transferrin as detected by an ELISA assay with a cut off of 10 ng/ml).
The issue of potency (a measure of the strength of the effect that the cells will produce) is not well defined at this time, and is often not easily separated from efficacy (the ability of the cell product to produce a desired or intended result). As it relates to bone regeneration, potency and efficacy are best demonstrated by in vivo transplantation of the cells with an appropriate scaffold into immunocompromised mice [5, 10]. Potency could be defined as the amount of bone formed by the transplanted cells based on a scoring system that we have previously developed [15]. It could also include a measure of the number of cells required to form a complete bone/marrow organ [16]. However, because cells would be used immediately without freezing, this assay cannot be used for each lot of cells generated due to the length of time that it takes (~4 months), and it was not required by the FDA after our validation runs. It is clear that there is a need for markers that would faithfully predict the potency of SSCs/BMSCs that could be applied rapidly at the time of lot release. For this reason, we have also employed microarray analyses to define the transcriptomic signature for each lot of cells generated, along with cells generated at different passages [17, 18]. We will continue to study these signatures in comparison to the results of the in vivo transplantation assay. As for efficacy, preliminary proof is provided by use of autologous cells in pre-clinical animal studies and is provided in the IND that is under development. While the FDA prefers large animals for bone regeneration, rats can sometimes be considered.
Stability of the products is tested after thawing (time 0), and at 2, 4, and 6 hrs after suspension of the cells to the desired concentration, and is performed by determining the total cell number and viability (trypan blue exclusion). For sterility, cells are tested in the NIH Clinical Center Department of Laboratory Medicine (CLIA certified) for aerobic and anaerobic bacteria (Bactec Plus, Becton Dickinson, Sparks, MD), for mycoplasma (PCR), and for endotoxin (LAL Pyrogent-5000 assay, Biowhittaker, Inc., Walkersville, MD) (Table 1). The safety requirement relates not only to evidence of sterility, but also to the use of appropriate pre-clinical studies that demonstrate safety in the use of the cells for a specific clinical application as provided in the INDs.
If the cell lot meets all of the requirements described above, it given a certificate of analysis (CoA) and the lot is released for use. At least three vials of cells are created for retention for further testing if needed. Of note, when cells are used directly (no cryopreservation), the results of sterility testing are not available at the time of release, thus requiring an action plan should abnormal results be obtained, which is included in the INDs.
Manufacturing quality control and quality assurance
The manufacturing quality control and quality assurance program for BMSCs is identical to that applicable to all products manufactured in Cell Processing Section, Department of Transfusion Medicine at the NIH Clinical Center, and is far too extensive to describe here, but is available upon request. StemLab software (STEMSOFT Software Inc., Vancouver, British Columbia, Canada) is used to track all components including cells, equipment, reagents, supplies, inventory, personnel and methods.
Further considerations
There is no doubt that the culture conditions employed to expand BMSCs impacts on their biological activities. Numerous methods have been reported with many variables starting with the acquisition of bone marrow, how it is processed prior to plating, medium components, plating density and passaging, to list just a few. We have spent considerable effort over the years to optimize all of the steps required for the successful ex vivo expansion of biologically active BMSCs, guided by the results of the in vivo transplantation assay, the gold standard by which to evaluate the osteogenic differentiation capacity of BMSCs. Using this assay allows for assessment not only of the ability of the cells to reform bone, but to reform a bone/marrow organ, an indicator that the SSC within the BMSC population has been maintained. Unfortunately, due to the length of time it takes to do this assay, in vitro assays are more often used, although they are not always predictive of in vivo outcomes. This is not to say that our procedure cannot be improved upon, but only to say that there is an assay that strictly defines the biological activity of the cells in an in vivo setting. For sure, development of predictive markers would mark a major step forward.
From our studies, we have learned a number of things that pertain directly to the cell processing procedure that we have developed. The volume of bone marrow aspirate must be controlled to avoid dilution of marrow with peripheral blood, which can impact on the growth of BMSCs in primary cultures. Often density gradient centrifugation is used to partially separate hematopoietic cells from BMSCs, but due to their highly adhesive nature, this step can result in a significant loss of cells. We have also optimized the level of lot-selected, non-heat-inactivated FBS that is needed for optimal cell growth [19, 20]. This medium component can have a profound effect on SSCs/BMSCs (as can be evidenced by colony forming efficiency assays using different lots of FBS) and is probably the most variable component between different laboratories. In addition, many laboratories use growth factors to increase the proliferation rate; however, this can have detrimental effects on maintenance of SSCs within the BMSC population (see [20] as an example). Furthermore, the cells are not immortal, and while they can be expanded significantly, extensive passaging results in a change in their transcriptome [18], which most certainly impacts on their function. Along these lines, there is a current effort to develop a strategy to generate a “MSC” reference material to which different laboratories could compare their cell preparations, based on a number of properties [21]. However, “MSCs” are derived from many different tissues and while they share common markers, their differentiation capacities are quite different. It is not clear that having a reference material would be useful when comparing “MSCs” from one tissue to another. However, attempts to standardize BMSC preparations specifically would not be unwarranted in terms of bone regeneration.
Lastly, while optimized for expansion of BMSCs, it should be noted that the infrastructure and the basic process that we have developed is amenable to the ex vivo expansion of adherent populations of cells from other types of tissues that may be useful in engineering of specific tissues that are associated with bone (muscle, tendon, meniscus, to name a few). What would be required is a clinical protocol for donor evaluation and acquisition of tissue under GMP conditions, and establishment of a GMP compliant method for liberating cells from a tissue, selection and characterization, and culture conditions. From that point on, the basic process would be the same.
In summary, we have developed an FDA-approved Drug Master File for the ex vivo expansion of bone marrow stromal cells using a modified semi-closed system. After expansion, these cells are comparable to those grown in the traditional setting to tissue culture flasks based on cell surface markers, and the formation of a bone/marrow organ upon in vivo transplantation. The manufacturing process includes in process testing at multiple steps, and post processing testing to ensure safety of the cell product to the highest degree possible. It is highly likely that the process will change as reagents and technologies improve, especially with development of new markers that can predict potency and efficacy, but the basics are in place.
Acknowledgments
The authors would like to acknowledge all of the effort of the members in the Cell Processing Section and the Department of Transfusion Medicine, NIH CC who worked on developing the cell manufacturing procedure, and Sharon Mavroukakis, (MAVS RESEARCH & SUPPORT, LLC) for the development of all regulatory documents that were required. The NIH Bone Marrow Transplantation Center is supported by the Intramural Research Programs of NIDCR, NCI, NHLBI, NIAID, NIAMS, NIBIB, NINDS, and the CC, all of the Intramural Research Program of the NIH, DHHS.
References
- 1.Robey PG. Cell sources for bone regeneration: the good, the bad, and the ugly (but promising) Tissue Eng Part B, Reviews. 2011;17:423–30. doi: 10.1089/ten.teb.2011.0199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Friedenstein AJ, Piatetzky-Shapiro II, Petrakova KV. Osteogenesis in transplants of bone marrow cells. J Embryol Exp Morphol. 1966;16:381–90. [PubMed] [Google Scholar]
- 3.Bianco P, Riminucci M, Gronthos S, Robey PG. Bone marrow stromal stem cells: nature, biology, and potential applications. Stem Cells. 2001;19:180–92. doi: 10.1634/stemcells.19-3-180. [DOI] [PubMed] [Google Scholar]
- 4.Friedenstein AJ, Chailakhyan RK, Latsinik NV, Panasyuk AF, Keiliss-Borok IV. Stromal cells responsible for transferring the microenvironment of the hemopoietic tissues. Cloning in vitro and retransplantation in vivo. Transplantation. 1974;17:331–40. doi: 10.1097/00007890-197404000-00001. [DOI] [PubMed] [Google Scholar]
- 5.Krebsbach PH, Kuznetsov SA, Satomura K, Emmons RV, Rowe DW, Robey PG. Bone formation in vivo: comparison of osteogenesis by transplanted mouse and human marrow stromal fibroblasts. Transplantation. 1997;63:1059–69. doi: 10.1097/00007890-199704270-00003. [DOI] [PubMed] [Google Scholar]
- 6.Sacchetti B, Funari A, Michienzi S, Di Cesare S, Piersanti S, Saggio I, Tagliafico E, Ferrari S, Robey PG, Riminucci M, Bianco P. Self-renewing osteoprogenitors in bone marrow sinusoids can organize a hematopoietic microenvironment. Cell. 2007;131:324–36. doi: 10.1016/j.cell.2007.08.025. [DOI] [PubMed] [Google Scholar]
- 7.Bianco P, Cao X, Frenette PS, Mao JJ, Robey PG, Simmons PJ, Wang CY. The meaning, the sense and the significance: translating the science of mesenchymal stem cells into medicine. Nat Med. 2013;19:35–42. doi: 10.1038/nm.3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sabatino M, Ren J, David-Ocampo V, England L, McGann M, Tran M, Kuznetsov SA, Khuu H, Balakumaran A, Klein HG, Robey PG, Stroncek DF. The establishment of a bank of stored clinical bone marrow stromal cell products. J Transl Med. 2012;10:23. doi: 10.1186/1479-5876-10-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stroncek DF, Sabatino M, Ren J, England L, Kuznetsov SA, Klein HG, Robey PG. Establishing a Bone Marrow Stromal Cell Transplant Program at the National Institutes of Health Clinical Center. Tissue Eng Part B Rev. 2014 doi: 10.1089/ten.teb.2013.0529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kuznetsov SA, Krebsbach PH, Satomura K, Kerr J, Riminucci M, Benayahu D, Robey PG. Single-colony derived strains of human marrow stromal fibroblasts form bone after transplantation in vivo. J Bone Miner Res. 1997;12:1335–47. doi: 10.1359/jbmr.1997.12.9.1335. [DOI] [PubMed] [Google Scholar]
- 11.Schallmoser K, Bartmann C, Rohde E, Reinisch A, Kashofer K, Stadelmeyer E, Drexler C, Lanzer G, Linkesch W, Strunk D. Human platelet lysate can replace fetal bovine serum for clinical-scale expansion of functional mesenchymal stromal cells. Transfusion. 2007;47:1436–46. doi: 10.1111/j.1537-2995.2007.01220.x. [DOI] [PubMed] [Google Scholar]
- 12.Selvaggi TA, Walker RE, Fleisher TA. Development of antibodies to fetal calf serum with arthus-like reactions in human immunodeficiency virus-infected patients given syngeneic lymphocyte infusions. Blood. 1997;89:776–9. [PubMed] [Google Scholar]
- 13.Dos Santos F, Andrade PZ, Boura JS, Abecasis MM, da Silva CL, Cabral JM. Ex vivo expansion of human mesenchymal stem cells: a more effective cell proliferation kinetics and metabolism under hypoxia. J Cell Physiol. 2010;223:27–35. doi: 10.1002/jcp.21987. [DOI] [PubMed] [Google Scholar]
- 14.Bianco P, Robey PG, Simmons PJ. Mesenchymal stem cells: revisiting history, concepts, and assays. Cell Stem Cell. 2008;2:313–9. doi: 10.1016/j.stem.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mankani MH, Kuznetsov SA, Avila NA, Kingman A, Robey PG. Bone formation in transplants of human bone marrow stromal cells and hydroxyapatite-tricalcium phosphate: prediction with quantitative CT in mice. Radiology. 2004;230:369–76. doi: 10.1148/radiol.2302011529. [DOI] [PubMed] [Google Scholar]
- 16.Mankani MH, Kuznetsov SA, Robey PG. Formation of hematopoietic territories and bone by transplanted human bone marrow stromal cells requires a critical cell density. Exp Hematol. 2007;35:995–1004. doi: 10.1016/j.exphem.2007.01.051. [DOI] [PubMed] [Google Scholar]
- 17.Ren J, Jin P, Sabatino M, Balakumaran A, Feng J, Kuznetsov SA, Klein HG, Robey PG, Stroncek DF. Global transcriptome analysis of human bone marrow stromal cells (BMSC) reveals proliferative, mobile and interactive cells that produce abundant extracellular matrix proteins, some of which may affect BMSC potency. Cytotherapy. 2011;13:661–74. doi: 10.3109/14653249.2010.548379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ren J, Stroncek DF, Zhao Y, Jin P, Castiello L, Civini S, Wang H, Feng J, Tran K, Kuznetsov SA, Robey PG, Sabatino M. Intra-subject variability in human bone marrow stromal cell (BMSC) replicative senescence: molecular changes associated with BMSC senescence. Stem Cell Res. 2013;11:1060–73. doi: 10.1016/j.scr.2013.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kuznetsov SA, Friedenstein AJ, Robey PG. Factors required for bone marrow stromal fibroblast colony formation in vitro. Br J Haematol. 1997;97:561–70. doi: 10.1046/j.1365-2141.1997.902904.x. [DOI] [PubMed] [Google Scholar]
- 20.Kuznetsov SA, Mankani MH, Bianco P, Robey PG. Enumeration of the colony-forming units-fibroblast from mouse and human bone marrow in normal and pathological conditions. Stem Cell Res. 2009;2:83–94. doi: 10.1016/j.scr.2008.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Viswanathan S, Keating A, Deans R, Hematti P, Prockop D, Stroncek DF, Stacey G, Weiss DJ, Mason C, Rao MS. Soliciting Strategies for Developing Cell-Based Reference Materials to Advance MSC Research and Clinical Translation. Stem Cells Dev. 2014 doi: 10.1089/scd.2013.0591. [DOI] [PMC free article] [PubMed] [Google Scholar]