

Abstract
Thymic atrophy occurs during normal aging, and is accelerated by exposure to chronic stressors that elevate glucocorticoid levelsand impair the naïve T cell output. The orexigenic hormone ghrelin was recently shown to attenuate age-associated thymic atrophy. Here, we report that ghrelin enhances the proliferation of murine CD4+ primary T cells and a CD4+ T-cell line. Ghrelin induced activation of the ERK1/2 and Akt signaling pathways, via upstream activation of phosphatidylinositol-3-kinase and protein kinase C, to enhance T-cell proliferation. Moreover, ghrelin induced expression of the cell cycle proteins cyclin D1, cyclin E, cyclin-dependent kinase 2 (CDK2) and retinoblastoma phosphorylation. Finally, ghrelin activated the above-mentioned signaling pathways and stimulated thymocyte proliferation in young and older mice in vivo.
Keywords: Ghrelin, T-cells, stress, glucocorticoids, proliferation, signaling, thymus
Introduction
The thymus is required for the generation of T cells, a developmental process that is maintained throughout adolescence and early adulthood, and then progressively declines with advancing age [1, 2]. The thymus and developing thymocytes are highly sensitive to both physiological and pathological stressors, such as infections, immunosuppressive treatments, surgery, anxiety/depression and malnutrition, which can result in the apoptosis of developing thymocytes [3–7]. Such stressors result in increased production of glucocorticoids (GCs) [8–10]. Excessive production of GCs as a result of hypothalamus-pituitary-adrenal (HPA) axis hyperactivation also occurs in inflammatory conditions and aging [5, 11]. GCs have been established as major regulators of thymocyte and mature lymphocyte apoptosis [4, 8, 9]. The synthetic GC, dexamethasone, is routinely used to suppress immune functions in organ and bone marrow transplant recipients, in autoimmune and systemic inflammatory disorders and in the treatment of malignancies [13]. Adverse effects of these GCs on thymic activity and integrity have been established and the underlying alterations of T cell signaling pathways that regulate cell activation and proliferation are being elucidated [5, 11, 12].
Several hormones and cytokines are involved in GC-mediated thymic atrophy, and seem to influence thymocyte survival, proliferation and gene expression [4, 13]. Here, we focused on the hormone ghrelin — an acylated 28-amino-acid polypeptide that is secreted by the X/A-like enteroendocrine cells of the stomach and plays a major role in the regulation of food intake, adiposity and energy homeostasis [13, 14]. Activated ghrelin has an n-octanoyl group covalently linked to the hydroxyl group of the serine 3 (Ser3) residue, which is critical for its binding to the G-protein coupled growth hormone secretagogue receptor-1a (GHS-R1a) [15]. Activation of GHS-R1a by ghrelin in somatotroph cells of the pituitary gland induces GH release through enhanced phospholipase C (PLC) and protein kinase C (PKC) activation and intracellular calcium mobilization [16]. Moreover, ghrelin mediates diverse biologic functions beyond its effects on the central nervous system [17]. It increases cell proliferation, and inhibits apoptosis of cardiomyocytes, endothelial cells and enterocytes [18], and it is expected that additional functions of this potent hormone will be identified in the near future, given the wide distribution of GHS-Rs in a variety of tissues and cell types [18,19].
GHS-R1a-mediated mobilization of calcium is one of the best characterized intracellular events resulting from GHS-R1a activation [20]. Growth hormone (GH) secretagogues stimulate intracellular calcium mobilization mediated via the activation of PLC through the GTP-binding protein (Gαq11) [21]. PLC cleaves the membrane lipid phosphatidylinositol 4,5 diphosphate (PIP2) into diacylglycerol (DAG) and inositol (1,4,5) triphosphate (IP3). IP3 represents one arm of this cascade and mediates the release of calcium from the endoplasmic reticulum. DAG activates PKC, which in turn phosphorylates multiple protein substrates resulting in specific phenotypic responses of the cells [22]. The pro-proliferative activity of ghrelin may involve the activation of the downstream mitogen-activated protein kinases (MAPK) and Akt (also known as protein kinase B) in several cell types [19,23,24]. In Chinese hamster ovary cells engineered to overexpress GHS-R1a, ghrelin administration activated ERK1/2 via a PLC- and PKCε-dependent pathway [21]. Similarly, the mitogenic effect of ghrelin on endothelial cells and 3T3-L1 preadipocytes is mediated by the PI3K/Akt and MAPK pathways [25,26].
Recent studies have demonstrated a potential role for ghrelin in influencing the immune system and inflammatory responses [1,3,19,27,28]. Ghrelin receptors are present in several leukocyte subsets, and immune cells (such as T cells, monocytes, dendritic cells) can also produce ghrelin [3,5]. Moreover, exogenous administration of ghrelin can ameliorate both acute and chronic inflammatory conditions [3]. However, little is known about the effect of GHS-R1a activation on signal transduction pathways in immune cells, in general, and on the proliferation and activation of T cells, in particular.
Here, we report that ghrelin partially reverses dexamethasone-induced thymocyte ablation and increases thymocyte cell proliferation in vivo. Analysis of the signaling pathways associated with the proliferative effects of ghrelin revealed that GHSR-1a ligation results in the activation of the PI3-kinase, PLC, Akt and MAP kinase signaling pathways in both primary murine CD4+ T cells and murine CD4+ T hybridoma cells stably expressing the GHS-R1a receptor. Ghrelin-mediated activation of these pathways results in an enhancement of T-cell proliferation and activation, influences cell cycle progression upon TCR ligation, and enhances recovery from glucocorticoid-mediated thymic atrophy. These findings advance our understanding of the mechanisms through which ghrelin protects against thymic involution, with implications for aging, chronic stress and associated diseases.
Materials and Methods
Reagents
All supplements for cell culture were obtained from Invitrogen (Carlsbad, CA). Recombinantrat ghrelin (acylated ghrelin) was purchased from GenScript (Piscataway, NJ), and rat des-octanoyl ghrelin was from Tocris Bioscience (Ellisville, MO). The PI3-kinase inhibitor Wortmannin, the ERK inhibitor PD98059, the p38 inhibitor SB203580, the JNK inhibitor SP600125 and the PLC inhibitor U73122 were obtained from EMD Chemicals (Gibbstown, NJ). Antibodies against phosphorylated forms of Akt (D9E), ERK1/2 (E10), p38, JNK, Cyclin D (92G2), Rb and the various PKC family members were purchased from Cell Signaling Technology (Beverly, MA). The GHS-R1a antibody for immunostaining was obtained from Phoenix Pharmaceuticals, Inc. (Burlingame, CA). Alexa-Fluor 488-conjugated goat anti-rabbit antibody was purchased from Invitrogen. The antibodies specific for GHS-R1a (H-80), Cyclin E (E-4), CDK2 (D-12), CDK4 (C-22) and actin (I-19) used for Western blotting were purchased from Santa Cruz Biotechnology Inc. (Santa Cruz, CA). Stripping buffer for membrane reprobing was obtained from Thermo Fisher Scientific Inc. (Waltham, MA). Pertussis toxin was purchased from Sigma-Aldrich (St. Louis, MO). The MTS assay kit for cell proliferation was purchased from Promega (Leiden, the Netherlands).
Animals and experimental protocol
C57BL/6 male mice (8–10 weeks old) were purchased from The Jackson Laboratories (Bar Harbor, ME) and experiments were performed under a protocol approved by the National Institute on Aging Animal Care and Use Committee. The mice were maintained under a 12 h light and 12 h dark cycle. Mice were injected i.p. with 100 μg/kg ghrelin or des-acyl ghrelin or 0.9% saline (control) every 12-h for 1 or 3 days. In some aged mouse experiments, mice were infused with PBS or ghrelin using osmotic minipumps as previously described [1]. In the BrdU incorporation studies, 2 h prior to euthanasia the treated mice were injected i.p. with 100 mg/kg BrdU (Sigma). The thymus was subsequently isolated and cut in half for immunohistological examination. Half of the thymus was fixed in 10% neutral buffered formalin and paraffin-embedded. Deparaffinized sections (5–10 μm) were stained with diaminobenzidine (Vector Laboratories, Burlingame, CA) and analyzed by light microscopy. As a negative control for the primary T cell proliferation studies, CD4+ T cells derived from GHS-R null mice (generously donated by Dr. Roy Smith, The Scripps Research Institute, Jupiter, FL) were utilized as described previously [1, 27,28].
Primary T cell preparation
Eight week-old mice were euthanized and single-cell suspensions were made of the spleen or thymuses in cold Hank’s balanced saline solution (HBSS) with 1% fetal bovine serum (FBS) (Invitrogen). After the removal of contaminating red blood cells using ACK lysis buffer (Quality Biological, Inc., Gaithersburg, MD), CD4+ T cells were isolated using the mouse CD3+ CD4+ T-cell enrichment column (R&D System, Minneapolis, MN) or using mouse CD4+ T cell isolation kit (Miltenyi Biotec, Bergisch Gladbach, Germany) and kept in RPMI supplemented with 10% FBS (Invitrogen) and 50 μM β-ME under 5% CO2 at 37°C.
GHS-R1a-transfected CD4+ T-cell hybridoma line
A GHS-R1a transfected CD4+ T cell line called D0.11.10 was generated and utilized for the majority of cell signaling analyses examined in this study. Total RNA was isolated from the murine pituitary gland and cDNA was prepared using SuperScript III first-Strand Synthesis System for RT-PCR and amplified by PCR using Bam HI overhang primers as described previously [29]. PCR-products were directly cloned into pcDNA3.1/V5-His TOPO TA vector, which was subsequently and stably transfected in the CD4+ T-helper cell hybridoma line, D0.11.10. These transfected cells were selected for and grown in G418 (Calbiochem)-supplemented medium containing RPMI 1640 with 10% FBS, 1% penicillin/streptomycin, 4% HEPES and 1% sodium pyruvate in a humidified atmosphere of 5% CO2/95% room air. These lines were subsequently subcloned and individual transfected clones were isolated and examined for expression. Optimal subclones expressing GHS-R1a were isolated and utilized in these studies (Figure 1).
Figure 1. Acylated ghrelin administration induces thymocyte subset proliferation in dexamethasone-treated mice.

C57BL/6 mice were injected with acylated ghrelin at 100 μg/kg over a 24 h time period in the presence or absence of dexamethasone after which the thymocytes were examined for thymocyte subset numbers and the number of BrdU-positive subset cells using flow cytometric analysis as described in the Methods. Each group included 5 mice. The data are expressed as either (A) the absolute thymocyte subset numbers (×106 cells), or (B) BrdU-positive cells (×106 cells). Values represent the mean and SEM of determinations made in three independent experiments. *P<0.05, ** P<0.01.
Confocal microscopy
D0.11.10 cells (1×105) were placed on slides using a cytospin centrifuge (Thermo Fisher Scientific, Asheville, NC) for 3 min at 800 rpm after which the cells were fixed with 4% formaldehyde in PBS for 20 min. The slides were subsequently blocked with 0.5% bovine serum albumin for 1 h and then stained with rabbit anti-GHS-R1a antibody overnight at 4°C. After incubation, the cells were subsequently incubated with Alexa-Fluor 488-conjugated goat-anti-rabbit antibody and the recombinant cholera toxin subunit B-conjugated Alexa Fluor® 594 for 1 h. After washing the slides three times with PBS, the ProLong Gold anti-fade reagent (Invitrogen) was added to the slides, which were then coverslipped and dried overnight at room temperature. Fluorescence microscopy was performed using a Carl Zeiss LSM 510 confocal microscope (Oberkochen, Germany). Alexa-Fluor-488 and Alexa-Fluor-594 were excited with an Ar (488 nm) or He-Ne (543 nm) laser and emission was filtered using narrow band LB 505–530 nm and the LP 560 nm filters, respectively. Images were then analyzed using Carl Zeiss LSM image software 3.0.
Cell proliferation assay
Cells were plated (3.0 × 103 cells per well) in 96-well plates and stimulated with recombinant ghrelin or des-acylated ghrelin for specified time periods at 37°C in a humidified, 5% CO2 atmosphere. After incubation, 20 μl of MTS reagent (CellTiter 96® Aqueous one solution cell proliferation assay, Promega) was added to the cultures. After 1 h incubation, the intensity of the absorbance was quantified using an ELISA microplate reader at an absorbance of 592 nm. The data are expressed as % proliferation (+/− SEM).
Immunoblot analysis
Protein lysates were prepared from T cells by lysing them in RIPA buffer containing a protease inhibitor cocktail tablet (Roche) and phosphatase inhibitors (Sigma), and then incubated on ice for 10 min, followed by centrifugation at 15,000 g for 15 min at 4°C. Protein concentrations were subsequently determined and 30 μg of each sample were separated using SDS–PAGE and then transferred onto PVDF membranes. The membranes were subsequently blocked in a TBS-T buffer (10 mmol/L Tris-HCl [pH 7.5], 150 mmol/L NaCl, and 0.05% Tween 20) containing 5% skimmed milk powder for 1 h, after which the membrane was incubated with individual primary antibodies at 4°C overnight. After washing with a TBS-T buffer, the membrane was then incubated with horseradish peroxidase-coupled secondary antibodies for 1 h at room temperature. Blotting detection was subsequently conducted using an enhanced ECL detection system (Amersham Biosciences, Buckinghamshire, UK).
Cell cycle analysis by propidium iodide (PI) staining
T cells were plated at 1 × 106 cells per well in 12-well plate for 16 h at 37°C. After treatment with 10 nM ghrelin, the cells were incubated for the designated time periods, and then washed twice and suspended into 70% ethanol for 30 min at 4°C. Cells were subsequently washed once, and suspended in 500 μl of PI solution (25 μg/ml PI, 0.1 mg/ml of RNase A in PBS) and then incubated for 30 min in darkness. The cells were analyzed by flow cytometric analysis using a FACScan (Becton Dickinson, San Jose, CA), followed by data analysis using MultiCycle (Phoenix Flow Systems, San Diego, CA).
Real-time PCR analysis
One half to one microgram of RNA was purified and quantitated from each sample and made into cDNA with the iScript cDNA synthesis kit (BioRad, Hercules, CA). One microliter of each cDNA sample was then used to measure quantity using the SYBR Green PCR master mix (Applied Biosystems) and reactions were run on the 7500 fast or 7300 PCR system (Applied Biosystems). The results were normalized to 18S using the QuantumRNA universal 18S (Ambion, Austin, TX) and were also used to determine relative quantities. The GHS-R primers utilized in this study were described previously [30].
PKC activity assay
In order to determine the effect of ghrelin on PKC activity, we measured PKC activity in cell lysates. Samples were prepared from T cells by lysing them in RIPA buffer after which the lysates were centrifuged at 15,000 g for 15 min at 4°C. These supernatants were assayed using the PKC Kinase Activity Assay Kit (EKS-420A; Stressgen Bioreagents, Victoria, BC, Canada). Samples were assayed in triplicate.
Statistical analysis
The data are presented as the mean ± SEM from three or more independent experiments. All statistical significance was determined by ANOVA using the Statistical Analysis System (SAS, Cary, NC). Comparisons between two groups were performed using Student’s t-test. Pairwise comparisons for data with multiple time points or treatment concentrations were done using Duncan’s multiple range test. A value of P<0.05 was considered statistically significant.
Results
Ghrelin induces thymocyte proliferation in dexamethasone-treated mice
Dexamethasone (Dex), a potent synthetic member of the glucocorticoid class of steroid drugs can mimic the effects of endogenous GCs by inducing thymic ablation through the programmed cell death of thymocytes, and in particular of the immature double-positive (DP) subset [2,31]. We first examined the ability of ghrelin infusion to promote a restoration of thymocyte numbers and proliferation after Dex treatment. Thymocyte proliferation was increased already at day 1 following combined Dex and ghrelin treatment as compared to Dex treatment alone (Figure 1B). Ghrelin also increased the absolute numbers of DP thymocytes in the Dex-treated mice (Figure 1A). Ghrelin and the saline vehicle control failed to induce significant changes in cell numbers and proliferation when administered to mice that had not received any Dex treatment. These in vivo data suggest that ghrelin promotes thymocyte proliferation and survival [1].
Ghrelin-induced proliferation of T cells is both Akt- and Erk1/2-dependent
To examine the signaling pathways that act downstream of GHS-R1a in T cells, we transfected the murine D0.11.10 CD4+ T cell line with GHS-R1a. GHS-R1a mRNA expression was 140-fold higher in GHS-R1a-transfected D0.11.10 CD4+ T cells than in control pcDNA-transfected cells, following normalization for 18S expression (Figure 2A). This increased receptor expression corresponded to higher GHS-R1a protein levels (Figure 2B). Moreover, GHS-R1a was found to be biologically active in D0.11.10 CD4+ T cells, as treatment with ghrelin resulted in a significant increase in phospho-ERK levels in GHS-R1a-transfected but not in pcDNA-transfected cells (Figure 2C). Previous studies from our laboratory demonstrated that GHS-R1a localizes within lipid rafts upon T cell activation and that this association might be necessary for optimal GHS-R1a activity [28]. In our examination of the D0.11.10 transfectants under laser confocal microscopy, we found that the GHS-R1a receptor was expressed constitutively on the surface of the GHS-R1a transfected cells and localized within the ganglioside GM1 lipid raft domain (Figure 2D).
Figure 2. Expression of GHS-R1a in the murine CD4+ T cells and a CD4+ GHSR1a-transfected T cell hybridoma line, D0.11.10.

D0.11.10 cells transfected with either the pcDNA control vector alone or the vector containing the GHS-R1a gene were processed for analysis of either GHS-R1a mRNA levels by real time RT-PCR (A) or GHS-R1a protein levels by immunoblot analysis (B). Values represent the mean and SEM of determinations made in three independent experiments. *P < 0.05, **P < 0.01 compared to control (EL4) cells. (C) The transfected cells were examined for responsiveness to ghrelin through the examination of phosphorylated ERK levels. The pcDNA- and GHS-R1a-transfected D0.11.10 cells were stimulated with 10 nM ghrelin for 7 min after which cell lysates were examined by immunoblot for pERK levels. Experiments were repeated 3 times and the representative results are shown. The numbers under each blot are intensity of the ERK band relative to that of the β-actin band. (D) To assess the cell surface expression of GHS-R1a, the GHS-R-transfected D0.11.10 cells (1 × 105) were co-stained using rabbit anti-GHS-R1a antibody (red) and fluorescently tagged recombinant cholera toxin subunit B (green) which binds selectively to the membrane lipid raft marker ganglioside GM1. Areas of co-localization are indicated by yellow color in the overlay image. (E) Cells (3 × 103) were treated 10 nM ghrelin or vehicle in 96-well plates for various time intervals after which cell proliferation was examined using the MTS method. The data are expressed as % cell proliferation and values represent the mean and SEM of determinations made in four independent experiments. *P < 0.05 **P < 0.01, compared to 0 h; ##P<0.05, ‡‡P<0.01 compared to the pcDNA value at the same time point. ††P<0.01 compared to the values of each of the other groups at the same time point.
Given previous findings demonstrating the proliferative effects of ghrelin in tumor cells and several types of normal cells [3,19,23,24], we next examined the effects of ghrelin on the proliferative response of GHS-R1a-transfected D0.11.10 cells. GHS-R1a-transfected D0.11.10 cells stimulated with ghrelin exhibited significantly greater proliferation compared to the non-treated cells and the pcDNA-transfected D0.11.10 cells (Figure 2E). The ghrelin-mediated effects on cell proliferation were GHSR-1a-specific, as the GHSR-1a selective antagonist, D-Lys-GHRP-6, significantly inhibited the ghrelin-mediated pro-proliferative effects on D0.11.10 cells (data not shown). These data suggested that these transfected cell lines would be valuable in examining the signaling effects of ghrelin on murine T cells and potential roles for ghrelin in T cell proliferative and activation responses.
The ERK and Akt signaling pathways are known to play important roles in cellular proliferation [32]. To evaluate the role of the Akt, Erk1/2 and PI-3-kinase signaling pathway(s) on ghrelin activation of murine T cells, a kinetic analysis was performed examining the ability of ghrelin (10 nM) to stimulate GHS-R1-transfected D0.11.10 cells for various time periods (Figure 3A). Ghrelin induced Akt and ERK 1/2 phosphorylation within 30 seconds after stimulation and this activity was sustained for 15–30 minutes, with maximal increases in levels of both phosphorylated kinases being observed at 3 minutes. Ghrelin also induced phosphorylation of c-Jun N-terminal kinase 1/2 (JNK1/2) and p38, both of which are known to play a role in cellular activation and proliferation of lymphocytes [33]. A dose-response analysis was then performed with ghrelin concentrations ranging between 1 and 10 nM, and maximum Akt and ERK activation was shown to occur 7 minutes after treatment with the highest ghrelin concentration(Figure 3B). Thus, a ghrelin concentration of 10 nM was used in the majority of the subsequent experiments in this study.
Figure 3. Kinetic and dose response analysis of the effects of ghrelin on Akt and ERK1/2 phosphorylation in D0.11.10 cells.
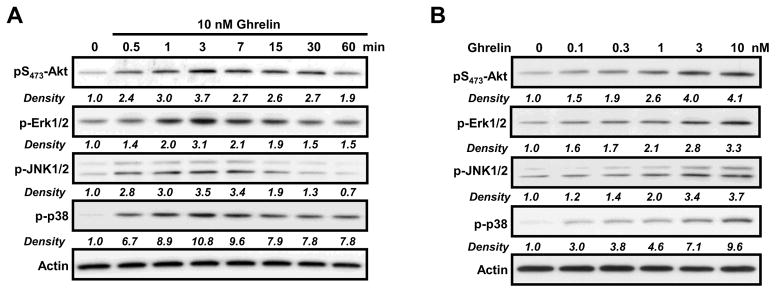
D0.11.10 GHS-R1a-transfected T cells (2 × 106) were incubated for the indicated time periods with ghrelin at 10 nM (A) or with increasing concentrations of ghrelin for 7 min (B), after which the cells were lysed and extracted proteins were examined by immunoblot analysis using antibodies specific for phosphorylated residues of Akt, ERK1/2, JNK1/2 and P38. Experiments were repeated 4–5 times and representative blots are shown. The density of each band was measured by Multi Gauge V3.1 software (Fujifilm, Tokyo, Japan) and normalized to the intensity of the β-actin band for the same sample.
The pro-proliferative effect of ghrelin on GHS-R1a- transfected D0.11.10 T cells was significantly inhibited by the PI3-kinase inhibitor wortmannin, as well as by the ERK1/2 inhibitor PD98059 (Figure 4A). These results suggested that ghrelin stimulates T cell proliferation in a PI3-kinase-, Akt- and Erk-dependent manner. This is supported by the data in Figure 4B, which show that wortmannin inhibits ghrelin-induced Akt phosphorylation and ERK activation in a concentration-dependent manner. Thus, the ability of ghrelin to activate both Akt and ERK1/2 is highly dependent on PI3-kinase activity. Wortmannin treatment at concentrations as low as 0.03 μM almost completely abrogated ghrelin-induced phosphorylation of Akt and ERK1/2. None of the concentrations of wortmannin applied in these studies were found to be toxic, and no inhibitory responses were observed using vehicle alone (data not shown). Interestingly, GHS-R1a-transfected D0.11.10 T cells that had been pre-treated with various doses of PD98059 (a MEK1 inhibitor that has been shown to act as a highly selective inhibitor of MEK1 activation and the MAP kinase cascade) revealed that only ghrelin-mediated ERK1/2, but not Akt, phosphorylation was influenced by MEK1 inhibition, suggesting that ghrelin-induced Akt phosphorylation was independent of ERK1/2 activation (Figure 4B).
Figure 4. Ghrelin enhances D0.11.10 cell proliferation and activation via PI-3-kinase, Akt and ERK1/2 pathways.

(A) D0.11.10 cells (3 × 103) were pretreated with wortmannin (0.3 μM), PD98059 (10 μM), SB203580 (10 μM) or SP600125 (10 μM) or 0.1% DMSO vehicle control (CTR) for 30 min prior to treatment with 10 nM ghrelin. After incubation for 24 or 48 hours, the plates were examined for cell proliferation using MTS assay. The data are expressed as the percentage of cell number compared to the CTR group at time 0. Values represent the mean and SEM of determinations made in three independent experiments. *P<0.05, **P < 0.01 compared to 0 h (control); #P< 0.05, ##P < 0.01 compared to ghrelin treatment. (B) D0.11.10 cells (2 × 106) were cultured with 10 nM ghrelin in the presence or absence of various concentrations of wortmannin or PD98059 for 30 min after which the cells were lysed and examined for Akt and ERK1/2 phosphorylation via immunoblot analysis. These blots are representative of results obtained in 5 separate experiments.
The addition of SB203580, a potent inhibitor of the p38-MAP kinase pathway, had no significant inhibitory effects on ghrelin-induced T cell proliferation. However, it should be pointed out that the addition of SB203580 alone enhanced cell proliferation making data interpretation difficult. Moreover, the addition of SP600125, a selective inhibitor of JNK1/2, resulted in significant albeit modest effects on ghrelin-induced T cell proliferation (Figure 4A). These data suggest that although ghrelin stimulates the phosphorylation of JNK1/2 and p38 in GHS-R1a-transfected D0.11.10 cells, these kinases may not play a major role in ghrelin-mediated proliferation in GHS-R1a- transfected D0.11.10 T cells.
The GHS-R mediated Akt and Erk1/2 activation requires PLC, but not PTX-sensitive G proteins
GHS-R1a is a G-protein coupled receptor, and ghrelin has previously been shown to activate ERK1/2 through a Gα(i)-dependent pathway in several cell types [34,35]. In an effort to determine if G-protein signals associated with GHS-R1a mediated ghrelin-induced proliferation of T cells, we pretreated the cells for 24 hours with pertussis toxin (PTX, 100 ng/ml), which inactivates the heterotrimeric G-proteins coupled to the GPCRs, and then subsequently examined its effects on ghrelin-mediated T-cell proliferation. The results in Figure 5A reveal that pre-treatment with pertussis toxin failed to inhibit ghrelin-induced proliferation at 24 or 48 h. This is further supported by the lack of any effect of pertussis toxin addition on ghrelin-mediated Akt and ERK1/2 phosphorylation (Figure 5B). In contrast, U73122 (1 μM), an inhibitor of phospholipase C (PLC)-dependent processes, suppressed both ghrelin-induced cell proliferation (at 24 h and 48 h) and Akt and ERK1/2 phosphorylation in a concentration-dependent manner (Figure 5A and B). As Gαq is known to activate PLC [23], these results suggest that ghrelin increases cell proliferation via a Gαq- and PLC-dependent pathway.
Figure 5. Evidence that the effects of ghrelin on T cell proliferation and Akt and ERK1/2 phosphorylation are mediated by PLC.

D0.11.10 cells (3×103) were incubated in the presence or absence of ghrelin (10 nM) for the indicated time periods with pertussis toxin (PTX; 100 ng/ml), U73122 (1 μM), BAPTA-AM or vehicle alone. (A) Cell proliferation was examined at 24 and 48 h as determined by the MTS assay. The data are expressed as % cell proliferation, and the values represent the mean and SEM of determinations made in three independent experiments. *P<0.05, **P<0.01 compared to 0 h (control); #P<0.05, ##P<0.01 compared to ghrelin treatment. (B) GHS-R1a-transfected D0.11.10 cells (2 × 106) were incubated with different concentrations of PTX or U73122 in the presence or absence of ghrelin at 10 nM for 7 min after which the cultures were lysed and examined by immunoblot analysis using antibodies specific for phospho-Akt and ERK1/2. (C) Cells were pre-incubated with 0, 3, 10 and 30 μM BAPTA-AM for 1 h prior to ghrelin treatment for 7 min, at which time cells were harvested and processed for immunoblot analysis. Experiments were repeated 4–5 times and representative blots are shown. The numbers under each blot are the intensity of the indicated band relative to that of the β-actin band.
The activation of PLC stimulates the conversion of PIP2 to IP3 and DAG, resulting in the release Ca2+ from intracellular stores and activation of PKC, respectively. To determine if the effects of ghrelin on Akt and Erk1/2 phosphorylation depends on the release of intracellular calcium, T cells were treated with the Ca2+ chelator, BAPTA-AM (10 μM), and the effects of ghrelin on Akt, ERK 1/2 and PKC activation were determined (Figure 5C). Similarly with the inhibitory effects of wortmannin (Figure 3), treatment of T cells with BAPTA-AM significantly inhibited ghrelin-induced Akt and Erk1/2 phosphorylation (Figure 5C), as well as PKC activation, suggesting that ghrelin-induced activation of T cells involves the release of intracellular Ca2+ and the activation of PKC.
PKCγ and PKCζ are specifically activated by ghrelin
Previous reports have demonstrated that ligation of GHS-R1a with ghrelin or receptor-specific agonists results in an increase in intracellular Ca2+ levels, following Ca2+ release from IP3–sensitive stores [36]. This Ca2+ release subsequently leads to the activation of various PKC isoforms. Using a pan anti-phosphorylated PKC antibody, we observed that ghrelin indeed induces PKC phosphorylation in GHS-R1a-transfected D0.11.10 T cells cells (Figure 5C). The PKC isoforms PKCγ and PKCζ were specifically phosphorylated upon treatment of GHS-R1a-transfected D0.11.10 T cells with acylated ghrelin (Figure 6A and 6B). Little or no effect of ghrelin was observed for the PKC isoforms α, β, μ, δ, or θ (Figure 6A). As expected, stimulation of the cells with the phorbol ester PMA resulted in the phosphorylation of many of the PKC isoforms. Moreover, the data shown in Figure 6B also reveal that the kinetics of ghrelin-induced phosphorylation of PKCγ and PKCζ are similar to the kinetics of ghrelin-induced Akt and ERK1/2 phosphorylation.
Figure 6. Ghrelin induces the activation of the PKC isoforms PKCγ and PKCξ, which are required for ghrelin-induced activation of Akt and ERK1/2.

(A, B) D0.11.10 GHS-R1a-transfected cells (2 × 106) were treated with ghrelin (10 nM) for the indicated time periods after which the cells were lysed and examined by Western blot analysis for several of the phosphorylated PKC isoforms. Phorbol 12-myristate 13-acetate (PMA, 50 ng/ml) was utilized in these studies as a positive control as it is capable of activating several forms of PKC. Actin levels were included to demonstrate that equivalent quantities of protein were loaded in each lane. (C) D0.11.10 cells were pretreated with various PKC inhibitors for 30 min after which the cells were treated with ghrelin (10 nM) or 50 ng/ml of PMA for 7 min. After incubation, cell lysates were prepared and Akt and ERK1/2 phosphorylation were examined. Experiments were repeated 5 times and representative blots are shown. The numbers under each blot are the intensity of the indicated band relative to that of the β-actin band. (D) PKC activity of the particulate fraction was measured using the Assay Designs non-radioactive PKC activity ELISA assay kit as described in Materials and Methods. The values are the mean and SEM for three independent experiments. *p<0.05, **P<0.01 compared to PBS control. #P<0.05, ##P<0.01.
To determine if PKC activation is involved in the ghrelin-induced activation of Akt and ERK1/2, we treated the GHS-R1a-transfected D0.11.10 T cells with the PKC inhibitors Gö6983 (10 μM) and GF109203X (2 μM). Gö6983 inhibits PKCα, PKCβ, PKCγ, PKCδ and PKCζ, whereas GF109203X inhibits PKCα, PKCβ, PKCγ, PKCδ and PKCε. We found that phosphorylation of both phospho-Akt and –ERK1/2 was inhibited using either of the PKC inhibitors (Figure 6C and 6D). Together, these results suggest that ghrelin-induced T-cell proliferation and the activation of Akt and ERK1/2 is associated with the activation of PKCγ and PKCζ.
Ghrelin promotes expression of cell cycle proteins
When cells enter the cycle from G0, Cyclin D1 is a key protein involved in phosphorylation and consequent inactivation of the cell cycle inhibitor retinoblastoma protein (Rb) [37]. Both of these cell cycle regulatory proteins are regulated by the ERK1/2 and Akt signaling pathways [38]. Based on our cell proliferation and protein phosphorylation findings, we examined the effects of ghrelin on the expression of cell-cycle regulatory proteins at various times after ghrelin treatment. Total cyclin D1 protein levels were significantly increased by ghrelin, with significant effects being observed 2 h post treatment and maximal levels at 48 h, corresponding to the optimal time period for ghrelin-induced proliferation in GHS-R1a-transfected D0.11.10 T cells (Figure 7A). Similar increases in ghrelin-induced protein expression were observed for cyclin E (between 2 and 24 h) and CDK2 (between 24 and 48 h). In addition, ghrelin also induced the phosphorylation of Rb at S807/811, but not at S780, within 1 h (Figure 7A). No significant effects were observed on CDK4 expression in response to ghrelin treatment.
Figure 7. Analysis of ghrelin-induced expression of cell-cycle proteins and entry in D0.11.10 cells expressing GHS-R1a.

(A) D0.11.10 cells (2 × 106) were treated with ghrelin (10 nM) for various time intervals after which cell lysates were prepared and subsequently examined by immunoblot analysis using antibodies specific for the phosphorylated forms of Rb or the total proteins for cyclin D1, cyclin E, CDK 2 and CDK4. Experiments were repeated 4 times and representative blots are shown. The numbers under each blot are the intensity of the indicated band relative to that of the β-actin band. (B) D0.11.10 cells (1 × 106) were treated with ghrelin (10 nM) for 12 h after which the cells were fixed with 70% ethanol for an additional 16 h, after which the cells were stained with 500 μl of PI staining buffer. The cells were analyzed by flow cytometry for cell-cycle progression. Values are the mean +/− SEM of three independent experiments. *P<0.05.
To determine if ghrelin can influence cell-cycle progression in the GHS-R1a-expressing D0.11.10 T cells, cell-cycle analysis was performed using propidium iodide (PI) staining of cells at 6 and 12 h post ghrelin stimulation. As shown in Figure 7B, the PI-staining demonstrated a lower proportion of cells in G1 phase (~55.6%) compared with the control cells (~60.2%) at 12 h after ghrelin treatment, while ghrelin-treated cells demonstrated a higher proportion of cells in S phase compared to control cells at the 12 h time period (Figure 7B).
Ghrelin induces primary murine T cell proliferation in a GHS-R1a-specific manner
To establish the physiological relevance of the signaling pathways through which ghrelin promotes the proliferation of GHS-R1a-transfected D0.11.10 T cells, we next examined the ability of acylated ghrelin and desacyl ghrelin (which cannot bind GHS-R1a), to promote the proliferation of T cells from C57BL/6 mice in concert with T-cell receptor crosslinking. T cells were treated with acylated ghrelin or desacyl ghrelin on plates coated with anti-CD3 and CD28 monoclonal antibodies. T cell proliferation was analysed after 3 days of stimulation. Acylated ghrelin induced a dose-dependent increase in anti-CD3/CD28-mediated CD4+ T cell proliferation, whereas deacyl ghrelin failed to enhance T cell proliferation (Figure 8A). By contrast, both acylated ghrelin and desacyl ghrelin failed to influence T cell proliferation when using GHS-R-deficient mouse CD4+ T cells (Figure 8C). These data indicate that the effect of ghrelin on CD4+ T cell proliferation specifically depends on the GHS-R1a receptor.
Figure 8. Ghrelin stimulation of primary CD4+ T cells augments TCR-mediated proliferation and the phosphorylation of Akt or Erk1/2.

(A, B) Purified murine CD4+ T cells were isolated from C57BL/6 mice and treated with various concentrations of acylated ghrelin or desacyl ghrelin (100 nM) or in media alone on plates coated with anti-CD3 (1 μg/ml) and CD28 (2 μg/ml) mAbs. The cells (1 × 105) were incubated for 3 days after which cell proliferation was examined using the MTS assay. (C) CD4+ T cells were isolated from wild-type control mice and GHS-R knockout mice (GHS-R KO) and cultured in the presence or absence of acylated ghrelin (100 nM) or desacyl ghrelin (100 nM) on anti-CD3/CD28 mAb-coated plates and were examined on days 1 and 3 for T cell proliferation. The data for panels A and B are expressed as % proliferation. *P<0.05, **P<0.01 compared to PBS-treated mice. #P<0.05, ##P<0.01. values represent the mean and SEM of determinations made in three independent experiments. (D) Purified murine CD4+ T cells (5 × 106) were treated with acylated ghrelin (100 nM) for the specified time periods after which the cells were lysed and examined by immunoblot analysis for phospho-Akt, ERK1/2, PKC and PKCγ. Experiments were repeated 5 times and representative blots are shown. The numbers under each blot are the intensity of the indicated band relative to that of the β-actin band.
Similarly to GHS-R1a-transfected D0.11.10 T cells, purified murine CD4+ T cells treated with 100 nM of acylated ghrelin demonstrated an increase in Akt, ERK1/2, PKC and PKCγ phosphorylation starting at 5 min of ghrelin treatment (Figure 8D). Many attempts were made to examine the effects of ghrelin in combination with anti-CD3/CD28 crosslinking on these signaling molecules. However, although some modest enhancement in Erk1/2 signaling was noted in cells treated with both ghrelin and anti-CD3/anti-CD28 (Supplemental Figure 2), no effects were observed on Akt activation. Thus, the contribution of ghrelin to the TCR-mediated signaling process remains unclear. Additional work is underway examining additional signaling pathways and the role these may have in the anti-inflammatory and positive proliferative/activation effects on CD4+ T cells.
Next, we examined the effects of ghrelin on thymocyte proliferation in vivo. Mice were injected i.p. with 100 μg/kg acylated ghrelin every 12 h for either 1 or 3 days. Control mice were injected with the same volume of 0.9% saline. Two hours prior to euthanasia, all mice were injected i.p. with 100 mg/kg BrdU, and the thymus was subsequently examined for the number of BrdU-positive cells at day 1 and day 3 of ghrelin treatment. The results in Figure 9A reveal a significant, albeit modest, increase in the numbers of BrdU-positive cells on day 3 in the ghrelin-treated group, as compared to saline-treated mice, suggesting that ghrelin can promote thymocyte proliferation in vivo. Only modest changes in proliferating cells were observed at 24 h, with no effect on proliferation in sham control mice. Moreover, similarly to what was observed with GHS-R1a-transfected D0.11.10 T cells and primary CD4+ T cells, thymocytes derived from mice that had been treated with 100 μg/kg acylated ghrelin every 12 h for 1 day demonstrated an increase in cyclin D1 expression and in phosphorylation of Rb (at S807/811, but not at S780Rb), Akt and ERK1/2 (Figure 9B).
Figure 9. In vivo administration of ghrelin results in increased thymocyte proliferation and ghrelin receptor signaling in C57BL/6 mice.

(A) C57BL/6 mice (11 week) were injected with acylated ghrelin at 100 μg/kg and the thymuses were examined on day 3 using 5-bromo-2′-deoxyridine (BrdU) staining. The number of BrdU-positive cells was examined in the thymic sections from each animal (n = 5) and the data are expressed as BrdU-positive cells (+/− SEM). **P<0.01. (B) Thymocytes derived from two control and two ghrelin-treated mice over a 24 h time period were examined for Erk1/2, Akt, Rb and cyclin D1 levels by immunoblot analysis.
Finally, the effects of ghrelin on thymocyte proliferation were investigated in middle-aged (12 month-old) and aged (22 month-old) mice. Mice were treated with acylated ghrelin or with PBS for 2 weeks using osmotic mini-pumps, and then thymocyte numbers were assessed. Aged and middle-aged mice demonstrated a significant increase in thymocyte cell numbers in response to acylated ghrelin infusion when compared to PBS sham control mice (Supplemental Table 1). These findings support our previous observations [1,3,5] and suggest that ghrelin can be used as a therapeutic tool for facilitating thymocyte restoration and proliferation in aging and stressed animals.
Discussion
Our findings demonstrate that ghrelin exerts a direct effect on the activation and proliferation of murine T cells in a PI3K-Akt- and ERK1/2-dependent manner (Figure 10). It was previously reported that ghrelin can activate ERK1/2 via a Gq-dependent pathway that involves PI3K/Akt [39]. The activation of this pathway mediates the inhibitory effect of ghrelin on endothelial cell apoptosis [39]. Similar findings have been obtained with cardiomyocytes, neuronal cell populations, 3T3-L1 cells and several tumor cell types [19,23,24,40,41].
Figure 10. GHS-R1a-mediated ghrelin signaling in murine CD4+ T cells.

Ligation of acylated ghrelin to the GHS-R1a receptor results in the activation of the kinases PI3K/AKT and MAPK, which \ play important roles in T cell activation and proliferation. PLCβ is activated through GHS-R1a signaling via the activation of Gαq associated subunits resulting in the phosphorylation of PI3K and AKT. Phosphorylation of AKT at Thr-473/308 by PI3K results in the activation of protein kinase activity. PLCβ activates protein kinase C (PKC) γ and ζ and induces the phosphorylation of Erk1/2 resulting in the phosphorylation of cyclin D1 and E1, which play an important role in cell cycle progression. AKT also regulate the Erk1/2 and the activation of cyclin D1 and E1 and the downstream cyclin D1 associated kinases (CDKs). Phosphorylation of Ser795 and Ser807/811in Rb is thought to be mediated by the CDK4/6 and CDK2. Rb binds to the E2F-1 transcription factor preventing it from interacting with the cell’s transcription machinery. However, upon phosphorylation, E2F mediates the transactivation of target genes that encode proteins that help to facilitate the G1/S transition and S-phase. Thus, ghrelin receptor signaling influences T cell activation and promotes T cell proliferation directly (as observed with T-cell hybridoma cells) or possibly via costimulatory pathways (as observed with primary CD4+ T cells). Ghrelin may counteract the adverse effects of chronic stress and aging on T cell proliferation and survival.
Here, we show that ligation of acylated ghrelin to the GHSR-1a receptor on T cells results in the activation of the kinases PI3K/Akt and MAPK, which are known to play an important role in T cell activation and proliferation. PLCβ is activated through GHS-R1a signaling via the activation of Gαq associated subunits resulting in the phosphorylation of PI3K and Akt. Phosphorylation of Akt at Thr-473/308 by PI3K results in increased activity of PKCγ and PKCζ. The ghrelin-induced activation of Akt and Erk1/2 also results in the induction of cyclin D1 and cyclin E1 expression and activation of the downstream cyclin D1-associated kinases (CDKs), which play important roles in cell cycle progression. Overall, these data support a role for ghrelin signaling in T cell activation and proliferation directly (as observed in D0.11.10 cells) or possibly via costimulatory pathways (as observed in primary CD4+ T cells). It should be noted that although our cell culture experiments show that ghrelin directly simulates T cell proliferation through the ghrelin receptor, we cannot rule out the possibility that in vivo ghrelin impacts thymocyte and T cell proliferation also indirectly, for example through stimulation of growth hormone production.
The involvement of ERK, Akt and PI3K pathways in the proliferative response of T cells to ghrelin does not exclude the possible involvement of a receptor type other than a G-protein-coupled receptor. Recent findings suggest the existence of active acylated ghrelin and desacyl-ghrelin binding sites on proteins other than GHS-R1a [19,42]. For example, activation of ERK and PI3K/Akt pathways was reported to be mediated not only by acylated ghrelin via GHS-R1a, but also by desacyl ghrelin via another unidentified receptor, and this effect could not be blocked by the addition of the GHS-R1a antagonist, (D-Lys3)-GHRP-6 [43]. In another study, desacyl ghrelin was reported to activate Ca2+-activated K+ channels in mesenteric vascular endothelial cells via a GHS-R1a-independent mechanism [42]. However, the latter ghrelin signaling mechanism was not evident in T cells, as we observed no discernable effect of desacyl ghrelin on T cell proliferation.
Ghrelin differentially affects cell growth depending on the cell type. Many endocrine and non-endocrine neoplastic cells have been shown to express ghrelin and GHS-R1a, suggesting that this hormone plays a role in the autocrine/paracrine regulation of tumor growth and/or metastasis [44]. In thyroid and breast cell lines, ghrelin inhibits serum-induced proliferation [45], while in other cell types, ghrelin stimulates cellular proliferation [3,5,36,46]. Ghrelin stimulates proliferation of vascular endothelial cells [24], pancreatic β-cells [25], cardiomyocytes [47], adipocytes [41], adrenal cortical cells [48] and prostate cancer cells [45]. On the other hand, ghrelin inhibits the proliferation of aortic smooth muscle cells [49] and human umbilical vein endothelial cells [50]. Interestingly, whereas we found here that ghrelin enhanced the proliferation of peripheral CD4+ murine T cells upon activation with anti-CD3/CD28 mAb and thymic murine T cells upon in vivo ghrelin administration, it has been reported that ghrelin can also inhibit the proliferation of splenic T cells when they are stimulated with anti-CD3 [51]. These differential effects of ghrelin on T cell proliferation may depend upon the niche in which the T cells reside, the state of immune cell activation and the activation stimuli used to promote cell proliferation. In addition to affecting T cell proliferation, we recently demonstrated that ghrelin may act to inhibit pro-inflammatory cytokine expression [27] and Th17 development [28], possibly shifting T cell responses towards regulatory phenotypes. The latter effects of ghrelin would be expected to suppress inflammation and, as our data suggest, may contribute to the preservation of the thymic T cell population by ghrelin during aging and in the presence of GCs.
Stimulation of proliferation of thymic T cells by ghrelin, via the kinase cascades established in the present study, may contribute to previously reported effects of fasting/dietary energy restriction and stress responses on the thymus and T cells, as well as the actions of ghrelin in maintaining thymus T cell numbers and immune function during aging (Table 1) [1,3,27]. Dietary energy restriction, which typically results in increased levels of circulating acylated ghrelin, has been reported to preserve T cell numbers during aging in rhesus monkeys [52] and can reduce age-related thymic involution in mice [53]. Caloric restriction enhanced the responsiveness of splenic T cells to concanavalin A consistent with the ability of caloric restriction to sustain immune function during aging [54]. Interestingly, similar to ghrelin, caloric restriction inhibited the proliferation of splenic T cells [55], while increasing the numbers of T cells in the thymus [53]. A large-scale transcriptome analysis of the thymus in young, middle-age and old mice that had been maintained on either a usual diet or caloric restriction revealed that many age-related changes in gene expression are attenuated or prevented by caloric restriction [56]. Although the role of ghrelin in the latter effects of caloric restriction on the aging thymus has not been established, the elevation of ghrelin levels during caloric restriction suggests a potential role for ghrelin and the signaling pathways identified in the present study in preservation of T cells by caloric restriction. Moreover, our findings that ghrelin treatment for 24 h also significantly restored the thymocyte loss induced by glucocorticoid treatment (Figure 1) suggests that ghrelin could be a potential therapeutic tool for the treatment of conditions of physiological stress that promote thymic involution, such as adrenal activation, energy restriction, inflammation, chemotherapeutic ablation [3,5]. A greater understanding of the interactions between metabolic mediators and hormones with immune cell may reveal novel signaling pathways that influence immune function and inflammation as well as define some of mechanisms by which the various neuroendocrine and immune systems interact with each other under conditions of disease, stress and aging.
Supplementary Material
Figure S1. (A) Cell proliferation in response to ghrelin was examined using cells transfected to express GHR-R1a and as described in the Materials and Methods and shown in Figure 1, Cells were pretreated with either saline vehicle alone or D-[Lys3]GHRP-6 (20 uM) for 30 min after which the cells were cultured in the presence or absence of ghrelin (10 nM) or vehicle in 96-well plates for 3 days at which time cell proliferation was evaluated using the MTS assay. The data are expressed as % cell proliferation (SEM). *P<0.05, **P<0.01. These data demonstrate that the ghrelin-mediated effects on D0.11.10 proliferation are mediated through the GHS-R1a receptor and are not non-specific. (B) Cell proliferation in response to ghrelin was examined using vector alone or GHS-R1a-transfected D0.11.10 cells and cell proliferation was examined at various points over a 48hr time period. The data are expressed as % cell proliferation (SEM). In all cases, the addition of ghrelin to the GHS-R1a-transfected cells resulted in significantly greater proliferation in comparison to the vector-transfected cells or the GHS-R1a-transfected cells cultured alone in media. *P<0.05, **P<0.01. (C) While the vector-transfected cells do indeed expression endogenous GHS-R1a on their surface (see Figure 1), they do not proliferate in response to ghrelin as effectively or significantly over various time periods. All data presented here are representatives of 3 independent experiments.
Figure S2. The effects of ghrelin stimulation of primary CD4+ T cells in the presence or absence of TCR and CD28 crosslinking. Similar to other figures, T cells were treated with acylated ghrelin (10 and 100 nM) for the specified time periods after which the cells were lysed and examined by immunoblot analysis for the combined effects on activation induced for phospho-AKT and ERK1/2 levels. The results demonstrate that while a slight augmentation in ERK1/2 phosphorylation was observed using a combination of ghrelin and TCR crosslinking, the effect was modest versus ghrelin treatment alone. Moreover, the high degree of AKT phosphorylation in response to CD3/CD28 crosslinking made the examination of the effects of ghrelin difficult (even at various time points), while ghrelin treatment alone resulted in some modest effects on both ERK1/2 and AKT signaling.
Ghrelin enhances the cellularity of thymuses in 6- or 22-month old C57BL/6 mice. Ghrelin or PBS infusion for 2 weeks via subcutaneous osmotic mini-pumps into middle aged (12 m) or aged (18 m) mice induced a significant increase in total thymocyte numbers. Each group included 5 mice.
Highlights.
Ghrelin enhances the proliferation of murine CD4+ primary T cells and a CD4+ T-cell line.
Ghrelin stimulated the ERK1/2 and Akt signaling pathwaysto enhance T-cell proliferation.
Ghrelin induces the cell cycle proteins cyclin D1, E, CDK2 and phospho-Rb.
Ghrelin stimulates thymocyte proliferation in young and older mice.
Acknowledgments
We thank Gary Collins and Arnell Carter for technical assistance. This work was supported by the Intramural Research Program of the National Institute on Aging, National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Taub DD, Longo DL. Insights into thymic aging and regeneration. Immunological reviews. 2005;205:72–93. doi: 10.1111/j.0105-2896.2005.00275.x. [DOI] [PubMed] [Google Scholar]
- 2.Taub DD, Murphy WJ, Longo DL. Rejuvenation of the aging thymus: growth hormone-mediated and ghrelin-mediated signaling pathways. Curr Opin Pharmacol. 2010;10:408–424. doi: 10.1016/j.coph.2010.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Berczi I, Quintanar-Stephano A, Kovacs K. Neuroimmune regulation in immunocompetence, acute illness, and healing. Ann N Y Acad Sci. 2009;1153:220–239. doi: 10.1111/j.1749-6632.2008.03975.x. [DOI] [PubMed] [Google Scholar]
- 4.Baatar D, Patel K, Taub DD. The effects of ghrelin on inflammation and the immune system. Mol Cell Endocrinol. 2011;340:44–58. doi: 10.1016/j.mce.2011.04.019. [DOI] [PubMed] [Google Scholar]
- 5.Billard MJ, Gruver AL, Sempowski GD. Acute endotoxin-induced thymic atrophy is characterized by intrathymic inflammatory and wound healing responses. PLoS One. 2011;6(3):e17940. doi: 10.1371/journal.pone.0017940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pazirandeh A, Jondal M, Okret S. Glucocorticoids delay age-associated thymic involution through directly affecting the thymocytes. Endocrinology. 2004;145:2392–2401. doi: 10.1210/en.2003-1660. [DOI] [PubMed] [Google Scholar]
- 7.Belkaya S, et al. Dynamic modulation of thymic microRNAs in response to stress. PLoS One. 2011;6(11):e27580. doi: 10.1371/journal.pone.0027580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gomez-Sanchez CE. Glucocorticoid production and regulation in thymus: of mice and birds. Endocrinology. 2009;150:3977–3979. doi: 10.1210/en.2009-0615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Qiao S, Okret S, Jondal M. Thymocyte-synthesized glucocorticoids play a role in thymocyte homeostasis and are down-regulated by adrenocorticotropic hormone. Endocrinology. 2009;150:4163–4169. doi: 10.1210/en.2009-0195. [DOI] [PubMed] [Google Scholar]
- 10.Chen Y, Qiao S, Tuckermann J, Okret S, Jondal M. Thymus-derived glucocorticoids mediate androgen effects on thymocyte homeostasis. FASEB J. 2010;24:5043–5051. doi: 10.1096/fj.10-168724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Qiao S, Chen L, Okret S, Jondal M. Age-related synthesis of glucocorticoids in thymocytes. Exp Cell Res. 2008;314:3027–3035. doi: 10.1016/j.yexcr.2008.06.014. [DOI] [PubMed] [Google Scholar]
- 12.Herold MJ, McPherson KG, Reichardt HM. Glucocorticoids in T cell apoptosis and function. Cell Mol Life Sci. 2006;63:60–72. doi: 10.1007/s00018-005-5390-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dixit VD, et al. Ghrelin promotes thymopoiesis during aging. J Clin Invest. 2007;117:2778–2790. doi: 10.1172/JCI30248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen C-Y, Asakawa A, Fujimiya M, Lee S-D, Inui A. Ghrelin gene products and the regulation of food intake and gut motility. Pharmacol Rev. 2009;61:430–481. doi: 10.1124/pr.109.001958. [DOI] [PubMed] [Google Scholar]
- 15.Davenport AP, et al. International Union of Pharmacology. LVI. Ghrelin receptor nomenclature, distribution, and function. Pharmacol Rev. 2005;57:541–546. doi: 10.1124/pr.57.4.1. [DOI] [PubMed] [Google Scholar]
- 16.Yamazaki M, et al. Ghrelin-induced growth hormone release from isolated rat anterior pituitary cells depends on intracellullar and extracellular Ca2+ sources. J Neuroendocrinol. 2004;16:825–831. doi: 10.1111/j.1365-2826.2004.01237.x. [DOI] [PubMed] [Google Scholar]
- 17.Tesauro M, Schinzari F, Caramanti M, Lauro R, Cardillo C. Cardiovascular and metabolic effects of ghrelin. Curr Diabetes Rev. 2010;6:228–235. doi: 10.2174/157339910791658871. [DOI] [PubMed] [Google Scholar]
- 18.Baldelli R, Bellone S, Broglio F, Ghigo E, Bona G. Ghrelin: a new hormone with endocrine and non-endocrine activities. Pediatr Endocrinol Rev. 2004;2:8–14. [PubMed] [Google Scholar]
- 19.Veldhuis JD, Bowers CY. Integrating GHS into the ghrelin system. Int J Pept. 2010 doi: 10.1155/2010/879503. pii: 879503. Epub 2010 Mar 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Malagón MM, et al. Intracellular signaling mechanisms mediating ghrelin-stimulated growth hormone release in somatotropes. Endocrinology. 2003;144:5372–5380. doi: 10.1210/en.2003-0723. [DOI] [PubMed] [Google Scholar]
- 21.Mousseaux D, et al. Regulation of ERK1/2 activity by ghrelin-activated growth hormone secretagogue receptor 1A involves a PLC/PKCvarepsilon pathway. Br J Pharmacol. 2006;148:350–365. doi: 10.1038/sj.bjp.0706727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mladenov MI, Hristov KL, Duridanova DB. Ghrelin suppression of potassium currents in smooth muscle cells of human mesenteric artery. Gen Physiol Biophys. 2006;25:333–338. [PubMed] [Google Scholar]
- 23.Granata R, et al. Acylated and unacylated ghrelin promote proliferation and inhibit apoptosis of pancreatic beta-cells and human islets: involvement of 3′,5′-cyclic adenosine monophosphate/protein kinase A, extracellular signal-regulated kinase 1/2, and phosphatidyl inositol 3-Kinase/Akt signaling. Endocrinology. 2007;148:512–529. doi: 10.1210/en.2006-0266. [DOI] [PubMed] [Google Scholar]
- 24.Rossi F, et al. Ghrelin induces proliferation in human aortic endothelial cells via ERK1/2 and PI3K/Akt activation. Peptides. 2008;29:2046–2051. doi: 10.1016/j.peptides.2008.07.002. [DOI] [PubMed] [Google Scholar]
- 25.Li A, Cheng G, Zhu Gh, Tarnawski AS. Ghrelin stimulates angiogenesis in human microvascular endothelial cells: Implications beyond GH release. Biochem Biophys Res Commun. 2007;353:238–243. doi: 10.1016/j.bbrc.2006.11.144. [DOI] [PubMed] [Google Scholar]
- 26.Lodeiro M, Theodoropoulou M, Pardo M, Casanueva FF, Camiña JP. c-Src regulates Akt signaling in response to ghrelin via beta-arrestin signaling-independent and -dependent mechanisms. PLoS One. 2009;4(3):e4686. doi: 10.1371/journal.pone.0004686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dixit VD, et al. Ghrelin inhibits leptin- and activation-induced proinflammatory cytokine expression by human monocytes and T cells. J Clin Invest. 2004;114:57–66. doi: 10.1172/JCI21134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dixit VD, et al. Reduction of T cell-derived ghrelin enhances proinflammatory cytokine expression: implications for age-associated increases in inflammation. Blood. 2009;113:5202–5205. doi: 10.1182/blood-2008-09-181255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marrack P, et al. The major histocompatibility complex-restricted antigen receptor on T cells. II. Role of the L3T4 product. J Exp Med. 1983;158:1077–1091. doi: 10.1084/jem.158.4.1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sun Y, Wang P, Zheng H, Smith RG. Ghrelin stimulation of growth hormone release and appetite is mediated through the growth hormone secretagogue receptor. Proc Natl Acad Sci U S A. 2004;101:4679–4684. doi: 10.1073/pnas.0305930101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cidlowski JA, et al. The biochemistry and molecular biology of glucocorticoid-induced apoptosis in the immune system. Recent Prog Horm Res. 1996;51:457–490. [PubMed] [Google Scholar]
- 32.Okamoto N, Tezuka K, Kato M, Abe R, Tsuji T. PI3-kinase and MAP-kinase signaling cascades in AILIM/ICOS- and CD28-costimulated T-cells have distinct functions between cell proliferation and IL-10 production. Biochem Biophys Res Commun. 2003;310:691–702. doi: 10.1016/j.bbrc.2003.09.065. [DOI] [PubMed] [Google Scholar]
- 33.Rincón M, Pedraza-Alva G. JNK and p38 MAP kinases in CD4+ and CD8+ T cells. Immunol Rev. 2003;192:131–142. doi: 10.1034/j.1600-065x.2003.00019.x. [DOI] [PubMed] [Google Scholar]
- 34.Dezaki K, Kakei M, Yada T. Ghrelin uses Galphai2 and activates voltage-dependent K+ channels to attenuate glucose-induced Ca2+ signaling and insulin release in islet beta-cells: novel signal transduction of ghrelin. Diabetes. 2007;56:2319–2327. doi: 10.2337/db07-0345. [DOI] [PubMed] [Google Scholar]
- 35.Dimitrova DZ, et al. Ghrelin signaling in human mesenteric arteries. J Physiol Pharmacol. 2010;61:383–390. [PubMed] [Google Scholar]
- 36.Hattori N. Expression, regulation and biological actions of growth hormone (GH) and ghrelin in the immune system. Growth Horm IGF Res. 2009;19:187–197. doi: 10.1016/j.ghir.2008.12.001. [DOI] [PubMed] [Google Scholar]
- 37.Zukerberg LR, et al. Expression of the retinoblastoma protein in low-grade B-cell lymphoma: relationship to cyclin D1. Blood. 1996;88:268–276. [PubMed] [Google Scholar]
- 38.Liang J, Slingerland JM. Multiple roles of the PI3K/PKB (Akt) pathway in cell cycle progression. Cell Cycle. 2003;2:339–345. [PubMed] [Google Scholar]
- 39.Camiña JP, Lodeiro M, Ischenko O, Martini AC, Casanueva FF. Stimulation by ghrelin of p42/p44 mitogen-activated protein kinase through the GHS-R1a receptor: role of G-proteins and beta-arrestins. J Cell Physiol. 2007;213:187–200. doi: 10.1002/jcp.21109. [DOI] [PubMed] [Google Scholar]
- 40.Baldanzi G, et al. Ghrelin and des-acyl ghrelin inhibit cell death in cardiomyocytes and endothelial cells through ERK1/2 and PI 3-kinase/AKT. J Cell Biol. 2002;159:1029–1037. doi: 10.1083/jcb.200207165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kim MS, et al. The mitogenic and antiapoptotic actions of ghrelin in 3T3-L1 adipocytes. Mol Endocrinol. 2004;18:2291–2301. doi: 10.1210/me.2003-0459. [DOI] [PubMed] [Google Scholar]
- 42.Moazed B, Quest D, Gopalakrishnan V. Des-acyl ghrelin fragments evoke endothelium-dependent vasodilatation of rat mesenteric vascular bed via activation of potassium channels. Eur J Pharmacol. 2009;604:79–86. doi: 10.1016/j.ejphar.2008.10.032. [DOI] [PubMed] [Google Scholar]
- 43.Chung H, Seo S, Moon M, Park S. Phosphatidylinositol-3-kinase/Akt/glycogen synthase kinase-3 beta and ERK1/2 pathways mediate protective effects of acylated and unacylated ghrelin against oxygen-glucose deprivation-induced apoptosis in primary rat cortical neuronal cells. J Endocrinol. 2008;198:511–521. doi: 10.1677/JOE-08-0160. [DOI] [PubMed] [Google Scholar]
- 44.Nikolopoulos D, Theocharis S, Kouraklis G. Ghrelin: a potential therapeutic target for cancer. Regul Pept. 2010;163:7–17. doi: 10.1016/j.regpep.2010.03.011. [DOI] [PubMed] [Google Scholar]
- 45.Jeffery PL, Herington AC, Chopin LK. Expression and action of the growth hormone releasing peptide ghrelin and its receptor in prostate cancer cell lines. J Endocrinol. 2002;172:R7–11. doi: 10.1677/joe.0.172r007. [DOI] [PubMed] [Google Scholar]
- 46.Korbonits M, Goldstone AP, Gueorguiev M, Grossman AB. Ghrelin--a hormone with multiple functions. Front Neuroendocrinol. 2004;25(1):27–68. doi: 10.1016/j.yfrne.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 47.Isgaard J, Barlind A, Johansson I. Cardiovascular effects of ghrelin and growth hormone secretagogues. Cardiovasc Hematol Disord Drug Targets. 2008;8(2):133–137. doi: 10.2174/187152908784533676. [DOI] [PubMed] [Google Scholar]
- 48.Mazzocchi G, et al. Ghrelin enhances the growth of cultured human adrenal zona glomerulosa cells by exerting MAPK-mediated proliferogenic and antiapoptotic effects. Peptides. 2004;25(8):1269–1277. doi: 10.1016/j.peptides.2004.05.011. [DOI] [PubMed] [Google Scholar]
- 49.Rossi F, et al. Ghrelin inhibits contraction and proliferation of human aortic smooth muscle cells by cAMP/PKA pathway activation. Atherosclerosis. 2009;203(1):97–104. doi: 10.1016/j.atherosclerosis.2008.06.015. [DOI] [PubMed] [Google Scholar]
- 50.Díaz-Lezama N, et al. Ghrelin inhibits proliferation and increases T-type Ca2+ channel expression in PC-3 human prostate carcinoma cells. Biochem Biophys Res Commun. 2010;403(1):24–29. doi: 10.1016/j.bbrc.2010.10.100. [DOI] [PubMed] [Google Scholar]
- 51.Xia Q, et al. Effects of ghrelin on the proliferation and secretion of splenic T lymphocytes in mice. Regul Pept. 2004;122(3):173–178. doi: 10.1016/j.regpep.2004.06.016. [DOI] [PubMed] [Google Scholar]
- 52.Messaoudi I, et al. Delay of T cell senescence by caloric restriction in aged long-lived nonhuman primates. Proc Natl Acad Sci U S A. 2006;103(51):19448–19453. doi: 10.1073/pnas.0606661103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yang H, Youm Y-H, Dixit VD. Inhibition of thymic adipogenesis by caloric restriction is coupled with reduction in age-related thymic involution. J Immunol. 2009;183(5):3040–3052. doi: 10.4049/jimmunol.0900562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pahlavani MA, Vargas DM. Influence of aging and caloric restriction on activation of Ras/MAPK, calcineurin, and CaMK-IV activities in rat T cells. Proc Soc Exp Biol Med. 2000;223(2):163–169. doi: 10.1046/j.1525-1373.2000.22322.x. [DOI] [PubMed] [Google Scholar]
- 55.Varady KA, et al. Modified alternate-day fasting regimens reduce cell proliferation rates to a similar extent as daily calorie restriction in mice. FASEB J. 2008;22(6):2090–2096. doi: 10.1096/fj.07-098178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lustig A, et al. Transcriptome analysis of age-, gender- and diet-associated changes in murine thymus. Cell Immunol. 2007;245:42–61. doi: 10.1016/j.cellimm.2007.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. (A) Cell proliferation in response to ghrelin was examined using cells transfected to express GHR-R1a and as described in the Materials and Methods and shown in Figure 1, Cells were pretreated with either saline vehicle alone or D-[Lys3]GHRP-6 (20 uM) for 30 min after which the cells were cultured in the presence or absence of ghrelin (10 nM) or vehicle in 96-well plates for 3 days at which time cell proliferation was evaluated using the MTS assay. The data are expressed as % cell proliferation (SEM). *P<0.05, **P<0.01. These data demonstrate that the ghrelin-mediated effects on D0.11.10 proliferation are mediated through the GHS-R1a receptor and are not non-specific. (B) Cell proliferation in response to ghrelin was examined using vector alone or GHS-R1a-transfected D0.11.10 cells and cell proliferation was examined at various points over a 48hr time period. The data are expressed as % cell proliferation (SEM). In all cases, the addition of ghrelin to the GHS-R1a-transfected cells resulted in significantly greater proliferation in comparison to the vector-transfected cells or the GHS-R1a-transfected cells cultured alone in media. *P<0.05, **P<0.01. (C) While the vector-transfected cells do indeed expression endogenous GHS-R1a on their surface (see Figure 1), they do not proliferate in response to ghrelin as effectively or significantly over various time periods. All data presented here are representatives of 3 independent experiments.
Figure S2. The effects of ghrelin stimulation of primary CD4+ T cells in the presence or absence of TCR and CD28 crosslinking. Similar to other figures, T cells were treated with acylated ghrelin (10 and 100 nM) for the specified time periods after which the cells were lysed and examined by immunoblot analysis for the combined effects on activation induced for phospho-AKT and ERK1/2 levels. The results demonstrate that while a slight augmentation in ERK1/2 phosphorylation was observed using a combination of ghrelin and TCR crosslinking, the effect was modest versus ghrelin treatment alone. Moreover, the high degree of AKT phosphorylation in response to CD3/CD28 crosslinking made the examination of the effects of ghrelin difficult (even at various time points), while ghrelin treatment alone resulted in some modest effects on both ERK1/2 and AKT signaling.
Ghrelin enhances the cellularity of thymuses in 6- or 22-month old C57BL/6 mice. Ghrelin or PBS infusion for 2 weeks via subcutaneous osmotic mini-pumps into middle aged (12 m) or aged (18 m) mice induced a significant increase in total thymocyte numbers. Each group included 5 mice.