

Abstract
Early heart development takes place through a complex series of steps, including the induction of cardiac mesoderm, formation of the cardiovascular progenitor cells and the commitment of cardiovascular lineage cells, such as cardiomyocytes (CMs), smooth muscle cells (SMCs) and endothelial cells (ECs). Recently, microRNAs, a family of endogenous, small non-coding RNAs, have been implicated as critical regulators at the posttranscriptional level in cardiogenesis as well as cardiovascular disease. Previous studies demonstrated that microRNA-1 (miR-1) could promote cardiac differentiation from mouse and human embryonic stem (ES) cells. However, the underlying mechanism largely remained unclear. We performed microRNA deep sequencing among human ES cells, ES cell derived-multipotent cardiovascular progenitors (MCPs), and MCP-specified CMs, ECs, and SMCs. A specific enrichment of miR-1 was found in CMs, not in SMCs or ECs, implying a key role of miR-1 in determining cardiovascular commitment from MCPs. When overexpressed in human pluripotent stem cells, miR-1 enhanced the expression of key cardiac transcriptional factors and sarcomeric genes. Importantly, we found miR-1 promoted CM differentiation and suppressed EC commitment from MCPs by modulating the activities of WNT and FGF signaling pathways. FZD7 and FRS2 were confirmed as miR-1 targets using luciferase reporter assay and western blot. Overall, this study reveals a switch role of miR-1 at early human cardiovascular commitment stage via suppressing both WNT and FGF signaling pathways.
Keywords: MicroRNA-1, Induced pluripotent stem cells, Cardiomyocyte, Multipotent Cardiovascular Progenitors
1. Introduction
Early heart development takes place through a complex series of steps, including the induction of cardiac mesoderm, formation of the cardiovascular progenitor cells and the commitment of cardiovascular lineage cells, including cardiomyocytes (CMs), smooth muscle cells (SMCs) and endothelial cells (ECs). Each step is precisely regulated at various levels, including transcriptional, posttranscriptional and posttranslational regulations. Recently, microRNAs (miRNAs), a family of endogenous, small non-coding RNAs, have been implicated as a critical regulator in cardiovascular development and pathological conditions at the posttranscriptional level [1–4]. Mature miRNAs form RNA-induced silencing complexes (RISC) with other proteins and interact with the 3′ untranslated region (UTR) of the target messenger RNAs to repress mRNA translation, or degrade mRNA transcripts [1]. The essential role of miRNA in early heart development was first revealed by a knockout study of Dicer, which is a miRNA processing enzyme. The early lethality in the Dicer mutant, which results in the complete depletion of mature miRNAs, indicated the crucial role of miRNAs in the early heart development [5]. Additionally, conditional Dicer deletion in adult mouse heart causes various cardiac defects and postnatal lethality [6]. Those studies led to the discovery of functional miRNAs, such as miRNA-1, miRNA-133, miRNA-499, miRNA-143/145, in vertebrate cardiogenesis [7–10].
Among all muscle-specific miRNAs, miRNA-1(miR-1) is among the most specifically and abundantly expressed miRNAs in heart and its function in vertebrate heart formation has been extensively studied [7, 11]. Overexpressing of miRNA-1 in mouse embryonic stem (ES) cells suppresses endoderm and ectoderm differentiation, and promotes CM differentiation [11]. miR-1 enhances the CM differentiation from human ES cells, as well as cardiomyocyte progenitor cells (CPCs) which were directly isolated from fetal human heart [11,12]. Although SRF has been identified as an upstream transcriptional activator of miR-1 in heart, little is known for the downstream targets of miR-1 in human heart development [7]. By recapitulating early mammalian heart development, we established a well-defined protocol supporting the efficient and reproducible development of multipotent cardiovascular progenitors (MCPs), as well as MCP-specified cardiomyocytes (CMs), endothelial cells (ECs), and smooth muscle cells (SMCs) from human pluripotent stem cells [13,14]. MCPs represent the earliest cardiovascular progenitor cells in human cardiogenesis. This system allows us to define the role of miR-1 during human heart development, especially at the stage when cardiovascular lineage cells are segregated from MCPs. In this study, we found that miR-1 promoted CM differentiation and suppressed EC formation from MCPs by modulating the activities of WNT and FGF signaling pathways. FZD7 and FRS2 were confirmed as miR-1 targets in WNT and FGF pathway respectively. Overall, this study reveals a switch role of miRNA-1 in controlling early stage human cardiovascular commitment.
2. Materials and methods
2.1 Animals
All of the animal studies conformed to the principles of the National Institutes of Health Guide for the Care and Use of Laboratory Animals ((NIH Publication No. 85–23, revised 1996)) and the Institutional Animal Care and Use Committee approved all protocols. CD1 mice were anaesthetized in a chamber with the introduction of 100% CO2 for 7–10 min Euthanasia was accomplished by cervical dislocation. Mouse embryos were dissected at E8.5 (embryonic day 8.5) to E8.75 with approximately 10–12 pairs of somites under a dissecting microscope (Leica). From the ventral side, the heart was dissected by cutting anteriorly at the truncus ateriosus and caudally at the two branches of the sinus venosus, followed with 0.1% Collagenase B (Roche) for 30 min at 37°C into single cells.
2.2 Cell culture and differentiation
Human S3 iPS cells and human RUES2 ES cells were maintained on irradiated mouse embryonic fibroblasts (MEF) and cultured in DMEM/F12 supplemented with 20% knock-out serum replacement (Invitrogen) with 10 ng/mL human basic fibroblast growth factor. Cardiac lineage differentiation was conducted as previously described [13,14] with the formation of embryoid bodies (EBs). EBs were treated with the following conditions: days 0–1, BMP4 (5 ng/mL); days 1–4, BMP4 (10 ng/mL), bFGF (5 ng/mL) and Activin A (2 ng/mL). MCPs were isolated using a FACSAria II cell sorter (BD Biosciences) as previously described. All cytokines were from R&D Systems. The FGFR antagonist, PD173074, was from Stemgent.
2.3 microRNA deep-transcriptome sequencing
Human RUES2 ES cells, RUES2-derived-MCPs, cardiomyocytes, smooth muscle cells and endothelial cells were isolated and enriched as previously described [14]. MicroRNA deep-transcriptome sequencing was performed with the collected five cell samples using a service from LC Sciences (Houston, TX) with the Illumina platform. Reads were mapped to human reference genome UCSC hg18 (ftp://igenome:G3nom3s4u@ftp.illumina.com/Homo_sapiens/UCSC/hg18) using Bowtie version 0.12.7 and Tophat version 1.3.2 (Trapnell et al., 2012). Expression intensity was estimated using Cufflinks version 1.2.1 as Fragment Per Kilobase of exon per Million reads (FPKM) (Trapnell et al., 2012). The FPKM values were normalized to 1 million of sum after adding pseudocounts (FPKM+1).
2.4 Plasmid construction and viral transduction
The mouse Pre-miR-1 with ~100bp flanking sequences on both sides of seed sequence was amplified from genomic DNA of mouse fibroblasts, using primers described in Supplementary Table SI. The PCR product was cloned into the lentivirus expression vector pcFEFW-IRES-EGFP, which is provided by Dr. Yi Sheng at University of Pittsburgh. The lentiviral plasmid containing miR-1, as well as the empty lentiviral plasmid were transfected into the 293T cells (ATCC) along with lentiviral packaging plasmids including pLP1, pLP2, and pLP/VSVG using the FuGENE® HD transfection reagent (Roche). Viral bearing supernatant was collected and cellular debris was removed by syringe filtering (0.45 μm pore size; Millipore). Human iPS cells (iPSCs) were transducted and the virus containing media were removed after 24 hrs. After 2 weeks, GFP positive cells were sorted out using a FACSAria II cell sorter (BD Biosciences) and expanded.
2.5 microRNA extraction and quantification
RNAs were extracted using miRNeasy Mini extraction kit (Qiagen). RNAs (1 μg) were reverse transcribed using qScript microRNA cDNA synthesis kit (Quanta Biosciences, Gaithersburg, MD, USA). Real-time quantitative PCR was to measure miRNA expression. MicroRNA-specific stem-loop primers used in the amplification reaction are listed in Supplementary Table SI. miRNA expression was normalized against the expression of SNORD44 small nucleolar RNA.
2.6 Quantitative RT-PCR
RNA was extracted using the Qiagen RNeasy Kit (Qiagen). cDNA was generated by using the high capacity RNA-to-cDNA kit (Applied Biosystems). PCR primers are listed in supplementary Table SI. PCR was performed on an Applied Biosystems 7900HT quantitative PCR system (Applied Biosystems) using Power SYBR Green (Applied Biosystems). Quantification of gene expression was based on the −ΔΔCt method and was normalized against the expression of Cyclophilin G.
2.7 Cell growth assay
Cell growth rate was determined by 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H -tetrazolium bromide (MTT; Merck KGaA) assay. Control and miRNA-1 overexpressing iPSCs were cultured in ES medium in 96-well plates. At indicated time points, MTT was added into the plates at 37°C and incubated for 3.5 h. The insoluble formazan product formed and was solubilized with DMSO (100 μl/well). The plates were read at a wavelength of 570nm by a microtiter plate reader (Promega).
2.8 Fluorescence-activated cell sorting (FACS)
EBs were harvested and trypsinized (0.25% trypsin-EDTA) for 5 minutes at 37°C. The dissociated single cells were fixed in 4% PFA for 10 minutes on ice, washed 3 times with PBS. Cells were blocked with 10% FBS and then incubated with primary antibody: anti-Troponin T (Thermo Scientific, Fremont, CA, USA) or anti-PECAM-1 (CD31; BD Biosciences), followed with an incubation of 1 hr at room temperature with APC-conjugated secondary antibodies (BD Biosciences). FACS analysis was carried out with a BD Accuri C6 flow cytometer (Becton Dickinson). Data were analyzed using FlowJo (Treestar).
2.9 MicroRNA target prediction and validation
Five algorithms: miRanda [15], TargetScan [16], PicTar [17], PITA [18] and ComiR [19] were used to predict potential miR-1 target genes and potential miR-1 binding sites in their 3′ untranslated regions (3′UTRs). These algorithms were selected because they provide complementary information. miRanda and PITA are based on thermodynamics; TargetScan on sequence matching and evolutionary conservation; whereas PicTar and ComiR can identify combinatorial miRNA targets. One novelty of ComiR is that it utilizes miRNA expression level when determining a target and it combines multiple prediction models in a support vector machine framework [20]. miR-1 binding sites, which were predicted from at least three of these algorithms were selected for further validation. 3′UTRs of predicted microRNA target genes (GJA1, FRS2, FZD7) were PCR amplified from human genomic DNA using primers listed in Supplementary Table SI and cloned into the dual-luciferase pmirGLO vector (Promega). HEK293T cells were transfected with 100 ng luciferase reporter plasmids with 20 nM pre-miR-1 or control oligo-nucleotides (Thermo Scientific) in each well using X-tremeGENE siRNA transfection reagent (Roche). Cells were harvested 24 hours post-transfection. The firefly luciferase activity in cell lysates was measured with a Dual-Glo dual luciferase reporter assay kit (Promega) on a Synergy H1 hybrid microplate reader (BioTek Instruments), and normalized with the activity of Renilla luciferase.
2.10 Western blot
Cells were trypsinized, collected, and protein was extracted using RIPA buffer (Cell Signaling) with a serine protease inhibitor, phenylmethanesulfonylfluoride (PMSF). Homogenates were centrifuged, supernatants were collected and stored at −80°C. Proteins were resolved on a 10% Mini-PROTEAN TGX precast SDS-PAGE gel (Bio-Rad) and transferred onto nitrocellulose membranes (Bio-Rad). The membranes were blocked with 10% non-fat dry milk in PBS for 2 hrs, followed with incubation with primary antibody: anti-FRS2 (Santa Cruz Biotechnology), anti-FZD7 (Millipore), or anti-GAPDH (Cell Signaling) overnight at 4°C. After 1 hr of incubation with anti-rabbit IgG HRP-conjugated secondary antibody (Cell Signaling), signal was detected using ECL reagents (Amersham Pharmacia Biotechnolog).
2.11 siRNA Transfection
To silence gene expression, the On-Target plus SMARTpool siRNA duplexes targeting human FRS2 and FZD7 genes were purchased from Dharmacon RNA Technologies (Lafayette). An ON-TARGET plus nontargeting siRNA (siCTR) was used as the negative control. MCPs were transferred to 24-well plates, transfected with 50 nM siRNA oligos using X-tremeGENE siRNA transfection reagent (Roche). After 24 hrs, media were removed and changed to fresh basal differentiation medium without any growth factor.
2.12 Statistics
All experiments were conducted independently for three times. Error bars represent s.d. Statistical significance was estimated with an ANOVA test and P-values were calculated using Student’s t-test.
3. Results
3.1 microRNA expression profiling during human cardiovascular development
In order to profile microRNA expression during human cardiovascular differentiation, we induce and isolated human ES cell-derived MCPs, as well as MCP-specified CMs, ECs, and SMCs using our previously published method (Figure 1A). Illumina microRNA deep-sequencing was next conducted with those samples. Figure 1B illustrates several signature microRNAs in each cell type. For example, miR-302-367 cluster is known to be enriched in undifferentiated ES cells [21]; miR-1, -133a, -499 in CMs [7–9]; miR-143 and -145 in SMCs [10]; let-7b in ECs [22]. It is interesting to note that miR-302-367 cluster is also highly expressed in human MCPs, suggesting its role for MCP induction. Unlike previous findings in mouse [10], miR-143 is highly expressed in both human CMs and SMCs, while miR-145 is more specific in SMCs. Of all miRNAs, miR-1 is of particular interest for its abundant and specific enrichment in CMs, but not in SMCs or ECs. This, together with the observation that miR-1 is highly conserved across species (Figure 1C), suggests an essential role of miR-1 in regulating human cardiovascular commitment from early stage heart progenitor cells. In order to evaluate the function of miR-1 during human cardiovascular development, an EGFP-miR-1 cassette was lentivirally transduced to normal human S3 induced pluripotent stem cells (iPSCs) [23](Figure 1D). A control S3 iPSC line was established by infecting S3 cells with the empty EGFP lentivirus. Using FACS, both GFP-miR-1 and GFP control iPSCs were isolated and expanded. Effective miR-1 overexpression in iPSCs was confirmed by the expression of GFP reporter, as well as the ~60 fold of miR-1 and ~10 fold of miR-1* expression increases in miR-1 overexpressing cells vs. control cells (Figure 1E).
Figure 1. miR-1 overexpression in human iPSCs.
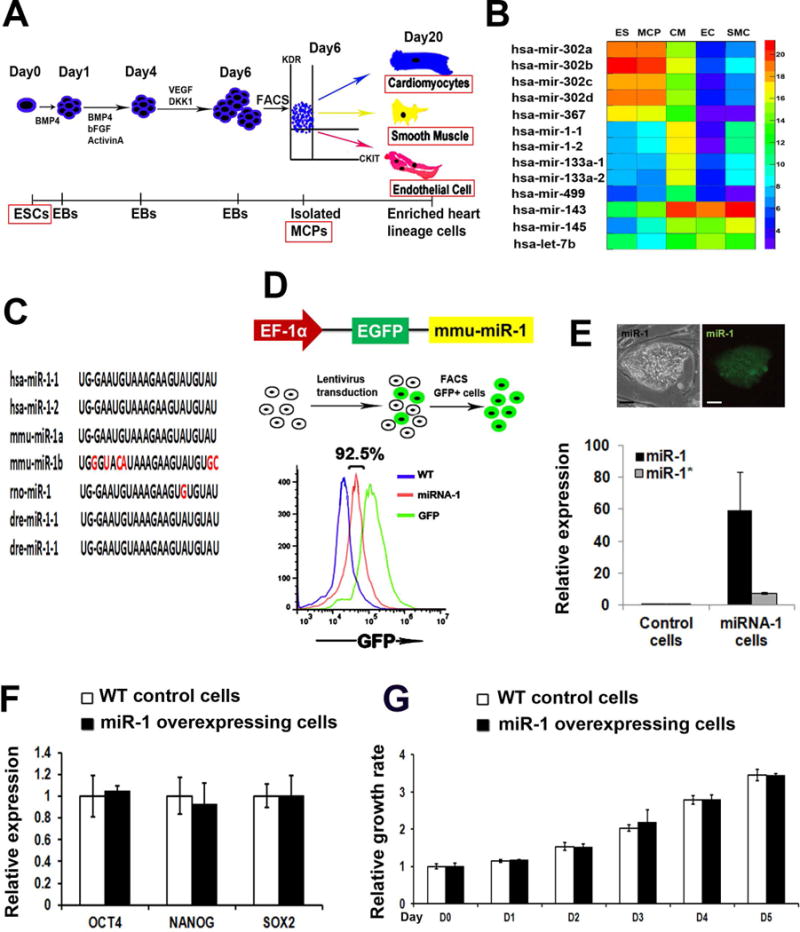
(A) A scheme of the cardiovascular differentiation of human embryonic stem and iPS cells. EBs, embryoid bodies; MCPs, multipotent cardiovascular progenitors. (B) A heat map of microRNA profiling of human ES, MCP, CM, SMC, and EC populations using deep-sequencing. (C) miR-1 seed sequences across multiple vertebrate species. Red color indicates the non-conserved nucleotides. (D) Schematic representation of miR-1 overexpression using a lentiviral EGFP reporter vector (up). GFP+ human iPSCs were isolated by FACS (middle), with over 90% iPSCs expressing GFP in both miR-1 overexpressing and GFP control cells (low). (E) A representative green GFP/miR-1 + iPS clone (up). Detection of miR-1 and miR-1* expressions in GFP-control and miR-1 overexpressing iPSCs (low) by quantitative RT-PCR (q-PCR). Scale bar, 50μm. q-PCR analysis of pluripotency marker genes, OCT4, NANOG, and SOX2 (F) and cell proliferation assay by MTT (G) in the wild type control and miR-1 overexpressing iPSCs.
To examine whether miR-1 overexpression influenced iPSC pluripotency, miR-1 overexpressing and control iPSCs were grown with human ES medium for 5 days, followed with q-RT-PCR analysis of pluripotent gene expression. Both iPSCs remained a classic morphology of pluripotent stem cells (Figure 1E) and a comparable expression level of pluripotent genes (Figure 1F). In addition, overexpression of miR-1 didn’t affect cellular physiology such as the proliferation index (Figure 1G). All these data indicate that the overexpression of miR-1 could not bypass pluripotency of human iPSCs to initiate cardiac differentiation.
3.2 miR-1 promotes cardiac differentiation of human iPS cells
Using our staged cardiovascular differentiation protocol with human pluripotent stem cells [13,14], we sought to examine the role of miR-1 in human early cardiovascular development. Figure 2A illustrates the whole differentiation strategy. Adding of bone morphogenetic protein 4 (BMP4), basic fibroblast growth factor (bFGF) and Activin A from day 0 to day 4 induced cardiovascular mesoderm formation as previously described [13,14], and no cytokines were added into the cell culture after day 4. Given the fact that MCPs are induced from human ES/iPS cells at day 6, this strategy allowed us to determine the role of miR-1 at human cardiovascular commitment stage. Firstly, we traced the dynamic ratios of beating embryoid bodies (EBs) from day 10 to day 20 of differentiation. Overexpression of miR-1 significantly increased the ratio of contracting EBs since day 12 when compared with either GFP-control or wild type iPSCs (Figure 2B). In addition, an increased ratio of cardiac Troponin T positive (CTNT+) cells was observed from the dissociated day 20 EBs with miR-1 overexpression than control (Figure 2C). CTNT is a marker of CMs [13]. All these data indicate that miR-1 could significantly promote CM differentiation from human pluripotent stem cells. Next, we collected EBs and profiled the temporal gene expressions of miR-1-iPSCs and control GFP-iPSCs from day 0 to day 20 of differentiation. As shown in Figure 2D, expression of pluripotency marker genes OCT4 and NANOG decreased during differentiation and was not detectable after day 8. miR-1 overexpression accelerated the decrease of OCT4. An enhanced expression of early primitive streak marker gene, T, was observed in miR-1 iPSCs [24]. Additionally, miR-1 overexpression promoted the expression levels of early stage cardiac transcription factors, NKX2.5, ISL1 and GATA4, as well as the late stage sarcomeric genes, CTNT, MYH7 and MYH6 [13,14].
Figure 2. miR-1 promotes CM differentiation of human iPSCs.
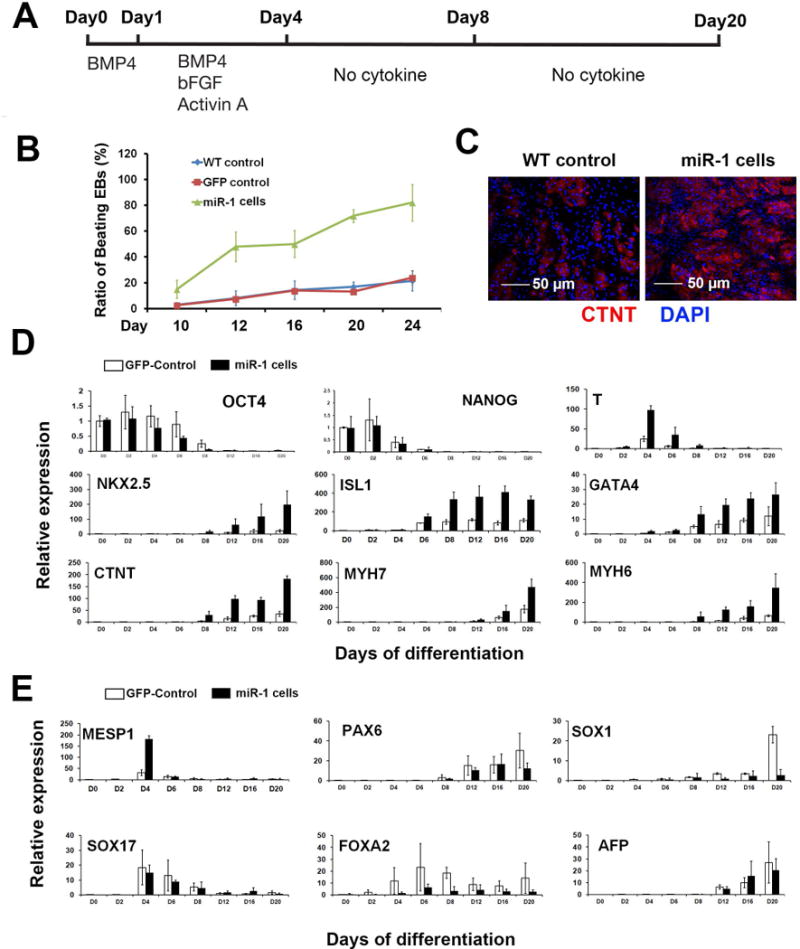
(A) A protocol outline describing the cardiovascular differentiation of iPSCs. (B) Ratios of beating EBs in WT control, GFP control and miR-1 iPSCs from day 10 to day 20 of differentiation. (C) A representative image of CTNT+ CMs from dissociated day 20 EBs. Scale bar, 50μm. (D) Dynamic gene expression profiling of GFP control and miR-1 iPSCs during cardiovascular differentiation by q-PCR. (E) q-PCR analysis of germ layer marker genes during cardiovascular differentiation. The ratio of ΔΔCT was normalized to the internal control Cyclophinin G, and fold change results were obtained by normalization to undifferentiated iPS cells of day 0. Error bars represent SEM of three independent experiments. * p<0.05.
Previously studies indicated that mouse miR-1 could promote mesoderm and cardiac differentiation of mouse ES cells [11]. In order to test whether miR-1 could impact the germ layer differentiation [25] in human, we compared the dynamic expressions of mesoderm marker, MESP1; ectoderm markers, PAX6, SOX1, SOX17 and endoderm markers, FOXA2 and AFP in miR-1 iPSCs vs. control iPSCs. Interestingly, the early expression of MESP1 was significantly improved by miR-1 overexpression at day 4, and FOXA2 expression was suppressed by miR-1 overexpression from day 2 until day 20 (Figure 2E). All these observations suggest that miR-1 could promote early human mesoderm formation from pluripotency.
3.3 miR-1 modulates WNT and FGF pathways
The isolation of MCPs from human pluripotent stem cells provides an ideal in vitro system to dissect the early segregation of cardiovascular lineage cells from the common progenitor population [13,14]. We, together with other groups, revealed that stage-specific inhibition of canonical WNT signaling is critical for CM differentiation [13, 14, 26, 27], and activation of FGF pathway induces EC and suppresses CM differentiation from human ES cells [13]. Therefore, we were interested to know whether miR-1 could promote CM differentiation from human MCPs via influencing the activities of WNT and FGF signaling pathways. To address this question, we added various pathway modulators into the cell cultures from day 4 of differentiation and quantified the ratio of beating EBs at day 20 (Figure 3A), respectively. As shown in Figure 3B, adding dickkopf homolog 1 (DKK1), a WNT inhibitor, significantly promoted the ratio of beating EBs in control iPSCs (20% vs. 60%, DKK1: 0 vs.100 ng/ml) as well as in miR-1 iPSCs from 70% to 80%. Importantly, we observed that adding of Wnt3A, a WNT ligand, in control iPSCs completely suppressed CM differentiation (2% beating EBs) with the concentration of 25 ng/ml. However, the same dose of Wnt3A could not suppress CM differentiation in miR-1 overexpressing iPSCs, with approximately 30% beating EBs at day 20. Furthermore, around 10% beating EBs in miR-1 iPSCs was observed with the treatment of 50 ng/ml Wnt3A, while only 1% beating EBs was found in control iPSCs under the same condition. These data indicate that miR-1 could antagonize the suppressor role of Wnt3A, which activates WNT signal, in CM differentiation. A similar approach was utilized to study the impact of miR-1 on FGF pathway (Figure 3C). Adding of DKK1 together with PD173074, a chemical FGFR antagonist [28], synergistically improved the ratio of beating EBs in control iPSCs at day 20. Administrant of PD173074 alone also increased CM differentiation with a dose-dependent manner. In addition, 10% beating EBs were observed in control cells with the presence of bFGF (10ng/ml), while 30% beating EBs remained in miR-1 iPSCs with the same treatment. This indicates the antagonist role of miR-1 in bFGF induced FGF signaling, which suppresses CM differentiation. Lastly, we quantified the dynamic ratios of CTNT+ cells and CD31+ cells in control and miR-1 cells, which were treated without any factor from day 4 (Figure 3A). Overexpression of miR-1 prominently increased the ratio of CTNT+ cells and suppressed the differentiation of CD31+ cells after day 12. CD31 is a surface marker of ECs [14]. Taken together we found miR-1 could promote CM formation by suppressing both WNT and FGF pathways, as well as the EC differentiation from human iPSCs.
Figure 3. miR-1 modulates WNT and FGF pathways.
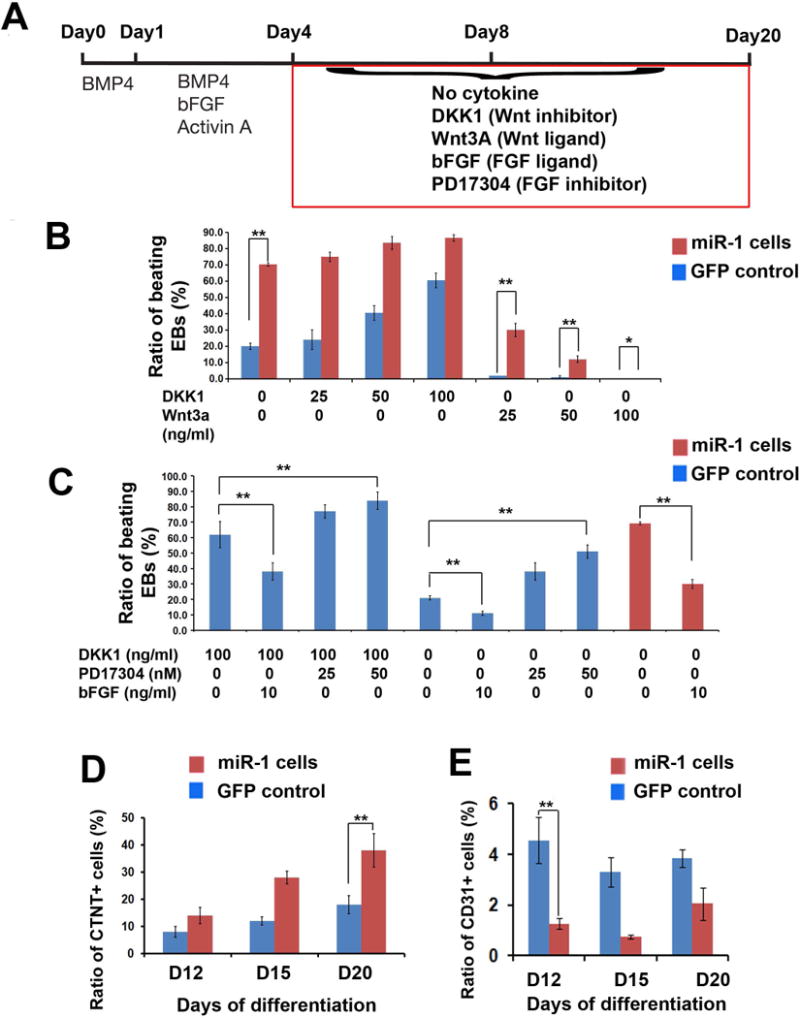
(A) An outline describing the administration of pathway modulators after day 4 of differentiation. (B) Quantification of beating EBs at day 20 with the modulation of WNT pathway from day 4 in both GFP control and miR-1 iPSCs. (C) Quantification of beating EBs at day 20 with the modulation of FGF pathway from day 4 in both GFP control and miR-1 iPSCs. (D) Quantification of CTNT+ CMs and CD31+ ECs in in both GFP control and miR-1 iPSCs during cardiovascular differentiation. * p>0.05. ** p<0.01.
3.4 FZD7 and FRS2 are direct targets of miR-1
We next sought to identify the target genes of miR-1 in WNT and FGF pathways. miR-1 target was predicted using five algorithms as described in Methods. Both FZD7 and FRS2 were identified as putative targets of miR-1 with confident scores (Figure 4A). FZD7 transduces extracellular signals into cytoplasm to activate canonical WNT pathway [29]. FRS2 is a lipid-anchored Grb2-binding protein, which functions importantly to transduce signals from FGFR to the Ras/MAPK signaling pathway [30]. The dynamic expression analysis of those two genes demonstrated that the peak expressions of FZD7 and FRS2 during cardiac differentiation started from day 6 when MCPs were formed, and miR-1 overexpression significantly suppressed the expressions of FZD7 and FRS2 after day 6 (Figure 4B). These implied that miR-1 could directly target to those two genes. In order to validate the targets, miR-1-binding sites within 3′ untranslated region (UTR) of FZD7 and FRS2 were cloned (Figure 4A). To validate those putative binding sites, we generated luciferase reporter constructs with insertion of the wild type (wt) and mutated miR-1 binding sites (mt) in 3′UTR of FZD7 and FRS2. Each mutant construct had 2 mismatch nucleotides in the seed region of the predicted sites (Figure 4A). A reporter construct containing 3′ UTR of GJA1, which has been demonstrated as a direct target of miR-1 [31], was included as the positive control. As shown in Figure 4C and 4D, a decreased luciferase activity was observed with the co-transfection of premiR-1 mimic oligonucleotides and the dual–luciferase reporter plasmids containing wt binding sites of GJA1, FZD7 and FRS2. However, no decrease was observed when co-transfection of premiR-1 mimic with mutated binding sites. All these indicate that seed sequences of putative binding sites were specifically recognized by miR-1. Next, western blot was utilized to detect the FZD7 and FRS2 protein levels in iPSCs with miR-1 overexpression. Quantitative histogram of Figure 4D indicated the decreased protein levels in iPSCs with miR-1 overexpression. Taken together, our results demonstrate that FZD7 and FRS2 are direct targets of miR-1.
Figure 4. Identification of FZD7 and FRS2 as targets of miR-1.
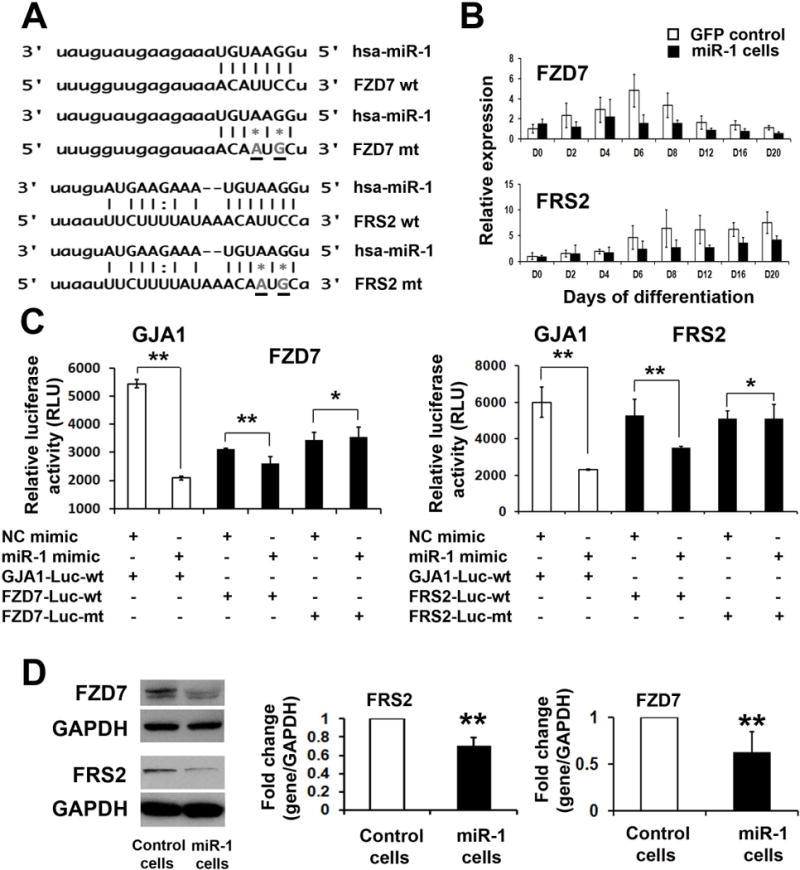
(A) The predicted binding sites of miR-1 in the 3′UTR of human FZD7 and FRS2 genes. The core miR-1 binding sequences in FZD7 and FRS2 were mutated by site-directed mutagenesis (grey stars). (B) Dynamic expression of FZD7 and FRS2 during cardiovascular differentiation of GFP control and miR-1 iPSCs by q-PCR. (C) Luciferase reporter assay was performed by co-transfection of dual-luciferase reporter constructs containing the wild type or mutated putative binding sites with pre-miR-1 or control oligonucleotides into HEK-293T cells. GJA1, a known target of miR-1, served as a positive control. Luciferase activity was determined 24 hours after transfection. (D) Western blot analysis of protein levels of FZD7 and FRS2 in GFP control and miR-1 iPSCs. Quantification of protein level was normalized to GAPDH. * p>0.05. ** p<0.01.
3.5 Functional validation of FZD7 and FRS2 at MCP specification stage
In order to define the role of FZD7 and FRS2 at cardiovascular commitment stage of human heart development, MCPs were obtained from day 6 differentiation of human iPSCs as previously described [14]. Both FZD7 and FRS2 were then knocked down in MCPs separately or synergistically using siRNAs. We transfected siRNAs to MCP-formed re-aggregates and monolayers, followed with the culture condition containing no cytokines. Transfected cells were sampled on day 12 and 20 of differentiation. We observed an increased ratio of CTNT+ CMs with treatment of siFZD7 and siFRS2. siFZD7 and siFRS2 co-transfection significantly promoted CMs formation from MCPs in both 3 dimensional (3D, MCPS formed re-aggregates) and 2D (monolayers) conditions (Figure 5A, B). Overall, these results indicate that synergistic knock-down of FZD7 and FRS2 promotes CM commitment from human MCPs.
Figure 5. Knock-down of FZD7 and FRS2 promotes CM commitment.
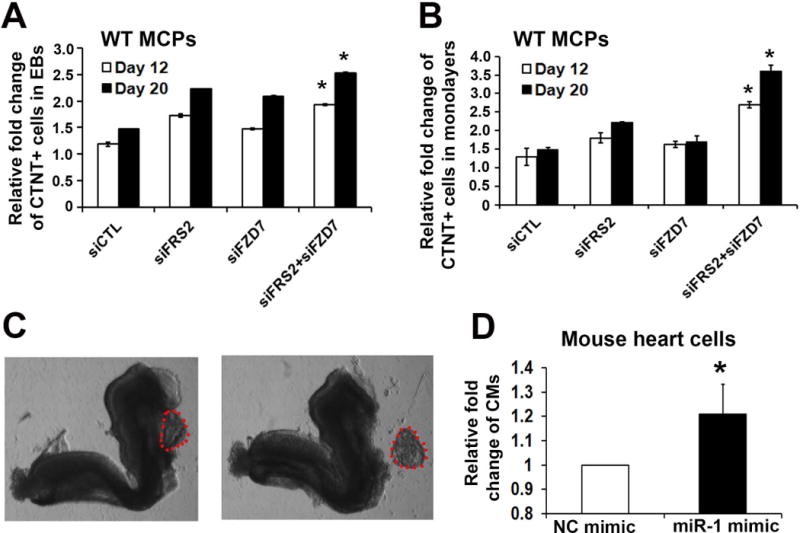
siRNAs targeting to FZD7 and FRS2, as well as a non-specific control siRNA (siCTL) were transfected into MCP formed re-aggregates (A) or monolayers (B). Cells were harvested on day 12 and day 20 of differentiation, with CM ratio analyzed by FACS. (C) A representative image showing the dissecting of primitive heart (red dots included area) from mouse E8.5 embryo. (D) Transfection of pre-miR-1 mimic into primitive mouse heart cells and quantification of CM differentiation 10 days later. * p<0.05.
3.6 miR-1 promotes CM differentiation of mouse primitive heart cells
In order to evaluate the role of miR-1 in mouse, E8.5 mouse primitive hearts were dissected and dissociated. premiR-1 mimic oligonucleotide was next transfected into the dissociated hearts. After 10 days, we observed that miR-1 transfection could improve ratio of CM than control oligonucleotide. Taken together, this study reveals a new switch role of miR-1 in promoting human CM differentiation from MCPs by targeting FZD7 and FRS2 to simultaneously suppress WNT and FGF pathways (Figure 6).
Figure 6. Schematic for the role of miR-1 in early human heart development.
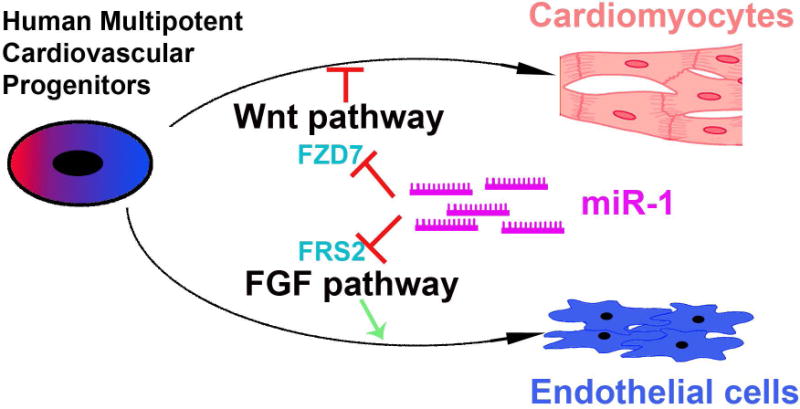
miR-1 promotes human CM differentiation and suppresses EC formation from human MCPs by targeting FZD7 and FRS2 to simultaneously suppress both WNT and FGF signaling pathways.
4. Discussion
Previous studies have elucidated the key role of signaling pathways, such as WNT, BMP and FGF, in regulating early stage heart development at transcriptional level [25, 27]. The temporal regulation of signaling pathways has been found essential during the early steps of heart formation, including induction of cardiac mesoderm, formation of the cardiovascular progenitor cells and commitment of cardiovascular lineage cells. In addition, each step is precisely regulated at posttranscriptional and posttranslational levels, where miRNA has been implicated as a critical regulator. Recently, accumulated evidences indicated that miRNA could regulate various bioprocesses by targeting signaling pathways [32]. For example, miRNA-34 decreases cancer cell proliferation through suppressing a set of conserved targets in a network of WNT pathway [33]. Therefore, these observations raised an interesting question whether miRNA could modulate signaling pathway to regulate cardiovascular development.
WNT signaling is one of the most important signaling pathways involved in early heart development [26]. Stage-specific regulation of WNT signaling is critical for early primitive streak formation, as well as CM commitment from cardiovascular progenitor cells at a later stage [34]. In this study, we observed an antagonistic effect of miR-1 on Wnt3A induced WNT activity, indicating that miR-1 promotes CM generation by suppressing WNT pathway. In addition, we confirmed FZD7 as a new miR-1 target in early human heart formation. This is consistent with previous observation that down regulation of FZD7 could inactivate WNT signaling [35] and FZD7 mutant exhibited cardiac structural defects [36].
Early cardiovascular commitment includes the specification of MCPs towards CMs, SMCs or ECs. In mouse, CMs and SMCs could be derived from bi-potential Nkx2.5+ heart progenitors, while ECs could not [37]. This indicates the early segregation of ECs with muscle lineages in cardiogenesis. A key role of FGF pathway was found in promoting EC differentiation from human ES cell derived MCPs [13], as well as angiogenesis in animals [38]. Interestingly, it has been demonstrated that FGFs interfere with muscle regulatory factors to suppress the expression of muscle specific miRNAs including miR-1 [39], and miR-1 negatively regulates angiogenesis during zebrafish development [40]. In addition, a cross-talk between WNT and FGF pathways has been suggested in the formation of anterior heart field (AHF) [41]. Loss of WNT signaling in the AHF led to cardiac structural defect with a concomitant loss of FGF signaling. All those evidences suggest an interaction of miR-1 with FGF and WNT in cardiovascular development. In this study, we found miR-1 suppressed EC commitment from MCPs by targeting FRS2, which mediates the signal transduction from FGF receptors to down–stream molecules [30]. In conclusion, we uncovered a switch role of miR-1 in determining CM vs. EC commitment in early human heart formation via the synergistic suppressing of both WNT and FGF pathways.
In a previous study, Ivey et al. indicated that both miR-1 and miR-133 enhanced mesoderm development as well as suppressed the ectoderm and endoderm specification in mouse [11]. In this study, we showed that miR-1 overexpression could significantly enhance the expression of early mesoderm-related genes, T and MESP1 at day 4 of differentiation before MCPs are formed, implying a key role of miR-1 in inducing mesoderm formation in human heart development. These observations are controversy to previous findings that WNT and FGF pathways are required for early mesoderm formation [25, 42], suggesting the existence of undefined miR-1 target genes, which could regulate early germ layer patterning in mammalian. Our ongoing study is to uncover the underlying mechanism of miR-1 in the early germ layer formation stage.
Supplementary Material
Acknowledgments
Funding
This work was supported by the University of Pittsburgh start-up and American Heart Association (AHA) Grant (#11SDG5580002) to LY, by NIH grant R01LM009657 to PVB and by the National Science Council (Taiwan) Grant (#NSC100-2917-I-564-015) to TYL, by National Center for Research Resources of National Institutes of Health Grant (RR031300) to YS. AA was supported by grant UL1RR024153 to the Center for Translational Science (CTSI), University of Pittsburgh School of Medicine.
Footnotes
Conflict of interests
None declared
References
- 1.Bushati N, Cohen SM. microRNA functions. Annu Rev Cell Dev Biol. 2007;23:175–205. doi: 10.1146/annurev.cellbio.23.090506.123406. [DOI] [PubMed] [Google Scholar]
- 2.Thum T, Catalucci D, Bauersachs J. MicroRNAs: novel regulators in cardiac development and disease. Cardiovasc Res. 2008;79:562–70. doi: 10.1093/cvr/cvn137. [DOI] [PubMed] [Google Scholar]
- 3.Cordes KR, Srivastava D. MicroRNA regulation of cardiovascular development. Circ Res. 2009;104:724–32. doi: 10.1161/CIRCRESAHA.108.192872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Latronico MV, Condorelli G. MicroRNAs and cardiac pathology. Nat Rev Cardiol. 2009;6:419–29. doi: 10.1038/nrcardio.2009.56. [DOI] [PubMed] [Google Scholar]
- 5.Bernstein E, Kim SY, Carmell MA, Murchison EP, Alcorn H, Li MZ, et al. Dicer is essential for mouse development. Nat Genet. 2003;35:215–7. doi: 10.1038/ng1253. [DOI] [PubMed] [Google Scholar]
- 6.da Costa Martins PA, Bourajjaj M, Gladka M, Kortland M, van Oort RJ, Pinto YM, et al. Conditional dicer gene deletion in the postnatal myocardium provokes spontaneous cardiac remodeling. Circulation. 2008;118:1567–76. doi: 10.1161/CIRCULATIONAHA.108.769984. [DOI] [PubMed] [Google Scholar]
- 7.Zhao Y, Ransom JF, Li A, Vedantham V, von Drehle M, Muth AN, et al. Dysregulation of cardiogenesis, cardiac conduction, and cell cycle in mice lacking miRNA-1-2. Cell. 2007;129:303–17. doi: 10.1016/j.cell.2007.03.030. [DOI] [PubMed] [Google Scholar]
- 8.Liu N, Bezprozvannaya S, Williams AH, Qi X, Richardson JA, Bassel-Duby R, et al. microRNA-133a regulates cardiomyocyte proliferation and suppresses smooth muscle gene expression in the heart. Genes Dev. 2008;22:3242–54. doi: 10.1101/gad.1738708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wilson KD, Hu S, Venkatasubrahmanyam S, Fu JD, Sun N, Abilez OJ, et al. Dynamic microRNA expression programs during cardiac differentiation of human embryonic stem cells: role for miR-499. Circ Cardiovasc Genet. 2010;3:426–35. doi: 10.1161/CIRCGENETICS.109.934281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cordes KR, Sheehy NT, White MP, Berry EC, Morton SU, Muth AN, et al. miR-145 and miR-143 regulate smooth muscle cell fate and plasticity. Nature. 2009;460:705–10. doi: 10.1038/nature08195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ivey KN, Muth A, Arnold J, King FW, Yeh RF, Fish JE, et al. MicroRNA regulation of cell lineages in mouse and human embryonic stem cells. Cell Stem Cell. 2008;2:219–29. doi: 10.1016/j.stem.2008.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sluijter JP, van Mil A, van Vliet P, Metz CH, Liu J, Doevendans PA, et al. MicroRNA-1 and -499 regulate differentiation and proliferation in human-derived cardiomyocyte progenitor cells. Arterioscler Thromb Vasc Biol. 2010;30:859–68. doi: 10.1161/ATVBAHA.109.197434. [DOI] [PubMed] [Google Scholar]
- 13.Yang L, Soonpaa M, Edler E, Roepke T, Kattman S, Kennedy M, et al. Human cardiovascular progenitors develop from a KDR+ ES cell-derived population. Nature. 2008;453:524–8. doi: 10.1038/nature06894. [DOI] [PubMed] [Google Scholar]
- 14.Lin B, Kim J, Li Y, Pan H, Carvajal-Vergara X, Salama G, et al. High-purity enrichment of functional cardiovascular cells from human iPS cells. Cardiovasc Res. 2012;95:327–35. doi: 10.1093/cvr/cvs185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.John B, Enright AJ, Aravin A, Tuschl T, Sander C, Marks DS. Human MicroRNA targets. PLoS Biol. 2004;2:e363. doi: 10.1371/journal.pbio.0020363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lewis BP, Shih IH, Jones-Rhoades MW, Bartel DP, Burge CB. Prediction of mammalian microRNA targets. Cell. 2003;115:787–98. doi: 10.1016/s0092-8674(03)01018-3. [DOI] [PubMed] [Google Scholar]
- 17.Krek A, Grun D, Poy MN, Wolf R, Rosenberg L, Epstein EJ, et al. Combinatorial microRNA target predictions. Nat Genet. 2005;37:495–500. doi: 10.1038/ng1536. [DOI] [PubMed] [Google Scholar]
- 18.Kertesz M, Iovino N, Unnerstall U, Gaul U, Segal E. The role of site accessibility in microRNA target recognition. Nat Genet. 2007;39:1278–84. doi: 10.1038/ng2135. [DOI] [PubMed] [Google Scholar]
- 19.Coronnello C, Benos PV. ComiR: Combinatorial microRNA target prediction tool. Nucl Acids Res. 2013 doi: 10.1093/nar/gkt379. accepted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Coronnello C, Hartmaier R, Arora A, Huleihel L, Pandit KV, Bais AS, Butterworth M, Kaminski N, Stormo GD, Oesterreich S, Benos PV. Novel modeling of combinatorial miRNA targeting identifies SNP with potential role in bone density. Plos Comput Biol. 2012;8:e1002830. doi: 10.1371/journal.pcbi.1002830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Miyoshi N, Ishii H, Nagano H, Haraguchi N, Dewi DL, Kano Y, et al. Reprogramming of mouse and human cells to pluripotency using mature microRNAs. Cell Stem Cell. 2011;8:633–8. doi: 10.1016/j.stem.2011.05.001. [DOI] [PubMed] [Google Scholar]
- 22.Repetto E, Briata P, Kuziner N, Harfe BD, McManus MT, Gherzi R, et al. Let-7b/c enhance the stability of a tissue-specific mRNA during mammalian organogenesis as part of a feedback loop involving KSRP. PLoS Genet. 2012;8:e1002823. doi: 10.1371/journal.pgen.1002823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Carvajal-Vergara X, Sevilla A, D’Souza SL, Ang YS, Schaniel C, Lee DF, et al. Patient-specific induced pluripotent stem-cell-derived models of LEOPARD syndrome. Nature. 2010;465:808–12. doi: 10.1038/nature09005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wilson V, Beddington R. Expression of T protein in the primitive streak is necessary and sufficient for posterior mesoderm movement and somite differentiation. Dev Biol. 1997;192:45–58. doi: 10.1006/dbio.1997.8701. [DOI] [PubMed] [Google Scholar]
- 25.Gadue P, Huber TL, Nostro MC, Kattman S, Keller GM. Germ layer induction from embryonic stem cells. Exp Hematol. 2005;33:955–64. doi: 10.1016/j.exphem.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 26.Zhang KK, Kamp J, Palecek SP. Robust cardiomyocyte differentiation from human pluripotent stem cells via temporal modulation of canonical Wnt signaling. Proc Natl Acad Sci U S A. 2012;109:E1848–57. doi: 10.1073/pnas.1200250109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Marvin MJ, Di Rocco G, Gardiner A, Bush SM, Lassar AB. Inhibition of Wnt activity induces heart formation from posterior mesoderm. Genes Dev. 2001;15:316–27. doi: 10.1101/gad.855501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kumar SB, Narasu L, Gundla R, Dayam R, J A R P S Fibroblast growth factor receptor inhibitors. Curr Pharm Des. 2013;19:687–701. [PubMed] [Google Scholar]
- 29.Pinson KI, Brennan J, Monkley S, Avery BJ, Skarnes WC. An LDL-receptor-related protein mediates Wnt signalling in mice. Nature. 2000;407:535–8. doi: 10.1038/35035124. [DOI] [PubMed] [Google Scholar]
- 30.Kouhara H, Hadari YR, Spivak-Kroizman T, Schilling J, Bar-Sagi D, Lax I, et al. A lipid-anchored Grb2-binding protein that links FGF-receptor activation to the Ras/MAPK signaling pathway. Cell. 1997;89:693–702. doi: 10.1016/s0092-8674(00)80252-4. [DOI] [PubMed] [Google Scholar]
- 31.Yang B, Lin H, Xiao J, Lu Y, Luo X, Li B, et al. The muscle-specific microRNA miR-1 regulates cardiac arrhythmogenic potential by targeting GJA1 and KCNJ2. Nat Med. 2007;13:486–91. doi: 10.1038/nm1569. [DOI] [PubMed] [Google Scholar]
- 32.Inui M, Martello G, Piccolo S. MicroRNA control of signal transduction. Nat Rev Mol Cell Biol. 2010;11:252–63. doi: 10.1038/nrm2868. [DOI] [PubMed] [Google Scholar]
- 33.Kim NH, Kim HS, Kim NG, Lee I, Choi HS, Li XY, et al. p53 and microRNA-34 are suppressors of canonical Wnt signaling. Sci Signal. 2011;4:ra71. doi: 10.1126/scisignal.2001744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gessert S, Kuhl M. The multiple phases and faces of wnt signaling during cardiac differentiation and development. Circ Res. 2010;107:186–99. doi: 10.1161/CIRCRESAHA.110.221531. [DOI] [PubMed] [Google Scholar]
- 35.Yang L, Wu X, Wang Y, Zhang K, Wu J, Yuan YC, et al. FZD7 has a critical role in cell proliferation in triple negative breast cancer. Oncogene. 2011;30:4437–46. doi: 10.1038/onc.2011.145. [DOI] [PubMed] [Google Scholar]
- 36.Yu H, Ye X, Guo N, Nathans J. Frizzled 2 and frizzled 7 function redundantly in convergent extension and closure of the ventricular septum and palate: evidence for a network of interacting genes. Development. 2012;139:4383–94. doi: 10.1242/dev.083352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wu SM, Fujiwara Y, Cibulsky SM, Clapham DE, Lien CL, Schultheiss TM, et al. Developmental origin of a bipotential myocardial and smooth muscle cell precursor in the mammalian heart. Cell. 2006;127:1137–50. doi: 10.1016/j.cell.2006.10.028. [DOI] [PubMed] [Google Scholar]
- 38.Cross MJ, Claesson-Welsh L. FGF and VEGF function in angiogenesis: signalling pathways, biological responses and therapeutic inhibition. Trends Pharmacol Sci. 2001;22:201–7. doi: 10.1016/s0165-6147(00)01676-x. [DOI] [PubMed] [Google Scholar]
- 39.Stahlhut C, Suarez Y, Lu J, Mishima Y, Giraldez AJ. miR-1 and miR-206 regulate angiogenesis by modulating VegfA expression in zebrafish. Development. 2012;139:4356–64. doi: 10.1242/dev.083774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sweetman D, Goljanek K, Rathjen T, Oustanina S, Braun T, Dalmay T, Munsterberg A. Specific requirements of MRFs for the expression of muscle specific microRNAs, miR-1, miR-206 and miR-133. Dev Biol. 2008;321:491–9. doi: 10.1016/j.ydbio.2008.06.019. 2012. [DOI] [PubMed] [Google Scholar]
- 41.Cohen ED, Wang Z, Lepore JJ, Lu MM, Taketo MM, Epstein DJ, et al. Wnt/beta-catenin signaling promotes expansion of Isl-1-positive cardiac progenitor cells through regulation of FGF signaling. J Clin Invest. 2007;117:1794–804. doi: 10.1172/JCI31731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ciruna B, Rossant J. FGF signaling regulates mesoderm cell fate specification and morphogenetic movement at the primitive streak. Dev Cell. 2001;1:37–49. doi: 10.1016/s1534-5807(01)00017-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.