

Abstract
The goals of the present study were to determine 1) the properties of the nicotinic acetylcholine receptor (nAChR) currents in rat cutaneous DRG neurons; 2) the impact of nAChR activation on the excitability of cutaneous DRG neurons; and 3) the impact of inflammation on the density and distribution of nAChR currents among cutaneous DRG neurons. Whole cell patch clamp techniques were used to study retrogradely labeled DRG neurons from naïve and complete Freund's adjuvant inflamed rats. Nicotine-evoked currents were detectable in ~70% of the cutaneous DRG neurons, where only one of two current types, fast or slow currents based on rates of activation and inactivation, was present in each neuron. The biophysical and pharmacological properties of the fast current were consistent with nAChRs containing an α7 subunit while those of the slow current were consistent with nAChRs containing α3/β4 subunits. The majority of small diameter neurons with fast current were IB4- while the majority of small diameter neurons with slow current were IB4+. Preincubation with nicotine (1 μM) produced a transient (1 min) depolarization and increase in the excitability of neurons with fast current and a decrease in the amplitude of capsaicin-evoked current in neurons with slow current. Inflammation increased the current density of both slow and fast currents in small diameter neurons and increased the percentage of neurons with the fast current. With the relatively selective distribution of nAChR currents in putative nociceptive cutaneous DRG neurons, our results suggest that the role of these receptors in inflammatory hyperalgesia is likely to be complex and dependent on the concentration and timing of acetylcholine release in the periphery.
Keywords: nociceptor sensitization, inflammatory pain, ligand-gated ion channel, voltage-clamp, current clamp
Despite a number of studies on the subject, there is still no consensus on the role of nAChR signaling in sensory neurons. Activation of nAChRs on afferent terminals was initially assumed to be pro-nociceptive, since activation of cationic channels by acetylcholine drive membrane depolarization (Steen and Reeh, 1993, Lang et al., 2003). Consistent with this assumption, there is evidence for nicotine-induced activation and sensitization of nociceptive afferents (Steen and Reeh, 1993, Bernardini et al., 2001). More recently, however, there is evidence that low concentrations of agonist not only desensitize neurons to subsequent nAChR activation, but also desensitize nociceptive neurons to the prototypical algogen, capsaicin (Fucile et al., 2005). Further, there is evidence that α7 selective agonists are analgesic, at least in part via a mechanism in the periphery (Loram et al., 2012). There is also evidence that the algesic effects of nicotine may be mediated via the activation of TRPA1 rather than nAChRs (Talavera et al., 2009), raising doubt as to whether any of the nociceptive actions of nicotine are mediated through nAChRs. However, this last study relied on the use of heterologous expression systems and mouse DRG neurons, and it is becoming increasingly clear that there are species differences in the density and distribution of nAChRs, where, for example, currents are detected in >60% of rat DRG neurons (Genzen et al., 2001, Dube et al., 2005, Hone et al., 2012) but less than 20% of mouse DRG neurons (Talavera et al., 2009, Albers et al., 2014). Thus, it remains to be determined whether nAChRs underlie the algesic vs analgesic actions of nicotine on cutaneous afferents.
There is also uncertainty as to the role of nAChR signaling in the presence of tissue injury. While not extensively studied, the majority of published studies have focused on changes in the presence of nerve injury. In these studies, there appears to be a decrease in nAChR current (Dube et al., 2005). Unless this is a feedback-inhibitory mechanism that counters a number of well-described changes that contribute to an increase in afferent excitability, for a decrease in nAChRs to contribute to neuropathic pain, one would have to conclude that nAChRs normally function to decrease afferent excitability. However, we have recently observed in the mouse that the neurotrophic factor artemin drives an increase in nAChR subunit expression which is associated with an increase in both the density and distribution of nAChR currents in mouse sensory neurons (Albers et al., 2014). Furthermore, given evidence that artemin is increased in peripheral tissue in the presence of inflammation, and artemin-induced hyperalgesia can be attenuated with peripheral administration of the nAChR antagonist hexamethonium (Albers et al., 2014), our results suggest that in the presence of inflammation, nAChR signaling may be pro-nociceptive. Given recent evidence that nicotine attenuates the increase in the excitability of colonic afferents following inflammation of the colon (Abdrakhmanova et al., 2010), this may also be an issue of target of innervation.
The purpose of the present study was therefore to further clarify the role of nAChR activation in nociceptive signaling in sensory neurons in the absence and presence of inflammation. Nicotine-evoked currents were recorded and the impact of nicotine on excitability was assessed using patch clamp methodology in DiI labeled cutaneous DRG neurons from naïve and CFA inflamed rats.
Experimental Procedures
Animals
Adult male Sprague Dawley rats (Harlan-Sprague Dawley, Indianapolis, IN) weighing between 250 and 350 g were used for all experiments. Rats were housed two per cage in the University of Pittsburgh AAALAC approved animal facility on a 12:12 light: dark schedule with food and water ad libitum. All procedures were approved by the University of Pittsburgh Institutional Animal Care and Use Committee and performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. All efforts were made to minimize the number of animals used and their suffering.
Labeling and inflammation
DRG neurons that innervate glabrous skin of the rat hind paw were retrogradely labeled with 1,1’-Dioctadecyl-3,3,3’,3’-tetramethylindocarbocyanine perchlorate (DiI, 17 mg/ml in DMSO and saline), which was injected (3-5 sites at 3-2 μL/site) with a 30g needle 14-17 days prior to electrophysiological recording (Lu and Gold, 2008). Complete Freud's adjuvant (CFA, Sigma-Aldrich, St Louis MO; mixed 1:1 with saline), was injected (100 μL) into the site previously injected with DiI. Inflamed DRG neurons were studied 72 hours after CFA injection. Both DiI and CFA were injected under isofluorane-induced anesthesia.
Preparation of isolated DRG neurons
Prior to tissue harvest, rats were deeply anesthetized with a subcutaneous injection (1 ml/kg) of a cocktail containing ketamine (55 mg/ml), xylaxine (20 mg/ml) and acepromazine (5.5 mg/ml). L4 and L5 DRG were harvested bilaterally. DRG were trimmed of connective tissue, enzymatically treated and mechanically dispersed as previously described (Lu et al., 2006). Isolated neurons were plated on poly-lysine coated coverslips and electrophysiology experiments were performed 2–8 h after plating.
Electrophysiology
Prior to study, neurons were incubated for 10 min in the bath solution used for electrophysiological recording. Whole cell current or voltage data were recorded in a bath solution containing (in mM): NaCl 130, KCl 3, CaCl2 2.5, MgCl2 0.6, HEPES 10, glucose 10; pH 7.4 (adjusted with Tris-base), 325 mOsm (adjusted with sucrose), to which FITC conjugated isolectin B4 (IB4) had been added to a final concentration of 5 μg/ml. Neurons were then placed in a recording chamber continuously superfused with bath solution at room temperature. Retrogradely labeled neurons were identified under epifluoresence illumination. The cell body diameter was determined with a calibrated eyepiece retical. Neurons were considered IB4+ if a clear ring of IB4 staining was visible on the plasma membrane (Stucky and Lewin, 1999). At the end of each experiment, capsaicin sensitivity was assessed with a 4 sec application of capsaicin (500 nM) (Lopshire and Nicol, 1997). Neurons were considered capsaicin responsive (CAP+), if the application of capsaicin resulted in an inward current >20% above the greatest deflection in holding current observed with application of capsaicin vehicle (~10 pA). A neuron was considered a putative nociceptor if it had a small cell body diameter (<30 μm, (Lawson, 2002)), was IB4+ (Fang et al., 2006) and/or CAP+ (Schmelz et al., 2000). Of note, while IB4 is generally used to identify a subpopulation of nociceptive afferents devoid of the neuropeptides substance P and calcitonin gene-related peptide (non-peptidergic neurons), we (Lu et al., 2006) and others (Petruska et al., 2000) have previously observed that there is a majority of both IB4+ and IB4− small diameter neurons that are CAP+. Thus, while these appear to be distinct subpopulations of nociceptive afferents, we do not distinguish among them with our use of the term “putative nociceptor”.
Voltage- and current-clamp data were acquired using conventional whole cell patch clamp techniques with an Axopatch 200B amplifier (Molecular Devices, Sunnyvale CA) controlled with a PC running pClamp Software (V 10.3, Molecular Devices). Current and voltage traces were sampled at 5-10 kHz and filtered at 1-2 kHz. Patch electrodes were pulled from borosilicate glass (WPI, Sarasota FL) on a horizontal puller (Sutter Inst. Novato CA), and were 2-5 MΩ when filled with an electrode solution containing (in mM): K-methanesulfonate 110, KCl 30, NaCl 5, CaCl2 1, MgCl2 2, HEPES 10, EGTA 11, Mg-ATP 2, Li-GTP 1, pH 7.2 (adjusted with Tris-base), 310 mOsm (adjusted with sucrose). Whole cell current or voltage data were recorded in the bath solution described above. The junction potential associated with all test solutions was less than 5 mV and therefore not corrected. Series resistance compensation was >80%. Whole-cell capacitance and series resistance were compensated with amplifier circuitry. Neurons were held at −60 mV when nicotine-evoked current was recorded. For current clamp recording a ramp (250 ms) and hold (250 ms) depolarizing current injection protocol was used to assess the effect of nicotine on neuronal excitability which could be quantified with changes in current threshold, action potential threshold and the number of action potentials evoked in response to the depolarizing current injection (Gold et al., 1996). The magnitude of the current injected was increased by 100 pA steps until an action potential was evoked during the ramp. Once determined, this protocol was used to stimulate neurons every 20 sec. After establishing a stable baseline (6-8 sweeps), 1 or 60 μM nicotine was applied for 5 min and data were collected during and after nicotine application.
Nicotine, cytisine, capsaicin and allyl isothiocyanate (AITC) were applied with a piezo-driven perfusion system (Warner Instruments, Hamden CT) with the tip of the tube 10-13 μm away from the cell. Subpopulations of DRG neurons were defined by cell body size, binding of the plant lectin IB(4) and responsiveness to the algogenic compound capsaicin (CAP).
Data were analyzed with pClamp software in combination with SigmaPlot (Systat, Chicago, IL). Current density was determined by dividing peak-evoked current by membrane capacitance (as determined with a 5 mV voltage step prior to compensation). Concentration-response data were fitted with a Hill equation to generate estimates of peak current (efficacy), the concentration 50% of peak (EC50, potency), and the Hill coefficient.
Behavioral testing
Behavioral measures were made using a plantar testing apparatus (IITC, Woodland Hills, CA) in a blinded manner in the University of Pittsburgh Rodent Behavior Analysis Core. Animals were acclimatized for 1h per day in the thermal testing apparatus for 2 days prior to testing. Animals were placed individually in a small plexiglass enclosure with a heated glass floor set to 32°C. A focused light beam was applied to the ventral surface of the hindpaw and the latency to paw withdrawal determined. This was repeated with an interstimulus interval of 3 min and the mean of three tests was used as the latency at each test time.
To assess the contribution of nAChR activation to inflammatory hypersensitivity, nociceptive behavior was assessed before and after an IP injection (20 mg/kg) of hexamethoniun or saline. While we were interested in the role of nAChRs on peripheral terminals, systemic administration of the nAChR antagonist was used rather than a local injection because in preliminary experiments, the intradermal injection of saline produced a transient decrease in thermal withdrawal latency (not shown), likely as a result of the needle stick, and because hexamethonium is not thought to cross the blood brain barrier (Malin et al., 1997). The dose of hexamthonium used was 4 (Rueter et al., 2003)to 10 (Dickson et al., 2010) times higher than that used previously, to ensure the concentration in the peripheral tissue would be high enough to detect an effect. The experimenter collecting the behavioral data was blinded to the contents of the IP injection.
Chemicals and drugs
AITC, capsaicin, cytisine, HC-0300310031(HC-030031), nicotine, hexamethonium, mecamylamine, methyllycaconitine (MLA) were purchased from Sigma-Aldrich (St. Louis, MO). Stock solutions of capsaicin (10 mM) and mecamylamine (10 mM) were made in 100% ethanol; stock solutions of AITC (100 mM), cytisine (100 mM) and HC-030031 (100 mM) were made in DMSO. A stock solution of MLA (1mM) was made in water. On the day of recording all stock solutions were diluted in bath solution so as to keep the final DMSO concentration less than 0.1%. Nicotine was made on the experiment day with bath solution and used at concentrations between 1 μM and 1 mM. The α7 nAChR subunit selective antagonist MLA was used at a concentration of 20 nM (Genzen et al., 2001). The TRPA1 selective antagonist HC-030031 was used at a final concentration of 10μM based on results from previous studies (Sculptoreanu et al., 2010). The α3β4 nAChR subunit selective agonist cytisine was applied at a concentration of 100 μM (Genzen et al., 2001). The TRPA1 selective agonist AITC was applied at a final concentration of 100 μM based on results from previous studies (Zhang et al., 2014). The TRPV1 selective agonist capsaicin was used at a final concentration of 500 nM (Lu et al., 2006).
Statistics
All pooled data are presented as mean ± standard error of the mean. Student's T-test was used for two-group comparisons. Two-way ANOVA was used to assess the impact of cell body diameter, IB4+ binding or capsaicin sensitivity on the density of current types. Two-way repeated measures ANOVA was used to assess the impact of nicotine on excitability and the impact of inflammation on the concentration-response relationship for the nicotine evoked current, with the Holm-Sidak test used for post-hoc comparisons. A Chi-square test was used to assess the distribution of current types among subpopulations of neurons. p < 0.05 was considered statistically significant.
Results
Analysis of nicotine-evoked currents in cutaneous DRG neurons from naïve rats
Nicotine evokes two distinct current types in cutaneous DRG neurons
A total of 141 DRGs neurons retrogradely labeled from the glabrous skin of the hindpaw from 13 naïve rats were studied. In an initial set of experiments, neurons were challenged with 300 μM nicotine from a holding potential of −60 mV. Three populations of cutaneous neurons were observed: 1) those unresponsive to nicotine, 2) those in which nicotine-evoked a slowly activating and slowly inactivating current (slow current, Fig. 1A), and 3) those in which nicotine-evoked a rapidly activating and rapidly inactivating current (fast current, Fig. 1B). The activation rate (the time from the start to peak) of slow current was an order of magnitude slower than that of the fast current (Figure 1Aa vs 1Bb; 134 ± 8 ms (n =21) vs 10.6 ± 1.1 ms (n=15)). While nicotine was applied for 500 ms in the initial screen of nicotine sensitivity, a 4 second application was required to more accurately assess the presence of inactivation of the slow current (Figure 1Ac). The decay of the slow current in response to a 4 sec application of nicotine could be well fitted with a single exponential revealing a time constant of decay of 1.83±0.40 s (n=9). The fast current was completely inactivated during the 500 ms application of nicotine with a decay that was well fitted with a double exponential, revealing time constants of 16.3 ± 1.9 ms and 61 ± 10.6 ms (n=15, Figure 1Bc).
Figure 1. Two types of nicotine-evoked currents observed in DRG neurons from naïve rats and their concentration-response curves.

Currents were evoked with 300 μMnicotine applied for 500 ms. A. Representative trace showing slow current activated (Ab) and inactivated (Ac) slowly. B. Representative trace showing fast current activated (Bb) and inactivated (Bc) rapidly. The activation rate (the time from the start to peak) of the slow current (Ab) was an order of magnitude slower than that of fast current (Bb). The decay of the slow current in response to a 4 sec application of nicotine could be well fitted with a single exponential (Ac) revealing a time constant of decay of 1.83±0.40 s (n=9). The decay of the fast current was well fitted with a double exponential (Bc) revealing time constants of 16.3 ± 1.9 ms (T1) and 61 ± 10.6 ms (T2; n=15). C. DRG neurons were challenged with increasing concentrations of nicotine ranging from 10 to 1000 μM at an inter-stimulus interval of 3 minutes. Typical concentration responses of fast (left traces) and slow current (right traces) are shown. Note that a more slowly activated current emerged at 1mM nicotine in neurons with fast current (bottom left trace). D. Pooled data fitted with a Hill equation to estimate the EC50 indicate that while slow current density was significantly greater than that of the fast current, the peak evoked fast current was achieved at lower concentrations of nicotine than that of slow currents (EC50 was 61 μM for slow current (n=26 cells) vs 38 μM for fast current (n=16 cells). Inset: Repeated application of 300 μM nicotine at 3 min interval did not change the current amplitude of either the slow or fast current.
Potency and efficacy of the slow and fast current
To confirm that 300 μM nicotine would enable the detection of all nicotine responsive cutaneous neurons as well as determine the potency and efficacy of nicotine-evoked currents, a subpopulation of neurons were challenged with increasing concentrations of nicotine ranging from 10 to 1000 μM (Figure 1C). Based on preliminary data indicating that responses to 300 μM nicotine were stable when nicotine was applied at an inter-stimulus interval of 3 minutes (Figs. 1D inset), this interval was used in this second series of experiments. Consistent with results obtained with 300 μM nicotine, current evoked was either slow or fast (Fig. 1C). Concentration-response data for each neuron was fitted with the modified Hill equation in order to obtain EC50 and Hill coefficients (n). The pooled data from 26 neurons with slow current and 16 neurons with fast current (Fig. 1D) indicated that while slow current density was significantly greater than that of the fast current, the peak of evoked fast currents was achieved at lower concentrations of nicotine than that of slow currents: the EC50 was 38 μM for the fast current and 61 μM for the slow current (Fig. 1D). The concentration response curves for both the slow and fast current confirmed that 300 μM nicotine was saturating and therefore would enable detection of all neurons with a fast or slow nicotine-evoked current.
Pooling data from the initial set of experiments, the concentration response experiments and the subsequent pharmacological experiments, a fast or slow current was observed in 68% (96 of 141) of cutaneous neurons. Of these, slow current was detected in 47 neurons and fast current was detected in 33 neurons. The amplitude of slow current was 1090 ± 265 pA (range 50-10380 pA) while that of the fast current was 260 ± 53 pA (range 50-1325 pA).
Of note, a very slow current was observed in response to the highest concentration of nicotine used (1 mM) in a subpopulation of cutaneous neurons (37 of 50, Fig. 1C). This current was present in a subpopulation of neurons with fast (n=10) or slow (n=14) current as well as neurons (n=13) with neither fast nor slow current. This very slow current was further characterized in experiments described below.
Pharmacological properties of slow and fast currents
We next performed a series of pharmacological experiments to further characterize the slow and fast nicotine-evoked currents in cutaneous neurons. Nicotine (300 μM) was applied before and then three minutes after antagonist application. Both slow and fast currents were blocked by the non-selective nAChR antagonists mecamylamine (Mec) (30 μM, n = 7 and 6 for slow and fast currents, respectively) and hexamethonium (Hex) (100 μM, n = 6 and 5 for slow and fast currents, respectively) (Figs. 2A, 2B). In contrast, the α7 subunit preferring antagonist methyllycaconitine citrate (MLA, 20 nM), completely blocked the fast current (n = 10) but did not affect the slow current (n = 5; Fig. 2C). Finally, to further implicate the contribution of β4 subunit containing nAChRs to the slow current, the relatively β4-subunit selective agonist cytisine (100 μM) was applied to neurons previously challenged with 100 μM nicotine. In 5 of 5 neurons with slow current, the cytosine-evoked current was larger than the nicotine-evoked current (mean = 140 ± 4 % of nicotine-evoked current). In contrast, in 4 of 4 neurons with a fast current, the cytosine-evoked current was significantly smaller than the nicotine-evoked current (mean = 21 ± 5 %, Fig. 2D).
Figure 2. Pharmacological properties of the slow and fast current.

Slow and fast currents were evoked with 300 μM nicotine for 500 ms (indicated by bars above each trace) before and after the application of (A) mecamylamine (MEC, 30 μM), a nAChRs antagonist, (B) hexamethonium (Hex, 100 μM), another general nAChRs antagonist, and (C) methyllycaconitine citrate (MLA), an α-7 subunit selective antagonist. Neurons with slow and fast nicotine-evoked currents were also challenged with cytisine (100 μM) for 500 ms, which selectively activated slow currents, consistent with properties of α3-β4 subunit containing receptors (D).
Distribution of fast and slow currents among subpopulations of cutaneous DRG neurons
To begin to assess the extent to which nicotinic currents in cutaneous neurons contribute to nociceptive processing, we determined the distribution of slow and fast currents among putative nociceptive cutaneous neurons defined by a small cell body diameter (< 30 μm), IB4 binding and capsaicin sensitivity (Lu et al., 2006). Because capsaicin sensitivity was assessed after the analysis of nicotine sensitivity was completed, capsaicin sensitivity was determined in a subpopulation of those in which IB4 binding was assessed. Of the cutaneous neurons with nicotine evoked currents, four were four were negative for IB4 (IB4−) and unresponsive to capsaicin (Cap−), four were IB4− and responsive to capsaicin (Cap+), seven were positive for IB4 binding (IB4+) and Cap-, and 31 were IB4+ and Cap+. A greater proportion of neurons with fast current were IB4− while a greater proportion of neurons with slow current were IB4+ (Fig. 3A, p<0.05, Chi square test). There was no detectable difference between small and medium diameter cutaneous neurons with respect to the presence of fast or slow currents (Fig. 3B; p>0.05, Chi square test). In contrast to this distribution, both fast and slow current density was greater in IB4− neurons than that in IB4+ neurons (Fig. 3C, p<0.01, two-way ANOVA), and slow current density was greater in medium rather than small diameter neurons (Fig. 3D, p<0.01, two-way ANOVA ). Finally, there was no detectable difference between capsaicin responsive (Cap+) and unresponsive (Cap−) neurons with respect to either the prevalence or the current density of either slow or fast currents (Table 1, p>0.05).
Figure 3. Slow and fast current are differentially distributed in different subpopulations of DRG neurons.

Data in (A) and (B) are plotted as a percentage of the total number of neurons studied in each group. Slow and fast current are differentially distributed among IB4+ and IB4− neurons (A, p<0.05, chi-square test), but not in small (<30μm) and medium diameter (>32 μm) neurons (B, p>0.05, chi-square test). In addition to the differences in the distribution of current types among subpopulations of cutaneous DRG neurons, there were also differences in the current density among subpopulations of neurons (C and D). Density data were analyzed with a two-way ANOVA. With a significant interaction between cell body size and current type (D), post-hoc comparisons were made within subpopulations defined by cell body size and within current type. ** p<0.01.
Table 1.
Nicotine-evoked current density in subpopulations of cutaneous neurons
| Subpopulation | Slow – Naïve | Slow – Inflamed | Fast – Naïve | Fast – Inflamed | Very Slow – Naïve | Very Slow – Inflamed |
|---|---|---|---|---|---|---|
| Small | 17.7±2.6 (n=37) | 31.6±5.4** (n=8) | 4.6±1.1 (n=22) | 8.4±1.6* (n=15) | 41±7.8 (30) | 23.1±3.7 (21) |
| Medium | 65.7±22.6 (n=6) | 66.2±35.4 (n=5) | 8.9±2.2 (n=7) | 13.8±7.3 (n=8) | 52±15.8 (7) | 33.0±14.2 (10) |
| IB4+ | 24.8±5.5 (n=27) | 27.7±4.2 (n=14) | 3.7±1.3 (n=16) | 4.1±0.8 (n=10) | 31.7±5.5 (23) | 22.1±3.3 (17) |
| IB4- | 59±37.7 (n=3) | 205.9 (n=1) | 13.5±3.7 (n=6) | 14.7±4.7 (n=12) | 61.7±15 (14) | 32.0±11.4* (14) |
| Cap+ | 27.2±7.1 (n=20) | 35.5±5.5 (n=6) | 5.0±1.2 (n=15) | 9.6±3.0 (n=19) | 38.5±6.0 (26) | 21.3±3.3 (19) |
| Cap- | 45.6±18.8 (n=6) | 61.1±29.1 (n=6) | 10.8±7.4 (n=3) | 15.1 [1.7,28.8] (n=2) | 54.8±18.6 (4) | 24.6±6.7 (7) |
Slow and Fast currents were evoked with 300 μM nicotine. Very slow currents were evoked with 1 mM nicotine. Data are mean ± SEM of current density (pA/pF), except for the group of two Cap- neurons where the actual current density for the two neurons is indicated in brackets. Numbers in parentheses are the number of neurons in each group. Comparisons were made for each subpopulation of neurons between neurons from naïve and inflamed rats within each current type. Data were not corrected for multiple comparisons.
is p < 0.05
is p < 0.01.
Nicotine-induced desensitization of slow and fast current
There is evidence that nicotine, at concentrations considerably lower than those needed for receptor activation, can drive receptor desensitization (Giniatullin et al., 2005). To assess nicotine-induced densitization of nAChRs in cutaneous DRG neurons, neurons were challenged with 60 μM nicotine every 10 min during which they were continually perfused with concentrations of nicotine ranging from 10 nM to 10 μM (Fig. 4A). Data from each neuron was analyzed as a percent decrease in the magnitude of the current evoked with 60 μM nicotine in response to the desensitizing concentrations of nicotine. The impact of time-dependent changes in current magnitude was assessed in 3 control neurons challenged with 60 μM nicotine every 10 min in the absence of nicotine: the current decrease observed in these neurons by the 4th application of nicotine was less than 8% relative to the first application of nicotine. In contrast, there was a concentration dependent inhibition of the nicotine (60 μM) evoked slow current (n=5, Figs. 4A). Pooled data indicate that the IC50 for desensitization of the slow current was 1.9 ± 0.3 μM (Fig. 4B).
Figure 4. Nicotine-induced desensitization of slow and fast currents.

(A) Typical recordings of neuron challenged with 60 μM nicotine every 10 min as they were continually superfused with concentrations of nicotine ranging from 10 nM to 10 μM. There was a concentration-dependent inhibition of the nicotine (60 μM)-evoked slow current. (B) Data from each neuron was analyzed as % Decrease from baseline peak evoked (60 μM nicotine) current in response to the desensitizing concentration of nicotine. Pooled data from 5 neurons indicated that the IC50 for inhibition of the slow current was 1.9 ± 0.3 μM. (C) Fast current desensitization was assessed using 1μM and 3μM nicotine. A 10 min application of 1μM or 3μM of nicotine caused reduction of the fast current evoked with 60 μM nicotine. (D) Pooled data show 1μM induced 46% (n=5) and 3μM induced 60% (n=4) reduction of the 60 μM evoked fast current.
Since the IC50 of nicotine for rat α7 mediated fast current desensitization was reported around 1μM (Giniatullin et al., 2005), instead of applying a series of concentrations, we assessed the fast current desensitization only with 1μM and 3μM nicotine. 10 min application of 1μM and 3μM of nicotine induced 46% (n=5) and 60% (n=4) reduction of 60 μM evoked fast current, respectively (Fig. 4C, 4D) which suggested that the IC50 for nicotine-induced desensitization of the fast current is between 1μM and 3μM.
Nicotine-evoked current in cutaneous DRG neurons does not involve activation of TRPA1
Based on previous results from mouse trigeminal ganglion neurons and heterologous expression of human TRPA1 suggesting that nicotine can activate TRPA1 channels (Talavera et al., 2009), we determined the extent to which TRPA1 may contribute to the nicotine-evoked current in rat DRG neurons. We first assessed the impact of the putative TRPA1 selective antagonist HC-030031. Consistent with results from previous studies (Sculptoreanu et al., 2010), HC-030031 (10 μM) completely blocked current evoked with the TRPA1 selective agonist AITC (100 μM, Fig. 5A, n=3). HC-030031 had no detectable influence on the slow current (Fig. 5B, n=4) evoked by nicotine (300 μM) in cutaneous DRG neurons. However, fast currents were completely blocked by HC-030031 (Fig. 5C, n=8). To investigate a possible interaction between TRPA1 and the nicotine-induced fast current, we assessed the effect of MLA (20 nM) on AITC-evoked current. MLA, applied for 3 min prior to the application of AITC, had a marginal influence on AITC-evoked current (Fig. 5D, n=3). To further assess the possibility that TRPA1 contributed to the fast current evoked by nicotine in cutaneous neurons, a group of neurons were first challenged with nicotine (300 μM) and then AITC (100 μM) 4 minutes later (Figure 5E-H). Of the 20 neurons tested, the proportion of neurons in which both fast nicotine-evoked and AITC-evoked currents were detected was comparable to the proportion of neurons in which either current was detected alone (Fig. 5H). Slow currents were also detected in 7 of the 20 neurons tested, and of these, only 3 were present in neurons responsive to AITC. Of note, in 3 of the neurons tested with fast current in which there was no detectable AITC-evoked current, HC-030031 was still able to completely block the fast current (not shown). To further assess the possibility that TRPA1 contributed to the nicotine-evoked fast current, nicotine-evoked currents were recorded in cutaneous DRG neurons from TRPA1 null mutant mice (n=2 mice). The fast current was still observed in 4 of 48 DRG neurons from TRPA1 null mutant mice and the prevalence of the fast current was comparable with that of DRG neurons from wild type mice (fast current was observed in 13 of 74 neurons (Albers et al., 2014)). Furthermore, the fast current in the neurons from these TRPA1 null mutant mice was still blocked by HC-030031. Taken together, these results suggest that HC-030031 has an “off-target” effect on α7 subunit containing nAChRs.
Figure 5. The TRPA1 antagonist HC-0300310031 blocks nicotine-evoked fast current.

(A) HC-030031 (10 μM) completely blocked current evoked with the TRPA1 selective agonist AITC (100 μM). (B) HC-030031 had no detectable influence on the slow current evoked by nicotine (300 μM) in cutaneous DRG neurons. (C) However, the fast current was completely blocked by HC-030031. (D) Application of MLA (20 nM), which blocks the fast current, did not influence the AITC-evoked current. (E-H) Neurons were first challenged with nicotine (300 μM) and then AITC (100 μM) 4 min later. (E) A neuron with fast current but no AITC-evoked current; (F) A neuron with AITC-evoked current but no fast current; (G) A neuron with both currents. (H) Pooled data from 20 neurons show the proportions of these 3 populations of neurons are similar.
As noted above, in addition to the slow and fast current, a very slow current was observed in 74% (37/50) of cutaneous DRG neurons in response to 1 mM nicotine. Because the activation kinetics of the very slow current was comparable to the currents activated by AITC, we next sought to assess the possibility that TRPA1 contributed to this very slow nicotine-evoked current. Cutaneous neurons were pretreated with either the TRPA1 antagonist HC-030031 (10 μM, n=4, Fig. 6A), or the nAChR antagonist mecamylamine (50 μM, n=4, Fig. 6B) or the α7 nAChR specific antagonist MLA (20 nM, n=4, Fig. 6C) 3 min before nicotine (1mM) on neurons with the very slow current. None of these antagonists had a detectable influence on the very slow current (Fig. 6).
Figure 6. Pharmacological properties of the very slow current.

Currents were evoked with 1 mM nicotine for 500 ms. Neurons with the very slow current were pretreated with either the TRPA1 antagonist HC-030031 (A) or the nAChR antagonist 30 μM mecamylamine (B) or the alpha-7 nAChR specific antagonist 20 nM MLA (C) 3 min before nicotine. None of these antagonists had a detectable influence on the very slow current.
Impact of nicotine on excitability in cutaneous DRG neurons
With evidence that nAChR agonists may be both pro- and anti-nociceptive, we next sought to determine the impact of nicotine on the excitability of cutaneous neurons. The effects of both activating (60 μM) and desensitizing (1 μM) concentrations of nicotine were tested on the excitability of neurons assessed with depolarizing current injection. Excitability was assessed before, during the 5 min application of nicotine, and then for 5 minutes after nicotine application. There was no detectable influence of nicotine (1 μM) on resting membrane potential or excitability in neurons with slow current as illustrated in a plot of the time course of changes in resting membrane potential and the number of action potentials evoked with a ramp and hold protocol (Inset to Fig. 7A, D & E), in slow current neurons treated with 1 μM nicotine (n=6, Fig 7A) or vehicle (n=9, Fig. 7B, D & E). As expected, however, application of 60 μM nicotine to slow current neurons resulted in membrane depolarization and an increase in excitability (Fig. 7C). Depolarization was 13.8 ± 2.7 mV at 20 seconds and 3.6±2.1mV at 1 minute after the application of nicotine and the increase in the number of evoked action potentials was 270 ± 99% at 20 second and 70 ± 33% at 1 minute after the application of nicotine (n=5, Fig. 7C, F and G). These changes were significant (p<0.05, two-way repeated measures ANOVA) compared with control (vehicle treated) neurons (n=6).
Figure 7. Impact of 1 or 60 μM nicotine on the excitability of cutaneous DRG neurons with slow current.

Nicotine (300 μM)-evoked current was recorded at the end of the recording to determine whether the neuron has fast or slow current. All data in this figure were from neurons with slow current. A. Data from a neuron in which 1 μM nicotine was applied for 5 minutes. Resting membrane potential (dotted line) and excitability (voltage traces) was assessed every 20 sec with a ramp (250 ms) and hold (250 ms) depolarizing current injection protocol (shown above the first voltage trace). In this and subsequent panels, voltage traces (a, b, c, and d) were collected at the times indicated on the membrane potential plot. B. Data from a neuron in which vehicle was applied for 5 min, during which a time-dependent hyperpolarization and decrease in excitability was observed. C. Data from a neuron in which 60 μM nicotine was applied for 5 min. The transient depolarization was associated with a dramatic, but transient increase in excitability. D. Pooled resting membrane potential (MP Change) data from neurons challenged with 1 μM nicotine or vehicle (control). E. Pooled excitability data, as manifest by the percent change in the number of action potentials evoked during the ramp and hold protocol (% change in firing), for the same group of neurons as plotted in D. F. Pooled resting membrane potential (MP Change) data from neurons challenged with 60 μM nicotine or vehicle (control). G. Pooled excitability data, plotted as in E, for the neurons in F. There was a time-dependent change both in control and experimental neurons (p<0.001, repeated two way ANOVA). MP change for each time point was calculated as (the average of MP from 3 sweeps in each min- the average of last 3 sweeps of baseline). * p<0.05.
In fast current neurons, 1 μM nicotine resulted in a small but significant (p<0.05, two-way repeated measures ANOVA) membrane potential depolarization (2.7±0.7mV, n= 25) and increase in excitability (103±27% increase in action potentials), over the first but not subsequent minutes of application relative to time dependent changes observed in control neurons (n=10, Fig. 8A, D and E), relative to vehicle treated control neurons (n = 10, Fig 8B, D & E). Interestingly, the impact of 60 μM nicotine was comparable to that of 1 μM nicotine with respect to membrane potential depolarization (2.8±0.8 mV) and the increase in action potential generation (61±51% increase) in fast current neurons (n=12) at 20 sec post nicotine application (n=12, Fig. 8C, F and G), and these changes were significant (p<0.05, two-way repeated measures ANOVA) compared with vehicle treated control neurons (n=10).
Figure 8. Impact of 1 or 60 μM nicotine on excitability in cutaneous DRG neurons with fast current.

Data were collected and analyzed as in Figure 7 except that all neurons had a fast nicotine-evoked current. A. Data from a neuron in which nicotine (1 μM) was applied for 5 minutes resulting in a transient depolarization (dotted line) and an increase in action potential generation in response to the ramp and hold current injection (voltage traces). B. Resting membrane potential and excitability were relatively stable in neurons in which vehicle was applied for 5 minutes. C. Data from a neuron in which 60 μM nicotine was applied for 5 minutes, resulting in a transient depolarization that was associated with a transient increase in excitability. D. Nicotine-evoked current was recorded at the end of the recording to determine if the cell exhibited a fast current. (A) In one control neuron (grey) for which no nicotine was applied, there was no significant time dependent change of MP (top figure) and firing (bottom figure). (B) In one fast current neuron application of 1μM nicotine for 5 min evoked transient depolarization (10 mV, upper) and more firing (bottom) during the first one min. (C) In another fast current neuron application of 60 μM nicotine for 5 min evoked depolarization and more firing. Pooled resting membrane potential (D) and excitability (E) data from neurons treated with 1 μM nicotine or vehicle (control) were analyzed with a two-way ANOVA which confirmed the transient depolarization and increase in excitability associated with nicotine application were significant. Comparable results were obtained from analysis of pooled membrane potential (F) and excitability (G) data from neurons treated with 60 μM nicotine. * p<0.05.
Nicotine suppresses capsaicin-evoked currents in slow but not fast current cutaneous DRG neurons
As there is at least one report in the literature suggesting that antinociceptive actions of nicotine may be secondary to the suppression of TRPV1 activity (Fucile et al., 2005), we next sought to determine the impact of nicotine application on capsaicin-evoked currents in cutaneous DRG neurons with slow or fast currents. The magnitude of the capsaicin (500 nM applied for 4 sec)-evoked current was significantly larger in neurons with fast current than those with slow current (Fig. 9: 273 ± 42 pA/pF (n=10) vs 101 ± 32 pA/pF (n=15), p<0.001). In fast current neurons, neither 1 μM (n=9) nor 60 μM (n=13) nicotine applied for 5 minute prior to the application of capsaicin had a detectable influence on the amplitude of the capsaicin current relative to control treated neurons (p>0.05, Fig. 9A, C). However, in slow current neurons, both concentrations significantly reduced capsaicin-induced current (p<0.05, Fig. 9B, C).
Figure 9. The impact of nicotine on capsaicin responses in cutaneous DRG neurons with fast or slow current.

Neurons were preincubated 5 min with either 1 or 60 μM nicotine and then capsaicin (500 nM applied for 4 sec) evoked inward currents were recorded. At the end of recording 300 μM nicotine was applied to determine if the cell exhibited a fast or slow current. The same protocol was used in control neurons but without nicotine preincubation. A. Capsaicin-evoked currents in a cutaneous neuron with fast nicotine-evoked current pre-incubated with 1 μM or 60μM nicotine for 5 min prior to capsaicin application. B. Capsaicin-evoked currents in a cutaneous neuron with slow nicotine-evoked current pre-incubated with 1 μM or 60μM nicotine for 5 min prior to capsaicin application. C. Pooled peak capsaicin-evoked current density from DRG neurons with fast and slow current; note that capsaicin-evoked inward current density in slow current control neurons was much smaller than in fast current control neurons (101±32 (n=15) vs 273±42 (n=10), p<0.001). * p<0.05.
Inflammation increases slow and fast current density in small DRG neurons
Given evidence that artemin expression is increased in the presence of inflammation and our recent evidence that nicotine-evoked current density is increased in artemin over-expressing mice (Albers et al., 2014), we assessed the impact of inflammation on the density and distribution of nicotine-evoked currents among cutaneous DRG neurons.
A new type of current is present after inflammation
Nicotine-evoked currents were detected in 70.5% (48/68) of cutaneous DRG neurons from 7 inflamed rats, which included slow current in 37.5% (18/48) and fast current in 52% (25/48) of the responsive neurons. A new or emergent current, with activation and inactivation kinetics between those of the fast and slow currents (Table 2) was present in 10.4% (5/48) of cutaneous neurons from inflamed rats (Fig. 10A, B) with at average density of 3.7 ± 1.1 pA/pF). While there was no increase in the proportion of neurons responsive to nicotine (~70%), the proportion of neurons with fast current (Fast or New) was significantly (p < 0.05, Chi square test) increased relative to the proportion of neurons with slow current (which was significantly decreased). Because of the rare occurrence of the new current, we did not characterize its pharmacological properties.
Table 2.
Kinetics of nicotine evoked currents
| Current Type | Time to Peak (ms) | Decay τ1 (ms) | Decay τ2 (ms) |
|---|---|---|---|
| Fast (n = 10) | 9 ± 0.8 | 16 ± 1.9 | 67 ± 9 |
| Slow (n = 14) | 157 ± 8 | 2257 ± 504Ψ | |
| Emergent (n = 5) | 48 ± 20 | 322 ± 79 |
The Emergent current was the current with intermediate kinetics that was detected in a subpopulation of cutaneous neurons from inflamed rats. Numbers in parenthesis are the number of neurons studied.
Current decay was only detected in a subpopulation of neurons with a slow current (n = 5).
Figure 10. Impact of inflammation on nicotine-evoked currents in cutaneous DRG neurons.

A. A current with unique biophysical properties was observed in a subpopulation of neurons from inflamed rats. The distinct kinetic properties can be easily seen when plotted with typical fast and slow currents scaled to the same amplitude for comparison. B. While there was no increase in the proportion of neurons responsive to nicotine (~67%), the proportion of neurons with fast current (Fast or New) was significantly (p < 0.05) increased relative to the proportion of neurons with slow current. Pooled data are from 7 naïve and 7 inflamed rats. C. Typical 300 μM nicotine-evoked slow currents in neurons from naïve (black) and inflamed (grey) rats suggest that inflammation is associated with an increase in current density. D. Pooled concentration-response data indicate the increase in slow current density is significant with no change in EC50 (55 vs. 59 μM). E. Typical 300 μM nicotine-evoked fast currents in neurons from naïve (black) and inflamed (grey) neurons also suggest that inflammation is associated with an increase in density. F. Pooled concentration-response data from fast current neurons are consistent with this suggestion indicating that the increase in fast current density is significant with no change in EC50 (47 vs. 41 μM). Data were analyzed with a mixed design two-way ANOVA. Post-hoc (Holm-Sidak) tests were performed if there was a significant interaction between experimental group (naïve vs inflamed) and nicotine concentration, where results of comparisons of the current density evoked at each concentration are shown: * is p < 0.05 and ** is p < 0.01.
Fast and slow current density is increased in small cutaneous neurons from inflamed rats
The density of both fast and slow currents was increased in cutaneous neurons from inflamed rats relative to that in neurons from naïve rats (Fig. 10C, D for slow current and Fig. 10E, F for fast current). This increase in density was not associated with a shift in the kinetics of activation (Fig. 11A, B for slow current and Fig. 11D, E for fast current) or inactivation (Fig. 11C for slow current and Fig. 11F for fast current) or the concentration response function (Fig. 10D for slow and Fig. 10F for fast): EC50 values for the slow current was 59 μM (n = 23) for naive vs 55 μM for inflamed (n = 11) neurons, respectively, and for the fast current, 47 μM for naïve (n = 14) vs 41 μM (n = 10) for inflamed, respectively. Changes in current density were only detected in small diameter cutaneous neurons, as differences in current density in medium diameter neurons or in other subpopulations were not significant (Table 1). Interestingly, the density of the very slow current tended to be reduced in other subpopulations (Table 1) from inflamed rats, and reached significance in IB4− neurons (p<0.05).
Figure 11. Inflammation does not change the kinetics of the fast and slow current.

To confirm that inflammation is primarily associated with an increase in current density rather than a change in biophysical properties, the kinetic properties of slow and fast nicotine-evoked currents were analyzed in greater detail. Typical slow (Aa) and fast (Ba) currents in neurons from naïve (black) and inflamed (grey) rats are scaled and overlayed. Pooled data for slow current activation (Ab) and inactivation (Ac) and fast current activation (Bb) and inactivation (Bc) confirm the absence of an impact of inflammation on the kinetics of nicotine-evoked current activation or inactivation.
The nAChR antagonist hexamethonium does not block thermal hyperalgesia induced by CFA
Because the increase in nicotine-evoked current density was restricted to putative nociceptive neurons, this increase may contribute to the hypersensitivity observed in the presence of persistent inflammation. To test this possibility, we assessed the impact of systemic hexamethonium (Hex, 20 mg/kg IP from a solution of 20 mg/ml) administration on the thermal hypersensitivity observed 3 days after CFA injection into the hindpaw. Systemic administration of Hex was used because it is not thought to cross the blood brain barrier and because of preliminary experiments suggesting that a subcutaneous injection of saline alone produced a transient thermal hypersensitivity (likely due to the hindpaw needle stick) comparable to that associated with CFA. No detectable influence of Hex was detected in either naïve (not shown) or CFA inflamed rats compared to naïve and CFA inflamed rats that received saline (Fig. 12).
Figure 12. Hexamethonium (Hex) did not block CFA-induced thermal hyperalgesia.
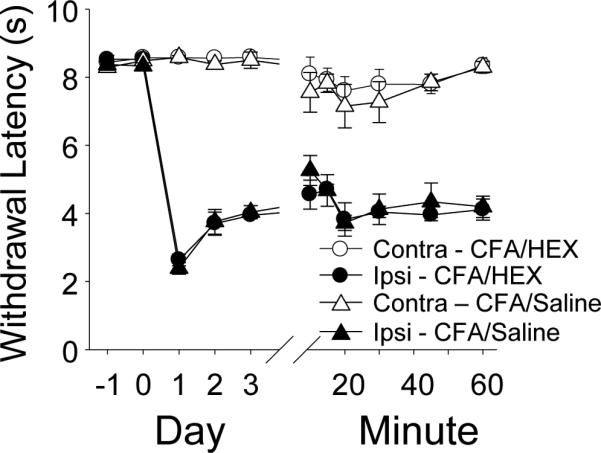
Thermal withdrawal latency was assessed in rats (n = 6 per group) once a day before and after intraplantar CFA injection, and then on the 3rd day after CFA, every 10 min after an injection hexamethonium (Hex, 20 mg/kg IP) or saline. Data plotted are for the hindpaw ipsilateral (ipsi) and contralateral (contra) to the site of inflammation.
Discussion
The purpose of the present study was to further clarify the role of nAChR activation in nociceptive signaling in sensory neurons in the absence and presence of inflammation. The main observations were: (1) Nicotine-evoked currents were present in most cutaneous DRG neurons (70%), including those with and without properties of putative nociceptors; (2) Two kinetically and pharmacologically distinct currents were found, slow and fast current, whose expression pattern was mutually exclusive; (3) The two currents were differentially distributed in IB4+ and IB4− neurons but not in Cap+ and Cap− neurons; (4) Despite the sensitivity of the fast current to the TRPA1 antagonist HC-030031, neither current appeared to be mediated by activation of TRPA1 channels; (5) Both low (1μM) and high (60 μM) nicotine concentrations produced a transient (~1 min) increase in the excitability of putative nociceptive cutaneous neurons with fast current, while only high concentrations of nicotine had a comparable effect in slow current neurons; (6) Neither low, nor high concentrations of nicotine influenced capsaicin-evoked currents in fast current neurons, while both concentrations significantly reduced capsaicin-evoked currents in slow current neurons; (7) Inflammation increased the current density of both slow and fast current in small diameter neurons and the percentage of neurons with the fast current; (8) Systemic hexamethonium had no detectable influence on inflammatory thermal hyperalgesia. Our results support the suggestion that while there are likely to be acute pro-nociceptive actions of nicotine, nAChR signaling in sensory neurons is complex where the impact on nociceptive processing will be concentration and time dependent.
We suggest the fast nAChR current in cutaneous DRG neurons was carried by homomeric α7 nAChRs while the slow current was carried by heteromeric (most likely α3β4) nAChRs. In support of the role for α7 channels in the fast current were the observations that 1) the activation and inactivation kinetics of the current were similar to that of heterologously expressed rat homomeric α7 nAChRs (Gopalakrishnan et al., 1995); 2) the potency of nicotine for both the activation (EC50 of ~ 38 μM) and desensitization (IC50 ~ 1.5 μM) of fast currents in cutaneous DRG neurons was similar to that of heterologously expressed rat homomeric α7 nAChRs (90 μM and 1.3 μM respectively (Giniatullin et al., 2005)); and 3) the current was blocked by MLA (20 nM). While MLA has been shown to block 6 subunit containing nAChRs (Rasmussen et al., 2014), the IC50 is considerably greater than the 20 nM used in the present study. In addition, our data argue against a potential role for α9 containing receptors, also shown to be sensitive to MLA block, because nicotine has been shown to antagonize α9 containing receptors (Verbitsky et al., 2000).
Evidence in support of the slow current mediated by α3β4 includes: 1) cytisine, a full β4 agonist, had greater efficacy than nicotine for slow current activation (Genzen et al., 2001); 2) the slow currents were completely blocked with MEC; and 3) the activation (EC50 ~61 μM ) and desensitization (IC50 ~1.9 μM ) of the slow current by nicotine, was similar to that reported with heterologously expressed rat α3β4 nAChRs (62 μM and 1.15 μM respectively) (Giniatullin et al., 2005). α4β2-like currents were also reported in rat DRG neurons (Genzen et al., 2001), however, the EC50 and IC50 obtained in our study are much higher than those of heterologously expressed α4β2 nAChRs (15 μM and 0.061 μM respectively for α4β2). Nevertheless, additional analysis will be needed to confirm the subunit composition of the channels underlying the slow current.
The biophysical and pharmacological properties of the nAChR currents described in the present study were consistent with currents described in previous studies of unlabeled DRG neurons (Genzen et al., 2001, Dube et al., 2005, Rau et al., 2005, Hone et al., 2012). Nevertheless, the homogeneity of the currents presents in cutaneous neurons was striking, particularly so in light of RT-PCR and immunohistochemical data suggesting the presence of 8 α (2-10) and 4 β (2-4) subunits in rat DRG neurons (Genzen et al., 2001, Haberberger et al., 2004). There are several possible explanations for the apparently narrow distribution of nAChR subtypes in cutaneous neurons and the more widespread pattern of subunits detected in DRG. First, it is possible that subunits are differently distributed within sensory neurons such that some subunits are present and functional in the cell body, while others are preferentially trafficked to other parts of the neuron. Arguing against this possibility, however, is the observation that there are no changes in current properties even after 24 h in culture (data not shown) despite evidence that it is possible to detect other channels that are trafficked out of the cell body within this same time period (Lee et al., 2012). A second possibility is that other subunit containing receptors are present in other cell types. Consistent with this possibility is the observation that the rate of fast current inactivation described by other investigators in unlabeled DRG neurons is slower than that of the fast current described here (Dube et al., 2005, Rau et al., 2005, Hone et al., 2012). Potentially more important is the observation that while fast and slow currents were never observed in the same cutaneous neuron, others have described the presence of both fast and slow currents in the same neuron (Fucile et al., 2005). Finally, a third possibility is that receptors with several subunit combinations contribute to the currents observed in the present study, a possibility that could be confirmed with a more extensive panel of pharmacological tools.
The observation that nAChR currents were differentially distributed among subpopulations of cutaneous DRG neurons defined by cell body diameter, IB4 binding and capsaicin sensitivity highlights not only the heterogeneity of sensory neurons in general, but of “putative nociceptive” cutaneous neurons in particular. The observation that nAChR current types and density were differentially distributed among subpopulations of cutaneous DRG neurons defined by IB4 binding but not capsaicin sensitivity, also highlights a potentially important species difference. That is, in contrast to the mouse, where IB4 binding and capsaicin sensitivity define essentially non-overlapping subpopulations of nociceptive neurons (Zwick et al., 2002), capsaicin sensitivity is detected in large subpopulations of both IB4+ and IB4− neurons (Petruska et al., 2000, Lu et al., 2006, Lu and Gold, 2008). With evidence that at least a subpopulation of capsaicin sensitive afferents may signal itch, it will be important to further classify the subpopulations of neurons responsive to nicotine to better predict the contribution of nAChR signaling to sensory processing.
Nevertheless, the differential distribution of fast and slow currents between IB4− and IB4+ neurons raises the possibility that these currents not only influence the pattern of activity impinging on the central nervous system, but engage different nociceptive circuits. That is, not only does IB4 label relatively distinct populations of neurons that have distinct termination patterns in both the periphery and spinal cord dorsal horn (Braz et al., 2005, Zylka et al., 2005), they also appear to engage dorsal horn neurons that project to different brain areas: IB4+ neurons appear to drive dorsal horn neurons that project to the brainstem and thalamus, and have, therefore been suggested to convey sensory discriminative aspects of pain; IB4− neurons appear to drive dorsal horn neurons that project to brain areas including the amygdala and hypothalamus, and have therefore been suggested to convey affective components of pain (Braz et al., 2005). Thus, changes in the relative pattern of activity in these circuits may contribute to changes in perception on persistent noxious stimulation.
Our data argue against the suggestion that TRPA1 underlies nicotine-induced irritation of skin (Talavera et al., 2009). Both fast and slow currents were blocked by hexamethonium, a nAChR antagonist that does not appear to block TRPA1 (Talavera et al., 2009). While there were neurons responsive to the TRPA1 agonist AITC, in which either fast or slow currents were also detected, the overlap was far from perfect with a significant fraction of neurons with fast or slow nicotine-evoked current that were unresponsive to AITC. The slow current was not blocked by the TRPA1 antagonist, HC-030031. And while the fast current was blocked by HC-030031, fast current that was blocked by HC-030031 was present in DRG neurons from TRPA1 null mutant mice, arguing that HC-030031 blocks nAChR currents rather than the possibility that the fast current is carried by TRPA1. Interestingly, despite the fact that a nicotine-evoked current was detectable with activation kinetics comparable to that of TRPA1, this very slow current in cutaneous DRG neurons was not sensitive to HC-030031. Thus, our data not only support the suggestion that nicotine-evoked currents are carried by nAChR channels, but suggest nicotine is unable to activate TRPA1 channels in rat sensory neurons, even at relatively high concentrations. Given our recent data indicating that TRPA1 is unlikely to contribute significantly to nicotine-evoked currents in mouse DRG neurons (Albers et al., 2014), a species difference is unlikely to account for the results of the present study and those of Talavera and colleagues. This leaves only methodological differences in the preparation of the neurons prior to study, or the duration of nicotine application (2 min in the Talavera study) to account for the differences in the conclusion as to the contribution of TRPA1 to the actions of nicotine. Minimally, however, while heterologous expression of nAChR would help confirm the off-target effects of HC-030031, our results suggest that interpretation of results obtained with HC-030031 should be made with caution.
The nicotine concentration in the blood of smokers and patients with a transdermal nicotine patch is relatively low (Lawson et al., 1998). And while higher concentrations of nicotine are needed to potentiate the capsaicin-evoked release of neuropeptides from the oral mucosa (Dussor et al., 2003), there is evidence that nAChR in the airway may be particularly sensitive to the excitatory actions of nicotine (Kichko et al., 2013). In contrast, there is evidence to suggest that relatively low concentrations of nicotine achieved by smokers and patch users are able to suppress hyperexcitability of colonic dorsal root ganglia neurons in experimental colitis. This suppression appears to be mediated by an α7 nAChR, and has been suggested to account for the therapeutic efficacy of the nicotine patch for the treatment of pain in colitis patients (Abdrakhmanova et al., 2010). That 1μM nicotine produced only a transient increase in the excitability of cutaneous DRG neurons from naïve rats as well as those three days after the induction of inflammation (not shown), with no evidence of a decrease in excitability suggests that the impact of nAChR activation on neuronal excitability depends on the context (i.e., the other ion channels present in the membrane) in which the nAChR are functioning. Consistent with this suggestion, we have described marked differences between colonic and cutaneous neurons with respect to both baseline electrophysiological properties as well as the mechanisms underlying the increase in excitability associated with the actions of inflammatory mediators (Gold and Traub, 2004).
On the other hand, the nAChR agonists, epibatidine and ABT-594 are thought to produce their analgesic effects via activation of α4β2 and to a lesser extent α3β4 nAChRs (Donnelly-Roberts et al., 1998). That we only observed an increase in the excitability of cutaneous neurons with slow current suggests that the analgesic actions of these compounds are not mediated via the regulation of the excitability of the peripheral terminals of cutaneous afferents. However, the observation that both 1 μM and 60 μM nicotine on neurons with slow current results in the suppression of capsaicin-evoked current, suggests that suppression of TRPV1 may underlie the analgesic actions of these compounds.
It is tempting to speculate that the inflammation-induced increase in both the density and distribution of fast currents and the density of slow currents would contribute to the hypersensitivity observed in the presence of inflammation. However, our excitability data clearly indicate that the response to nAChR activation is more complex, where fast current activation may contribute to transient increases in excitability, while slow current activation may contribute to both an increase in excitability per se and a decrease in TRPV1 mediated nociceptive signaling. Furthermore, the relative decrease in the proportion of neurons with slow current would serve to mitigate the more persistent impact of the slow current on excitability, yet also attenuate the slow current-induced decrease in TRPV1 signaling. Thus, it is not surprising that we were unable to detect a change in nociceptive threshold following systemic administration of hexamethonium. Nevertheless, given the differential distribution of the fast and slow currents and the evidence that these currents may engage different nociceptive circuits, it is possible that more sophisticated nociceptive tests, such as the conditioned place preference test (King et al., 2009), may be necessary to tease apart the relative contribution of peripheral nAChR activation on nociceptive signaling. It is also possible, as suggested previously, that peripheral nAChR signaling plays a relatively minor role on the modulation of nociceptive signaling, and that receptors on the central terminals of nociceptive afferents or on neurons in the dorsal horn play a more prominent role.
Highlights.
- Nicotine-evoked α7-like and α3β4-like currents are in distinct populations constituting ~70% of all cutaneous DRG neurons.
- Nicotine (1 μM) transiently increases α7-like neuron excitability and decreases capsaicin current in α3β4-like neurons.
- Nicotine (60 μM) transiently increases the excitability of neurons with both α7-like and α3β4-like current.
- Inflammation alters the distribution and increases the density of α3β4-like and α7-like currents in cutaneous neurons.
- The impact of nicotine on pain and hypersensitivity will depend on both concentration and timing of acetylcholine release.
Acknowledgements
This works was supported by NIH Grant #NS033730 to KMA
Abbreviations
- AITC
allyl isothiocyanate
- DRG
Dorsal root ganglion
- Cap
capsaicin
- DiI
1,1’-Dioctadecyl-3,3,3’,3’-tetramethylindocarbocyanine perchlorate
- IB4
isolectin B4
- MLA
methyllycaconitine citrate
- nAChR
nicotinic acetylcholine receptor
- TRPA1
transient receptor potential ankyrin type 1
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author's Contributions: XLZ: study design, acquisition of data, analysis and interpretation of data and writing of manuscript. KMA: study conception and design, analysis and interpretation of data, writing of manuscript. MSG: study conception and design, acquisition of data, analysis and interpretation of data, writing of manuscript. All authors read and approved the final manuscript.
References
- Abdrakhmanova GR, AlSharari S, Kang M, Damaj MI, Akbarali HI. {alpha}7-nAChR-mediated suppression of hyperexcitability of colonic dorsal root ganglia neurons in experimental colitis. Am J Physiol Gastrointest Liver Physiol. 2010;299:G761–768. doi: 10.1152/ajpgi.00175.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albers KM, Zhang XL, Diges CM, Schwartz ES, Yang CI, Davis BM, Gold MS. Artemin growth factor increases nicotinic cholinergic receptor subunit expression and activity in nociceptive sensory neurons. Mol Pain. 2014;10:31. doi: 10.1186/1744-8069-10-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardini N, Sauer SK, Haberberger R, Fischer MJ, Reeh PW. Excitatory nicotinic and desensitizing muscarinic (M2) effects on C-nociceptors in isolated rat skin. J Neurosci. 2001;21:3295–3302. doi: 10.1523/JNEUROSCI.21-09-03295.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braz JM, Nassar MA, Wood JN, Basbaum AI. Parallel “pain” pathways arise from subpopulations of primary afferent nociceptor. Neuron. 2005;47:787–793. doi: 10.1016/j.neuron.2005.08.015. [DOI] [PubMed] [Google Scholar]
- Dickson SL, Hrabovszky E, Hansson C, Jerlhag E, Alvarez-Crespo M, Skibicka KP, Molnar CS, Liposits Z, Engel JA, Egecioglu E. Blockade of central nicotine acetylcholine receptor signaling attenuate ghrelin-induced food intake in rodents. Neuroscience. 2010;171:1180–1186. doi: 10.1016/j.neuroscience.2010.10.005. [DOI] [PubMed] [Google Scholar]
- Donnelly-Roberts DL, Puttfarcken PS, Kuntzweiler TA, Briggs CA, Anderson DJ, Campbell JE, Piattoni-Kaplan M, McKenna DG, Wasicak JT, Holladay MW, Williams M, Arneric SP. ABT-594 [(R)-5-(2-azetidinylmethoxy)-2-chloropyridine]: a novel, orally effective analgesic acting via neuronal nicotinic acetylcholine receptors: I. In vitro characterization. J Pharmacol Exp Ther. 1998;285:777–786. [PubMed] [Google Scholar]
- Dube GR, Kohlhaas KL, Rueter LE, Surowy CS, Meyer MD, Briggs CA. Loss of functional neuronal nicotinic receptors in dorsal root ganglion neurons in a rat model of neuropathic pain. Neurosci Lett. 2005;376:29–34. doi: 10.1016/j.neulet.2004.11.020. [DOI] [PubMed] [Google Scholar]
- Dussor GO, Leong AS, Gracia NB, Kilo S, Price TJ, Hargreaves KM, Flores CM. Potentiation of evoked calcitonin gene-related peptide release from oral mucosa: a potential basis for the pro-inflammatory effects of nicotine. Eur J Neurosci. 2003;18:2515–2526. doi: 10.1046/j.1460-9568.2003.02935.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang X, Djouhri L, McMullan S, Berry C, Waxman SG, Okuse K, Lawson SN. Intense isolectin-B4 binding in rat dorsal root ganglion neurons distinguishes C-fiber nociceptors with broad action potentials and high Nav1.9 expression. J Neurosci. 2006;26:7281–7292. doi: 10.1523/JNEUROSCI.1072-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fucile S, Sucapane A, Eusebi F. Ca2+ permeability of nicotinic acetylcholine receptors from rat dorsal root ganglion neurones. J Physiol. 2005;565:219–228. doi: 10.1113/jphysiol.2005.084871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genzen JR, Van Cleve W, McGehee DS. Dorsal root ganglion neurons express multiple nicotinic acetylcholine receptor subtypes. J Neurophysiol. 2001;86:1773–1782. doi: 10.1152/jn.2001.86.4.1773. [DOI] [PubMed] [Google Scholar]
- Giniatullin R, Nistri A, Yakel JL. Desensitization of nicotinic ACh receptors: shaping cholinergic signaling. Trends Neurosci. 2005;28:371–378. doi: 10.1016/j.tins.2005.04.009. [DOI] [PubMed] [Google Scholar]
- Gold MS, Dastmalchi S, Levine JD. Co-expression of nociceptor properties in dorsal root ganglion neurons from the adult rat in vitro. Neuroscience. 1996;71:265–275. doi: 10.1016/0306-4522(95)00433-5. [DOI] [PubMed] [Google Scholar]
- Gold MS, Traub RJ. Cutaneous and colonic rat DRG neurons differ with respect to both baseline and PGE2-induced changes in passive and active electrophysiological properties. J Neurophysiol. 2004;91:2524–2531. doi: 10.1152/jn.00866.2003. [DOI] [PubMed] [Google Scholar]
- Gopalakrishnan M, Buisson B, Touma E, Giordano T, Campbell JE, Hu IC, Donnelly-Roberts D, Arneric SP, Bertrand D, Sullivan JP. Stable expression and pharmacological properties of the human alpha 7 nicotinic acetylcholine receptor. Eur J Pharmacol. 1995;290:237–246. doi: 10.1016/0922-4106(95)00083-6. [DOI] [PubMed] [Google Scholar]
- Haberberger RV, Bernardini N, Kress M, Hartmann P, Lips KS, Kummer W. Nicotinic acetylcholine receptor subtypes in nociceptive dorsal root ganglion neurons of the adult rat. Autonomic neuroscience : basic & clinical. 2004;113:32–42. doi: 10.1016/j.autneu.2004.05.008. [DOI] [PubMed] [Google Scholar]
- Hone AJ, Meyer EL, McIntyre M, McIntosh JM. Nicotinic acetylcholine receptors in dorsal root ganglion neurons include the alpha6beta4* subtype. FASEB J. 2012;26:917–926. doi: 10.1096/fj.11-195883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kichko TI, Lennerz J, Eberhardt M, Babes RM, Neuhuber W, Kobal G, Reeh PW. Bimodal concentration-response of nicotine involves the nicotinic acetylcholine receptor, transient receptor potential vanilloid type 1, and transient receptor potential ankyrin 1 channels in mouse trachea and sensory neurons. J Pharmacol Exp Ther. 2013;347:529–539. doi: 10.1124/jpet.113.205971. [DOI] [PubMed] [Google Scholar]
- King T, Vera-Portocarrero L, Gutierrez T, Vanderah TW, Dussor G, Lai J, Fields HL, Porreca F. Unmasking the tonic-aversive state in neuropathic pain. Nat Neurosci. 2009;12:1364–1366. doi: 10.1038/nn.2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang PM, Burgstahler R, Sippel W, Irnich D, Schlotter-Weigel B, Grafe P. Characterization of neuronal nicotinic acetylcholine receptors in the membrane of unmyelinated human C-fiber axons by in vitro studies. J Neurophysiol. 2003;90:3295–3303. doi: 10.1152/jn.00512.2003. [DOI] [PubMed] [Google Scholar]
- Lawson GM, Hurt RD, Dale LC, Offord KP, Croghan IT, Schroeder DR, Jiang NS. Application of serum nicotine and plasma cotinine concentrations to assessment of nicotine replacement in light, moderate, and heavy smokers undergoing transdermal therapy. Journal of clinical pharmacology. 1998;38:502–509. doi: 10.1002/j.1552-4604.1998.tb05787.x. [DOI] [PubMed] [Google Scholar]
- Lawson SN. Phenotype and function of somatic primary afferent nociceptive neurones with C-, Adelta- or Aalpha/beta-fibres. Exp Physiol. 2002;87:239–244. doi: 10.1113/eph8702350. [DOI] [PubMed] [Google Scholar]
- Lee KY, Charbonnet M, Gold MS. Upregulation of high-affinity GABA(A) receptors in cultured rat dorsal root ganglion neurons. Neuroscience. 2012;208:133–142. doi: 10.1016/j.neuroscience.2012.01.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopshire JC, Nicol GD. Activation and recovery of the PGE2-mediated sensitization of the capsaicin response in rat sensory neurons [In Process Citation]. J Neurophysiol. 1997;78:3154–3164. doi: 10.1152/jn.1997.78.6.3154. [DOI] [PubMed] [Google Scholar]
- Loram LC, Taylor FR, Strand KA, Maier SF, Speake JD, Jordan KG, James JW, Wene SP, Pritchard RC, Green H, Van Dyke K, Mazarov A, Letchworth SR, Watkins LR. Systemic administration of an alpha-7 nicotinic acetylcholine agonist reverses neuropathic pain in male Sprague Dawley rats. J Pain. 2012;13:1162–1171. doi: 10.1016/j.jpain.2012.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu SG, Gold MS. Inflammation-induced increase in evoked calcium transients in subpopulations of rat dorsal root ganglion neurons. Neuroscience. 2008;153:279–288. doi: 10.1016/j.neuroscience.2008.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu SG, Zhang X, Gold MS. Intracellular calcium regulation among subpopulations of rat dorsal root ganglion neurons. J Physiol. 2006;577:169–190. doi: 10.1113/jphysiol.2006.116418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malin DH, Lake JR, Schopen CK, Kirk JW, Sailer EE, Lawless BA, Upchurch TP, Shenoi M, Rajan N. Nicotine abstinence syndrome precipitated by central but not peripheral hexamethonium. Pharmacol Biochem Behav. 1997;58:695–699. doi: 10.1016/s0091-3057(97)90006-x. [DOI] [PubMed] [Google Scholar]
- Petruska JC, Napaporn J, Johnson RD, Gu JG, Cooper BY. Subclassified acutely dissociated cells of rat DRG: histochemistry and patterns of capsaicin-, proton-, and ATP-activated currents. J Neurophysiol. 2000;84:2365–2379. doi: 10.1152/jn.2000.84.5.2365. [DOI] [PubMed] [Google Scholar]
- Rasmussen AH, Strobaek D, Dyhring T, Jensen ML, Peters D, Grunnet M, Timmermann DB, Ahring PK. Biophysical and pharmacological characterization of alpha6-containing nicotinic acetylcholine receptors expressed in HEK293 cells. Brain Res. 2014;1542:1–11. doi: 10.1016/j.brainres.2013.10.024. [DOI] [PubMed] [Google Scholar]
- Rau KK, Johnson RD, Cooper BY. Nicotinic AChR in subclassified capsaicin-sensitive and -insensitive nociceptors of the rat DRG. J Neurophysiol. 2005;93:1358–1371. doi: 10.1152/jn.00591.2004. [DOI] [PubMed] [Google Scholar]
- Rueter LE, Kohlhaas KL, Curzon P, Surowy CS, Meyer MD. Peripheral and central sites of action for A-85380 in the spinal nerve ligation model of neuropathic pain. Pain. 2003;103:269–276. doi: 10.1016/s0304-3959(02)00455-4. [DOI] [PubMed] [Google Scholar]
- Schmelz M, Schmid R, Handwerker HO, Torebjork HE. Encoding of burning pain from capsaicin-treated human skin in two categories of unmyelinated nerve fibres. Brain 123 Pt. 2000;3:560–571. doi: 10.1093/brain/123.3.560. [DOI] [PubMed] [Google Scholar]
- Sculptoreanu A, Kullmann FA, Artim DE, Bazley FA, Schopfer F, Woodcock S, Freeman BA, de Groat WC. Nitro-oleic acid inhibits firing and activates TRPV1- and TRPA1-mediated inward currents in dorsal root ganglion neurons from adult male rats. J Pharmacol Exp Ther. 2010;333:883–895. doi: 10.1124/jpet.109.163154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steen KH, Reeh PW. Actions of cholinergic agonists and antagonists on sensory nerve endings in rat skin, in vitro. Journal of Neurophysiology. 1993;70:397–405. doi: 10.1152/jn.1993.70.1.397. [DOI] [PubMed] [Google Scholar]
- Stucky CL, Lewin GR. Isolectin B(4)-positive and -negative nociceptors are functionally distinct. J Neurosci. 1999;19:6497–6505. doi: 10.1523/JNEUROSCI.19-15-06497.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talavera K, Gees M, Karashima Y, Meseguer VM, Vanoirbeek JA, Damann N, Everaerts W, Benoit M, Janssens A, Vennekens R, Viana F, Nemery B, Nilius B, Voets T. Nicotine activates the chemosensory cation channel TRPA1. Nat Neurosci. 2009;12:1293–1299. doi: 10.1038/nn.2379. [DOI] [PubMed] [Google Scholar]
- Verbitsky M, Rothlin CV, Katz E, Elgoyhen AB. Mixed nicotinic-muscarinic properties of the alpha9 nicotinic cholinergic receptor. Neuropharmacology. 2000;39:2515–2524. doi: 10.1016/s0028-3908(00)00124-6. [DOI] [PubMed] [Google Scholar]
- Zhang X, Koronowski KB, Li L, Freeman BA, Woodcock S, de Groat WC. Nitro-oleic acid desensitizes TRPA1 and TRPV1 agonist responses in adult rat DRG neurons. Exp Neurol. 2014;251:12–21. doi: 10.1016/j.expneurol.2013.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwick M, Davis BM, Woodbury CJ, Burkett JN, Koerber HR, Simpson JF, Albers KM. Glial cell line-derived neurotrophic factor is a survival factor for isolectin B4-positive, but not vanilloid receptor 1-positive, neurons in the mouse. J Neurosci. 2002;22:4057–4065. doi: 10.1523/JNEUROSCI.22-10-04057.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zylka MJ, Rice FL, Anderson DJ. Topographically distinct epidermal nociceptive circuits revealed by axonal tracers targeted to Mrgprd. Neuron. 2005;45:17–25. doi: 10.1016/j.neuron.2004.12.015. [DOI] [PubMed] [Google Scholar]